

Abstract
The transporter protein Large-neutral Amino Acid Transporter 1 (LAT-1, SLC7A5) is responsible for transporting amino acids such as tyrosine and phenylalanine as well as thyroid hormones, and it has been exploited as a drug delivery mechanism. Recently its role in cancer has become increasingly appreciated, as it has been found to be up-regulated in many different tumor types, and its expression levels have been correlated with prognosis. Substitution at the meta position of aromatic amino acids has been reported to increase affinity for LAT-1; however, the SAR for this position has not previously been explored. Guided by newly refined computational models of the binding site, we hypothesized that groups capable of filling a hydrophobic pocket would increase binding to LAT-1, resulting in improved substrates relative to parent amino acid. Tyrosine and phenylalanine analogs substituted at the meta position with halogens, alkyl and aryl groups were synthesized and tested in cis-inhibition and trans-stimulation cell assays to determine activity. Contrary to our initial hypothesis we found that lipophilicity was correlated with diminished substrate activity and increased inhibition of the transporter. The synthesis and SAR of meta-substituted phenylalanine and tyrosine analogs is described.
Keywords: SLC7A5, Amino Acid, Negishi coupling, Transporter Substrate, Transporter Inhibitor
Graphical Abstract

The Large-neutral Amino Acid Transporter 1 (LAT-1, SLC7A5) has received considerable attention as both a drug delivery mechanism to access the BBB;1-5 and for its role in cancer, where it is up-regulated in many different types of human tumor cells, including prostate6, colorectal7, non-small-cell lung cancer (NSCLC)8 and glioblastoma multiforme (GBM)9, 10. Furthermore LAT-1 expression has been correlated with poor prognosis for cancer patients,11-14 and tumor growth can be repressed by use of LAT-1 inhibitors15-19 presumably due to a cancer cell's increased nutritional demands.
A challenge for using LAT-1 in drug delivery is the problem of transporter saturation by endogenous amino acids, which have plasma concentrations significantly higher than their Michaelis constant (Km) value.20 One approach to solving this problem would be to develop drugs or prodrugs with increased affinity (or reduced Km values) for LAT-1 in comparison to the Km values of natural amino acid substrates (Km values of 11-210 μM)20, while at the same time not sacrificing maximal velocity Vmax. It has been shown that the substitution pattern for aromatic amino acids can have a dramatic effect on LAT-1 activity, with meta substitution significantly improving binding affinity relative to other positions.21, 22 And though transporter substrates are desirable for drug delivery applications, potent LAT-1 inhibitors could also be useful as metabolic blockers in cancer. To date, the LAT-1 SAR for substituted aromatic amino acids has only received a cursory exploration, with significant gaps remaining to be filled. As part of our objective to identify improved transporter substrates, we sought to expand the current understanding of the effect of substitution at the meta position for a series of phenylalanine and tyrosine analogs.
The atomic structure of the human LAT-1 is not known; however, we have developed a homology model of LAT-1 based on a structure of a related transporter, the arginine-agmatine transporter AdiC from E. coli.23 The model can rationalize amino acid selectivity among amino acid transporters, and virtual screening against this model followed by experimental testing identified previously unknown LAT-1 ligands.24 Based on our previously published LAT-1 model23, which was subsequently refined (supplementary material), as well as those models developed by others25, 26 we hypothesized that meta substitution on the aromatic ring of phenylalanine and tyrosine with lipophilic groups capable of filling a hydrophobic pocket (PA, Figure 3) in the LAT-1 binding site might lead to an increase in LAT-1 binding and an improvement in substrate activity.
Figure 3.

Comparison of methyl substituted phenylalanine isomers.
To evaluate substrate activity, compounds were tested in two different cell-based assays, using a LAT-1 overexpressing cell line generated by transfection of HEK cells with human LAT-1 cDNA.24 HEK-LAT1 cells were used to identify ligands for LAT-1 using both cis-inhibition and trans-stimulation assays.24 cis-Inhibition involves competition of test compound with a radiolabeled amino acid substrate (e.g. [3H]-gabapentin) for LAT-1 mediated cellular uptake. Though the cis-inhibition assay can identify agents which interact with LAT-1, it is exclusively used to identify inhibitors, not substrates of the transporter. To determine whether a compound was a substrate, a trans-stimulation assay27 was performed. This assay exploits LAT-1's alternating access mechanism28, 29 by pre-loading cells with [3H]-gabapentin followed by incubation with test compound. We choose [3H]-gabapentin as a probe substrate due to its selectivity against other transporters.29 The efflux rate of radiolabeled amino acid in the presence of the test compound is compared to the efflux rate in the absence of the test compound and with both positive and negative control amino acids (leucine and arginine, respectively) and parent amino acids (phenylalanine and tyrosine) to assess whether a test compound is a LAT-1 substrate. Substrates enhance the efflux rate of the radiolabeled amino acid compared with its efflux rate in the absence of the substrate.
To prepare compounds 29 and 39, protected L-phenylalanine 1 and L-tyrosine 2 were subjected to a Suzuki coupling30 with phenylboronic acid followed by deprotection according to Scheme 1. Attempts to introduce a methyl group at R2 using Suzuki coupling with methyl boronic acid were unsuccessful,31 and NMR analysis of the reaction mixture for conversion of 2 to 5 indicated low yield (<25%) of desired product (not isolated).
Scheme 1.

Synthesis of compounds 29 and 39. Reagents and conditions: (a) PhB(OH)2, Na2CO3, Pd(OAc)2, P(o-tolyl)3, DME/H2O (6:1), 80 °C, 3: 63%, 4: 52%, 5: not isolated; (b) H2 (balloon), Pd/C; (c) 1:1 6N HCl (aq)/1,4-dioxane, 29: 35%, 39: 25% (prep. HPLC purification).
In order to prepare the desired alkyl-substituted amino acids of Figure 1, we decided to examine the Negishi coupling32 as an alternative to the Suzuki reaction. Initial attempts to perform Negishi coupling with diethyl zinc as a test case on 1 or 2 using conditions described for a protected amino acid33 gave poor yields (<20%) of the desired 3-ethyl substituted phenylalanine or tyrosine derivative. A literature search revealed no examples of Negishi coupling reactions between iodo-substituted phenylalanine or tyrosine analogs (e.g. 1 or 2) and alkyl zinc reagents. Apparently, Negishi coupling of aromatic amino acids has been limited to substitution with cyano34 or aromatic rings35. We hypothesized that the desired Negishi coupling might give a better yield if both the carbamate “N-H” and carboxylic acid of 6 and 7 were masked, and this prompted us to explore an oxazolidinone36, 37 protecting group (Scheme 2). To our knowledge, this synthetic method is a new approach for obtaining meta-substituted phenylalanine derivatives. In addition to serving the dual purpose of protecting both the carbamate and carboxylic acid, the oxazolidinone was easily removed under acidic conditions to simultaneously remove the Boc group giving the meta substituted amino acids 13, 25 and 28. Unfortunately the desired ethyl substituted tyrosine 35 was not obtained using this route due to failure to deprotect the benzyl ether of 13 presumably caused by poisoning of the palladium catalyst used in hydrogenolysis.38
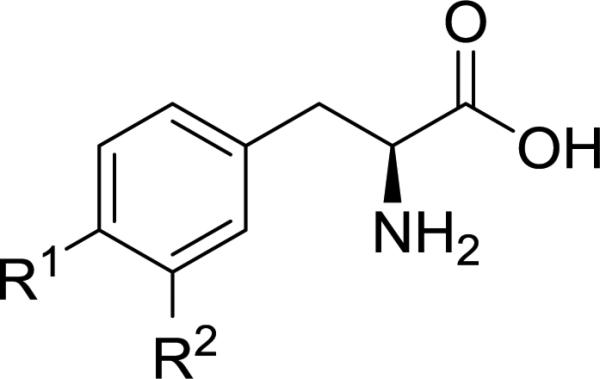
Figure 1.

The effect of meta substituents (R2) on exchange efflux rate and uptake of [3H]-gabapentin in HEK-hLAT1 cells for (a) L-phenylalanine (R1 = H) and (b) L-tyrosine (R1 = OH) analogs. Data represents the mean of at least three experiments. Positive controls L-leucine, L-phenylalanine, and L-tyrosine are known LAT-1 substrates; and negative control L-arginine is not.20 The left ordinate indicates efflux rate (fmol/min) in which compounds were tested at 200 μM for their ability to cause efflux of [3H]-gabapentin from preloaded HEK-hLAT1 cells. Average SD ± 0.2 fmol/min. The right ordinate indicates % inhibition in which compounds were tested at 200 μM for their ability to inhibit uptake of [3H]-gabapentin into HEK-hLAT1 cells. Data is presented as % inhibition relative to background signal in the absence of an inhibitor. Average SD ± 2%.
Scheme 2.

Synthesis of compounds 25 and 28. Reagents and conditions: (a) K2CO3, CH2Br2, CH3CN, 100 °C, 8: 43%, 9: 32%; (b) Et2Zn, PdCl2(dppf)-DCM, THF, rt, 18 h, 10: 74%, 11: 60%, 12: 62%; (c) 1:1 6N HCl (aq)/1,4-dioxane, 60°C, 25: 60%, 28: 38%, 13: 29%; (d) H2 (balloon), Pd/C, no reaction.
As deprotection of tyrosine intermediate 13 (Scheme 2) was problematic, and moreover, we desired bulky alkyl groups at R2 (i.e. isopropyl and tert-butyl) that could be problematic using the cross coupling strategy of Scheme 2 (e.g. conversion of 8 to 10), we explored the convergent approach depicted in Scheme 3 as an alternative. Conversion of commercially available alkyl iodide 14 to Jackson's organozinc 1539, 40 followed by an in situ Negishi coupling41 to give 17a-f was successfully performed. Though this route added additional synthetic steps to obtain noncommercial meta substituted aryl iodides42 16c-f, we found Scheme 3 to be a robust approach for preparing several of the desired amino acid analogs of Figure 1 (i.e. 26, 27, 34–36, and 40).
Scheme 3.

Synthesis of compounds 26, 27, 34–36, and 40. Reagents and conditions: (a) Zn, 1,2-dibromoethane, TMSCl, DMF, 60 °C for 30 min to activate zinc, then sonication with 14 for 30 min; (b) Pd2dba3, P(o-tolyl)3, DMF, rt, 42-57%; (c) H2 (balloon), Pd(OH)2/C, rt; (d) 1:1 6N HCl (aq)/1,4-dioxane, 60 °C, 1h, 28-91% (purification by prep. HPLC or crystallization).
Though tyrosine analogs 37 and 38 (R2 = tert-butyl and benzyl, respectively) could potentially be synthesized using the approach of Scheme 3, we employed a different method43 for these two compounds, taking advantage of the reactivity of tyrosine toward electrophilic aromatic substitution. Use of tert-butyl alcohol as a source of tert-butyl cation gave almost exclusively regioisomer 37, whereas, the analogous reaction with benzyl alcohol gave a mixture of products, presumably resulting from di- and tri-benzylation.44, 45 Fortunately, the desired product 38 was easily separated from the other isomers by preparative HPLC.
Tyrosine and phenylalanine analogs synthesized in Schemes 1-4, were evaluated in cis-inhibition and trans-stimulation assays (Figure 1).24 Halogen size for phenylalanine derivatives 20–22 correlated with an increase in substrate activity (blue bars, Figure 1), but this trend failed to hold at iodide 23, as a significant decrease was observed (Figure 1a). In contrast, a consistent trend of increasing % inhibition (yellow bars, Figure 1) with size was observed for the halogens. A more noticeable trend in activity was observed for alkyl- and aryl-substituted phenylalanines (24–29). Though methyl (24) was found to be slightly better than hydrogen (i.e. parent Phe), larger, more lipophilic groups (benzyl and phenyl in 28 and 29, respectively) lead to loss of substrate activity, but a rise in % inhibition. cLogP values46 indicate that the lipophilicity of iodo (23: −0.43) lies in between an ethyl (25: −0.53) and isopropyl group (26: −0.13). This order roughly matches what is observed with activity. For comparison sake, compounds 28 and 29 have substantially higher cLogP values (+0.51 and +0.33, respectively), and they both exhibited 98% inhibition of [3H]-gabapentin in the cis assay. IC50 values for compounds 28 and 29 were determined to be 7.3 and 6.6 μM, respectively. This level of transporter inhibition is comparable to lipophilic thyroid hormones (e.g. triiodothyronine), which also demonstrated poor substrate activity that correlated with lipophilicity.25 However, clearly other factors besides lipophilicity impact activity as methyl analog 24's cLogP value (−1.1) lies in between fluoro (20: −1.4) and chloro (21: −0.84), yet 24 had the greatest substrate activity of all the tested compounds (efflux rate = 3.8 fmol/min).
Scheme 4.

Synthesis of compounds 37 and 38. Reagents and conditions: (a) H3PO4, R2OH, 65 °C, 3d (purification by prep. HPLC).
Surprisingly, a different trend was apparent for halogenated tyrosines (30–33, Figure 1b), where the best substrate contained a meta fluoro substituent (30). And there was a decrease in activity going from chloro (31) to bromo (32), with a slight, but significant increase in substrate activity from bromo (32) to iodo (33) that was inversely related to % inhibition. At first glance the observed activities for the halogenated tyrosines appear difficult to rationalize; however, there is a notable correlation of substrate activity with published experimental pKa values for ortho-halophenols (F: 8.73, Cl: 8.51, Br: 8.39, and I: 8.46)47. It is unclear to us what role, if any, tyrosine's phenolic OH might have on interaction with the transporter, and this correlation could be a coincidence. As with the phenylalanine analogs, there was a general trend toward decrease in substrate activity and rise in % inhibition as alkyl and aryl substituents became more lipophilic. One exception to the trend was isopropyl-substituted tyrosine 36, which demonstrated markedly greater substrate activity than the corresponding isopropyl phenylalanine 26. Whether this is due to having reached some optimal balance between the overall polarity of the phenol ring and the size and/or lipophilicity of the isopropyl group (36: cLogP = −1.0) or some other factor is uncertain. Since tyrosine has been shown to be an inferior LAT-1 substrate to phenylalanine in both our experiments and by others20, presumably due to its polar phenolic hydroxyl group causing a drop in Km, we were pleasantly surprised to see that most of the tyrosine derivatives benefited from substitution, with meta-methyltyrosine 34 having comparable activity to the positive control L-leucine (3.3 vs. 3.5 fmol/min, respectively). Compound 38 (R2 = benzyl), one of the poorer tyrosine substrates gave an IC50 value of 9.1 μM, that was comparable in potency to 28 and 29. An attempt to mask the polarity of the phenol hydroxy by converting it to a methoxy group, while maintaining the preferred meta methyl substituent (40, Figure 2) resulted in a slight decrease in substrate activity relative to its comparator 34.
Figure 2.

Effect of replacing tyrosine's phenolic hydroxy with a methoxy group.
It was previously shown that substitution at phenylalanine's meta position resulted in increased LAT-1 activity and greater brain uptake relative to the para position, though the corresponding ortho isomer was not tested.21, 48 To verify this trend for the current series, we tested both the ortho- and para-methylphenylalanine analogs (Figure 3: 41 and 42, respectively). Both isomers had identical efflux rates (2.7 fmol/min), consistent with their being inferior substrates relative to the meta isomer (24: 3.8 fmol/min). Interestingly, the ortho isomer (41) demonstrated the greatest % inhibition (98%) of the three compounds.
We used the newly discovered substrates and inhibitors from Figure 1 to refine our models23, 24 with homology modeling and ligand docking as previously described49 (supplementary data). From this refined model and consideration of our SAR data, we propose that large lipophilic groups such as phenyl in 29 are causing an increase in binding affinity (lower Ki) due to hydrophobic interactions with a subpocket PA (Figure 4) at the expense of decreased transport capacity (lower Vmax) resulting in compounds being inhibitors rather than substrates. Future work will be aimed at identifying functional groups and substitution patterns on amino acid scaffolds to promote a dipole-dipole interaction with residues in the binding site (e.g. with S338) with the idea of finding an optimal balance between Km and Vmax in order to improve substrate activity.
Figure 4.

Predicted mode of interaction of inhibitor 29. Docking pose of inhibitor 29 in the substrate binding site of LAT-1. Inhibitor 29 is predicted to interact with the hydrophobic subpocket PA, which likely contributes to the increased affinity of this compound.
We have demonstrated that meta substitution of tyrosine or phenylalanine with large lipophilic moieties would likely not be beneficial for designing prodrugs, as they would tend to be inhibitors rather than substrates. However, lipophilic amino acids such as 29 might be a starting point for optimization to obtain potent LAT-1 inhibitors that could be useful as cancer probes and possibly as drugs, assuming potential metabolic liabilities with universally inhibiting LAT-1 could be addressed.15, 50 Taken together the SAR trends for these meta substituted aromatic amino acids suggest a correlation of substituent lipophilicity with substrate activity up to some limit, and once passed this limit increasing the lipophilicity causes a reversal in activity, as compounds trend toward being inhibitors rather than substrates. Our observations are consistent with those made earlier by Uchino.25 The definition of this lipophilicity limit likely varies in a complex manner with the properties of other ring substituents and their effect on both substrate Km and Vmax.
Supplementary Material
Acknowledgements
EA, KF, and LH all thank the University of Nebraska at Kearney (UNK) University Research Fellows (URF) Program for financial support. EA also thanks the UNK Summer Student Research Program (SSRP). This work was supported by the UNK Research Services Council (RSC) and the Nebraska EPSCoR Undergraduate Research Experiences (URE) Program (to AAT). We appreciate OpenEye Scientific Software Inc. for granting us access to its high-performance molecular modeling applications through its academic license program. This work was supported in part by the National Institutes of Health grant R01 GM108911 (to AS and CC), by the Department of Defense grant W81XWH-15-1-0539 (to AS and CC), and by the National Institutes of Health's National Institute of General Medical Sciences grant U01 GM61390 (to AAZ, HCC, LL and KMG).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Supplementary data
Full experimental details, compound characterization data, cell assay data in tabular format, IC50 curves, and model refinement and ligand docking description can be found in the online version.
References and Notes
- 1.Gynther M, Laine K, Ropponen J, Leppanen J, Mannila A, Nevalainen T, Savolainen J, Jarvinen T, Rautio J. J. Med. Chem. 2008;51:932. doi: 10.1021/jm701175d. [DOI] [PubMed] [Google Scholar]
- 2.Killian DM, Hermeling S, Chikhale PJ. Drug Deliv. 2007;14:25. doi: 10.1080/10717540600559510. [DOI] [PubMed] [Google Scholar]
- 3.Walker I, Nicholls D, Irwin WJ, Freeman S. Int. J. Pharm. 1994;104:157. [Google Scholar]
- 4.Peura L, Malmioja K, Huttunen K, Leppänen J, Hämäläinen M, Forsberg MM, Rautio J, Laine K. Pharm. Res. 2013;30:2523. doi: 10.1007/s11095-012-0966-3. [DOI] [PubMed] [Google Scholar]
- 5.Rautio J, Gynther M, Laine K. Ther. Deliv. 2013;4:281. doi: 10.4155/tde.12.165. [DOI] [PubMed] [Google Scholar]
- 6.Yanagisawa N, Satoh T, Hana K, Ichinoe M, Nakada N, Endou H, Okayasu I, Murakumo Y. Cancer Biomark. 2015;15:365. doi: 10.3233/CBM-150486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ebara T, Kaira K, Saito J, Shioya M, Asao T, Takahashi T, Sakurai H, Kanai Y, Kuwano H, Nakano T. Anticancer Res. 2010;30:4223. [PubMed] [Google Scholar]
- 8.Takeuchi K, Ogata S, Nakanishi K, Ozeki Y, Hiroi S, Tominaga S, Aida S, Matsuo H, Sakata T, Kawai T. Lung Cancer. 2010;68:58. doi: 10.1016/j.lungcan.2009.05.020. [DOI] [PubMed] [Google Scholar]
- 9.Nawashiro H, Otani N, Uozumi Y, Ooigawa H, Toyooka T, Suzuki T, Katoh H, Tsuzuki N, Ohnuki A, Shima K, Shinomiya N, Matsuo H, Kanai Y. Brain Tumor Pathol. 2005;22:89. doi: 10.1007/s10014-005-0188-z. [DOI] [PubMed] [Google Scholar]
- 10.Kobayashi K, Ohnishi A, Promsuk J, Shimizu S, Kanai Y, Shiokawa Y, Nagane M. Neurosurgery. 2008;62:493. doi: 10.1227/01.neu.0000316018.51292.19. [DOI] [PubMed] [Google Scholar]
- 11.Yazawa T, Shimizu K, Kaira K, Nagashima T, Ohtaki Y, Atsumi J, Obayashi K, Nagamori S, Kanai Y, Oyama T, Takeyoshi I. Am. J. Transl. Res. 2015;7:1126. [PMC free article] [PubMed] [Google Scholar]
- 12.Kaira K, Sunose Y, Arakawa K, Ogawa T, Sunaga N, Shimizu K, Tominaga H, Oriuchi N, Itoh H, Nagamori S, Kanai Y, Segawa A, Furuya M, Mori M, Oyama T, Takeyoshi I. Br. J. Cancer. 2012;107:632. doi: 10.1038/bjc.2012.310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kaira K, Nakamura K, Hirakawa T, Imai H, Tominaga H, Oriuchi N, Nagamori S, Kanai Y, Tsukamoto N, Oyama T, Asao T, Minegishi T. Am. J. Transl. Res. 2015;7:1161. [PMC free article] [PubMed] [Google Scholar]
- 14.Shimizu A, Kaira K, Kato M, Yasuda M, Takahashi A, Tominaga H, Oriuchi N, Nagamori S, Kanai Y, Oyama T, Asao T, Ishikawa O. Melanoma Res. 2015;25:399. doi: 10.1097/CMR.0000000000000181. [DOI] [PubMed] [Google Scholar]
- 15.Yun D-W, Lee SA, Park M-G, Kim J-S, Yu S-K, Park M-R, Kim S-G, Oh J-S, Kim CS, Kim H-J, Kim J-S, Chun HS, Kanai Y, Endou H, Wempe MF, Kim DK. J. Pharm. Sci. 2014;124:208. doi: 10.1254/jphs.13154fp. [DOI] [PubMed] [Google Scholar]
- 16.Rosilio C, Nebout M, Imbert V, Griessinger E, Neffati Z, Benadiba J, Hagenbeek T, Spits H, Reverso J, Ambrosetti D, Michiels JF, Bailly-Maitre B, Endou H, Wempe MF, Peyron JF. Leukemia. 2015;29:1253. doi: 10.1038/leu.2014.338. [DOI] [PubMed] [Google Scholar]
- 17.Ohkawa M, Ohno Y, Masuko K, Takeuchi A, Suda K, Kubo A, Kawahara R, Okazaki S, Tanaka T, Saya H, Seki M, Enomoto T, Yagi H, Hashimoto Y, Masuko T. Biochem. Biophys. Res. Commun. 2011;406:649. doi: 10.1016/j.bbrc.2011.02.135. [DOI] [PubMed] [Google Scholar]
- 18.Shennan DB, Thomson J. Oncol. Rep. 2008;20:885. [PubMed] [Google Scholar]
- 19.Wang Q, Holst J. Am. J. Cancer Res. 2015;5:1281. [PMC free article] [PubMed] [Google Scholar]
- 20.Smith QR. J. Nutr. 2000;130:1016S. doi: 10.1093/jn/130.4.1016S. [DOI] [PubMed] [Google Scholar]
- 21.Peura L, Malmioja K, Laine K, Leppanen J, Gynther M, Isotalo A, Rautio J. Mol. Pharm. 2011;8:1857. doi: 10.1021/mp2001878. [DOI] [PubMed] [Google Scholar]
- 22.Takada Y, Vistica DT, Greig NH, Purdon D, Rapoport SI, Smith QR. Cancer Res. 1992;52:2191. [PubMed] [Google Scholar]
- 23.Schlessinger A, Khuri N, Giacomini KM, Sali A. Curr. Top. Med. Chem. 2013;13:843. doi: 10.2174/1568026611313070007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Geier EG, Schlessinger A, Fan H, Gable JE, Irwin JJ, Sali A, Giacomini KM. Proc. Natl. Acad. Sci. U.S.A. 2013;110:5480. doi: 10.1073/pnas.1218165110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Uchino H, Kanai Y, Kim DK, Wempe MF, Chairoungdua A, Morimoto E, Anders MW, Endou H. Mol. Pharmacol. 2002;61:729. doi: 10.1124/mol.61.4.729. [DOI] [PubMed] [Google Scholar]
- 26.Ylikangas H, Peura L, Malmioja K, Leppanen J, Laine K, Poso A, Lahtela-Kakkonen M, Rautio J. Eur. J. Pharm. Sci. 2013;48:523. doi: 10.1016/j.ejps.2012.11.014. [DOI] [PubMed] [Google Scholar]
- 27.Fraga S, Serrao MP, Soares-da-Silva P. Eur. J. Pharmacol. 2002;441:127. doi: 10.1016/s0014-2999(02)01416-4. [DOI] [PubMed] [Google Scholar]
- 28.Forrest LR, Kramer R, Ziegler C. Biochim. Biophys. Acta. 2011;1807:167. doi: 10.1016/j.bbabio.2010.10.014. [DOI] [PubMed] [Google Scholar]
- 29.Dickens D, Webb SD, Antonyuk S, Giannoudis A, Owen A, Radisch S, Hasnain SS, Pirmohamed M. Biochem. Pharmacol. 2013;85:1672. doi: 10.1016/j.bcp.2013.03.022. [DOI] [PubMed] [Google Scholar]
- 30.Knoer S, Laufer B, Kessler H. J. Org. Chem. 2006;71:5625. doi: 10.1021/jo060704c. [DOI] [PubMed] [Google Scholar]
- 31.Methyl trifluoroborinate pioneered by Molander was not tested, as alternate methods (Scheme 3) were used to prepare the reported compounds.
- 32.Johansson Seechurn CC, Kitching MO, Colacot TJ, Snieckus V. Angew. Chem. Int. Ed. Engl. 2012;51:5062. doi: 10.1002/anie.201107017. [DOI] [PubMed] [Google Scholar]
- 33.Voss ME, Ralph JM, Xie D, Manning DD, Chen X, Frank AJ, Leyhane AJ, Liu L, Stevens JM, Budde C, Surman MD, Friedrich T, Peace D, Scott IL, Wolf M, Johnson R. Bioorg. Med. Chem. Lett. 2009;19:1245. doi: 10.1016/j.bmcl.2008.12.077. [DOI] [PubMed] [Google Scholar]
- 34.Cohen DT, Buchwald SL. Org. Lett. 2015;17:202. doi: 10.1021/ol5032359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sidduri A, Tilley JW, Lou J, Tare N, Cavallo G, Frank K, Pamidimukkala A, Choi DS, Gerber L, Railkar A, Renzetti L. Bioorg. Med. Chem. Lett. 2013;23:1026. doi: 10.1016/j.bmcl.2012.12.026. [DOI] [PubMed] [Google Scholar]
- 36.Karmakar S, Mohapatra DK. Synlett. 2001:1326. [Google Scholar]
- 37.Sivaev IB, Bregadze VI. ARKIVOC (Gainesville, FL, U. S.) 2008:47. [Google Scholar]
- 38.Hydrogenolysis was performed with and without aq. HCl as an additive, and there was no reaction of starting material in either case. Heating to 50 °C with HCl additive resulted in mulitiple products forming, and desired product was not isolated.
- 39.Dexter CS, Jackson RFW, Elliott J. J. Org. Chem. 1999;64:7579. [Google Scholar]
- 40.Jackson RFW, Perez-Gonzalez M. Org. Synth. 2005;81:77. [Google Scholar]
- 41.Though a similar procedure reported by Espejo used CuBr-DMS, we found that this had no effect on the reported Negishi couplings with 15. Additionally we found that use of THF rather than DMF as solvent to form the organic zinc 15 gave considerable (>50%) elimination to the α,β-unsaturated ester.
- 42.Edgar KJ, Falling SN. J. Org. Chem. 1990;55:5287. [Google Scholar]
- 43.Placzek AT, Scanlan TS. Tetrahedron. 2015;71:5946. doi: 10.1016/j.tet.2015.05.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Erickson BW, Merrifield RB. J. Am. Chem. Soc. 1973;95:3750. doi: 10.1021/ja00792a046. [DOI] [PubMed] [Google Scholar]
- 45.Iselin B. Helv. Chim. Acta. 1962;45:1510. [Google Scholar]
- 46.cLogP values calculated using ChemBioDraw Ultra (version 14).
- 47.Jover J, Bosque R, Sales J. QSAR Comb. Sci. 2007;26:385. [Google Scholar]
- 48.Ylikangas H, Malmioja K, Peura L, Gynther M, Nwachukwu EO, Leppanen J, Laine K, Rautio J, Lahtela-Kakkonen M, Huttunen KM, Poso A. ChemMedChem. 2014;9:2699. doi: 10.1002/cmdc.201402281. [DOI] [PubMed] [Google Scholar]
- 49.Schlessinger A, Geier E, Fan H, Irwin JJ, Shoichet BK, Giacomini KM, Sali A. Proc. Natl. Acad. Sci. U.S.A. 2011;108:15810. doi: 10.1073/pnas.1106030108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wempe MF, Rice PJ, Lightner JW, Jutabha P, Hayashi M, Anzai N, Wakui S, Kusuhara H, Sugiyama Y, Endou H. Drug Metab. Pharmacokinet. 2012;27:155. doi: 10.2133/dmpk.dmpk-11-rg-091. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.