

Abstract
Background
Hulless barley, also called naked barley, is an important cereal crop worldwide, serving as a healthy food both for human consumption and animal feed. Nevertheless, it often suffered from drought stress during its growth and development, resulting in a drastic reduction in barley yields. Therefore, study on molecular mechanism of hulless barley drought-tolerance is very important for increasing barley production. To investigate molecular mechanism of barley drought-resistance, this study examined co-regulated mRNAs that show a change in expression pattern under early well water, later water deficit and finally water recovery treatments, and to identify mRNAs specific to water limiting conditions.
Results
Total of 853 differentially expressed genes (DEGs) were detected and categorized into nine clusters, in which VI and VIII were apparently up-regulated under low relative soil moisture content (RSMC) level. The majority of genes in these two clusters was relevant to abiotic stress responses in abscisic acid (ABA) dependent and independent signaling pathway, including NCED, PYR/PYL/RCAR, SnRK2, ABF, MYB/MYC, AP2/ERF family, LEA and DHN. In contrast, genes within clusters II and IV were generally down-regulated under water stress; cluster IX genes were up-regulated during water recovery response to both low and high RSMC levels. Genes in implicated in tetrapyrrole binding, photosystem and photosynthetic membrane were the most affected in cluster IX.
Conclusion
Taken together, our findings indicate that the responses of hulless barley to drought stress shows differences in the pathways and genes activated. Furthermore, all these genes displayed different sensitivities to soil water deficit and might be profitable for future drought tolerance improvement in barley and other crops.
Electronic supplementary material
The online version of this article (doi:10.1186/s12864-016-2685-3) contains supplementary material, which is available to authorized users.
Keywords: Hordeum vuglare, DGE, Transcriptome sequencing, Drought-responsive genes, ABA, Tetrapyrrole
Background
Hulless barley (Hordeum vuglare L. var. nudum Hook. f.) is one of the most important crops in China, especially in Tibet Plateau, for over half of the total food production. With caryopses that thresh free from the pales, hulless (naked) barley provides an attractive advantage for the human consumption [1]. It is cultivated in the valleys and in the higher land on Tibet. Drought-induced water deficit greatly affects crop growth and development, and endangers crop agronomic yields. Crops have also formed various molecular and physiological changes to prevent water deficit. Drought tolerance crops maintain turgor and continue metabolism in cells even at low water potential, mainly by protoplasmic tolerance, synthesis of osmolytes or/and compatible solutes [2]. Signal transduction molecules play important roles in this process by mediating the transmission of the stress signals via complex signal transduction pathways. Numerous fundamental molecular aspects of tetrapyrroles and abscisic acid (ABA) are available [3–5]. Tetrapyrroles are the active cores of some compounds with crucial biochemical roles in living systems, such as chlorophyll, heme, siroheme and phytochromobilin [6]. Heme biosynthesis is transcriptionally responsive to reactive oxygen species (ROS)-mediated stress signaling in Arabidopsis [7]. ABA is a vital stress-responsive phytohormone sensitive to these cellular changes, particularly to the loss of turgor [8]. The ABA signal transduction pathway comprises the ABA-bound pyrabactin resistance/regulatory component of ABA receptor (PYR/RCAR) proteins [9], type 2C protein phosphatases (PP2C) [10], and SNF1-related kinases (SnRK2) [11], NCED [12], ABF [13], MYB/MYC [14], AP2/ERF family [15], LEA [16] and DHN [17]. In Arabidopsis, the molecular drought response mechanism can be divided into ABA-dependent and ABA-independent pathways [18]. In soybean, ABA treatment influences the expression of drought response genes [19].
Breeding for drought tolerance is particularly challenging because of the genetic complexity of this trait. On the one hand, hulless barley has a complex diploid genome (HH), with the genome size of 5000 Mb, larger than that of human [20]. On the other hand, drought tolerance has been well documented to result from cooperative interactions among multiple morphological, physiological, and biochemical characters. Different genotypes may have diverse responses to drought stress [18, 21, 22]. Therefore, efficient improvement requires an indepth understanding of the gene expression regulation mechanisms in response to drought stress.
Many researchers have analyzed Tibetan hulless barley, and many genes associated with drought stress responses in plants are known. Honsdorf et al. [23] has detected the drought tolerance QTL in wild barley, while Chen et al. [24] has analyzed grain development and nutrient storage in Tibetan hulless barley. However, little has been known about display the changes of gene expression in Tibetan hulless barley during the whole drought response process. In the study, we used transcriptome-seq to identify differentially expressed genes in response to drought stress in leaf tissue of a Tibetan hulless barley drought-resistibility cultivar, which were grown in different levels of relative soil moisture content (RSMC) and water recovery. This analysis serves as a reference for future studies on Tibetan hulless barley response to various stresses, such as to drought, cold and salt.
Results
Analysis of transcriptome-seq data
In the present study, cDNA libraries were constructed from leaves harvested at eight days after drought stress and during rehydration with three biological replicates, and then sequenced using the Illumina HiSeq™ 2000 platform. After cleaning and checking the read quality, we obtained almost 53.99 billion 200 bp paired-end clean reads. Among the clean reads, 100 % had quality scores at the Cycle Q20 level (a base quality greater than 20 and an error probability of 0.01). The statistics of sequencing sample and data were in Table 1.
Table 1.
Statistics of transcriptome sequencing data in different samples
| Genotype | Samples | Raw Reads (M) | Clean Reads (M) | Q20 (%) | GC (%) |
|---|---|---|---|---|---|
| Himalaya 10 | A1 | 75.61 | 67.79 | 96.64 | 55.83 |
| Himalaya 10 | B2 | 75.11 | 67.95 | 96.82 | 54.84 |
| Himalaya 10 | C3 | 75.84 | 64.22 | 95.22 | 55.57 |
| Himalaya 10 | D4 | 75.15 | 66.79 | 96.74 | 55.45 |
| Himalaya 10 | E5 | 73.23 | 65.40 | 96.69 | 56.21 |
| Himalaya 10 | F6 | 75.41 | 69.19 | 97.53 | 54.47 |
| Himalaya 10 | G7 | 74.46 | 68.07 | 97.49 | 55.49 |
| Himalaya 10 | H8 | 73.41 | 66.20 | 97.37 | 56.19 |
| Himalaya 10 | I9 | 69.88 | 64.24 | 97.37 | 56.63 |
The clean reads of each sample (A1-I9) were mapped to the full gene set of Tibetan hulless barley. The 9 transcriptome data could map to 28,077 genes, which covered 71.6 % of the whole gene-set.
DEGs analysis and validation of hulless barley sequencing data
Samples A1, D2, C3, D4, E5 and F6 were divided into two groups: one includes A1, B2 and C3, which is under water-sufficient conditions; the other consists of D4, E5 and F6, which is under withholding water conditions. Then, the comparison analysis between the two groups was performed use Noiseq method, and 313 DEGs were finally obtained. The pairwise comparison was also executed among samples F6, G7, H8 and I9. We filtered genes whose RPKM value are below five in all four samples and removed genes that the frequency of occurrence is less than three in pairwise comparison, and then we got 632 DEGs.
Among these DEGs, genes with distinctly changed expression profiling were confirmed using qPCR (Fig. 1). The results of this experiment were basically consistent with RNA-seq data.
Fig. 1.
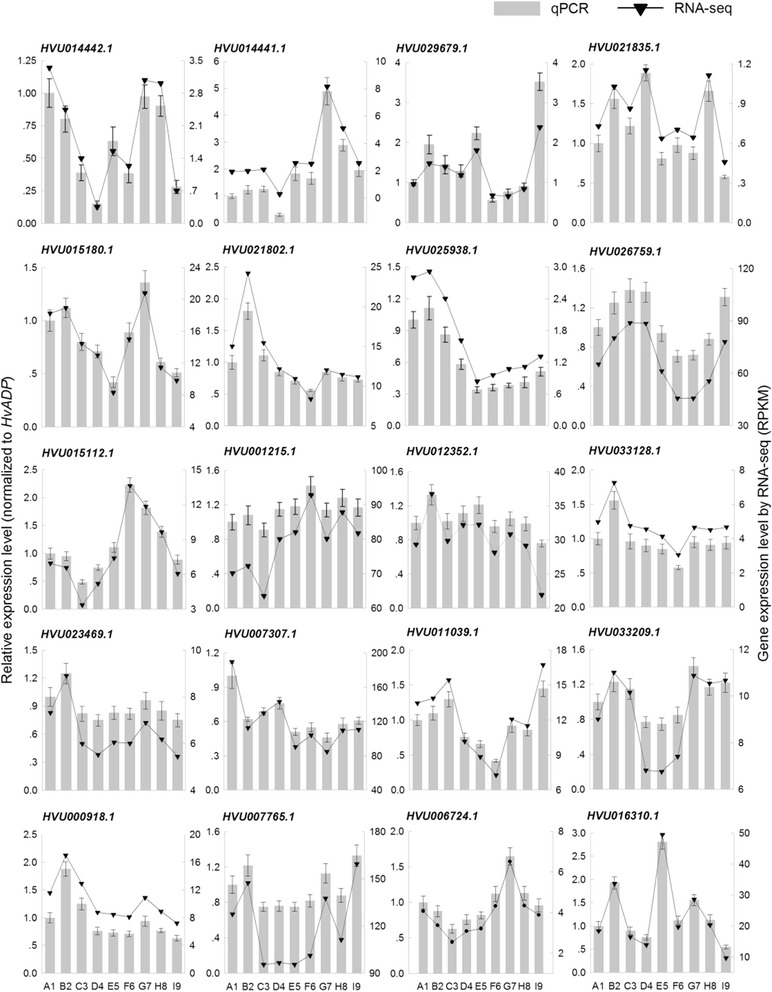
Expression of 20 differentially expressed genes in response to drought stress treatment. Vertical bar charts with simple error bars (left y-axis) represent quantitation of 20 genes transcripts in nine samples, using qPCR. Values are means ± SE (n = 3). Line and scatter plot (right y-axis) represents transcript abundance (RPKM) of nine samples for each gene detected by RNA-seq. The A1-F6 indicates the relative soil moisture content (RSMC) of 33.4, 27.5, 21.1, 15.5, 9.8 and 4.8 %, respectively, and G7-I9 indicates the 2 h, 4 h and 8 h after water recovery from 4.8 % to 33.4 %, respectively
Expression patterns and cluster analysis DEGs
The expression patterns and cluster analysis were conducted by Mev v4.7.4 software with K-Means clustering method and Pearson correlation as distance calculation method. The number of clusters is set to nine. Gene expression pattern for 853 potential drought-resistant related genes clustering into nine clusters (Fig. 2). 93 Genes in cluster IV were obviously down-regulated in low RSMC level, while 108 genes in cluster VI and 119 genes in cluster VIII were both up-regulated. Interestingly, 98 genes in cluster IX were up-regulated in the early phase of recovery process from sample G7 to H8 (Fig. 3). Furthermore, cluster IX shows up-regulation profile both in high and low RSMC level which indicates stress response to abnormal water content in soil. The other genes expression for the cluster IV, VI, VII, and VIII showed down-regulated pattern under drought stress (Additional file 1: Figure S1).
Fig. 2.
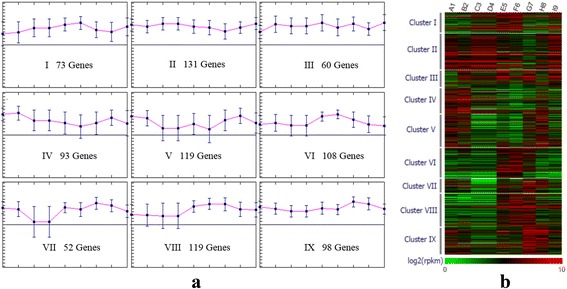
Gene expression pattern for 853 potential drought-resistant related genes clustering into nine clusters. a The horizontal axis shows the nine samples A1, B2, C3, D4, E5, F6, G7, H8, I9, while the vertical axis shows the mean value of log-transformed RPKM value for genes in clusters. Error bars were presented for each sample in each cluster. b Gene expression pattern for all 853 DEGs. The columns show the nine samples A1, B2, C3, D4, E5, F6, G7, H8, I9, while the rows show the log-transformed RPKM values
Fig. 3.

Gene expressions for the cluster IX showing down-regulated pattern under drought stress. The columns show the nine samples A1, B2, C3, D4, E5, F6, G7, H8, I9, while the rows show the log-transformed RPKM values of 98 genes in cluster IX, respectively. Hierarchical clustering of expression pattern for genes in was shown at the left of heat-map figure
GO and KEGG analyze DEGs
The combination of above two strategies results in a final 853 DEGs. We then conducted GO annotation and KEGG pathway analysis of these 853 genes. The assigned functions of these genes covered a broad range of GO categories (Fig. 4). Under the cellular component category, the parts of membrane-bounded organelle, cytoplasmic membrane-bounded vesicle, intrinsic to membrane, cytoplasmic part, intracellular part, plastid part, chloroplast part were prominently represented. Under the category of molecular function, the parts of catalytic activity, transferase activity, transferring phosphorus-containing groups, ion binding, cation binding, oxidoreductase activity represented the majorities of the category. For the biological process category, many genes were classified into the oxidation-reduction process, protein phosphorylation, phosphorus metabolic process, phosphate-containing compound metabolic process, regulation of biological process, biological regulation, transport, and cellular ketone metabolic process. GO enrichment of nine clusters were illustrated in Fig. 5. Of the listed twenty-one GO terms, majority GO terms fell into cluster IX. Tetrapyrrole binding, photosystem and photosynthetic membrane were the most affected in cluster IX. Only one GO term was included in cluster I and cluster VIII, respectively. GO terms focused on cellular component category, whereas DEGs were almost balanced distribution in three categories.
Fig. 4.
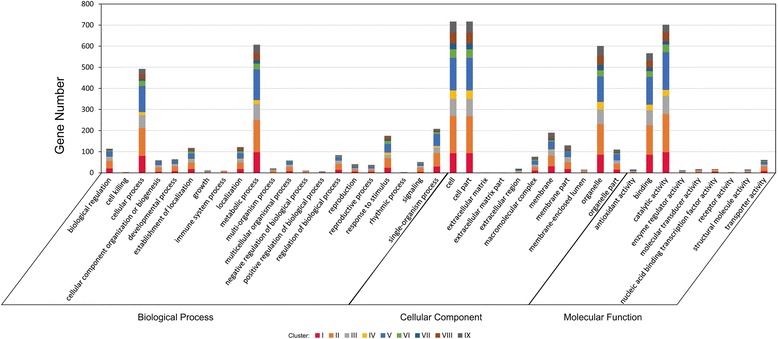
GO annotation of these 853 genes in each category. X-axis indicates GO terms, while the Y-axis indicates the percent of DGEs
Fig. 5.

GO terms distribution in nine clusters (I ~ IX). Each column indicated each cluster, and each row indicated GO degree of enrichment. Red indicated greater enrichment, whereas the grey indicated lower enrichment
KEGG pathway analysis showed that these genes were mainly located in RNA transport, mRNA surveillance pathway, plant hormone signal transduction, defense-related gene induction, and glycerophospholipid metabolism pathway (Table 2).
Table 2.
KEGG pathway of the 853 potential drought resistance related genes
| Pathway | Gene Number | Pathway ID |
|---|---|---|
| Metabolic pathways | 218 | ko01100 |
| RNA transport | 134 | ko03013 |
| Biosynthesis of secondary metabolites | 118 | ko01110 |
| mRNA surveillance pathway | 109 | ko03015 |
| Plant hormone signal transduction | 56 | ko04075 |
| Plant-pathogen interaction | 53 | ko04626 |
| Glycerophospholipid metabolism | 40 | ko00564 |
| Endocytosis | 37 | ko04144 |
| Ether lipid metabolism | 35 | ko00565 |
| Starch and sucrose metabolism | 26 | ko00500 |
| Phenylpropanoid biosynthesis | 24 | ko00940 |
| Cysteine and methionine metabolism | 23 | ko00270 |
| Pyrimidine metabolism | 23 | ko00240 |
| Purine metabolism | 20 | ko00230 |
| Galactose metabolism | 20 | ko00052 |
| Spliceosome | 19 | ko03040 |
| RNA polymerase | 17 | ko03020 |
| Circadian rhythm – plant | 17 | ko04712 |
| Flavonoid biosynthesis | 15 | ko00941 |
| Pentose and glucuronate interconversions | 15 | ko00040 |
| Amino sugar and nucleotide sugar metabolism | 14 | ko00520 |
| Stilbenoid, diarylheptanoid and gingerol biosynthesis | 14 | ko00945 |
| Fructose and mannose metabolism | 14 | ko00051 |
| Glycolysis/Gluconeogenesis | 13 | ko00010 |
| Photosynthesis - antenna proteins | 13 | ko00196 |
| Phenylalanine metabolism | 12 | ko00360 |
| Protein processing in endoplasmic reticulum | 11 | ko04141 |
| Carotenoid biosynthesis | 11 | ko00906 |
| Limonene and pinene degradation | 10 | ko00903 |
| Cyanoamino acid metabolism | 10 | ko00460 |
Drought defense-related genes and pathways
We also analyzed the genes related to ABA-dependent and independent signaling pathway of drought stress responses, including NCED, PYR/PYL/RCAR, SnRK2, ABF, MYB/MYC, and AP2/ERF transcription factors. The typical ABA-dependent and independent signaling pathways responsive to drought stress was illustrated in Fig. 6a. The pathways were described by Mustilli et al. [25] and Yoshida et al. [26]. Almost all genes were up-regulated except PP2C down-regulated. Dehydration first induced the expression of NCED in chloroplast and Ca2+ accumulation in nucleus. ABA biosynthesis induction of NCED triggered PYR/PYL/RCAR and MYB/MYC. PYR/PYL/RCAR located both in nucleus and the outside blocked the expression of SnRK2 by inhibiting PP2C synthesis. SnRK2 located in cytoplasm may accelerate the expression of SLAC1 but repress the expression of KAT1 located in cell wall, and then result in stomatal closure. This was one response formation in cytoplasm to drought. AP2/ERF caused by Ca2+ accumulation, ABF acted by SnRK2, and MYB/MYC induced by ABA together promoted the expression of LEA and DHN in nucleus, and finally mediated drought response. Figure 6b exhibited the expression pattern of representative genes allocated in Fig. 6a mentioned pathways. The expression of NCED dramatically decreased during water recovery. PYR/PYL/RCAR, PP2C, and MYC still kept a relative balance expression. The expression of MYB, LEA, DHN rapidly increased when RSMC reached 9.8 %. All genes presented an abundant expression during the RSMC maintained from 9.8 % to 4.8 %.
Fig. 6.
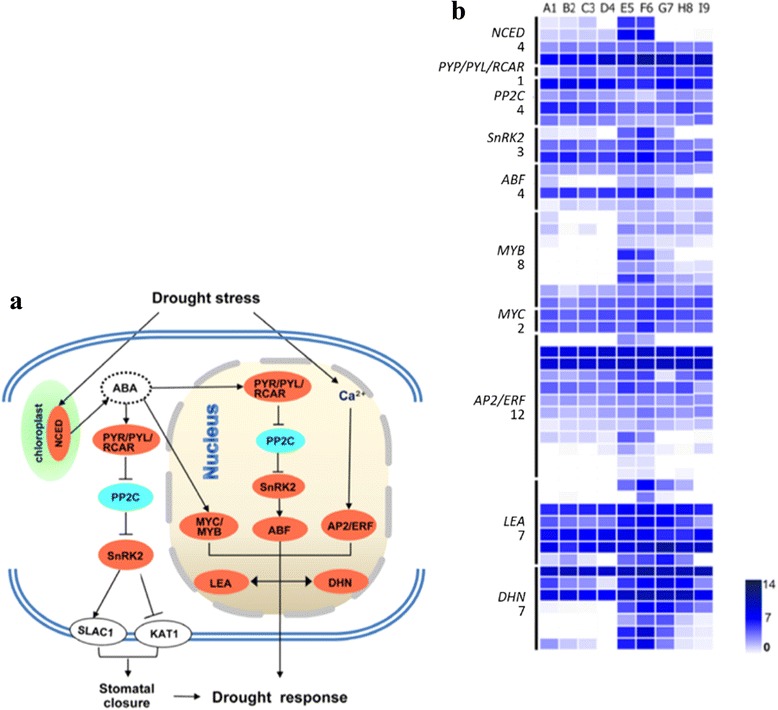
Expression of stress adaptation related genes. a, The typical ABA-dependent and independent signaling pathways responsive to drought stress. Red and blue ellipses indicate up-regulated and down-regulated genes identified by transcriptomes under drought stress, respectively. a was described by Mustilli et al. [22] and Yoshida et al. [23]. b, The expression pattern of representative genes allocated in above mentioned pathways. The expression levels were assessed by log2-transformed RPKM values. The A1-F6 indicates the relative soil moisture content (RSMC) of 33.4, 27.5, 21.1, 15.5, 9.8 and 4.8 %, respectively, and G7-I9 indicates the 2 h, 4 h and 8 h after water recovery from 4.8 % to 33.4 %, respectively. NCED, 9-cis-epoxycarotenoid dioxygenase; SnRK2, SNF1-related protein kinase 2; ABF, AREB/ABF, MYB/MYC, AP2/ERF, transcription factors, LEA, late embryogenesis abundant protein; DHN, dehydrin. The number below each gene indicates the member of each gene family
Discussion
Drought, salinity and cold are the major environmental factors impacting on survival and productivity of hulless barley in Tibetan Plateau of China. Barley is known to be relatively tolerant to abiotic stresses among the major cereal crops and, thus, is often grown in more marginal sites [27]. Tibetan hulless barley cultivar, Himalaya 10, has developed a strong tolerance and adaptation to drought deficit. Using high-throughput RNA sequencing technology, we compared in detail the transcriptional differences and overlap between different levels of RSMC and water recovery, and display the changes of gene expression in Tibetan hulless barley during the whole drought response process.
In this study, the mRNA of Himalaya 10 with good drought tolerance was sequenced using Illumina HiSeq™ 2000 in the manner of PE91, with Sera-mag Magnetic Oligo (dT) Beads. A clear bioinformatic map of mRNA involved in multiple biological processes was produced. As a result, 53.99 G clean reads were collected from nine samples under different RSMC and water recovery, which met the requirements for further analysis. Saturability analysis indicated a qualified coverage of most genes based on our data size. In addition, the clean reads of Q20 occupied over than 95 % of the total, suggesting high quality sequencing. TopHat package was used to blast the transripome data to the Tibetan hulless barley. It has been found that 71.6 % of the reads were mapped to the reference genome. Major mapping reads indicated reliable transcriptome data. These non-mapped tags most likely represent regions where the reference genome is incomplete [28], or there are allelic sequence differences between the reference genome and the cultivar Himalaya 10 used in this study. Another reason may be that RNA-seq data for reference genome annotation should represent all major tissue types, developmental stages and responses to abiotic and biotic stresses [29].
DGE global analysis provided a comprehensive dataset responding to drought stress in leaves of hulless barley seedlings. We identified nine clusters for all DGEs and coarsely assigned them to 31 functional categories (p <0.05) (Fig. 4). Interestingly, there were no other overlapping GO functional enrichments between clusters except catalytic activity, membrane, vesicle, indicating that these genes of different clusters were predicted to be involved in many plant biological processes, including defense [30]. These GO functions were enriched in IX cluster (Fig. 5). Tetrapyrrole binding, photosystem and photosynthetic membrane were the most affected in IX cluster. The former finding implied that tetrapyrroles were the structural backbone of chlorophyll and heme were essential for primary photochemistry, light harvesting, and electron transport [31]. Tetrapyrrole-binding proteins of cHBP1 and cHBP2 have properties suitable for tetrapyrrole carrier proteins [32]. Tetrapyrrole binding protein of genomes uncoupled 4 (GUN4) regulates chlorophyll synthesis and plastid-to-nucleus signal transduction by binding both the product and the substrate of Mg-chelatase, an enzyme that produces magnesium-protoporphyrin IX (Mg-Proto) [33]. Tetrapyrrole biosynthesis had recently been implicated in wilting avoidance [34, 35]. Heme mediated chloroplast-to-nucleus signalling upon drought stress [36, 37]. Enhanced tetrapyrrole biosynthesis was likely to confer drought tolerance via retrograde signaling and induction of drought-responsive gene expression [38].
Pathway enrichment analysis revealed 30 pathways were significantly affected by drought stress (Table 2). Plant hormone signal transduction pathway, for instance, ABA signal transduction pathway was a significant different during the whole drought response process in our study. Transcription factors of NCED, PYR/PYL/RCAR, PP2C, SnRK2 located in cytoplasm together lead to stomatal response and then transcription factors of PYR/PYL/RCAR, PP2C, SnRK2, ABF, MYB/MYC, AP2/ERF, LEA, and DHN located in nucleus, together mediated drought response (Fig. 6a). Soil drying first inducted ABA release in roots and then was distributed throughout the plant via the transpiration stream [18]. ABA induced reduction of leaf growth rate and stomatal closure, which triggered stress proteins and various metabolites to protect cells against drought stress [4, 37] when ABA was produced endogenously via water deficit, and then plant tolerance to drought was increased [39–41]. Drought stress signals can also be generated by osmotic stress-induced Ca2+ expansion, which promoted Ca2+ channels and induces protein kinases and resulted in drought-responsive gene expression [42–44]. Du et al. [45] has compared DHN between wild barley and Tibetan hulless barley associated with drought stress resistance. Liang et al. [46] indicated that LEA genes (HVA1 and Dhn6) might participate in adaptive responses to water deficit in different ways in Tibetan hulless barley. Qian et al. [47] indicate that the differential HVA1 gene has a functional role in the dehydration tolerance in Tibetan hulless barley.
Conclusions
In summary, this study provided a comprehensive analysis of drought-responsive genes and transcriptome expression profiles of Tibetan hulless barley leaves by combined DGE, RNA-seq, and computational approaches. Our results revealed 853 potentially drought-responsive genes in Tibetan hulless barley. We analyzed the genes related to ABA-dependent and independent signaling pathway of drought stress responses. Additionally, we also observed the genes related to tetrapyrrole binding of drought stress responses. This result filled up drought-resistant related genes in Tibetan hulless barley in the available literature. Characterizing the components of these pathways will contribute to improve drought tolerance in Tibetan hulless barley.
Methods
Plant growth under drought condition
An elite hulless barley cultivar Himalaya 10, with good drought tolerance, is used for drought tolerant gene analysis. The RSMC of the original soil sample using for planting were measured and adjusted to 33.4 % by adding proper volume of water. The seedlings of Himalaya 10 were growing under same condition in a greenhouse with a temperature of 23 °C/15 °C (day/night) and a relative humidity of 10–20 %. Three plants from each pot at given condition were considered as biological replicates. Prior to drought stress treatment, these seedlings were well-watered by supplying with proper amount of water every two days to maintain the RSMC at 33.4 %. Different RSMCs at stable status were described by Zhang et al. [48] and Wang et al. [49]. Based on their descriptions, we weighted each pot twice at 8:00 a.m. and 8:00 p.m. Since the soil and plant of each pot was relatively constant, we only add certain water to maintain the RSMC of each pot. Irrigation was removed when the seedlings grew to two-and-a-half leaf stage (18 days after sowing) at six different levels by deficit altering supply water. Eight days after drought stress, leaf samples were harvested from all six groups. The drought stress levels of these groups were evaluated with their score of RSMC (33.4, 27.5, 21.1, 15.5, 9.8 and 4.8 %). After drought treatment, the remaining seedlings of 4.8 % RSMC group were rewatered to restore the RSMC to 33.4 %, leaf samples during rehydration (2 h, 4 h and 8 h after RSMC recovered to 33.4 %) were also collected (Fig. 7). All these leaf samples mentioned above were fast frozen in nitrogen immediately and stored at −80 °C for RNA-seq analysis respectively.
Fig. 7.
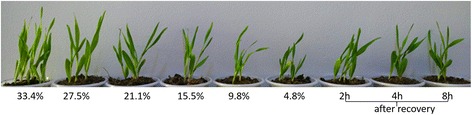
Himalaya 10 underwent a series of RSMCs (33.4, 27.5, 21.1, 15.5, 9.8 and 4.8 %) and rewatered conditions to 33.4 % for 2 h, 4 h and 8 h
RNA isolation and construction of hulless barley RNA-seq library
After the total RNA extraction and DNase I treatment, we used magnetic beads with Oligo (dT) to isolate mRNA. The mRNA molecules were fragmented into 200 bp and cDNAs were synthesized taking mRNA fragments as templates. The cDNA fragments were purified and resolved with EB buffer for end repair, single nucleotide A (adenine) addition and connection of adapters. After PCR amplification, Agilent 2100 Bioanaylzer and ABI StepOnePlus Real-Time PCR System were used in quantification and qualification of the sample library. At last, the 200 bp library was sequenced using Illumina HiSeq™ 2000 in the manner of PE91. The clean reads were obtained by filtering out adaptor-only reads, reads containing more than 5 % unknown nucleotides, and low-quality reads which the percentage of low quality bases (base quality ≤ 10) is more than 20 %.
Gene expression levels and identification of differentially expressed genes (DEG)
The gene expression was calculated by RPKM method (reads per kb per million reads) [50]. The RPKM method is able to eliminate the influence of different gene length and sequencing discrepancy on the calculation of gene expression, which could facilitate comparing the difference of gene expression among samples.
DEGs were identified between two samples in the method described below, which was intend to detect the significance of digital gene expression profiles [51]. The clean reads for gene A is x, x yields to the Poisson distribution. The total clean read numbers of the sample 1 and sample 2 are N1 and N2 respectively. Gene A holds x and y reads in sample1 and sample2. The probability of gene A expressed equally between two samples could be calculated by the following formula:
Also, the p value was corrected for false positive (type I errors) and false negative (type II errors) using FDR method [52]. We use FDR ≤ 0.001 and the absolute value of Log2Ratio ≥ 1 as the threshold to identify DEGs.
Gene Ontology (GO), pathway annotation and enrichment analyses were based on the Gene Ontology Database (http://www.geneontology.org/) [53] and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway (http://www.genome.jp/kegg/) [54], respectively. When we investigated pathways in which different genes were involved and enriched, q-value was used to aid identification according to the previous description [55].
DEGs and drought defense-related genes
Samples A1, D2, C3, D4, E5 and F6 were divided into two groups: one includes A1, B2 and C3, which is under water-sufficient conditions; the other consists of D4, E5 and F6, which is under withholding water conditions. Then, the comparison analysis between the two groups was performed use Noiseq method [56], and these DEGs were finally obtained. The pairwise comparison was also executed among samples F6, G7, H8 and I9. We filtered genes whose RPKM value are below five in all four samples and removed genes that the frequency of occurrence is less than three in pairwise comparison, and then we got those DEGs. The combination of above two strategies results in a final DEGs. We then conducted GO annotation and KEGG pathway analysis of these genes. The expression patterns and cluster analysis were conducted by Mev v4.7.4 software [57] with K-Means clustering method and Pearson correlation as distance calculation method.
qPCR analysis
To validate the results of the RNA-seq data, expression of the genes the same samples used for Transcriptome-seq analysis was performed by real-time quantitative reverse transcription-polymerase chain reaction (RT-PCR) with the fluorescent intercalating dye SYBRGreen in a detection system (Opticon 2; MJ Research, Waltham, USA), using a hulless barley gene (HvADP) as a standard control [58]. A two-step RT-PCR procedure was performed in the experiments. First, 2 μg of purified total RNA was reversely transcribed into cDNAs which were used as templates for PCR reactions using gene-specific primers (Table 3). Second, quantitative PCR was performed using PCR Master Mix (Toyobo, Osaka, Japan) according to the manufacturer’s instructions. Relative quantification of gene expression was determined using the comparative Ct method. To achieve optimal amplification, PCR conditions for each primer combination were optimized for annealing temperature, and PCR products were verified by melting curve analysis and confirmed on an agarose gel. Mean values and standard errors were calculated from three independent experiments with three biological replicates of hulless materials, and the data were normalized with the relative efficiency of each primer pair.
Table 3.
Primers of qRT-PCR assay used for eight RNA-seq libraries in this study
| Gene_id | Amplicon size (bp)a | Forward primer (5′- > 3′) | Reverse primer (5′- > 3′) |
|---|---|---|---|
| HVU014442.1 | 142 | ATTTCTTCGACTGGGGCCTG | ATGACCTTGCCGTCGATCTC |
| HVU014441.1 | 190 | CAGGAGCCTGAGTAGATGCG | AATGAGAGGCCGACCACAAG |
| HVU029679.1 | 194 | AAGAAACTGATCCGAGGCGG | ATTCGTCCGGCCCGTATTTT |
| HVU021835.1 | 180 | GCGGCTATATCCCACCTTCC | AAGAGCTGAGGTGAAGCGAC |
| HVU015180.1 | 208 | TGGAGAAACGGATCGAAGCC | TGCAGCCCAGCTAGAAAAGG |
| HVU021802.1 | 206 | CCTTCACCTCCAGGAACGTC | CACTCCGATTCCACTTGGCT |
| HVU025938.1 | 180 | TGTCACCGCTGAACCAATCA | TTCACTGGGATTCACGGACG |
| HVU026759.1 | 86 | GAAGTCGCGCGAATCTGTTC | GAGCAGAGCAGCCAGATCAA |
| HVU015112.1 | 215 | AAGATCCCAACAAGGCGAGG | CCTGGCTTGCTCCTCTTTGA |
| HVU001215.1 | 229 | AAGCATGCCGTCTTACACCA | GGCCATCGGAGAGTGCATTA |
| HVU012352.1 | 163 | TGGCGTGTCAGCTGAGATTT | GGCCGTGAAGGACCAAAAAC |
| HVU033128.1 | 214 | CCATCAGACTGTGGCGTCTT | GAACCCTCTGCGCATAGACA |
| HVU023469.1 | 244 | AGCACTACTACGGCACCAAC | CTGAAACCGGCGTACTCCTT |
| HVU007307.1 | 216 | AGATGGGGTGCGAACTTGAG | CAGGGACAGGACCATCCAAC |
| HVU011039.1 | 115 | GGTGATCATGCGTGTGTTCG | CTTCTTGGCCGAGTCCTTGT |
| HVU033209.1 | 234 | AGCCCAAACCTACCAAGCTC | ATCATAGCGCAGGAGCCATC |
| HVU000918.1 | 87 | GTAGCTATCCACGGTCACGG | TGGAACTGTAAGCGTCCACC |
| HVU007765.1 | 208 | CGCATGCTGATGGAAAAGGG | ATTGCACCGCACTCAACAAC |
| HVU006724.1 | 135 | TTTGTGTGGGGAGGTCGATG | CATGCACTCTTCGGTGACCT |
| HVU016310.1 | 245 | TGAGGATGAAGCGAGTGCAG | TGGGGACTAGCACGCAAAAT |
aThe amplicon lengths for all the genes are within 70–250 bp range to satisfy the requirement for the primer pairs used for real time RT-PCR analysis using SYBR green according to manufacturer’s guide
Declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Availability of supporting data
The transcriptome raw data of Tibetan hulless barley under drought stress have been deposited in the NCBI under the accession number PRJNA316037 (http://www.ncbi.nlm.nih.gov/bioproject/PRJNA316037).
Acknowledgments
This work was supported by grants from the National Program on Key Basic Research Project (2012CB723006), the National Science and Technology Support Program (2012BAD03B01), and the Tibet Autonomous Region Financial Special Fund (2014CZZX001).
Abbreviations
- ABA
abscisic acid
- ABF
ABRE binding factor
- ABRE
ABA-responsive element
- AP2/ERF
APETALA2/ethylene-responsive factor
- AREB
ABA-responsive element binding factor
- DEGs
differentially expressed genes
- DHN
dehydrin
- LEA
late embryogenesis abundant protein
- NCED
9-cis-epoxycarotenoid dioxygenase
- PP2C
type 2C protein phosphatases
- PYR/RCAR
pyrabactin resistance/regulatory component of ABA receptor
- qRT-PCR
quantative real-time PCR
- ROS
reactive oxygen species
- RSMC
relative soil moisture content
- RT-PCR
reverse transcriptase polymerase chain reaction.
- SnRK2
sucrose non-fermenting 1-related protein kinases2
Additional file
Gene expression for the cluster, IV (a), VI (b), VII (c), and VIII (d) showing down-regulated pattern under drought stress. The columns show the 9 samples A1, B2, C3, D4, E5, F6, G7, H8, I9, while the rows show the log-transformed RPKM values of 93 genes in cluster IV, l08 genes in cluster VI, 52 genes in cluster VII, and 119 genes in cluster VIII, respectively. Hierarchical clustering of expression pattern for genes in was shown at the left of heat-map figure. (DOCX 950 kb)
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
TN, XZ and LB conceived and designed the study. XZ, LB, ZW, HY, YW, QX and YT performed the analyses. XZ and LB wrote the manuscript with the participation of all authors. All authors have read and approved the manuscript.
References
- 1.Baik BK, Ullrich SE. Barley for food: characteristics, improvement, and renewed interest. J Cereal Sci. 2008;48(2):233–242. doi: 10.1016/j.jcs.2008.02.002. [DOI] [Google Scholar]
- 2.Nguyen HT, Babu RC, Blum A. Breeding for drought resistance in rice: physiology and molecular genetics considerations. Crop Sci. 1997;37(5):1426–1434. doi: 10.2135/cropsci1997.0011183X003700050002x. [DOI] [Google Scholar]
- 3.Tanaka R, Kobayashi K, Masuda T. Tetrapyrrole metabolism in Arabidopsis thaliana. Arabidopsis Book. 2011;9:e0145. doi: 10.1199/tab.0145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Umezawa T, Nakashima K, Miyakawa T, Kuromori T, Tanokura M, Shinozaki K, Yamaguchi-Shinozaki K. Molecular basis of the core regulatory network in ABA responses: sensing, signaling and transport. Plant Cell Physiol. 2010;51(11):1821–1839. doi: 10.1093/pcp/pcq156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lee SC, Luan S. ABA signal transduction at the crossroad of biotic and abiotic stress responses. Plant Cell Environ. 2012;35(1):53–60. doi: 10.1111/j.1365-3040.2011.02426.x. [DOI] [PubMed] [Google Scholar]
- 6.Tanaka R, Tanaka A. Tetrapyrrole biosynthesis in higher plants. Annu Rev Plant Biol. 2007;58:321–346. doi: 10.1146/annurev.arplant.57.032905.105448. [DOI] [PubMed] [Google Scholar]
- 7.Nagai S, Koide M, Takahashi S, Kikuta A, Aono M, Sasaki-Sekimoto Y, Ohta H, Takamiya K, Masuda T. Induction of isoforms of tetrapyrrole biosynthetic enzymes, AtHEMA2 and AtFC1, under stress conditions and their physiological functions in Arabidopsis. Plant Physiol. 2007;144:1039–1051. doi: 10.1104/pp.107.100065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schroeder JI, Kwak JM, Allen GJ. Guard cell abscisic acid signalling and engineering drought hardiness in plants. Nature. 2001;410:327–330. doi: 10.1038/35066500. [DOI] [PubMed] [Google Scholar]
- 9.Park SY, Fung P, Nishimura N, Jensen DR, Fujii H, Zhao Y, Lumba S, Santiago J, Rodrigues A, Chow TF, Alfred SE, Bonetta D, Finkelstein R, Provart NJ, Desveaux D, Rodriguez PL, McCourt P, Zhu JK, Schroeder JI, Volkman BF, Cutler SR. Abscisic acid inhibits type 2C protein phosphatases via the PYR/PYL family of START proteins. Science. 2009;324(5930):1068–1071. doi: 10.1126/science.1173041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ma Y, Szostkiewucz I, Korte A, Moes D, Yang Y, Christmann A, Grill E. Regulators of PP2C phosphatase activity function as abscisic acid sensors. Science. 2009;324(5930):1064–1068. doi: 10.1126/science.1172408. [DOI] [PubMed] [Google Scholar]
- 11.Melcher K, Ng LM, Zhou XE, Soon FF, Xu Y, Suino-Powell KM, Park SY, Weiner JJ, Fujii H, Chinnusamy V, Kovach A, Li J, Wang YH, Li JY, Peterson FC, Jensen DR, Yong EL, Volkman BF, Cutler SR, Zhu JK, Xu HE. A gate-latch-lock mechanism for hormone signaling by abscisic acid receptors. Nature. 2009;462:602–608. doi: 10.1038/nature08613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schwartz SH, Tan BC, Gage DA, Gage DA, Zeevaart JA, McCarty DR. Specific oxidative cleavage of carotenoids by VP14 of maize. Science. 1997;276(5320):1872–1874. doi: 10.1126/science.276.5320.1872. [DOI] [PubMed] [Google Scholar]
- 13.Choi H, Hong J, Ha J, Knag J, Kim SY. ABFs, a family of ABA-responsive element binding factors. J Biol Chem. 2000;275(3):1723–1730. doi: 10.1074/jbc.275.3.1723. [DOI] [PubMed] [Google Scholar]
- 14.Abe H, Urao T, Ito T, Seki M, Shinozaki K, Yamaguchi-Shinozaki K. Arabidopsis AtMYC2 (bHLH) and AtMYB2 (MYB) function as transcriptional activators in abscisic acid signaling. Plant Cell. 2003;15(1):63–78. doi: 10.1105/tpc.006130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang G, Chen M, Li L, Xu Z, Chen X, Guo J, Ma Y. Overexpression of the soybean GmERF3 gene, an AP2/ERF type transcription factor for increased tolerances to salt, drought, and diseases in transgenic tobacco. J Exp Bot. 2009;60(13):3781–3796. doi: 10.1093/jxb/erp214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hundertmark M, Hincha DK. LEA (Late Embryogenesis Abundant) proteins and their encoding genes in Arabidopsis thaliana. BMC Genomics. 2008;9:118. doi: 10.1186/1471-2164-9-118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Robertson M. Increased dehydrin promoter activity caused by HvSPY is independent of the ABA response pathway. Plant J. 2003;34(1):39–46. doi: 10.1046/j.1365-313X.2003.01697.x. [DOI] [PubMed] [Google Scholar]
- 18.Shinozaki K, Yamaguchi-Shinozaki K. Gene networks involved in drought stress response and tolerance. J Exp Bot. 2007;58(2):221–227. doi: 10.1093/jxb/erl164. [DOI] [PubMed] [Google Scholar]
- 19.Marcolino-Gomes J, Rodrigues FA, Fuganti-Pagliarini R, Bendix C, Nakayama TJ, Celaya B, Molinari HB, de Oliveira MC, Harmon FG, Nepomuceno A. Diurnal oscillations of soybean circadian clock and drought responsive genes. PLoS One. 2014;9(1):e86402. doi: 10.1371/journal.pone.0086402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.The International Barley Genome Sequencing Consortium. A physical, genetic and functional sequence assembly of the barley genome. Nature. 491:711–16.doi:10.1038/nature11543. [DOI] [PubMed]
- 21.Neumann PM. Coping mechanisms for crop plants in drought-prone environments. Ann Bot. 2008;101(7):901–907. doi: 10.1093/aob/mcn018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Deikman J, Petracek M, Heard JE. Drought tolerance through biotechnology: improving translation from the laboratory to farmers’ fields. Curr Opin Biotech. 2012;23(2):243–250. doi: 10.1016/j.copbio.2011.11.003. [DOI] [PubMed] [Google Scholar]
- 23.Honsdorf N, March TJ, Berger B, Tester M, Pillen K. High-throughput phenotyping to detect drought tolerance QTL in wild barley introgression lines. PLoS One. 2014;9(5):e97047. doi: 10.1371/journal.pone.0097047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen X, Long H, Gao P, Deng G, Pan Z, Liang J, Tang Y, Tashi N, Yu M. Transcriptome assembly and analysis of Tibetan hulless barley (Hordeum vulgare L. var. nudum) developing grains, with emphasis on quality properties. PLoS One. 2014;9(5):e98144. doi: 10.1371/journal.pone.0098144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mustilli AC, Merlot S, Vavasseur A, Fenzi F, Giraudat J. Arabidopsis OST1 protein kinase mediates the regulation of stomatal aperture by abscisic acid and acts upstream of reactive oxygen species production. Plant Cell. 2003;14(12):3089–3099. doi: 10.1105/tpc.007906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yoshida R, Hobo T, Ichimura K, Mizoguchi T, Takahashi F, Aronso J, Ecker JR, Shinozaki K. ABA-activated SnRK2 protein kinase is required for dehydration stress signaling in arabidopsis. Plant Cell Physiol. 2002;43:1473–1483. doi: 10.1093/pcp/pcf188. [DOI] [PubMed] [Google Scholar]
- 27.Jana S, Wilen RW. Breeding for abiotic stress tolerance in barley. In: Ashraf M, Harris PJC, editors. Abiotic stresses: plant resistance through breeding and molecular approaches. 1. New York, USA: Food Products Press; 2005. pp. 491–505. [Google Scholar]
- 28.Zeng X, Long H, Wang Z, et al. The draft genome of Tibetan hulless barley reveals adaptive patterns to the high stressful Tibetan Plateau. Proc Natl Acad Sci U S A. 2015;112(4):1095–1100. doi: 10.1073/pnas.1423628112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Consortium PGS, Xu X, Pan S, Cheng S, Zhang B, Mu D, Ni P, Zhang G, Yang S, Li R, et al. Genome sequence and analysis of the tuber crop potato. Nature. 2011;475:189–95. [DOI] [PubMed]
- 30.Shinozaki K, Yamaguchi-Shinozaki K, Seki M. Regulatory network of gene expression in the drought and cold stress responses. Curr Opin Plant Biol. 2003;6:410–417. doi: 10.1016/S1369-5266(03)00092-X. [DOI] [PubMed] [Google Scholar]
- 31.Papenbrock J, Grimm B. Regulatory network of tetrapyrrole biosynthesis--studies of intracellular signalling involved in metabolic and developmental control of plastids. Planta. 2001;213(5):667–681. doi: 10.1007/s004250100593. [DOI] [PubMed] [Google Scholar]
- 32.Takahashi S, Ogawa T, Inoue K, Masuda T. Characterization of cytosolic tetrapyrrole-binding proteins in Arabidopsis thaliana. Photochem Photobiol Sci. 2008;7(10):1216–1224. doi: 10.1039/b802588f. [DOI] [PubMed] [Google Scholar]
- 33.Larkin RM, Alonso JM, Ecker JR, Chory J. GUN4, a regulator of chlorophyll synthesis and intracellular signaling. Science. 2003;299:902–906. doi: 10.1126/science.1079978. [DOI] [PubMed] [Google Scholar]
- 34.Allen SM, Luck S, Mullen J, Sakai H, Sivasanker S, Tingey SV, Williams RW. Drought tolerant plants and related constructs and methods involving genes encoding ferrochelatases. United States Patent US8158859 B2. 2010. [Google Scholar]
- 35.Phung TH, Jung HI, Park JH, Kim JG, Back K, Jung S. Porphyrin biosynthesis control under water stress: sustained porphyrin status correlates with drought tolerance in transgenic rice. Plant Physiol. 2011;157:1746–1764. doi: 10.1104/pp.111.188276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Voigt C, Oster U, B€ornke F, Jahns P, Dietz KJ, Leister D, Kleine T. In-depth analysis of the distinctive effects of norflurazon implies that tetrapyrrole biosynthesis, organellar gene expression and ABA cooperate in the GUN-type of plastid signalling. Physiol Plant. 2010;138:503–519. doi: 10.1111/j.1399-3054.2009.01343.x. [DOI] [PubMed] [Google Scholar]
- 37.Woodson JD, Perez-Ruiz JM, Schmitz RJ, Ecker JR, Chory J. Sigma factor-mediated plastid retrograde signals control nuclear gene expression. Plant J. 2013;73:1–13. doi: 10.1111/tpj.12011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nagahatenna DSK, Langridge P, Whitford R. Tetrapyrrole-based drought stress signalling. Plant Biotechnol J. 2015;13:447–459. doi: 10.1111/pbi.12356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kuromori T, Mizoi J, Umezawa T, Yamaguchi-Shinozaki K, Shinozaki K. Drought stress signaling network. Mol Biol. 2014;2:383–409. [Google Scholar]
- 40.Atkinson CJ, Davies WJ, Mansfield TA. Changes in stomatal conductance in intact aging wheat leaves in response to abscisic-acid. J Exp Bot. 1989;40(9):1021–1028. doi: 10.1093/jxb/40.9.1021. [DOI] [Google Scholar]
- 41.Munns R, Sharp RE. Involvement of abscisic-acid in controlling plant-growth in soils of low water potential. Austra J Plant Physiol. 1993;20(5):425–437. doi: 10.1071/PP9930425. [DOI] [Google Scholar]
- 42.Schulz P, Herde M, Romeis T. Calcium-dependent protein kinases: hubs in plant stress signaling and development. Plant Physiol. 2013;163:523–530. doi: 10.1104/pp.113.222539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zou JJ, Wei FJ, Wang C, Wu JJ, Ratnasekera D, Liu WX, Wu WH. Arabidopsis calcium-dependent protein kinase CPK10 functions in abscisic acid-and Ca2+-mediated stomatal regulation in response to drought stress. Plant Physiol. 2010;154:1232–1243. doi: 10.1104/pp.110.157545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pei ZM, Murata Y, Benning G, Thomine S, Klusener B, Allen GJ, Grill E, Schroeder JI. Calcium channels activated by hydrogen peroxide mediate abscisic acid signalling in guard cells. Nature. 2000;406:731–734. doi: 10.1038/35021067. [DOI] [PubMed] [Google Scholar]
- 45.Du JB, Yuan S, Chen YE, Sun X, Zhang ZW, Xu F, Yuan M, Shang J, Lin HH. Comparative expression analysis of dehydrins between two barley varieties, wild barley and Tibetan hulless barley associated with different stress resistance. Acta Physiol Plant. 2011;33(2):567–574. doi: 10.1007/s11738-010-0580-0. [DOI] [Google Scholar]
- 46.Liang JJ, Deng GB, Long H, Pan ZF, Wang CP, Cai P, Xu D, Nima ZX, Yu MQ. Virus-induced silencing of genes encoding LEA protein in Tibetan hulless barley (Hordeum vulgare ssp. vulgare) and their relationship to drought tolerance. Mol Breeding. 2012;30(1):441–451. doi: 10.1007/s11032-011-9633-3. [DOI] [Google Scholar]
- 47.Qian G, Han ZX, Zhao T, Deng GB, Pan ZF, Yu MQ. Genotypic variability in sequence and expression of HVA1 gene in Tibetan hulless barley, Hordeum vulgare ssp. vulgare, associated with resistance to water deficit. Aust J Agr Res. 2007;58(5):425–431. doi: 10.1071/AR06300. [DOI] [Google Scholar]
- 48.Zhang H, Song GQ, Ji WQ, Hu YG. Gene induction by drought stress in wheat variety Xiaoyan 22 and their expression analysis. J Agr Biotechnol. 2009;17(4):670–676. [Google Scholar]
- 49.Wang DL, Ye WW, Wang JJ, Song LY, Fan WL, Cui YP. Constructing SSH library of cotton under drought stress and analysis of drought associated genes. Acta Agron Sinica. 2010;36(12):2035–2044. doi: 10.1016/S1875-2780(09)60087-0. [DOI] [Google Scholar]
- 50.Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods. 2008;5(7):621–628. doi: 10.1038/nmeth.1226. [DOI] [PubMed] [Google Scholar]
- 51.Audic S, Claverie JM. The significance of digital gene expression profiles. Genome Res. 1997;7(10):986–995. doi: 10.1101/gr.7.10.986. [DOI] [PubMed] [Google Scholar]
- 52.Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J Royal Stat Soc. 1995;57(1):289–300. [Google Scholar]
- 53.The Gene Onthology Consortium The gene ontology project in 2008. Nucleic Acids Res. 2008;36(suppl 1):440–444. doi: 10.1093/nar/gkm883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kanehisa M, Goto S. KEGG: Kyoto Encyclopedia of Genes andGenomes. Nucleic Acids Res. 2000;28(1):27–30. [DOI] [PMC free article] [PubMed]
- 55.Storey JD, Tibshirani R. Statistical significance for genomewide studies. Proc Natl Acad Sci U S A. 2003;100(16):9440–9445. doi: 10.1073/pnas.1530509100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Tarazona S, Garcia-Alcalde F, Dopazo J, Ferrer A, Conesa A. Differential expression in RNA-seq: a matter of depth. Genome Res. 2011;21:2213–2223. doi: 10.1101/gr.124321.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Mar JC, Wells CA, Quackenbush J. Defining an informativeness metric for clustering gene expression data. Bioinformatics. 2011;27(8):1094–1100. doi: 10.1093/bioinformatics/btr074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ferdous J, Li Y, Reid N, Langridge P, Shi BJ, Tricker PJ. Identification of reference genes for quantitative expression analysis of MicroRNAs and mRNAs in barley under various stress conditions. PLoS One. 2015;10(3):e0118503. doi: 10.1371/journal.pone.0118503. [DOI] [PMC free article] [PubMed] [Google Scholar]