

Abstract
Biopharmaceuticals have the potential to raise an immunogenic response in treated individuals, which may impact the efficacy and safety profile of these drugs. As a result, it is essential to evaluate immunogenicity throughout the different phases of the clinical development of a biopharmaceutical, including post-marketing surveillance. Although rigorous evaluation of biopharmaceutical immunogenicity is required by regulatory authorities, there is a lack of uniform standards for the type, quantity, and quality of evidence, and for guidance on experimental design for immunogenicity assays or criteria to compare immunogenicity of biopharmaceuticals. Moreover, substantial technological advances in methods to assess immune responses have yielded higher immunogenicity rates with modern assays, and limit comparison of immunogenicity of biopharmaceuticals outside of head-to-head clinical trials. Accordingly, research programs, regulatory agencies, and clinicians need to keep pace with continuously evolving analyses of immunogenicity. Here, we review factors associated with immunogenicity of biopharmaceuticals, potential clinical ramifications, and current regulatory guidance for evaluating immunogenicity, and discuss methods to assess immunogenicity in non-clinical and clinical studies. We also describe special considerations for evaluating the immunogenicity of biosimilar candidates.
Key Points
| Evaluation of immunogenicity is a key step in the development of all biopharmaceuticals, including biosimilars. |
| Lack of uniform standards for the type, quantity, and quality of evidence contributes to the challenges of assessing immunogenicity. |
| Immunogenicity assessment will continue to evolve as novel techniques emerge. |
Introduction
Biopharmaceuticals are therapeutics produced by a living organism, most often made by genetically engineering living bacterial, animal, or plant cells [1]. Biopharmaceuticals have advanced patient care by providing highly effective, targeted treatments for a number of life-threatening and chronic diseases, such as hematologic malignancies and solid tumors, as well as systemic immune-mediated diseases, such as rheumatoid arthritis (RA), inflammatory bowel disease, systemic lupus erythematosus, and psoriasis [2]. In contrast to small-molecule drugs, biopharmaceuticals are typically large and highly complex and, by their nature, require highly specialized techniques and long production times to manufacture.
Biosimilars are versions of the original biopharmaceutical molecule, with demonstrated similarity in structure, function, efficacy, and safety to the reference product. Development of biosimilars may increase treatment access and therapeutic options for patients and physicians, yielding savings and efficiencies for national healthcare systems and local providers.
The regulatory pathway for approval of biosimilar candidates [3, 4] is distinct from the pathway for generic versions of small-molecule drugs due to the fundamental intricacies of biopharmaceuticals as well as core differences in manufacturing processes. The US Food and Drug Administration (FDA) [5], European Medicines Agency (EMA) [6], and World Health Organization (WHO) [7] have issued guidelines for establishing biosimilarity. Other countries have also developed their own recommendations for biosimilar development, similar to those from the FDA, EMA, and WHO [3]. Although the nature and extent of supporting data differ somewhat among regulatory agencies, approval is based on the totality of the evidence. Several national academic organizations, such as societies, colleges, and associations, have also provided position statements on the approval processes for and clinical use of biosimilars [8–14]. These recommendations encourage stringent standards for biosimilar approval and post-marketing pharmacovigilance, and for extrapolation of efficacy and safety data for other indications for which the biosimilar has not been investigated in clinical trials.
Biopharmaceuticals, including biosimilars, have the potential to elicit an immunogenic response in treated individuals (immunogenicity), which may have an impact on the efficacy and safety profiles of the drug. Thus, it is critical that immunogenicity is evaluated throughout the various phases of clinical development and during post-marketing surveillance. Prevailing concepts and methods to detect immunogenicity have evolved over the past two decades. Herein, we review factors associated with immunogenicity, possible clinical ramifications, and current regulatory guidance for evaluating immunogenicity. We also discuss methods to assess the immunogenicity of biopharmaceuticals, including biosimilars, in non-clinical and clinical studies. In addition, we review the procedures for monitoring immunogenicity after regulatory approval in the context of dynamic standards and practices for evaluating immune responses.
Considerations for Assessing Immunogenicity of Biopharmaceuticals
Anti-Drug Antibodies (ADAs) and Clinical Adverse Effects
Immunogenicity is characterized by the presence of anti-drug antibodies (ADAs) detected in the circulation of either animals or humans after administration of a biopharmaceutical. ADAs that bind to the active site of a biopharmaceutical and may inhibit its activity are termed neutralizing antibodies. Non-neutralizing antibodies do not bind to the active site but may still produce important clinical consequences, such as reduced therapeutic efficacy by compromising bioavailability [15]. The generation of ADAs (neutralizing or non-neutralizing) is increasingly recognized as a mechanism explaining the reduced efficacy or therapeutic failure of some biopharmaceuticals [16]. This can occur by altered drug pharmacokinetics or, in other instances, the presence of neutralizing antibodies bound to active sites, thereby reducing drug activity [17, 18].
ADAs have been associated with safety risks that range from mild to life-threatening events. The presence of ADAs following administration of therapy does not necessarily translate into clinically meaningful consequences for efficacy or safety. In fact, adverse events associated with immunogenicity, such as hypersensitivity, are relatively uncommon [19–21]. In addition, neutralizing antibodies could cross-react with endogenous proteins, thus leading to deficiency syndromes [22, 23]. Therefore, unwanted immunogenicity can constitute a serious impediment to developing biopharmaceuticals.
Key Elements Influencing Immunogenicity
A multitude of factors can influence immunogenicity, which may be classified into three major categories: treatment-, patient-, and drug property-associated factors (Table 1). For example, treatment-associated factors include the route of administration (subcutaneous vs. intramuscular vs. intravenous) [24], duration of therapy (short-term vs. long-term), and frequency of administration (intermittent vs. continuous) [25], all of which may affect the likelihood of an immune response. Patient-associated factors include immune system function, which, when compromised, may lower the probability of mounting antibodies [26], disease state, and polymorphisms in major histocompatibility complex (MHC), which can affect the magnitude of T cell-dependent immune responses [27]. Drug property-associated factors include the degree to which a biopharmaceutical is humanized, glycosylation patterns, and removal or concealment of MHC epitopes by design, as well as issues that arise during the manufacturing process, such as the presence of impurities, aggregates, and contaminants [26, 28].
Table 1.
Factors influencing immunogenicity of biopharmaceuticals
| Category | Example |
|---|---|
| Treatment-associated | Mechanism of action |
| Route of administration | |
| Frequency of administration Duration of therapy | |
| Patient-associated | Disease type |
| Disease status | |
| Immune system function | |
| Genetic factors | |
| Concomitant disease | |
| Concomitant medications | |
| Prior exposure | |
| Prior sensitization | |
| Drug property-associated | Recombinant expression system |
| Post-translational protein modifications | |
| Impurities | |
| Contaminants | |
| Aggregates |
Immunogenicity Assessments
Overall Approach
Immunogenicity of biopharmaceuticals in clinical studies is evaluated using a stepwise approach (Fig. 1). First, a screening assay is employed to detect the presence of ADAs in treated patients. This is followed by confirmatory assays to determine the specificity of ADAs for the biopharmaceutical and eliminate false positives. For ADA-positive samples, characterization assays are conducted to determine the titer and type of ADAs, and bioassays or ligand-binding assays are used to identify neutralizing antibodies [29]. To evaluate the potential clinical impact of ADAs, immunogenicity assessments are conducted in conjunction with pharmacokinetic, safety, and efficacy assessments, with the totality of data considered.
Fig. 1.

Stepwise approach to assessing immunogenicity of biopharmaceuticals. ADA anti-drug antibody
ADA Screening Assays
A variety of techniques have been used to screen for ADAs in biopharmaceuticals, and methodologies have advanced considerably in the past two decades (Fig. 2). Commonly used assays include enzyme-linked immunosorbent assays (ELISAs), which may be conducted using a direct, indirect, or capture format; electrochemiluminescence (ECL) assays and antigen-binding tests, such as radioimmunoassays. Some of these assays can be modified to render them “drug tolerant” [25, 30, 31]. Because each format has relative strengths and weaknesses (reviewed in detail elsewhere [30–33]), there is no single assay appropriate for assessing the immunogenicity of all biopharmaceuticals. Selecting the optimal assay for ADA screening is a key consideration in biopharmaceutical development and must take into account the properties of the therapeutic to be tested.
Fig. 2.
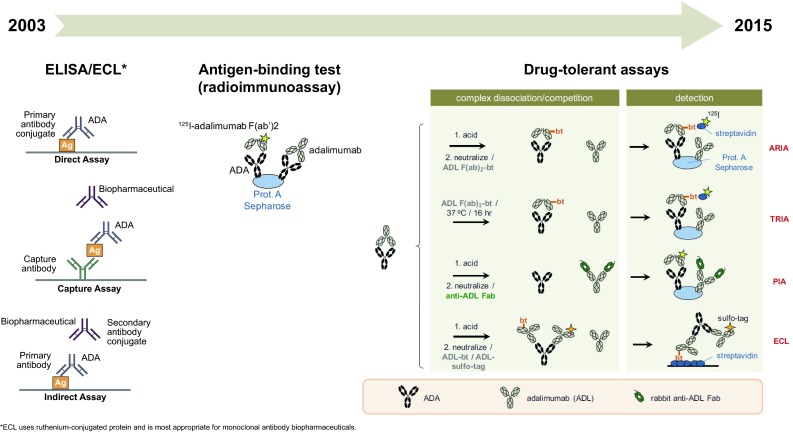
Evolution of immunogenicity assays for biopharmaceuticals and assessment of anti-adalimumab antibodies (reprinted from van Leeuwen et al. [43], © 2015, with permission from Elsevier and adapted from Thermo Fisher Scientific [Carlsbad, CA, USA]). ADA anti-drug antibody, ADL adalimumab, ARIA acid-dissociation radioimmunoassay, ECL electrochemiluminescence, ELISA enzyme-linked immunosorbent assay, PIA pH-shift anti-idiotype antigen-binding test, TRIA temperature-shift radioimmunoassay
Assay Types
Because of their convenience, ease, and high-throughput capability, ELISAs are frequently utilized for immunogenicity screening (Fig. 2) [32]. In a direct ELISA, ADAs from patient samples are captured by the biopharmaceutical, which has been immobilized on a plastic plate; the plate is washed several times and ADAs are then detected spectrophotometrically with a colorimetric labeled anti-immunoglobulin reagent. Due to the potential for cross-reactivity, direct ELISAs are not appropriate for therapeutic monoclonal antibodies [33, 34]. A limitation of the direct ELISA is that fixation of the biopharmaceutical to a plastic surface may alter its conformation and conceal epitopes, resulting in underestimation of ADAs. The indirect ELISA format circumvents this complication by first immobilizing antibodies on the plate to orient the biopharmaceutical. Disadvantages of the direct and indirect ELISA formats include false positives and high background noise due to non-specific binding as well as potential loss of low-affinity ADAs during washes. In a capture ELISA, ADAs are captured by immobilized biopharmaceutical and then detected using a conjugated version of the biopharmaceutical. This version is more selective and specific than the direct or indirect formats, but the possibility of losing low-affinity ADAs remains [33].
The ECL assay applies the same principles as an ELISA but makes use of a ruthenium-conjugated protein rather than an antibody for detection, and is therefore more suitable for testing monoclonal antibody biopharmaceuticals [33]. The dynamic range of the ruthenium-conjugated protein complexes also increases the sensitivity of the assay compared with ELISA. An example of an antigen-binding test is radioimmunoassay, in which ADAs from patient sera are captured by a sepharose-bound reagent, such as protein A, and detected with 125I-adalimumab (Fig. 2). This technique is more sensitive than ELISA but the use of radioactivity may deter its application by researchers. Surface plasmon resonance assays employ a sensor with immobilized biopharmaceutical, such that bound ADAs from patient samples produce a signal due to a change in mass. This method has the advantages of continuous measurements of ADAs and the ability to detect low-affinity ADAs and discern antibody isotypes, but may be less sensitive than the other assays described and cannot be used to perform high-throughput analyses [35].
Detection of ADAs in the assays may be confounded by drug interference. ADAs may be present but bound to excess biopharmaceutical and the ADA in this ADA–biopharmaceutical complex usually cannot be detected by ADA assays, resulting in false-negative results [36]. This may be a particular problem for biopharmaceuticals with a long half-life [32]. To overcome this potential limitation, novel drug-tolerant assays have been developed to detect both free and bound ADAs, as shown in the example of anti-adalimumab antibodies (Fig. 2). These include acid-dissociation radioimmunoassays (ARIAs), such as pH-shift anti-idiotype antigen-binding tests (PIA) [37–39], affinity capture elution assays [40, 41], homogenous mobility-shift assays [42], and temperature-shift radioimmunoassays (TRIAs) [43]. These assays use different techniques to dissociate ADA–biopharmaceutical complexes prior to ADA detection. For instance, in the PIA, bound ADAs are released by acid treatment and the samples are then neutralized before ADA screening [37]. As would be expected due to increased sensitivity, these newer assays have yielded higher rates of immunogenicity when directly compared with more traditional assays such as ELISA or radioimmunoassay [37, 43, 44].
Regardless of the technique utilized to screen for ADAs, proper validation of the methods is essential to ensure reproducible, consistent, and definitive results. This step should occur early during the course of clinical development of a biopharmaceutical and may require ongoing monitoring and modification during the pre-approval processes. As described in detail in previously published recommendations for immunoassays [31, 45], validation parameters should include cut-points, sensitivity, drug tolerance, specificity, precision, dilution, and reproducibility [45]. Due to a lack of agreement on the use of reference standards, the experimental systems for assessment of immunogenicity cannot be calibrated, and the assays are merely quasi-quantitative [31, 33]. Therefore, assays must include positive controls (e.g., samples of purified ADAs from a patient with characterized immunoglobulin levels) and negative controls (e.g., serum samples from untreated healthy individuals).
Analysis of ADA Samples
Cut-points are used to define thresholds and classify samples as ADA positive or negative. It is necessary to pre-define cut-points before immunogenicity evaluations are conducted in patient samples, and they are established based on the signals observed with negative controls. The sensitivity of the assay (the ADA concentration that produces a signal equal to the cut-point) will be determined by the positive control that is utilized; sensitivity is increased when higher-affinity positive controls are used [45]. Sensitivities of 250–500 ng/mL have been recommended for clinical studies [31]. Due to the potential issue of drug interference, described in Sect. 3.2.1, it is also critical to establish the limit of drug tolerance (the concentration of the biopharmaceutical that blocks detection of positive-control ADAs). As with the sensitivity parameter, the choice of positive control will impact the drug-tolerance threshold, with higher-affinity positive controls yielding lower tolerance [45].
Following the initial screening for ADAs, a confirmatory assay is performed and will employ the same techniques used for screening with the added step of drug competition/immunodepletion [46]. Samples are first incubated with an excess of the biotherapeutic test protein to saturate ADA-binding sites. If the antibodies detected during screening are indeed specific to the biopharmaceutical, this pre-incubation step will eliminate or diminish a positive signal in the subsequent confirmatory assay [29, 31]. A confirmatory cut-point (threshold for signal inhibition) is needed to eliminate false positives and is determined experimentally, preferably concurrently with estimation of the screening cut-point [45]. Characterization of the ADAs, titer, and immunoglobulin subtype may be performed in conjunction with the confirmatory assay [31].
Neutralizing Antibody Assays
Samples that have been verified as ADA positive should be tested for neutralizing antibodies using cell-based bioassays or competitive ligand-binding assays. The data output for cell-based assays is inhibition of biopharmaceutical activity, whereas the read-out for competitive ligand-binding assays is inhibition of target binding. Cell-based bioassays, which monitor the function of the biopharmaceutical in the presence of neutralizing antibodies, are recommended by the FDA [5]; these more accurately reflect the circumstances in treated patients and may yield insight into the potential clinical ramifications of immune responses to biopharmaceuticals. However, cell-based approaches may be laborious and difficult to develop and validate [33].
The mechanism of action of the biopharmaceutical will dictate the most appropriate assay to evaluate inhibition of biologic activity [47]. As with ADA screening assays, positive and negative controls should be included, and validation parameters should include specificity, drug tolerance, and cut-points [47].
Immunogenicity Assessments: A Practical Approach
In the clinical trial setting, patients should be screened for immunogenicity at baseline: those previously treated with a biopharmaceutical may have pre-existing ADAs, in which case stratified randomization based on treatment history may be considered. Moreover, multiple sampling timepoints to evaluate the kinetics of ADA development are critical in immunogenicity studies to differentiate pre-existing versus drug-induced immune responses and transient versus persistent immune responses.
The development of an immunogenicity risk assessment plan that will fulfill all requirements for the approval of biopharmaceuticals is a challenging process. Nevertheless, a risk-based approach, based on the characteristics of the biopharmaceutical and planned patient population, as well as known safety issues with products of the same class, has been proposed to design immunogenicity analyses in clinical studies [20, 24, 46].
Variability in Immunogenicity Assays: A Case Study in Adalimumab
Beyond the numerous biopharmaceutical and patient features that may influence immunogenicity, heterogeneity in the design and conduct of assays can manifest as substantial differences in immunogenicity rates, even when a single biopharmaceutical is tested in a particular patient population. Higher ADA detection rates with modern assays compared with data from past studies are due to the increased robustness and sensitivity of newer techniques, and should not be interpreted as heightened immunogenicity of biopharmaceuticals over time.
The above phenomenon is exemplified by immunogenicity studies of adalimumab, an antibody to tumor necrosis factor (TNF)-α, which has been approved to treat various inflammatory conditions [48]. In analyses conducted from 2003 to 2015 in patients with RA, ADA rates ranging from 1 to 66 % have been observed (Table 2). In studies using traditional assays, ADA rates <10 % were seen in this patient population by ELISA only [49–51]. It should be noted that ADA rates <1 % were observed in patients receiving concomitant methotrexate, which has been shown to reduce ADA formation [49, 50]. Conversely, higher rates of immunogenicity (13–29 %) were observed using antigen-binding tests, such as radioimmunoassays, in patients receiving methotrexate [43, 51–54]. In a direct comparison of these two methods in a cohort of 216 patients with RA treated with adalimumab, fewer ADA-positive samples were identified by bridging ELISA than by radioimmunoassay (7 vs. 13 %, respectively), and it was determined that the ELISA is more vulnerable to drug interference [51]. Predictably, drug-tolerant assays designed to detect both free and bound antibodies showed higher ADA rates (approximately two- to threefold higher) when directly compared with standard antigen-binding tests (Table 2) [43, 44].
Table 2.
Evaluation of adalimumab immunogenicity in patients with rheumatoid arthritisa
| Study description, number of adalimumab-treated patients | Timing of ADA analysis | ADA rate (%) | Immunogenicity assay |
|---|---|---|---|
| Double-blind, placebo-controlled study of adalimumab with concomitant methotrexate [49], n = 209 | 24 weeks of treatment | 1 | ELISA |
| Double-blind, placebo-controlled study of adalimumab with concomitant methotrexate [50], n = 419 | 52 weeks of treatment | 1 | ELISA |
| Double-blind, placebo-controlled study of adalimumab after prior DMARD [71], n = 434 | 26 weeks of treatment plus 6-month follow-up | 12 | ELISA |
| Cohort study of adalimumab [54], n = 121 | 28 weeks of treatment | 17 | Radioimmunoassay |
| Double-blind study of three doses of adalimumab in Japanese patients [72], n = 265 | 24 weeks of treatment plus 30-day follow-up | 37 | ELISA |
| Prospective observational cohort study [52], n = 249 | 28 weeks of treatment | 16 | Radioimmunoassay |
| Prospective cohort study of adalimumab [53], n = 272 | 3 years of treatment | 28 | Radioimmunoassay |
| Prospective observational cohort study [51], n = 216 | 28 weeks of treatment | 7 | ELISA |
| 13 | Radioimmunoassay | ||
| Prospective observational cohort study [44], n = 99 | 3 years of treatment | 29 | Radioimmunoassay |
| 54 | PIA antigen-binding test | ||
| Cohort study [43], n = 94 | 52 weeks of treatment | 14.9 | Radioimmunoassay |
| 66.0 | PIA antigen-binding test | ||
| 57.4 | Acid-dissociation radioimmunoassay | ||
| 51.1 | Temperature-shift radioimmunoassay | ||
| 57.4 | ECL-based assay |
ADA anti-drug antibody, DMARD disease-modifying anti-rheumatic drug, ECL electrochemiluminescence, ELISA enzyme-linked immunosorbent assays, PIA pH-shift anti-idiotype antigen-binding test
aIncludes studies of ≥ 50 patients receiving adalimumab
The broad range of immunogenicity rates observed in adalimumab-treated patients with RA, according to different assay types, as well as in different studies that used the same methodology, demonstrates the shortcomings of inter-trial comparisons of biopharmaceutical immunogenicity.
Evolving Standards and Practices for Assessing Immunogenicity of Biopharmaceuticals
Regulatory Guidelines
Evaluating the immunogenicity of biopharmaceuticals is mandatory for regulatory approval [16, 55, 56]. Specific guidance for evaluating immunogenicity of therapeutic protein products has been provided by the FDA and EMA [55, 57]; the EMA has also produced specific guidelines for assessing immune responses to therapeutic monoclonal antibodies [58].
Recommendations include development of assays to adequately detect and confirm the presence of ADAs and techniques to discern neutralizing and non-neutralizing antibodies, systematic collection of data from patients in clinical studies, and standardization of methods across studies for a given biopharmaceutical. Both the FDA and the EMA advise that data from immunogenicity assessments in animals are not necessarily indicative of immune responses in humans but can supplement the information obtained from preclinical toxicology studies. Furthermore, both agencies are inclined to accept the use of the one-assay approach to evaluate immunogenicity (see Sect. 7) [55, 57].
Post-Approval Surveillance of Immunogenicity
Because of strict eligibility criteria and short-term follow-up, results from immunogenicity analyses in clinical trials of biopharmaceuticals may not reflect real-world clinical experience. Indeed, it is necessary to continue the assessment of safety (including immunogenicity) following the approval of all biopharmaceuticals [57, 59]. Long-term follow-up of patients with RA treated with adalimumab [53] or infliximab [60] (another anti-TNF-α antibody) found that the presence of ADAs negatively impacts clinical response and leads to earlier treatment discontinuation. Moreover, clinical trials are unlikely to detect immune responses that result in severe or life-threatening, yet uncommon, safety issues. For example, a remarkable increase in the frequency of pure red-cell aplasia in patients administered recombinant erythropoietin due to generation of cross-reactive anti-erythropoietin antibodies was identified post approval; this coincided with a change in manufacturing process and formulation [61, 62]. Thus, ongoing monitoring of immunogenicity post approval is extremely valuable, and observational clinical and laboratory test databases from patients treated with biopharmaceuticals in routine clinical practice may provide further insight into the immunogenicity of these drugs. Guidelines from the FDA, EMA, and WHO recommend immunogenicity be considered in pharmacovigilance and risk management plans for all biopharmaceuticals, including biosimilars; however, only the EMA guidelines stipulate how immunogenicity should be addressed in post-approval surveillance strategies [5–7, 55–57].
A risk-based approach for evaluating the immunogenicity of biopharmaceuticals that has been suggested for clinical trials during drug development has also been proposed for post-approval safety monitoring plans [24, 46]. In preparation for pharmacovigilance strategies, however, it should be noted that suboptimal detection of ADAs in clinical studies may lead to underestimation of risk [5, 57]. According to the properties of the biopharmaceutical and the immunogenicity rates reported in pre-approval clinical trials, the risk management plan for unwanted immunogenicity may include additional clinical studies, retrospective analyses of real-world use, pharmacovigilance, and provisions in the prescribing information [63]. For biopharmaceuticals that produce persistent immune responses, there is the potential for ADA cross-reactivity in patients who switch to a biosimilar or new biopharmaceutical, and this should be recognized in the risk management plan.
Monitoring ADA levels in patients treated with biopharmaceuticals with a high risk of immunogenicity may be a useful component of post-approval registries to evaluate long-term immunogenic capacity. Moreover, it has been proposed that tracking levels of anti-TNF-α biopharmaceuticals and ADAs in patients with chronic inflammatory diseases may be useful for clinical decision-making, i.e., altering doses or switching to a different anti-TNF-α therapy [21]. Notwithstanding, a recent study suggests that adalimumab and etanercept serum ADA levels are not predictive of successful dose reduction or discontinuation in RA [64].
Additional Considerations for Biosimilar Immunogenicity Assessments
For approval of biosimilars, comparative immunogenicity testing with reference products in clinical studies is required by the FDA, EMA, and WHO [5–7]. Because even small differences in product quality (impurities and contaminants) may alter immunogenicity, demonstration of similar immunogenicity to the reference product is an important component of the biosimilar safety evaluation. Whereas the EMA and WHO do not require immunogenicity testing of biosimilars in preclinical animal studies, FDA guidance suggests that these analyses may provide useful data but are not obligatory. Nevertheless, investigations of biosimilar immunogenicity in animals are useful to determine titers of cross-reactive and neutralizing antibodies, and the data could contribute to the totality of the evidence evaluated by regulatory authorities for biosimilar approval [3].
Although historical immunogenicity data for the reference product should not be used for comparison with the biosimilar (due to more sensitive and specific assays being used today), this information may provide a guide to clinical developers to aid better assessment of biosimilar immunogenicity. Head-to-head comparisons of biosimilars and reference products are recommended [5, 6].
Comparative immunogenicity assessments may be accomplished using one-assay or two-assay methodology (Fig. 3) [63, 65]. In the one-assay approach, the biosimilar reagent is used to detect ADAs for both the biosimilar and the reference product. With this method, detection of biosimilar ADAs is optimized and comparison of biosimilar and reference product ADAs is facilitated but information on the immunogenicity of the reference product may be lost. In the two-assay approach, each assay will detect the amount of ADA either for the biosimilar or for the reference product. This method can yield a comprehensive analysis of biosimilar and reference product ADAs but requires extra time and resources. Each approach has merits and flaws (Fig. 3), and selection of the strategy to compare immunogenicity is an important consideration for comparative clinical trials of biosimilars. Because the intended purpose of biosimilar immunogenicity assessments is verification of comparable ADA rates with the reference product, rather than generation of new information about the reference product, a validated one-assay approach is generally recommended in order to minimize bias [58, 63, 66].
Fig. 3.

One-assay versus two-assay approach for anti-drug antibody screening of biosimilars. ADAs anti-drug antibodies
A predicament of biosimilar development is the inability to categorically designate a clinically meaningful difference in ADA rate that constitutes dissimilar immunogenicity of a biosimilar versus the reference product. Comparative evaluation of ADA rates alone may not fully elucidate differences in the immunogenicity profiles of biosimilars and reference biopharmaceuticals, due to biases in bioanalytical methods and inadequate comprehension of the effects of ADAs on short-term and long-term efficacy and safety [63]. As such, it is not practical to pre-define a difference in immunogenicity rates between biosimilars and reference products that is allowable to support biosimilarity [63]. Instead, ADA rates of biosimilars should be considered in the context of drug trough concentrations and efficacy and safety outcomes, which is consistent with guidelines from regulatory agencies to weigh the sum of evidence for biosimilar approval [5–7]. While it is clear that higher immunogenicity rates of a biosimilar versus the reference product would cast uncertainty about overall biosimilarity, the implications of lower immunogenicity are not well-defined. Reduced immunogenicity of a biosimilar would not prohibit its approval, but it also should not be translate as having enhanced efficacy or safety; additional subgroup analyses in patients with and without immune responses may be recommended by regulatory authorities to help inform the interpretation of efficacy results in clinical trials [6].
The EMA advises that a description of the potential safety concerns that may arise due to differences in the manufacturing process from the reference biopharmaceutical should be included in a biosimilar risk management plan [6]. Further, because immunogenicity may be affected by drug property-associated factors, such as differences in glycosylation patterns or aggregate formation, post-approval changes to the formulation or manufacturing process of the biosimilar may require additional analyses of immunogenicity, including re-validation of the assays [26, 28]. Similarly, extrapolation of indications for a biosimilar or plans for use in pediatric patients may require further assessments of immunogenicity, depending on patient-specific factors in the new population to be treated [26].
It is important here to distinguish biosimilars from non-comparable biopharmaceuticals (also known as ‘intended copies’ or biomimics), which may not have undergone a thorough evaluation of physicochemical and functional properties, efficacy and safety, including immunogenicity, against a reference product. Pre-existing legislation in some countries in Latin America and Asia have allowed copies of biopharmaceuticals to be approved without meeting the current high regulatory standards for biosimilarity established outside of Latin America and Asia (e.g., by the FDA, EMA, and WHO). Since these products may not have demonstrated comparable efficacy and safety to reference products, they are designated as intended copies instead of biosimilars [67]. An intended copy of rituximab was withdrawn by regulatory authorities in Mexico due to a lack of evidence on its biosimilarity and reports of anaphylactic reactions in patients who switched between the reference product and the intended copy. This was observed despite the fact that there was evidence that the incidence of peri-infusion reactions to the originator’s rituximab in the Mexican population was low [68].These findings underscore the importance of rigorous evaluation of safety, including immunogenicity assessments, prior to routine use of biosimilars. Biosimilarity designation is based on thorough, high-quality comparability of product attributes, including the variables that could influence immunogenicity.
Lessons Learned and Recommendations
Assessment of immunogenicity is an essential element of the development program for all biopharmaceuticals, including biosimilars. However, technological progress over the past two decades and inconsistent methodology among research groups limit comparison of the immunogenicity of biopharmaceuticals outside of head-to-head clinical trials. To address the heterogeneity of approaches, strong guidance from regulatory authorities on assessment of immunogenicity would be welcome. In the interim, both the American Association of Pharmaceutical Scientists and the Anti-Biopharmaceutical Immunization: Prediction and Analysis of Clinical Relevance to Minimize the Risk consortium have advocated strategies to align the investigation and understanding of immunogenicity of biopharmaceuticals [69, 70]. These two organizations recommend standardization of terminology and data reporting as well as linking the presence of ADAs with efficacy, safety, and pharmacokinetic endpoints to explore the clinical consequences of immune responses to biopharmaceuticals. These proposals may signify an initial step toward unifying the evaluation of immunogenicity.
Our understanding of immunogenicity and the methods to assess unwanted immune responses have advanced substantially since the introduction of biopharmaceuticals. The precision and sensitivity of immunogenicity assays have increased over time and will likely continue to improve. Accordingly, higher rates of immunogenicity are observed using modern assays. Physicians should be aware of these enhanced technologies and not presume that new biopharmaceuticals, including biosimilars, are more immunogenic. In fact, with the emergence of biosimilars and the requirement for comparisons to reference products in clinical trials, state-of-the-art technologies may yield new information on the immunogenicity of already approved biopharmaceuticals. We anticipate that immunogenicity analyses will continue to evolve as novel techniques emerge, such as evaluation of genetic and epigenetic biomarkers, and in silico prediction of immunogenicity, which may augment the predictive value of risk assessment in patients. Consequently, research programs, regulatory agencies, and clinicians will need to keep pace with progressively complex analyses of immunogenicity.
Acknowledgments
Medical writing support was provided by Neel Misra, MSc, and Joanna Bloom, PhD, of Engage Scientific Solutions, and funded by Pfizer.
Author contributions
All authors were involved in drafting the article and revising it critically for important intellectual content. All authors read and approved the final manuscript submitted for publication.
Compliance with Ethical Standards
Conflict of interest
CP has no competing interests to declare. GC-H has received consultancy fees from Amgen, AbbVie, AstraZeneca, Bayer, Boehringer Ingelheim, Eli Lilly, Janssen-Cilag, Laboratorios Liomont, Laboratorios Sophia, Medix, Merck Serono, Merck, Sharp and Dohme, Novartis, Pfizer, Roche, Sanofi, and UCB. IJ, DA, and CC are full-time employees of Pfizer.
Funding
This report was sponsored by Pfizer Inc.
References
- 1.Morrow T, Felcone LH. Defining the difference: what makes bologics unique. Biotechnol Healthc. 2004;1(4):24–29. [PMC free article] [PubMed] [Google Scholar]
- 2.Walsh G. Biopharmaceutical benchmarks 2010. Nat Biotechnol. 2010;28(9):917–924. doi: 10.1038/nbt0910-917. [DOI] [PubMed] [Google Scholar]
- 3.Wang J, Chow SC. On the regulatory approval pathway of biosimilar products. Pharmaceuticals (Basel). 2012;5(4):353–368. doi: 10.3390/ph5040353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bennett CL, Chen B, Hermanson T, Wyatt MD, Schulz RM, Georgantopoulos P, et al. Regulatory and clinical considerations for biosimilar oncology drugs. Lancet Oncol. 2014;15(13):e594–e605. doi: 10.1016/S1470-2045(14)70365-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.US Food and Drug Administration. Scientific considerations in demonstrating biosimilarity to a reference product: guidance for industry. SIlver Spring: U.S. Department of Health and Human Services, Center for Drug Evaluation and Research (CDER), Center for Biologics Evaluation and Research (CBER); 2015 (last update Apr 2015). http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM291128.pdf. Accessed 23 Mar 2015.
- 6.European Medicines Agency, Committee for Medicinal Products for Human Use (CHMP). Guideline on similar biological medicinal products containing biotechnology-derived proteins as active substance: non-clinical and clinical issues. London: EMA; 2013 (last update 3 June 2013). http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2013/06/WC500144124.pdf. Accessed 23 Mar 2015.
- 7.World Health Organization, Expert Committee on Biological Standardization. Geneva, 19 to 23 October 2009. Guidelines on evaluation of similar biotherapeutic products (SBPs). Geneva: WHO; 2009 (last update 19–23 Oct 2009). http://www.who.int/biologicals/areas/biological_therapeutics/BIOTHERAPEUTICS_FOR_WEB_22APRIL2010.pdf. Accessed 21 Apr 2015.
- 8.Espinosa Morales R, Diaz Borjon A, Barile Fabris LA, Esquivel Valerio JA, Medrano Ramirez G, Arce Salinas CA, et al. Biosimilar drugs in Mexico: position of the Mexican College of Rheumatology, 2012. Reumatol Clin. 2013;9(2):113–116. doi: 10.1016/j.reuma.2012.11.001. [DOI] [PubMed] [Google Scholar]
- 9.American College of Rheumatology. American College of Rheumatology position statement: biosimilars. Atlanta: ACR; 2015. http://www.rheumatology.org/Portals/0/Files/Biosimilars.pdf. Accessed 27 May 2015.
- 10.Barosi G, Bosi A, Abbracchio MP, Danesi R, Genazzani A, Corradini P, et al. Key concepts and critical issues on epoetin and filgrastim biosimilars. A position paper from the Italian Society of Hematology, Italian Society of Experimental Hematology, and Italian Group for Bone Marrow Transplantation. Haematologica. 2011;96(7):937–942. doi: 10.3324/haematol.2011.041210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fiorino G, Girolomoni G, Lapadula G, Orlando A, Danese S, Olivieri I. The use of biosimilars in immune-mediated disease: a joint Italian Society of Rheumatology (SIR), Italian Society of Dermatology (SIDeMaST), and Italian Group of Inflammatory Bowel Disease (IG-IBD) position paper. Autoimmun Rev. 2014;13(7):751–755. doi: 10.1016/j.autrev.2014.02.004. [DOI] [PubMed] [Google Scholar]
- 12.Arguelles-Arias F, Barreiro-de-Acosta M, Carballo F, Hinojosa J, Tejerina T. Joint position statement by “Sociedad Espanola de Patologia Digestiva” (Spanish Society of Gastroenterology) and “Sociedad Espanola de Farmacologia” (Spanish Society of Pharmacology) on biosimilar therapy for inflammatory bowel disease. Rev Esp Enferm Dig. 2013;105(1):37–43. doi: 10.4321/S1130-01082013000100006. [DOI] [PubMed] [Google Scholar]
- 13.Fonseca JE, Goncalves J, Araujo F, Cordeiro I, Teixeira F, Canhao H, et al. The Portuguese Society of Rheumatology position paper on the use of biosimilars. Acta Reumatol Port. 2014;39(1):60–71. [PubMed] [Google Scholar]
- 14.Mularczyk A, Gonciarz M, Bartnik W, Durlik M, Eder P, Gasiorowska A, et al. Biosimilar medicines—their use in the treatment of inflammatory bowel diseases. Position statement of the Working Group of the Polish National Consultant in Gastroenterology. Prz Gastroenterol. 2014;9(1):1–3. doi:10.5114/pg.2014.40842. [DOI] [PMC free article] [PubMed]
- 15.Bendtzen K. Anti-TNF-alpha biotherapies: perspectives for evidence-based personalized medicine. Immunotherapy. 2012;4(11):1167–1179. doi: 10.2217/imt.12.114. [DOI] [PubMed] [Google Scholar]
- 16.European Medicines Agency, European Agency for the Evaluation of Medicinal Products, Committee for Proprietary Medicinal Products (CPMP). Development pharmaceutics for biotechnological and biological products (CPMP/BWP/328/99). Annex to note for guidance on development of pharmaceutics (CPMP/QWP/155/96). London: EMA; 1999 (last update Apr 2000). http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500003603.pdf. Accessed 28 May 2015.
- 17.Pendley C, Schantz A, Wagner C. Immunogenicity of therapeutic monoclonal antibodies. Curr Opin Mol Ther. 2003;5(2):172–179. [PubMed] [Google Scholar]
- 18.Schellekens H. Bioequivalence and the immunogenicity of biopharmaceuticals. Nat Rev Drug Discov. 2002;1(6):457–462. doi: 10.1038/nrd818. [DOI] [PubMed] [Google Scholar]
- 19.Schellekens H. Immunogenicity of therapeutic proteins: clinical implications and future prospects. Clin Ther. 2002;24(11):1720–1740. doi: 10.1016/S0149-2918(02)80075-3. [DOI] [PubMed] [Google Scholar]
- 20.Shankar G, Pendley C, Stein KE. A risk-based bioanalytical strategy for the assessment of antibody immune responses against biological drugs. Nat Biotechnol. 2007;25(5):555–561. doi: 10.1038/nbt1303. [DOI] [PubMed] [Google Scholar]
- 21.Vincent FB, Morand EF, Murphy K, Mackay F, Mariette X, Marcelli C. Antidrug antibodies (ADAb) to tumour necrosis factor (TNF)-specific neutralising agents in chronic inflammatory diseases: a real issue, a clinical perspective. Ann Rheum Dis. 2013;72(2):165–178. doi: 10.1136/annrheumdis-2012-202545. [DOI] [PubMed] [Google Scholar]
- 22.Casadevall N, Nataf J, Viron B, Kolta A, Kiladjian JJ, Martin-Dupont P, et al. Pure red-cell aplasia and antierythropoietin antibodies in patients treated with recombinant erythropoietin. N Engl J Med. 2002;346(7):469–475. doi: 10.1056/NEJMoa011931. [DOI] [PubMed] [Google Scholar]
- 23.Li J, Yang C, Xia Y, Bertino A, Glaspy J, Roberts M, et al. Thrombocytopenia caused by the development of antibodies to thrombopoietin. Blood. 2001;98(12):3241–3248. doi: 10.1182/blood.V98.12.3241. [DOI] [PubMed] [Google Scholar]
- 24.Buttel IC, Chamberlain P, Chowers Y, Ehmann F, Greinacher A, Jefferis R, et al. Taking immunogenicity assessment of therapeutic proteins to the next level. Biologicals. 2011;39(2):100–109. doi: 10.1016/j.biologicals.2011.01.006. [DOI] [PubMed] [Google Scholar]
- 25.Jahn EM, Schneider CK. How to systematically evaluate immunogenicity of therapeutic proteins—regulatory considerations. N Biotechnol. 2009;25(5):280–286. doi: 10.1016/j.nbt.2009.03.012. [DOI] [PubMed] [Google Scholar]
- 26.Barbosa MD, Kumar S, Loughrey H, Singh SK. Biosimilars and biobetters as tools for understanding and mitigating the immunogenicity of biotherapeutics. Drug Discov Today. 2012;17(23–24):1282–1288. doi: 10.1016/j.drudis.2012.07.003. [DOI] [PubMed] [Google Scholar]
- 27.Barbosa MD, Celis E. Immunogenicity of protein therapeutics and the interplay between tolerance and antibody responses. Drug Discov Today. 2007;12(15–16):674–681. doi: 10.1016/j.drudis.2007.06.005. [DOI] [PubMed] [Google Scholar]
- 28.Liu PM, Zou L, Sadhu C, Shen WD, Nock S. Comparative immunogenicity assessment: a critical consideration for biosimilar development. Bioanalysis. 2015;7(3):373–381. doi: 10.4155/bio.14.311. [DOI] [PubMed] [Google Scholar]
- 29.Nechansky A, Kircheis R. Immunogenicity of therapeutics: a matter of efficacy and safety. Expert Opin Drug Discov. 2010;5(11):1067–1079. doi: 10.1517/17460441.2010.514326. [DOI] [PubMed] [Google Scholar]
- 30.Wadhwa M, Bird C, Dilger P, Gaines-Das R, Thorpe R. Strategies for detection, measurement and characterization of unwanted antibodies induced by therapeutic biologicals. J Immunol Methods. 2003;278(1–2):1–17. doi: 10.1016/S0022-1759(03)00206-0. [DOI] [PubMed] [Google Scholar]
- 31.Mire-Sluis AR, Barrett YC, Devanarayan V, Koren E, Liu H, Maia M, et al. Recommendations for the design and optimization of immunoassays used in the detection of host antibodies against biotechnology products. J Immunol Methods. 2004;289(1–2):1–16. doi: 10.1016/j.jim.2004.06.002. [DOI] [PubMed] [Google Scholar]
- 32.Yin L, Chen X, Vicini P, Rup B, Hickling TP. Therapeutic outcomes, assessments, risk factors and mitigation efforts of immunogenicity of therapeutic protein products. Cell Immunol. 2015;295(2):118–126. doi: 10.1016/j.cellimm.2015.03.002. [DOI] [PubMed] [Google Scholar]
- 33.Wadhwa M, Knezevic I, Kang HN, Thorpe R. Immunogenicity assessment of biotherapeutic products: an overview of assays and their utility. Biologicals. 2015;43(5):298–306. doi: 10.1016/j.biologicals.2015.06.004. [DOI] [PubMed] [Google Scholar]
- 34.Aarden L, Ruuls SR, Wolbink G. Immunogenicity of anti-tumor necrosis factor antibodies-toward improved methods of anti-antibody measurement. Curr Opin Immunol. 2008;20(4):431–435. doi: 10.1016/j.coi.2008.06.011. [DOI] [PubMed] [Google Scholar]
- 35.Vollmer N, Trombini F, Hely M, Bellon S, Mercier K, Cazeneuve C. Methodology to study polymers interaction by surface plasmon resonance imaging. MethodsX. 2015;2:14–18. doi: 10.1016/j.mex.2014.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wolbink GJ, Aarden LA, Dijkmans BA. Dealing with immunogenicity of biologicals: assessment and clinical relevance. Curr Opin Rheumatol. 2009;21(3):211–215. doi: 10.1097/BOR.0b013e328329ed8b. [DOI] [PubMed] [Google Scholar]
- 37.van Schouwenburg PA, Bartelds GM, Hart MH, Aarden L, Wolbink GJ, Wouters D. A novel method for the detection of antibodies to adalimumab in the presence of drug reveals “hidden” immunogenicity in rheumatoid arthritis patients. J Immunol Methods. 2010;362(1–2):82–88. doi: 10.1016/j.jim.2010.09.005. [DOI] [PubMed] [Google Scholar]
- 38.Patton A, Mullenix MC, Swanson SJ, Koren E. An acid dissociation bridging ELISA for detection of antibodies directed against therapeutic proteins in the presence of antigen. J Immunol Methods. 2005;304(1–2):189–195. doi: 10.1016/j.jim.2005.06.014. [DOI] [PubMed] [Google Scholar]
- 39.Sickert D, Kroeger K, Zickler C, Chokote E, Winkler B, Grenet JM, et al. Improvement of drug tolerance in immunogenicity testing by acid treatment on Biacore. J Immunol Methods. 2008;334(1–2):29–36. doi: 10.1016/j.jim.2008.01.010. [DOI] [PubMed] [Google Scholar]
- 40.Bourdage JS, Cook CA, Farrington DL, Chain JS, Konrad RJ. An affinity capture elution (ACE) assay for detection of anti-drug antibody to monoclonal antibody therapeutics in the presence of high levels of drug. J Immunol Methods. 2007;327(1–2):10–17. doi: 10.1016/j.jim.2007.07.004. [DOI] [PubMed] [Google Scholar]
- 41.Schmidt E, Hennig K, Mengede C, Zillikens D, Kromminga A. Immunogenicity of rituximab in patients with severe pemphigus. Clin Immunol. 2009;132(3):334–341. doi: 10.1016/j.clim.2009.05.007. [DOI] [PubMed] [Google Scholar]
- 42.Wang SL, Ohrmund L, Hauenstein S, Salbato J, Reddy R, Monk P, et al. Development and validation of a homogeneous mobility shift assay for the measurement of infliximab and antibodies-to-infliximab levels in patient serum. J Immunol Methods. 2012;382(1–2):177–188. doi: 10.1016/j.jim.2012.06.002. [DOI] [PubMed] [Google Scholar]
- 43.Bloem K, van Leeuwen A, Verbeek G, Nurmohamed MT, Wolbink GJ, van der Kleij D, et al. Systematic comparison of drug-tolerant assays for anti-drug antibodies in a cohort of adalimumab-treated rheumatoid arthritis patients. J Immunol Methods. 2015;418:29–38. doi: 10.1016/j.jim.2015.01.007. [DOI] [PubMed] [Google Scholar]
- 44.van Schouwenburg PA, Krieckaert CL, Rispens T, Aarden L, Wolbink GJ, Wouters D. Long-term measurement of anti-adalimumab using pH-shift-anti-idiotype antigen binding test shows predictive value and transient antibody formation. Ann Rheum Dis. 2013;72(10):1680–1686. doi: 10.1136/annrheumdis-2012-202407. [DOI] [PubMed] [Google Scholar]
- 45.Shankar G, Devanarayan V, Amaravadi L, Barrett YC, Bowsher R, Finco-Kent D, et al. Recommendations for the validation of immunoassays used for detection of host antibodies against biotechnology products. J Pharm Biomed Anal. 2008;48(5):1267–1281. doi: 10.1016/j.jpba.2008.09.020. [DOI] [PubMed] [Google Scholar]
- 46.Koren E, Smith HW, Shores E, Shankar G, Finco-Kent D, Rup B, et al. Recommendations on risk-based strategies for detection and characterization of antibodies against biotechnology products. J Immunol Methods. 2008;333(1–2):1–9. doi: 10.1016/j.jim.2008.01.001. [DOI] [PubMed] [Google Scholar]
- 47.Gupta S, Indelicato SR, Jethwa V, Kawabata T, Kelley M, Mire-Sluis AR, et al. Recommendations for the design, optimization, and qualification of cell-based assays used for the detection of neutralizing antibody responses elicited to biological therapeutics. J Immunol Methods. 2007;321(1–2):1–18. doi: 10.1016/j.jim.2006.12.004. [DOI] [PubMed] [Google Scholar]
- 48.AbbVie Inc. HUMIRA (adalimumab) prescribing information. North Chicago: AbbVie Inc.; 2014. http://www.rxabbvie.com/pdf/humira.pdf. Accessed 2 June 2015.
- 49.Weinblatt ME, Keystone EC, Furst DE, Moreland LW, Weisman MH, Birbara CA, et al. Adalimumab, a fully human anti-tumor necrosis factor alpha monoclonal antibody, for the treatment of rheumatoid arthritis in patients taking concomitant methotrexate: the ARMADA trial. Arthritis Rheum. 2003;48(1):35–45. doi: 10.1002/art.10697. [DOI] [PubMed] [Google Scholar]
- 50.Keystone EC, Kavanaugh AF, Sharp JT, Tannenbaum H, Hua Y, Teoh LS, et al. Radiographic, clinical, and functional outcomes of treatment with adalimumab (a human anti-tumor necrosis factor monoclonal antibody) in patients with active rheumatoid arthritis receiving concomitant methotrexate therapy: a randomized, placebo-controlled, 52-week trial. Arthritis Rheum. 2004;50(5):1400–1411. doi: 10.1002/art.20217. [DOI] [PubMed] [Google Scholar]
- 51.Hart MH, de Vrieze H, Wouters D, Wolbink GJ, Killestein J, de Groot ER, et al. Differential effect of drug interference in immunogenicity assays. J Immunol Methods. 2011;372(1–2):196–203. doi: 10.1016/j.jim.2011.07.019. [DOI] [PubMed] [Google Scholar]
- 52.Bartelds GM, de Groot E, Nurmohamed MT, Hart MHL, van Eede PH, Wijbrandts CA, et al. Surprising negative association between IgG1 allotype disparity and anti-adalimumab formation: a cohort study. Arthritis Res Ther. 2010;12(6). doi:10.1186/ar3208. [DOI] [PMC free article] [PubMed]
- 53.Bartelds GM, Krieckaert CL, Nurmohamed MT, van Schouwenburg PA, Lems WF, Twisk JW, et al. Development of antidrug antibodies against adalimumab and association with disease activity and treatment failure during long-term follow-up. JAMA. 2011;305(14):1460–1468. doi: 10.1001/jama.2011.406. [DOI] [PubMed] [Google Scholar]
- 54.Bartelds GM, Wijbrandts CA, Nurmohamed MT, Stapel S, Lems WF, Aarden L, et al. Clinical response to adalimumab: relationship to anti-adalimumab antibodies and serum adalimumab concentrations in rheumatoid arthritis. Ann Rheum Dis. 2007;66(7):921–926. doi: 10.1136/ard.2006.065615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.US Food and Drug Administration. Immunogenicity assessment for therapeutic protein products: guidance for industry. Silver Spring: U.S. Department of Health and Human Services, Center for Drug Evaluation and Research (CDER), Center for Biologics Evaluation and Research (CBER); 2014 (last update August 2014). http://www.fda.gov/downloads/drugs/guidancecomplianceregulatoryinformation/guidances/ucm338856.pdf. Accessed 28 May 2015.
- 56.World Health Organization. Guidelines on the quality, safety, and efficacy of biotherapeutic protein products prepared by recombinant DNA technology. Geneva: WHO; 2013. http://www.who.int/biologicals/biotherapeutics/rDNA_DB_final_19_Nov_2013.pdf. Accessed 28 May 2015.
- 57.European Medicines Agency, Committee for Medicinal Products for Human Use (CHMP). Guideline on immunogenicity assessment of biotechnology-derived therapeutic proteins. London: EMA; 2008. http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500003946.pdf. Accessed 28 May 2015.
- 58.European Medicines Agency, Committee for Medicinal Products for Human Use (CHMP). Guideline on immunogenicity assessment of monoclonal antibodies intended for in vivo clinical use. London: European Medicines Agency; 2012 (last update 1 Dec 2012). http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2012/06/WC500128688.pdf. Accessed 25 Nov 2015.
- 59.Ebbers HC, Crow SA, Vulto AG, Schellekens H. Interchangeability, immunogenicity and biosimilars. Nat Biotechnol. 2012;30(12):1186–1190. doi: 10.1038/nbt.2438. [DOI] [PubMed] [Google Scholar]
- 60.Pascual-Salcedo D, Plasencia C, Ramiro S, Nuno L, Bonilla G, Nagore D, et al. Influence of immunogenicity on the efficacy of long-term treatment with infliximab in rheumatoid arthritis. Rheumatology (Oxford) 2011;50(8):1445–1452. doi: 10.1093/rheumatology/ker124. [DOI] [PubMed] [Google Scholar]
- 61.Rosenberg AS. Immunogenicity of biological therapeutics: a hierarchy of concerns. Dev Biol (Basel) 2003;112:15–21. [PubMed] [Google Scholar]
- 62.Woodcock J, Griffin J, Behrman R, Cherney B, Crescenzi T, Fraser B, et al. The FDA’s assessment of follow-on protein products: a historical perspective. Nat Rev Drug Discov. 2007;6(6):437–442. doi: 10.1038/nrd2307. [DOI] [PubMed] [Google Scholar]
- 63.Chamberlain P. Assessing immunogenicity of biosimilar therapeutic monoclonal antibodies: regulatory and bioanalytical considerations. Bioanalysis. 2013;5(5):561–574. doi: 10.4155/bio.13.6. [DOI] [PubMed] [Google Scholar]
- 64.van Herwaarden N, Bouman CA, van der Maas A, van Vollenhoven RF, Bijlsma JW, van den Hoogen FH, et al. Adalimumab and etanercept serum (anti)drug levels are not predictive for successful dose reduction or discontinuation in rheumatoid arthritis. Ann Rheum Dis. 2015;74(12):2260–2261. doi: 10.1136/annrheumdis-2015-207814. [DOI] [PubMed] [Google Scholar]
- 65.Cai XY, Thomas J, Cullen C, Gouty D. Challenges of developing and validating immunogenicity assays to support comparability studies for biosimilar drug development. Bioanalysis. 2012;4(17):2169–2177. doi: 10.4155/bio.12.185. [DOI] [PubMed] [Google Scholar]
- 66.Chamberlain P. Multidisciplinary approach to evaluating immunogenicity of biosimilars: lessons learnt and open questions based on 10 years’ experience of the European Union regulatory pathway. Biosimilars. 2014;4:23–43. doi: 10.2147/BS.S50012. [DOI] [Google Scholar]
- 67.Scheinberg M, Castaneda-Hernandez G. Anti-tumor necrosis factor patent expiration and the risks of biocopies in clinical practice. Arthritis Res Ther. 2014;16(6):501. doi: 10.1186/s13075-014-0501-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Arredondo-Garza T, Majluf-Cruz A, Vela-Ojeda J, Mariscal-Ramirez I, Solis-Anaya L, Lopez-Gutierrez JR, et al. Peri-infusional adverse reactions to rituximab in patients with non-Hodgkin’s lymphoma. Arch Med Res. 2013;44(7):549–554. doi: 10.1016/j.arcmed.2013.09.011. [DOI] [PubMed] [Google Scholar]
- 69.Shankar G, Arkin S, Cocea L, Devanarayan V, Kirshner S, Kromminga A, et al. Assessment and reporting of the clinical immunogenicity of therapeutic proteins and peptides-harmonized terminology and tactical recommendations. AAPS J. 2014;16(4):658–673. doi: 10.1208/s12248-014-9599-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Rup B, Pallardy M, Sikkema D, Albert T, Allez M, Broet P, et al. ABIRISK Consortium. Standardizing terms, definitions and concepts for describing and interpreting unwanted immunogenicity of biopharmaceuticals: recommendations of the Innovative Medicines Initiative ABIRISK consortium. Clin Exp Immunol. 2015;181(3):385–400. doi: 10.1111/cei.12652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.van de Putte LB, Atkins C, Malaise M, Sany J, Russell AS, van Riel PL, et al. Efficacy and safety of adalimumab as monotherapy in patients with rheumatoid arthritis for whom previous disease modifying antirheumatic drug treatment has failed. Ann Rheum Dis. 2004;63(5):508–516. doi: 10.1136/ard.2003.013052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Miyasaka N. Clinical investigation in highly disease-affected rheumatoid arthritis patients in Japan with adalimumab applying standard and general evaluation: the CHANGE study. Mod Rheumatol. 2008;18(3):252–262. doi: 10.3109/s10165-008-0045-0. [DOI] [PMC free article] [PubMed] [Google Scholar]