

Abstract
Background
Reduced motor and sensory nerve amplitudes in critical illness polyneuropathy (CIP) are characteristic features described in electrophysiological studies and due to dysfunction of voltage-gated sodium channels. Yet, faulty membrane depolarization as reported in various tissues of critically ill patients may cause reduced membrane excitability as well. The aim of this study was to compare the pathophysiological differences in motor nerve membrane polarization and voltage-gated sodium channel function between CIP patients and critically ill patients not developing CIP during their ICU stay (ICU controls).
Methods
ICU patients underwent electrophysiological nerve conduction studies and were categorized as either ICU controls or CIP patients. Subsequently, excitability parameters were recorded as current-threshold relationship, stimulus-response behavior, threshold electrotonus, and recovery of excitability from the abductor pollicis brevis following median nerve stimulation.
Results
Twenty-six critically ill patients were enrolled and categorized as 12 ICU controls and 14 CIP patients. When compared to 31 healthy subjects, the ICU controls exhibited signs of membrane depolarization as shown by reduced superexcitability (p = 0.003), depolarized threshold electrotonus (p = 0.007), increased current-threshold relationship (p = 0.03), and slightly prolonged strength-duration time constant. In contrast, the CIP patients displayed a significantly reduced strength-duration time constant (p < 0.0001), which indicates an increased inactivation of voltage-gated sodium channels.
Conclusions
Abnormal motor nerve membrane depolarization is a general finding in critically ill patients whereas voltage-gated sodium channel dysfunction is a characteristic of CIP patients.
Electronic supplementary material
The online version of this article (doi:10.1186/s40635-016-0083-4) contains supplementary material, which is available to authorized users.
Keywords: Critical illness polyneuropathy, Critical illness myopathy, Motor nerve excitability, Intensive care unit acquired weakness, Sepsis
Background
Critically ill patients with systemic inflammatory response syndrome and multiple organ failure frequently develop muscle weakness due to critical illness myopathy and/or critical illness polyneuropathy (CIP) [1]. This weakness is caused by failure of muscle fibers and motor nerves to generate action potentials [2]. One of the primary affections is loss of membrane excitability [3, 4]. Loss of membrane excitability can either be caused by pathological membrane depolarization or by inactivation of voltage-gated sodium channels. So far, there are no in vivo studies in critically ill patients investigating these mechanisms. In an animal CIP model, inactivation of motor-nerve voltage-gated sodium channels was key in loss of membrane excitability [5]. On the other hand, faulty membrane depolarization was reported in various tissues of critically ill patients including muscle fibers, monocytes, and platelet mitochondria [6–8]. Importantly, motor-neuron excitability testing showed that CIP patients featured membrane depolarization after their discharge from the ICU [9]. However, membrane polarization in motor nerves of critically ill patients has received less attention. Moreover, the relationship between membrane depolarization and motor-nerve excitability in critically ill patients is poorly defined in general and particularly in CIP patients. This study tested the hypothesis that membrane depolarization is a general feature of critically ill patients, whereas inactivation of voltage-gated sodium channels is related to loss of membrane excitability in CIP patients, developing muscle weakness.
Methods
The institutional review board of the Charité approved this study (ISRCTN77569430), and written informed consent of legal proxies was obtained. We screened ICU patients requiring mechanical ventilation on three of five consecutive days within the first week. Conventional nerve conduction studies were performed within 14 days by portable 2-Channel Keypoint Medtronic equipment (Skovlunde, Denmark) [1]. We performed sensory and motor nerve conduction studies using surface electrodes as follows: sensory nerve conduction velocity and sensory nerve action potentials of the sural nerve/median nerve followed by motor nerve conduction velocity and compound muscle action potential after nerve stimulation of the median/peroneal/tibial nerve (neCMAP). Nerve conduction studies were categorized according to the normal values of the neurophysiological laboratory of the Charité. Electromyography was performed to assess spontaneous activity using concentric needle electrodes in the extensor digitorum communis muscle and tibialis anterior muscles. To assess compound muscle action potential after direct muscle stimulation (dmCMAP), we placed a conventional stimulating surface electrode longitudinally over the muscle fibers just proximal to the distal tendon insertion. For recordings of dmCMAP, we used disposable concentric needle electrodes (length 25 or 37 mm; diameter 0.46 mm) and stimulated the muscle with gradually increasing strength (from 10 to 100 mA) using pulses of 0.1 ms in a duration delivered at 1 Hz. The recording electrode was placed 15–50 mm proximal to the stimulating electrode, guided by a muscle twitch. If there was no twitch visible, the recording electrode was placed in four different directions in order not to miss small amplitudes. In cases were no responses were obtained, the muscle was assumed to be inexcitable. The responses evoked were measured peak to peak. Muscle fiber action potentials were recorded using filter settings between 500 Hz and 10 kHz. Whenever possible, we examined the tibialis anterior muscle in the lower limb and the extensor digitorum communis muscle in the upper limb. Critical illness myopathy was diagnosed according to standard criteria, including a dmCMAP of less than <3 mV [10]. CIP was diagnosed according to standard criteria, including reduced motor and sensory nerve amplitudes [10]. Pathological spontaneous activity and reduced neCMAP were classified as unspecific and unable to differentiate between myopathy or neuropathy since it is a typical finding in both features. Patients without any pathologic features in electrophysiological testing consistent with myopathy or polyneuropathy were classified as ICU control.
Patients featuring isolated CIM were not further evaluated. All CIP patients presented critical illness myopathy criteria in electrophysiological assessment as well. Limb temperature was kept at >32 °C during electrophysiological exams and during the excitability test.
An automated protocol (Threshold tracking; Qtrac version 28/10/2009; Institute of Neurology, Queen Square, London, TRONDF) was used to measure excitability parameters (current-threshold relationship, stimulus-response relationship, threshold electrotonus, and recovery of excitability) within 7 to 10 days after ICU admission [11]. This multiple excitability protocol assesses motor-nerve threshold following different conditioning stimuli [12]. The median nerve was stimulated with surface electrodes at the wrist, and motor action potentials were recorded from the abductor pollicis brevis muscle.
The current-threshold relationship (I/V) was measured with 1-ms pulses following sub-threshold polarizing currents of a 200-ms duration, which were altered in steps of 10 % between +50 % (depolarizing) and −100 % (hyperpolarizing) of the control threshold.
Stimulus-response relationship was generated using current impulses of 0.2 and 1 ms. The peak 1-ms response was used to calculate the target response (set at 40 % of the supramaximal CMAP response). The ratio between stimulus-response curves of both stimuli was used to calculate rheobase and strength-duration time constant (SDtc).
Threshold electrotonus (TE) was measured by altering nerve excitability using prolonged sub-threshold polarizing currents of 100 ms duration set at 40 % of the control threshold currents and is defined as threshold changes occurring in response to sub-threshold depolarizing and hyperpolarizing pulses [7, 8]. Finally, TE will pass a phase of sub-excitability following the depolarizing conditioning current (=TEd40 (undershoot)) and a phase of superexcitability following the hyperpolarizing conditioning current (=TEh40 (overshoot)).
The recovery of excitability following a supramaximal conditioning stimulus was tested at 18 conditioning test intervals, decreasing from 200 to 2 ms in geometric progression.
Excitability parameters of critically ill patients were matched with data assessed in 31 healthy subjects (age 36 ± 9.5 years).
Data analysis has been carried out by QtracP software (Qtrac version 28/10/2009; Institute of Neurology, Queen Square, London), additionally using the modeling software “MEMFIT” included in the QtracP software by Bostock and colleagues to simulate the threshold tracking data [13–15]. Lab results were recorded during threshold tracking assessments in the ICU. Potassium administration over the first 2 weeks was averaged as means per day.
Physical examination of muscle strength was conducted using the Medical Research Council (MRC) score (range: 0 = no muscle contraction to 5 = normal strength) at ICU discharge [16]. Whenever possible, we examined three muscles in each limb, including the triceps, biceps brachii, and extensor digitorum muscles in the upper limbs and rectus femoris, the tibialis anterior as well as the gastrocnemius muscles in the lower limbs, respectively. In case of hindered circumstances (e.g., unilateral or central paresis, bone fractures, or fixateur externe), the affected muscles where not included in our MRC results. MRC scores are presented as average muscle strength of the muscles examined.
Statistical analysis tests were computed by SPSS, Version 19, Copyright© SPSS, Inc., Chicago, IL 60606, USA. We conducted non-parametric tests using the Mann-Whitney test for two independent samples, Kruskal-Wallis test for three or more independent samples, and Fisher’s test (chi-square test) for qualitative data. In case of small samples, greater differences in sample sizes, large but unbalanced groups, data sets containing ties, or sparse data, tests were carried out in an exact version. For analysis of correlations between lab parameters and excitability, we used the Spearman correlation coefficients.
Results
Eight hundred seventy-four mechanically ventilated patients with a Sequential Organ Failure Assessment (SOFA) score ≥8 for three consecutive days in the first 5 days on ICU were screened in this prospective observational study. Eight hundred forty-one patients did not meet the inclusion criteria, and four patients were excluded from further analysis since they featured isolated CIM (Fig. 1). We enrolled and classified 26 ICU patients as either ICU controls (n = 12) or CIP patients (n = 14). Patient’s characteristics are shown in Table 1. Muscle strength was significantly reduced in the CIP patients compared to the ICU controls (p = 0.01) (Table1).
Fig. 1.
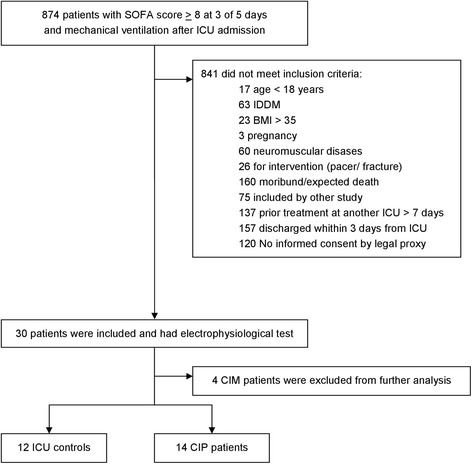
Study flow chart
Table 1.
Clinical characteristics
| ICU controls (n = 12) | CIP patients (n = 14) | p value | |
|---|---|---|---|
| Age (years) | 48.4 (11.4) | 53.2 (14.8) | 0.193 |
| Gender (m/f) | 8 / 4 | 11 / 3 | 0.66 |
| BMI (kg/m2) on admission | 29.6 (4.4) | 27.0 (4.7) | 0.25 |
| Diagnosis on admission: | |||
| ALI/ARDS | 2 | 10 | 0.011 |
| Sepsis | – | 2 | |
| Intracranial bleeding | 7 | 1 | |
| Multiple trauma | 2 | 1 | |
| Severe cardiac dysfunction/after resuscitation | 1 | – | |
| SAPS-II on admission | 39.9 (14.5) | 56.4 (17.8) | 0.027 |
| SOFA max. within ICU stay | 11.4 (4) | 13.2 (3.9) | 0.179 |
| MRC score (mean of 12 muscles assessed) at ICU discharge | 4.7 (0.4) | 3.1 (0.2) | 0.01 |
| ICU survival (yes/no) | 11/1 | 8/5 | 0.16 |
| ICU length of stay (days) | 27.3 (10.8) | 42.2 (30.5) | 0.347 |
p value compares ICU control patients versus CIP patients, (Mann-Whitney U/Fischer’s exact test). Values are shown as mean (SD) or as absolute numbers/%
BMI body mass index, ARDS acute respiratory distress syndrome, ALI acute lung injury, SAPS-II simplified acute physiology score, SOFA Sequential Organ Failure Assessment, MRC medical research council, ICU length of stay intensive care unit length of stay.
Significant differences of excitability parameters (Table 2) between the ICU controls and healthy subjects [reduced: superexcitability %, (p = 0.003); TEd40(10–20ms), (p = 0.007); S240 accommodation, (p = 0.006)] indicated membrane depolarization in the ICU controls. When compared to the healthy subjects, the CIP patients showed a different pathology [reduced: superexcitability %, (p < 0.0001); TEd40(10–20ms); (p < 0.0001); S240 accommodation, (p < 0.0001); TEh40(10–20 ms), (p = 0.028); late sub-excitability, (p < 0.0001); SDtc, (p < 0.0001)], which coincides with an inactivation of voltage-gated sodium channels. Significant differences between the ICU controls and the CIP patients were observed for SDtc (p = 0.03) and TEd40(10–20ms) (p = 0.002); S240 accommodation (p = 0.021) (Fig. 2a, b).
Table 2.
Excitability parameters
| Healthy subjects (n = 31) | ICU controls (n = 12) | CIP patients (n = 14) | p a value | p b value | p c value | |
|---|---|---|---|---|---|---|
| Temperature °C | 33.94 (1.3) | 34.5 (1.8) | 34.8 (1.5) | 0.399 | 0.075 | 0.705 |
| (1) Current/threshold relationship | ||||||
| Resting I/V slope | 0.58 (0.01) | 0.66 (0.04) | 0.63 (0.03) | 0.03 | 0.239 | 0.728 |
| Minimum I/V slope | 0.26 (0.01) | 0.28 (0.02) | 0.26 (0.02) | 0.536 | 0.781 | 0.406 |
| Hyperpol. I/V slope | 0.4 (0.01) | 0.31 (0.05) | 0.44 (0.08) | 0.225 | 0.263 | 0.079 |
| (2) Stimulus response/strength duration | ||||||
| Stimulus 50 % (mA) | 4.66 (0.27) | 5.52 (1.06) | 8.6 (1.21) | 0.282 | 0.001 | 0.075 |
| SDtc (ms) | 0.51 (0.02) | 0.56 (0.08) | 0.4 (0.07) | 0.759 | <0.0001 | 0.03 |
| Rheobase (mA) | 3.07 (0.22) | 3.97 (0.58) | 6.1 (0.87) | 0.081 | 0.002 | 0.095 |
| Stimulus-response slope | 6.39 (0.29) | 4.51 (0.51) | 4.61 (0.38) | 0.004 | 0.003 | 1 |
| (3) Threshold electrotonus (%) | ||||||
| TEd40 (10–20 ms) | 69.97 (1.69) | 64.8 (2.04) | 44.7 (11.1) | 0.007 | <0.0001 | 0.002 |
| TEh40 (10–20 ms) | −74.15 (1.76) | −76.92 (2.67) | −66.65 (3.74) | 0.841 | 0.028 | 0.051 |
| S240 accommodation | 24.99 (1.03) | 20.1 (1.33) | 14.77 (1.97) | 0.006 | <0.0001 | 0.021 |
| TEd40 (90–100 ms) | 45.19 (1.5) | 43.59 (1.64) | 44.29 (2.58) | 0.123 | 0.17 | 0.932 |
| TEh40 (90–100 ms) | −123.42 (3.37) | −121.91 (8.76) | −110.96 (7.61) | 0.862 | 0.222 | 0.379 |
| TEh40 overshoot | 15.88 (0.84) | 14.64 (1.42) | 9.39 (1.12) | 0.342 | <0.0001 | 0.004 |
| TEd40 undershoot | −18.75 (0.74) | −17.93 (1.38) | −11.58 (1.64) | 0.752 | 0.001 | 0.009 |
| (4) Recovery of excitability | ||||||
| RPR (ms) | 3.06 (0.08) | 2.98 (0.45) | 3.06 (0.18) | 0.874 | 0.8 | 0.968 |
| Superexcitability (%) | −25.69 (1.09) | −17.13 (2.74) | −14.39 (2.05) | 0.003 | <0.0001 | 0.403 |
| Superexcitability at 7 ms | −23.92 (1.19) | −15.7 (2.41) | −11.05 (1.8) | 0.001 | <0.0001 | 0.12 |
| Superexcitability at 5 ms | −26.4 (1.17) | −17.3 (2.85) | −14.46 (2.26) | 0.004 | <0.0001 | 0.501 |
| Sub-excitability (%) | 14.78 (0.91) | 11.8 (1.7) | 9.04 (0.92) | 0.114 | <0.0001 | 0.106 |
Values are given as mean ± SE
aHealthy subjects versus ICU control
bHealthy subjects versus CIP patients
cICU control versus CIP patients
Fig. 2.
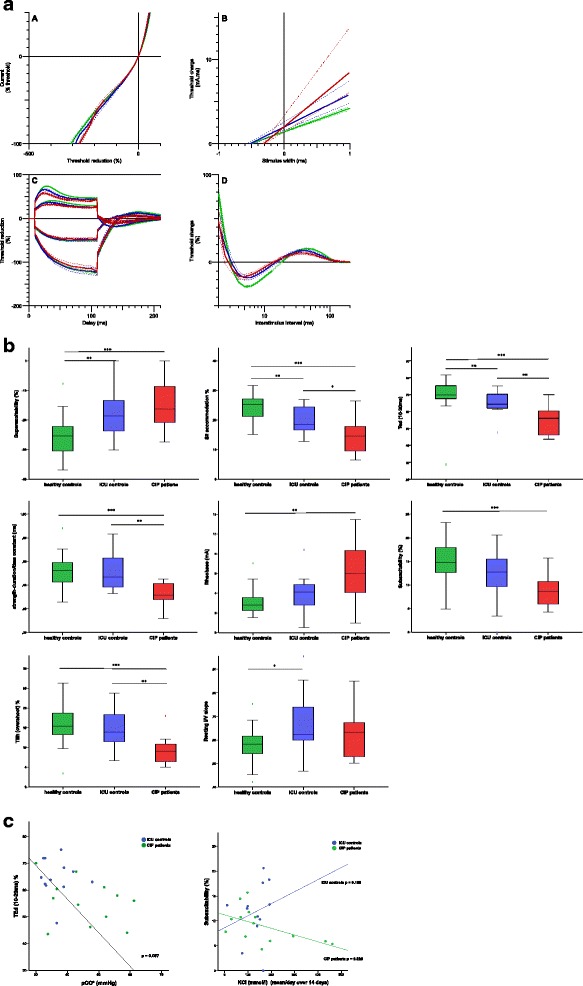
a Mean excitability data ± SE for 31 healthy subjects (n = 31, green curve), ICU controls (n = 12, blue curve), and CIP patients (n = 14, red curve). (1) I/V slope. (2) Stimulus-response relationship. (3) Threshold electrotonus. (4) Excitability recovery. b Correlation between healthy controls (n = 31, green boxes), ICU controls (n = 12, blue boxes), and CIP patients (n = 14, red boxes) for superexcitability %, TEd(10–20 ms), S2 accommodation, resting I/V slope, strength-duration time constant, rheobase, sub-excitability, and TEh (overshoot). Differences between groups are indicated as *p < 0.05; **p < 0.01; ***p < 0.0001. c Correlation between laboratory parameters and excitability parameters in ICU controls (n = 12) and CIP patients (n = 14). pCO2 and TEd(10–20 ms) (p = 0.007). KCl mmol/l infusion rate mean/day for the first 14 days on ICU and sub-excitability. Significant correlation in CIP patients (p = 0.026), contrary to ICU control patients (p = 0.159)
In the CIP patients, pCO2 (46.4 ± 9.8 versus 36.7 ± 4.8 mmHg; p = 0.009), HCO3 (29.4 ± 6.2 versus 23.8 ± 2.5 mmol/l; p = 0.031), and lactate (12.6 ± 4.8 versus 8.1 ± 2.6 mg/dl; p = 0.006) levels were significantly higher than in those of the ICU controls. Regarding all ICU patients, elevated pCO2 (p = 0.007) and lactate (p = 0.016) levels were significantly correlated with membrane depolarization [reduced TEd(10–20 ms), S240 accommodation] (Table 3/Fig. 2c). On average, the mean KCl administration was 131 mmol/l in ICU controls and 161 mmol/l in CIP patients (Additional file 1: Table S2). CIP showed a statistically significant negative correlation between reduced sub-excitability and elevated KCl administration. In contrast, ICU controls showed a direct correlation between increased sub-excitability and elevated KCl administration (Fig. 2c).
Table 3.
Correlation between selected excitability parameters and lab results
| pCO2 mmHg | HCO3 mmol/l | Lactate mg/dl | K mmol/l | Na mmol/l | |
|---|---|---|---|---|---|
| SDtc (ms) | 0.075 | 0.41 | 0.102 | 0.993 | 0.543 |
| Rheobase (mA) | 0.203 | 0.875 | 0.066 | 0.097 | 0.728 |
| TEd40 (10–20 ms) | 0.007** | 0.2 | 0.016* | 0.58 | 0.924 |
| S240 accommodation | 0.175 | 0.215 | 0.013* | 0.475 | 0.072 |
| Superexcitability % | 0.832 | 0.533 | 0.396 | 0.348 | 0.087 |
| Late sub-excitability % | 0.165 | 0.066 | 0.664 | 0.796 | 0.147 |
SD tc strength duration time constant, TE threshold electrotonus, RRP relative refractory period, HCO 3 bicarbonate
* p values < 0.05; ** p values < 0.01
To help interpret these different changes in critically ill patients, we ran the modeling software MEMFIT included in the QtracP software [12]. Using MEMFIT, sodium currents of motor axons were modeled using the voltage clamp data of Schwarz and colleagues [13]. Further empirical parameter adjustments were made to improve the fit of recovery of excitability, SDtc, TE, and current-threshold-relationship in the healthy controls [14]. We ran MEMFIT to get the best fit of data recorded in the ICU controls and the CIP patients. Excitability measurements of ICU controls where best modeled by a two fold increase of fast K+ currents (Additional file 2: Figure S1/Table 4), whereas in the CIP patients, the differences in comparison to healthy controls were best modeled by a twofold reduction of Na+ permeability (Additional file 3: Figure S2/Table 4).
Table 4.
MEMFIT results modeling excitability parameters for ICU controls and CIP patients
| ICU controls (%) | CIP patients (%) | |
|---|---|---|
| P Na p (%) (percent of persistent Na) | 51.87 | 80.4 |
| P Na N (nodal sodium permeability) | 46.37 | 72.49 |
| G Kf l (internodal fast K conductance) | 65.89 | 74.217 |
| G Kf N (nodal fast K conductance) | 57.71 | 73.52 |
| G Ks N (nodal slow K conductance) | 51.56 | 53.2 |
| I pump NI (pump currents) | 31.07 | 11.6 |
MEMFIT results modeling ICU controls and CIP patients excitability parameters showing discrepancy in percentage for sodium and potassium currents between healthy controls and ICU controls as well as healthy controls and CIP patients (please notice that in the modeling model, only the primary change is reliable)
Discussion
By performing motor-nerve excitability tests in critically ill patients during their early ICU stay, we are the first group to demonstrate in vivo-reduced membrane excitability related to Na+ channel inactivation in CIP patients. Contrary, the ICU control patients without CIP showed motor axon membrane depolarization.
Reduced membrane excitability in CIP patients during their ICU stay
Membrane depolarization as indicated by reduced superexcitability, TE, and elevated SDtc in motor nerve excitability testing has been proposed as the primary, pathological sign in the CIP patients after ICU discharge [9]. In contrast, we observed significantly reduced SDtc [beside reduced superexcitability, TE, and sub-excitability] indicating reduced membrane excitability. SDtc depends on passive membrane properties, as well as on voltage-dependent Na+ conductance [17, 18]. Possible explanations for reduced SDtc in motor axons could be structural changes, membrane hyperpolarization, or persistently decreased Na+ conductance [11, 17, 18]. Structural changes of the motor neurons are not likely, since nerve conduction velocity was normal and since it has been reported that biopsies of sensory nerves obtained from patients with CIP show no structural abnormalities [19]. Membrane hyperpolarization was not evident. We suggest that a decrease in persistent Na+ conductance accounts for the reduction of SDtc in CIP patients. This hypothesis is confirmed by modeling our excitability data for CIP patients. The twofold decrease of Na+ current in CIP patients is in line with a nerve excitability study of patients with puffer fish—TTX [Na+ channel blocker] intoxication [17]—where a decrease of Na+ currents by a factor of two accounted for reduced superexcitability, sub-excitability, and TE and increased SDtc.
Our study confirms that dysfunction of voltage-gated Na+ channels are involved in the pathomechanism of reduced motor nerve membrane excitability in CIP patients. This hypothesis is supported by an animal model of CIP describing increased inactivation of voltage-gated sodium channels as an important contributor to reduced excitability [3]. Voltage-gated Na+ channel inactivation can be impaired by oxidative stress and endotoxins [20, 21], which coincides with significant elevated pCO2 levels in our CIP patients group compared to the ICU controls.
Our study also confirms that increased inactivation of voltage-gated Na+ channels shown in an animal model of CIP [3] causes reduced membrane excitability, which is a trigger for the development of muscle weakness [2].
Reduced TEd(10–20 ms) and S240 accommodation indicate membrane depolarization and were correlated with elevated blood lactate concentrations in all patients. As increased blood lactate concentration is a marker for illness severity in critically ill patients [22], our results are in line with studies showing that membrane depolarization in critically ill patients is directly correlated with severity of illness [6–8]. However, further studies are needed to elucidate the molecular mechanisms of our observations.
In contrast to our study, Z’Graggen and colleagues observed membrane depolarization of motor nerves in CIP patients [9]. However, these patients were examined 2–3 weeks after their ICU stay. According to the study of Haeseler and colleagues, dysfunction of sodium channels is related to endotoxin levels (LPS), yet patients in the post intensive care period should feature endotoxin levels close to zero [21]. Moreover, dysfunction of sodium channels has been reported to be associated with oxidative stress. Likewise, oxidative stress should be resolved after the ICU stay [20]. These important differences explain the different findings in the study of Z’Graggen and our study.
Early changes in membrane excitability during the ICU stay are very likely and have been shown in muscle as early as 6 h after the onset of porcine fecal peritonitis [23]. However, due to different excitability examination setups in muscle, it was impossible to differentiate between sodium channel dysfunction and/or membrane depolarization.
K+ currents, sub-excitability, and membrane depolarization
According to the MEMFIT model, an increase in fast K+ currents causes the changes in the ICU controls. In the healthy subjects, elevated [K+]o causes an increase of K+ current over the membrane, as demonstrated by a decrease in superexcitability [24, 25]. Late sub-excitability is the best indicator of [K+]o from nerve excitability measurements, owing to the activation of slow K+ channels during the action potential [12]. But interestingly, even though K+ currents are elevated according to the model, there is reduced sub-excitability in critically ill patients. Furthermore, we did not find a correlation between [K+]o and superexcitability or late sub-excitability. Interestingly, elevated KCl infusion rates were correlated to reduced late sub-excitability (p = 0.026) in the CIP patients, the opposite of what we observed in the ICU controls and what has been shown in the healthy subjects [25]. These paradox findings may be related to instable [K+]o plasma levels in critically ill patients requiring potassium substitution in order to keep [K+]o plasma concentration within the normal range. Further, it needs to be mentioned, that changes in sub-excitability/superexcitability are related to local potassium levels which may be different from local potassium levels in healthy subjects due to poor microcirculation in critically ill patients [26]. The inverse relation between the ICU controls and the CIP patients may be related to the significantly elevated pCO2 concentrations in CIP patients which will cause higher intracellular H+ levels and could finally induce reduced K+ current. It has been shown that prolonged ischemia causes membrane depolarization in sensory nerves, correlating with a “paradoxical” reduction of K+ currents by increasing intracellular acidosis [27]; however, those findings are still a matter of discussion and need further exploration.
Limitations
Our study has limitations. All of our CIP patients featured concomitant critical illness myopathy, which may have influenced excitability results in motor axons. However, since conditioning currents applied in stimulus-response and current-threshold relationships and TE assessments would not be sensed by the innervated muscle fibers, these data should not have been affected by concomitant myopathy. Due to the explorative and non-confirmatory study design, we avoid to report a post hoc power analysis in the paper, as recommended by Hoenig and colleagues [28].
Conclusions
We confirmed previous in vitro findings in vivo, showing that inactivation of motor axon voltage-gated Na+ channels is the primary contributor of muscle weakness in CIP patients. Additionally, we provided evidence that abnormal membrane depolarization in motor axons is a general finding in ICU patients corresponding to illness severity. Moreover, we observed a paradoxical membrane depolarization related to K+ equilibrium that could be related to intracellular acidosis; however, this still needs to be evaluated in future studies.
Acknowledgements
We thank our patients and their consenting relatives. The Deutsche Forschungsgemeinschaft (DFG/KFO 192) supported this work. SWC and SK received support from the Deutsche Gesellschaft für Anästhesiologie und Intensivmedizin (DGAI). SWC and JF received funding from the Berlin Institute of Health (BIH), Twinning Research Grant (TRG 3).
Authors’ contributions
SK, JB, SWC, CS, and JF conceived and designed the experiments. SK, JB, KH, and SW performed the experiments. SK, JB, and JG analyzed the data. SWC and CS contributed to the materials/analysis tools. SK, JB, JF, FL and JG wrote the paper. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Tweet
Reduced motor nerve excitability in critical illness polyneuropathy is due to inactivation of Nav channels in vivo.
Take-home message
Patients with critical illness polyneuropathy feature reduced motor nerve excitability due to inactivation of Nav channels which is related to the development of muscle weakness. Abnormal motor nerve membrane depolarization is a general finding in critically ill patients and is correlated with illness severity.
Additional files
Electrophysiological data. neCMAP, nerve evoked compound muscle action potential amplitude; SNAP, sensory nerve action potential amplitude; dmCMAP, direct muscle evoked compound muscle action potential amplitude. p value compares ICU controls versus CIP patients (Mann-Whitney U). Values are given as mean ± SD. Table S2. Laboratory data p value compares ICU controls with CIP patients. pCO, partial arterial pressure of carbon dioxide; pO2, partial arterial pressure of oxygen; HCO3, bicarbonate; Na, sodium; K, potassium; Ca, calcium. (DOC 52 kb)
MEMFIT data showing best fit of current changes in % for ICU controls. GKfl, internodal fast K conductance; GKfN, nodal fast K conductance; GKsN, nodal slow K conductance, PNa p (%), percent of persistent Na; P Na N, nodal sodium permeability; GLkN, nodal leak conductance; IPumpNI, pump currents; GKsI internodal slow K conductance). (TIF 104 kb)
MEMFIT data showing best fit of current changes in % for critical illness polyneuropathy patients. GKfl, internodal fast K conductance; GKfN, nodal fast K conductance; GKsN, nodal slow K conductance; PNa p (%), percent of persistent Na; P Na N, nodal sodium permeability; GLkN, nodal leak conductance; IPumpNI, pump currents; GKsI, internodal slow K conductance). (TIF 112 kb)
References
- 1.Koch S, Spuler S, Deja M, et al. Critical illness myopathy is frequent: accompanying neuropathy protracts ICU discharge. JNNP. 2011;82:287–293. doi: 10.1136/jnnp.2009.192997. [DOI] [PubMed] [Google Scholar]
- 2.Weber-Carstens S, Koch S, Spuler S, Spies C, Bubser F, Wernecke K, Deja M. Non-excitable muscle membrane predicts ICU-acquired paresis in mechanically ventilated, sedated patients. Crit Care Med. 2009;37(9):2632–2637. doi: 10.1097/CCM.0b013e3181a92f28. [DOI] [PubMed] [Google Scholar]
- 3.Novak KR, Nardelli P, Cope TC, et al. Inactivation of sodium channels underlies reversible neuropathy during critical illness in rats. J Clin Invest. 2009;119:1150–1158. doi: 10.1172/JCI36570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rich MM, Bird SJ, Raps EC, et al. Direct muscle stimulation in acute quadriplegic myopathy. Muscle Nerve. 1997;20:665–73. doi: 10.1002/(SICI)1097-4598(199706)20:6<665::AID-MUS2>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- 5.Filatov G, Pinter MJ, Rich MM. Resting potential-dependent regulation of the voltage sensitivity of sodium channel gating in rat skeletal muscle in vivo. J Gen Physiol. 2005;126:161–172. doi: 10.1085/jgp.200509337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cunningham JN, Carter NW, Rector FC, Seldin DW. Resting transmembrane potential difference of skeletal muscle in normal subjects and severely ill patients. J Clin Invest. 1971;50:49–59. doi: 10.1172/JCI106483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Adrie C, Bachelet M, Vayssier-Taussat M, et al. Mitochondrial membrane potential and apoptosis peripheral blood monocytes in severe human sepsis. Am J Respir Crit Care Med. 2001;164:389–395. doi: 10.1164/ajrccm.164.3.2009088. [DOI] [PubMed] [Google Scholar]
- 8.Yamakawa K, Ogura H, Koh T, et al. Platelet mitochondrial membrane potential correlates with severity in patients with systemic inflammatory response syndrome. J Trauma Acute Care Surg. 2013;74:411–418. doi: 10.1097/TA.0b013e31827a34cf. [DOI] [PubMed] [Google Scholar]
- 9.Z’Graggen WJ, Lin CS, Howard RS, Beale RJ, Bostock H. Nerve excitability changes in critical illness polyneuropathy. Brain. 2006;129:2461–2470. doi: 10.1093/brain/awl191. [DOI] [PubMed] [Google Scholar]
- 10.Latronico B. Critical illness polyneuropathy and myopathy: a major cause of muscle weakness and paralysis. Lancet Neurol. 2001;10:931–941. doi: 10.1016/S1474-4422(11)70178-8. [DOI] [PubMed] [Google Scholar]
- 11.Bostock H, Cikurel K, Burke D. Threshold tracking techniques in the study of human peripheral nerve. Muscle Nerve. 1998;21:137–158. doi: 10.1002/(SICI)1097-4598(199802)21:2<137::AID-MUS1>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- 12.Kiernan MC, Bostock H. Effects of membrane polarization and ischaemia on the excitability properties of human motor axons. Brain. 2000;123:2542–2551. doi: 10.1093/brain/123.12.2542. [DOI] [PubMed] [Google Scholar]
- 13.Bostock H, Baker M, Reid G. Changes in excitability of human motor axons underlying post-ischaemic fasciculations: evidence for two stable states. J Physiol (Lond) 1991;441:537–557. doi: 10.1113/jphysiol.1991.sp018766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schwarz JR, Reid G, Bostock H. Action potentials and membrane currents in the human node of Ranvier. Pflugers Arch. 1995;430:382–392. doi: 10.1007/BF00374660. [DOI] [PubMed] [Google Scholar]
- 15.Bostock H, Rothwell JC. Latent addition in motor and sensory fibres of human peripheral nerve. J Physiol (Lond) 1997;498:277–294. doi: 10.1113/jphysiol.1997.sp021857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.British Medical Research Council . Aids to the examination of the peripheral nervous system. Memorandum No. 45. London: Her majesty’s Stationery Office; 1976. [Google Scholar]
- 17.Kiernan MC, Isbister G, Lin C, Burke D, Bostock H. Acute tetrodotoxin-induced neurotoxicity after ingestion of puffer fish. Ann Neurol. 2005;57:339–348. doi: 10.1002/ana.20395. [DOI] [PubMed] [Google Scholar]
- 18.Kanai K, Kuwabara S, Arai K, Sung JY, Ogawara K, Hattori T. Muscle cramp in Machado-Joseph disease. Altered motor axonal excitability properties and mexiletine treatment. Brain. 2003;126:965–973. doi: 10.1093/brain/awg073. [DOI] [PubMed] [Google Scholar]
- 19.Latronico N, Fenzi F, Recupero D, Guarneri B, Tomelleri G, Tonin P, De Maria G, Antonini L, Rizzuto N, Candiani A. Critical illness myopathy and neuropathy. Lancet. 1996;347:1579–1582. doi: 10.1016/S0140-6736(96)91074-0. [DOI] [PubMed] [Google Scholar]
- 20.Kassmann M, Hansel A, Leipold E, et al. Oxidation of multiple methionine residues impairs rapid sodium channel inactivation. Pflugers Arch. 2008;456(6):1085–1095. doi: 10.1007/s00424-008-0477-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Haeseler G, Foadi N, Wiegand E, et al. Endotoxin reduces availability of voltage-gated human skeletal muscle sodium channels at depolarized membrane potentials. Crit Care Med. 2008;36:1239–1247. doi: 10.1097/CCM.0b013e31816a02cf. [DOI] [PubMed] [Google Scholar]
- 22.Levy B, Gibot S, Franck P, Cravaisy A, Ballaert PE. Relation between muscle Na+K+ ATPase activity and raised lactate concentrations in septic shock: a prospective study. Lancet. 2005;365:871–875. doi: 10.1016/S0140-6736(05)71045-X. [DOI] [PubMed] [Google Scholar]
- 23.Ackermann KA, Bostock H, Brander L, et al. Early changes of muscle membrane properties in porcine faecal peritonitis. Crit Care. 2014;18:484. doi: 10.1186/s13054-014-0484-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kiernan MC, Walters RJL, Andersen KV, Taube D, Murray NM, Bostock H. Nerve excitability changes in chronic renal failure indicate membrane depolarization due to hyperkalaemia. Brain. 2002;125:1366–1378. doi: 10.1093/brain/awf123. [DOI] [PubMed] [Google Scholar]
- 25.Boerio D, Bostock H, Spescha R, Z’Graggen WJ. Potassium and the excitability properties of normal human motor axons in vivo. PLOSOne. 2014;9(6):e98262. doi: 10.1371/journal.pone.0098262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Beal AL, Deuser WE, Beilman GJ. A role for epinephrine in post-traumatic hypokalemia. Shock. 2007;27(4):358–363. doi: 10.1097/01.shk.0000245029.47106.db. [DOI] [PubMed] [Google Scholar]
- 27.Grosskreutz J, Lin C, Mogyoros I, Burke D. Changes in excitability indices of cutaneous afferents produced by ischaemia in human subjects. J Physiol. 1999;518(1):301–314. doi: 10.1111/j.1469-7793.1999.0301r.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hoenig JM, Heisey DM. The abuse of power. Am Stat. 2001;55(1):19–24. doi: 10.1198/000313001300339897. [DOI] [Google Scholar]