

Abstract
Background
Increasing numbers of patients (up to 40 %) with rheumatoid arthritis (RA) achieve remission, yet it remains to be elucidated whether this also normalizes their cardiovascular risk. Short-term treatment with TNF inhibitors lowers arterial wall inflammation, but not to levels of healthy controls. We investigated whether RA patients in long-term remission are characterized by normalized inflammatory activity of the arterial wall and if this is dependent on type of medication used (TNF-inhibitor versus nonbiological disease-modifying antirheumatic drugs (DMARDs)).
Methods
Arterial wall inflammation, bone marrow and splenic activity (index of progenitor cell activity) was assessed with 18F-fluorodeoxyglucose (18F-FDG) positron emission tomography/computed tomography (PET/CT) in RA patients in remission (disease activity score (DAS28) <2.6 for >6 months) and healthy controls. We performed ex vivo characterization of monocytes using flow cytometry and a transendothelial migration assay.
Results
Overall, arterial wall inflammation was comparable in RA patients (n = 23) in long-term remission and controls (n = 17). However, RA subjects using current anti-TNF therapy (n = 13, disease activity score 1.98[1.8–2.2]) have an almost 1.2-fold higher 18F-FDG uptake in the arterial wall compared to those using DMARDs (but with previous anti-TNF therapy) (n = 10, disease activity score 2.24[1.3–2.5]), which seemed to be predominantly explained by longer duration of their rheumatic disease in a multivariate linear regression analysis. This coincided with increased expression of pro-adhesive (CCR2) and migratory (CD11c, CD18) surface markers on monocytes and a concomitant increased migratory capacity. Finally, we found increased activity in bone marrow and spleen in RA patients using anti-TNF therapy compared to those with DMARDs and controls.
Conclusions
A subset of patients with RA in clinical remission have activated monocytes and increased inflammation in the arterial wall, despite the use of potent TNF blocking therapies. In these subjects, RA disease duration was the most important contributor to the level of arterial wall inflammation. This increased inflammatory state implies higher cardiovascular risk in these patients, who thus may require more stringent CV risk management.
Electronic supplementary material
The online version of this article (doi:10.1186/s13075-016-1008-z) contains supplementary material, which is available to authorized users.
Keywords: Rheumatoid arthritis, Cardiovascular disease, Inflammation, Imaging
Background
Patients with rheumatoid arthritis (RA) are characterized by an approximately twofold increase in cardiovascular disease (CVD) risk [1], which cannot be solely attributed to traditional CVD risk factors [2]. Thus, attention has shifted toward a direct pathogenic role of the systemic inflammatory state in RA. Although up to 40 % of RA patients will reach clinical (articular) remission following treatment [3], it remains to be established whether the increased CVD risk also disappears. Treatment with methotrexate (MTX) in RA patients resulted in a marked reduction in the inflammatory activity, in conjunction with a 21 % lower CVD event rate [4, 5]; yet mortality remains clearly increased compared to the general population [6, 7]. Considering the potent anti-inflammatory effects of immune-modulating biological therapies [8, 9], and in view of the central role of tumor necrosis factor (TNF) in atherogenesis [10], the introduction of TNF inhibitors holds the promise of further reducing this residual CVD burden, although definite outcomes are at present still controversial [11–13].
Supporting its potential role in reducing CVD, anti-TNF treatment was shown to have favorable effects on the arterial wall, with beneficial impact on intima media thickness (IMT) progression as well as arterial wall stiffness [14]. Short-term (8 weeks) TNF inhibition also significantly reduced arterial wall inflammation in patients with active RA, although it failed to completely normalize arterial inflammation to levels observed in control subjects [15]. Whether arterial inflammation can be further reduced during prolonged remission, with or without anti-TNF treatment, remains to be established.
In analogy to the central role of activated monocytes regulating synovial inflammation in RA [16], recent data substantiated a quite similar role for circulating innate immune cells driving arterial wall inflammation in atherosclerotic disease [17]. In experimental atherosclerosis models following an acute coronary event (ACS), increased mobilization of myelopoietic precursors from the bone marrow elicits inflammation of systemic atherosclerotic lesions, mediated by increased influx of these newly formed inflammatory monocytes [18]. However, whether RA-associated cell mobilization/activation contributes to arterial wall inflammation remains to be established.
In the present study, we assessed arterial wall inflammation in RA patients who were in stable clinical (articular) remission. Considering the above-mentioned direct role for TNF in atherosclerosis, we also evaluated the presence of potential drug-specific effects by categorizing RA subjects into those with either stable remission with anti-TNF therapy or stable remission without anti-TNF therapy (but with disease-modifying antirheumatic drugs (DMARDs). Moreover, we assessed monocyte phenotype and function, as well as bone marrow and splenic ‘metabolic’ activity (an index of progenitor cell activity) using 18F-fluorodeoxyglucose (18F-FDG) positron emission tomography (PET) with computed tomography (CT).
Methods
Study population
We performed a controlled cross-sectional cohort study in subjects with an established diagnosis of RA (based on the ACR/EULAR classification [19]), in remission (defined by disease activity score in 28 joints (DAS28) below 2.6 [20]) for more than 6 months. RA subjects were compared to healthy controls, matched for age and sex. Because of ethical constraints concerning radiation exposure, for the imaging studies, healthy controls were selected from a contemporaneous study using identical imaging protocols and performed on the same scanner. For the ex vivo studies, healthy controls (matched for age and sex) were recruited through advertisement. General exclusion criteria were medical history of CVD and the presence of diabetes. To enable comparison of different treatments, while minimizing confounding by indication, all patients were selected based on previous use of anti-TNF therapy. For patients currently on DMARDs, criteria for discontinuation of TNF inhibitors were: at least 6 months of treatment with stable use of concomitant DMARDs, remission based on DAS28 for at least 6 months. The study protocol was approved by the Institutional Review Board of the Academic Medical Center in Amsterdam. Written informed consent was obtained from each participant.
Baseline data collection
Fasting basal lipid levels, leukocyte count and differentiation, erythrocyte sedimentation rate (ESR) and C-reactive protein (CRP) were determined using standard laboratory procedures. Physical examination, including blood pressure was performed and medical and family history was recorded. In the RA subjects, disease severity was recorded at the time of visit using DAS28-ESR, combining swelling and tenderness in 28 joints, general wellbeing of the patient (visual analogue scale) and ESR levels [21]. Date of disease onset, anti-cyclic citrullinated peptide (anti-CCP), rheumatoid factor (RF) positivity and medication use were extracted from medical history.
18F-FDG PET/CT imaging
Fasting subjects underwent 18F-FDG PET/CT imaging on a PET/CT scanner (Philips, Best, the Netherlands) as previously described [22]. Imaging was initiated, 90 minutes post 18F-FDG infusion (250 MBq, 6.8 mCi), with a low-dose, noncontrast-enhanced CT scan for attenuation correction and anatomic co-registration, followed by PET imaging of the thoracic and splenic region.
Images were analyzed with dedicated software (OsiriX, Geneva, Switzerland; http://www.osirix-viewer.com). 18F-FDG uptake was assessed in the ascending aorta, bone marrow and spleen. In each artery five regions of interest (ROIs) were drawn. Maximum standardized uptake values (SUVmax) were averaged for each artery, and divided by the average venous background activity (SUVmean) to obtain the target-to-background ratio (TBRmax). Three consecutive segments with the highest uptake were used to determine the most diseased segment (MDS) [22]. The SUVmax values in the bone marrow and spleen were determined as follows; bone marrow activity was calculated as the average SUVmax of all imaged vertebrae, and splenic SUVmax was obtained as an average of the SUVmax obtained in axial, sagittal, and coronal planes, as previously described [23].
Ex vivo monocyte studies
Flow cytometry
Red blood cells were lysed with BD FSCSTM-lysis solution (BD Biosciences, San Diego, CA, USA). Peripheral blood mononuclear cell (PBMCs) were incubated with fluorchrome-labeled antibodies (Additional file 1: Table S1) for 15 minutes and washed with saline. Samples were analyzed using an FACSCalibur (Becton Dickinson, Franklin Lakes, NJ, USA). Delta median fluorescence intensity (MFI), determined with FlowJo software (version 5.4+; Tree Star, Ashland, OR, USA), was calculated by subtracting isotype MFI from MFI of the marker in the corresponding color.
Transendothelial migration
A transendothelial migration (TEM) assay was performed as described previously [24]. Briefly, after overnight stimulation with TNF-α (10 ng/ml), monocytes were added to primary human arterial endothelial cells (HAEC, Lonza, Baltimore, MD, USA), at a concentration of 1*106 cells/ml for 30 minutes at 37 °C, 5 % CO2 and then fixed with 3.7 % formaldehyde (Sigma-Aldrich, Zwijndrecht, the Netherlands). Images were recorded with a Zeiss Axiovert 200 microscope (Plan-apochromat 10x/0.45 M27 Zeiss-objective; Carl Zeiss Inc., Jena, Germany). Transmigrated monocytes were distinguished from adhered monocytes by their transitions from bright to dark morphology and quantified using the cell counter plugin (http://rsbweb.nih.gov/ij/plugins/cell-counter.html) in the Image-J software (http://rsb.info.nih.gov/nih-image/).
Statistical analysis
Data were analyzed using Prism version 5.0 (GraphPad Software, La Jolla, CA, USA) and SPSS version 21.0 (IBM Corp,. Armonk, NY, USA). Data are presented as mean ± standard deviation (SD), for normally distributed data or medians with interquartile range [IQR], for nonnormally distributed data unless stated otherwise. Depending on distribution all comparisons of subgroups were performed using Student’s t test or Mann-Whitney U test, and chi-square tests for categorical variables. Differences in TBR between the groups were assessed with a univariate analysis of covariance (ANCOVA) to account for known cardiovascular risk factors: age, gender and smoking status, and any value that differed at baseline. Univariate and multivariate linear regression (with body mass index (BMI), systolic blood pressure, current smoking and one additional explanatory parameter as covariates) was performed to explore the correlation between RA-specific characteristics and arterial wall inflammation (TBRmax). A p value of <0.05 was considered statistically significant.
Results
Baseline characteristics
For the PET/CT study, 23 consecutive RA patients in clinical remission for at least 6 months were included at the outpatient clinics of the Academic Medical Center (AMC) and the Amsterdam Rheumatology and Immunology Center, and compared to 17 age- and sex-matched healthy controls. RA subjects had an unfavorable CV risk profile with more smokers, higher blood pressure and higher BMI. Although CRP levels were generally low, they were higher in RA subjects compared to controls (RA: 18[13–30] mg/L vs control: 13[7–16] mg/L, p = 0.022) (Table 1).
Table 1.
Clinical characteristics of control subjects and RA patients
| Control (n = 17) | RA (n = 23) | p value | RA, DMARD (n = 10) | RA, anti-TNF (n = 13) | p value | |
|---|---|---|---|---|---|---|
| Sex, male/female | 7/10 | 8/15 | 0.680 | 3/7 | 5/8 | 0.637 |
| Age, years | 58 ± 13 | 58 ± 9 | 0.894 | 55 ± 11 | 60 ± 8 | 0.193 |
| Body mass index, kg/m2 | 22 ± 2 | 26 ± 6 | 0.005 | 25 ± 6 | 26 ± 4 | 0.368 |
| Smoking, yes/no | 0/17 | 8/23 | 0.022 | 3/7 | 4/13 | 0.708 |
| SBP, mmHg | 127 ± 13 | 143 ± 23 | 0.014 | 131 ± 22 | 151 ± 20 | 0.028 |
| DPB, mmHg | 78 ± 8 | 87 ± 15 | 0.036 | 83 ± 15 | 90 ± 14† | 0.270 |
| Total cholesterol, mmol/L | 5.4 ± 0.9 | 5.4 ± 0.9 | 1.000 | 5.3 ± 0.9 | 5.4 ± 0.9 | 0.613 |
| LDL cholesterol, mmol/L | 3.3 ± 0.9 | 3.6 ± 1.1 | 0.396 | 3.4 ± 0.8 | 3.7 ± 1.3 | 0.284 |
| HDL cholesterol, mmol/L | 1.7 ± 0.4 | 1.5 ± 0.5 | 0.135 | 1.6 ± 0.5 | 1.4 ± 0.4 | 0.152 |
| Triglycerides, mmol/L | 0.66[0.5–1.0] | 1.2[0.9–1.8] | 0.001 | 1.1[0.9–1.4] | 1.3[0.9–2.1] | 0.393 |
| CRP, mg/L | 13[7–16] | 18[13–30] | 0.022 | 16[12–48] | 19[14–30] | 0.888 |
Values are n (%), mean ± SD or median [IQR,] for skewed data
RA rheumatoid arthritis, DMARD disease-modifying antirheumatic drug, TNF tumor necrosis factor, SBP systolic blood pressure, DBP diastolic blood pressure, LDL low-density lipoprotein, HDL high-density lipoprotein, CRP C-reactive protein
Within the RA subjects, ten patients were currently using nonbiological DMARDs, after they successfully discontinued their anti-TNF therapy for more than 6 months, whereas 13 patients used anti-TNF therapy. Nine out of the 13 patients on anti-TNF treatment had a previous attempt to discontinue which failed, reaching remission again only upon re-introduction. Age, BMI and baseline lipid levels were comparable between groups. Systolic blood pressure was higher in patients using TNF inhibition (RA, DMARD: 131 ± 22; RA, TNF inhibition; 151 ± 20, p <0.05 versus all other groups) (Table 1). DAS28, CRP, ESR as well as percentage seropositive RA were equally distributed between the two groups, but disease duration and duration until first start of TNF inhibition was significantly longer in the RA, TNF-inhibition group. RA-specific parameters and type of RA medication, as well as concomitant cardiovascular medication are listed in Table 2.
Table 2.
Baseline – RA-specific characteristics
| RA, DMARD (n = 10) | RA, anti-TNF (n = 13) | p value | |
|---|---|---|---|
| ESR, mm/H | 9 [7–21] | 6 [6–17] | 0.689 |
| Disease activity score (DAS28) | 2.24 [1.3–2.5] | 1.98 [1.8–2.2] | 1.000 |
| Anti-CCP positive (%) | 7 (70) | 8 (61) | 0.673 |
| Rheumatoid factor positive (%) | 8 (80) | 7 (54) | 0.192 |
| Disease duration, years | 7.8 [4.0–10.2] | 13.3 [9.3–20.2] | 0.012 |
| Anti-TNF | n/a | n/a | |
| - Adalimumab | 10 | ||
| - Certolizumab | 1 | ||
| - Etanercept | 1 | ||
| Disease duration until start anti-TNF, years | 1.2 [1.0–1.7] | 5.6 [4.3–9.1] | 0.001 |
| Anti-TNF withdrawal attempts, yes/no | 10/0 | 9/4 | n/a |
| DMARD | n/a | ||
| - Methotrexate | 9 | 11 | |
| - Sulfasalazine | 1 | ||
| Other Rx | |||
| - Statin | 1 | 1 | 0.848 |
| - Antihypertensive | 5 | 5 | 0.580 |
Values are n (%), mean ± SD or median [IQR,] for skewed data
RA rheumatoid activity, DMARD disease-modifying antirheumatic drug, TNF tumor necrosis factor, ESR erythrocyte sedimentation rate, DAS28 disease activity score based on 28 joints, CCP cyclic citrullinated peptide
18F-FDG PET/CT
All parameters were adjusted for cardiovascular risk factors which differed between groups at baseline: systolic blood pressure, current smoking and BMI. Overall, RA subjects in stable remission had levels of arterial wall inflammation compared to matched controls (aortic TBRmax: RA; 2.3 ± 0.5 versus control; 2.1 ± 0.3, adjusted p = 0.251. Aortic TBRmds: RA; 23 ± 0.5 versus control; 22 ± 0.3 adjusted p = 0.331) (Fig. 1a, b).
Fig. 1.
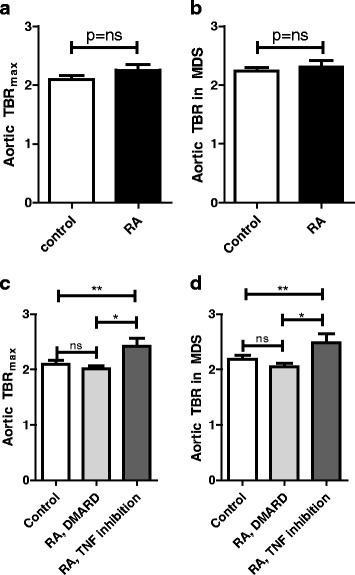
Increased arterial TBR in RA patients on anti-TNF therapy. Quantification of the 18F-FDG uptake as the maximum target-to-background ratio (TBR) (a, c) and TBR in the most diseased section (MDS) (b, d) in the ascending aorta revealed that RA subjects in stable remission have levels of arterial wall inflammation comparable to healthy controls (a, b). However, we found increased uptake in RA patients with anti-TNF treatment (n = 13) versus RA subjects on DMARDs (n = 10) and controls (n = 17). p values are adjusted for age, gender, BMI, current smoking and systolic blood pressure, * = p < 0.05, ** = p < 0.01. DMARD disease-modifying antirheumatic drug, MDS most diseased segment, RA rheumatoid arthritis, TBR target-to-background ratio, TNF tumor necrosis factor
Surprisingly, upon further assessment of subgroups, 18F-FDG uptake was significantly higher in RA subjects using anti-TNF therapy compared with the RA, DMARD group (with comparable disease activity, CRP and ESR levels) and control subjects, (aortic TBRmax: RA, TNF inhibition 2.42 ± 0.54 p = 0.004 versus controls. RA, DMARD; 2.02 ± 0.16, p = 0.011 versus RA, TNF inhibition and p = 0.107 versus controls. Similar for TBRmds) (Fig. 1c, d).
To assess which factors accounted for the difference in TBR, we performed a univariate and multivariate linear regression using CRP, ESR, DAS28 or disease duration and disease duration till start of TNF inhibition as response variables and TBRmax as the dependent variable. In the multivariate analyses, each of these predictors was corrected for the cardiovascular risk factors, which differed at baseline: BMI, systolic blood pressure (SBP) and current smoking (of which only smoking correlated to TBRmax, Additional file 1: Table S2). Disease duration and duration until start of TNF inhibition correlated with arterial wall inflammation (and in line, with biological use, data not shown), which remained significant after correction for potential confounders (Table 3).
Table 3.
Unadjusted and adjusted linear regression analysis with TBRmax as the dependent variable
| Characteristic | Unadjusted analyses | Adjusted analysesα | ||
|---|---|---|---|---|
| B (95 % CI) | p value | B (95 % CI) | p value | |
| ESR | 0.009(−0.029-0.048) | 0.602 | 0.024(−0.021-0.070) | 0.256 |
| CRP | −0.056(−0.272-0.159) | 0.584 | −0.075(−0.318-0.168) | 0.513 |
| DAS28 | 0.060(−0.623-0.744) | 0.850 | 0.087(−0.638-0.910) | 0.831 |
| Disease duration | 0.023(−0.003-0.050) | 0.084 | 0.038(0.002-0.074) | 0.040 |
| Disease duration until start of TNF inhibition | 0.064(0.015-0.113) | 0.015 | 0.081(0.004-0.158) | 0.040 |
Data are unadjusted coefficient (B) with 95 % confidence intervals (CI)
TBR max maximum target-to-background ratio, ESR erythrocyte sedimentation rate, CRP C-reactive protein, DAS28 disease activity score based on 28 joints
αAdjusted for BMI, systolic blood pressure and current smoking
Monocyte phenotype and function
We studied phenotype and function of circulating monocytes in RA patient subjects (including additional subjects to the imaging cohort: ten RA patients without anti-TNF therapy who were compared to 13 RA patients with anti-TNF therapy and 20 age- and sex-matched controls. All baseline and RA-specific characteristics were comparable to the imaging study (Additional file 1: Table S3 and Table S4). Analysis of surface markers showed increased expression of CCR2, CD11c and CD18 in RA subjects using anti-TNF therapy versus RA, DMARD and controls (Fig. 2a). Functionally, in a TEM assay, this resulted in a 1.5-fold increase of monocytes, which crossed the endothelial layer (transmigrated cells/mm2: controls 36 ± 24; RA, DMARD 41 ± 19 p = ns versus controls; RA, TNF inhibition 57 ± 17, p = <0.05 versus all other groups) (Fig. 2b, c).
Fig. 2.
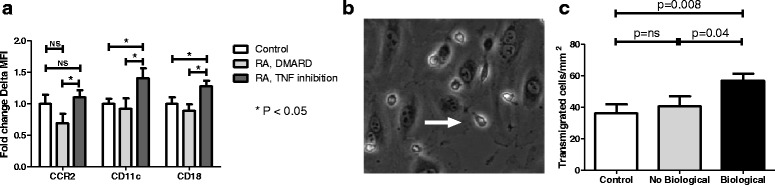
Ex vivo monocyte adhesive and migratory properties. Increased expression (represented as fold change of delta median fluorescent intensity) of monocyte surface markers CCR2, CD11c and CD18 in RA subjects with anti-TNF treatment (n = 20) versus RA patients with DMARDs (n = 16) and controls (n = 20) (a). Assessment of transendothelial migratory capacity showed a concomitant increase in number of transmigrated cells (represented as a white arrow in (b)) (c). Data are means + SEM (for each subject, transmigrated cells are calculated of independent counts of five frames of view). * = p < 0.05, ** = p < 0.01. DMARD disease-modifying antirheumatic drug, MFI median fluorescence intensity, RA rheumatoid arthritis, TNF tumor necrosis factor
Bone marrow and splenic metabolic activity in RA patients
Finally, we sought to investigate whether the increased presence of promigratory monocytes in RA subjects using anti-TNF therapy would coincide with increased 18F-FDG uptake in hematopoietic organs. Indeed, standard uptake values (SUVs) were significantly higher in RA patients using anti-TNF therapy in both bone marrow (BM) and spleen (BM SUVmax: control 2.44 ± 0.27; RA, DMARD 2.42 ± 0.24, p = 0.890 versus controls; RA, TNF inhibition 2.86 ± 0.66, p <0.05 versus all other groups. Splenic SUVmax: control 2.16 ± 0.45; RA, DMARD 2.20 ± 0.21, p = 0.817 versus controls; RA, TNF inhibition 2.65 ± 0.48, p = 0.01 versus all other groups) (Fig. 3).
Fig. 3.
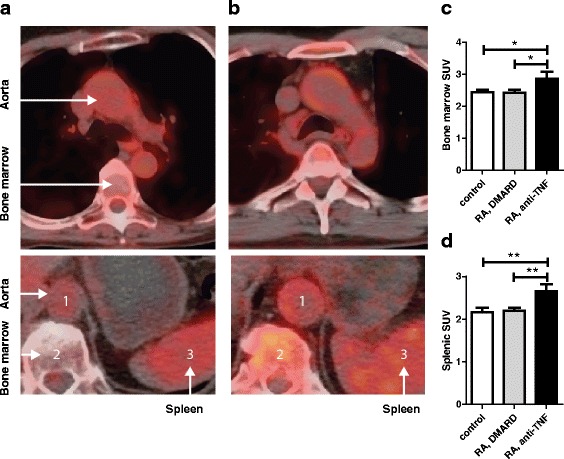
Bone marrow and splenic activity in RA patients. Cross-sectional 18F-FDG PET/CT images representing 18F-FDG uptake (red) in the aorta, bone marrow (top and bottom) and spleen (bottom) in a RA patient without (a) or with anti-TNF treatment (b). Quantification of maximum standard uptake values (SUVmax) showed increased activity in both bone marrow (c) and spleen (d) of RA patients with continued anti-TNF treatment versus RA patients without anti-TNF treatment and controls. * = p < 0.05, ** = p < 0.01. DMARD disease-modifying antirheumatic drug, RA rheumatoid arthritis, SUV standardized uptake values, TNF tumor necrosis factor
Discussion
In the present study, we show that, overall, RA subjects in long-term clinical remission do not have increased arterial wall inflammation compared to age- and sex-matched controls. However, contrary to the expectations, we observed a markedly increased inflammatory activity in the arterial wall, combined with an activated, promigratory phenotype of circulating monocytes in RA patients using anti-TNF therapy to maintain clinical remission, compared to RA patients using DMARDs, as well as age- and sex-matched healthy control subjects. The concomitantly increased 18F-FDG activity in hematopoietic organs (bone marrow and spleen) implies stimulated myelopoiesis contributing to the propagation of the systemic pro-inflammatory state in RA patients in remission using anti-TNF therapy.
Increased 18F-FDG uptake in the arterial wall is recognized as an independent predictive marker for future atherothrombotic events [25]. Our study shows that, despite the presence of an adverse CV risk profile, overall, RA subjects in remission had levels of 18F-FDG uptake comparable to age- and sex-matched controls. These data support the concept that effective control of inflammatory disease activity diminishes CV risk in RA subjects. However, the FDG uptake in the arterial wall was significantly elevated in RA patients using anti-TNF therapy, reaching levels previously associated with a significant increase in CV risk [25], whereas the signal in patients who used only DMARDs was comparable to that in age- and sex-matched controls. These data imply a persistent inflammatory drive in RA patients in need of continued anti-TNF therapy, despite clinical remission. From a cardiovascular point of view, stopping or tapering anti-TNF in these patients cannot be advocated as one could expect increased vessel wall inflammation, implying an increased CV risk. An obvious challenge for the future is to identify this subset of patients.
As TNF inhibitors were previously shown to effectively lower arterial wall inflammation [15], our findings are most likely explained by inherent differences between treatment groups. Patients with and without anti-TNF therapy were selected retrospectively thus, differences between these groups may reflect heterogeneity in ‘baseline’ disease severity, despite current clinical remission with low disease activity, ESR and CRP levels in both RA groups. To minimize confounding we only included patients who had previously been treated with TNF inhibitors. Moreover, in the group with ongoing anti-TNF therapy, 9 out of 13 patients had attempted to withdraw biological therapy, but failed due to clinical reactivation of articular symptoms (20 out of 20 for the ex vivo studies). Interestingly, Saleem et al. previously showed that short duration of disease symptoms is associated with successful withdrawal of TNF inhibitors [26]. Although we did not assess duration of active disease in the present study, total disease duration was longer in subjects who were on current TNF inhibition compared to the group who remained in remission after withdrawal of anti-TNF therapy, and moreover, duration of disease until first start of TNF inhibitors was also significantly longer. Notably, disease duration has previously been identified as an independent risk factor for CVD events [27, 28]. In our study, after correction for multiple confounders, disease duration and time until start of TNF inhibition were the only RA-specific characteristics that significantly correlated to TBR. These observations lend support to the concept that cumulative disease burden in RA may be particularly deleterious [29, 30], illustrating that its contribution to CVD risk cannot be captured in single measurement of disease activity. Eventually, a disease state can be reached where irrevocable changes in the joint [29, 31] or dysregulation of the immune response [26] persists, which may propagate inflammation in distant organs and cells, such as circulating monocytes, known to be key players in the chronic inflammatory state. Whether early use of biological therapies can prevent this [32] and thereby normalizing CVD risk, is a concept that warrants further investigation.
Activation markers of circulating monocytes in plasma predict the CV-event rate in patients [33]. Here, we found a significant increase of CCR2, CD11c and CD18 surface expression on monocytes from RA patients using anti-TNF therapy. Increased expression of CCR2, the receptor for monocyte chemo-attractant protein-1 (MCP-1) promotes chemotaxis [34], and murine studies corroborated that CCR2 is pivotal in the process of plaque formation [35]. The β2 integrin CD11c/CD18 serves as an adhesive ligand by interacting with endothelial VCAM-1, is important in monocyte recruitment and contributes to formation of atherosclerotic plaques [36]. The functional relevance of the increased activation status of circulating monocytes in RA patients using anti-TNF therapy was substantiated by the increased migratory capacity found in the ex vivo TEM assay. In patients with advanced atherosclerotic lesions we previously reported that activated circulating monocytes accumulated more avidly in arterial wall lesions with increased TBR activity on PET/CT [24]. In the present study, our observation in RA subjects on maintenance anti-TNF therapy suggests a similar process, where increased influx of activated plasma monocytes may contribute to increased arterial wall inflammation. Altogether, these data show that RA patients requiring continuation of anti-inflammatory biological treatment may have persistently increased ‘subclinical’ inflammatory activity in both the arterial wall and circulating innate immune cells.
The half-life of circulating monocytes is approximately 1–3 days [37]. This rapid turnover of inflammatory cells highlights the important role for their production sources, the hematopoietic tissues, to continuously supply new immune cells driving (arterial wall) inflammation. In RA patients on anti-TNF therapy, both bone marrow and spleen exhibited increased metabolic activity compared to RA subjects without anti-TNF therapy as well as matched controls. In patients’ post-acute coronary syndrome (ACS), bone marrow and splenic activity were recently found to correlate to arterial wall inflammation as well as to independently predict future CV events [23, 38]. Moreover, activity in BM and spleen correlates to expression of pro-inflammatory markers on circulating leukocytes [18, 23]. The present study reveals increased BM as well as splenic activity in RA patients with clinical quiescent disease under anti-TNF therapy. This elevated activity is associated with both arterial wall inflammation as well as cellular activation, substantiating a multilevel, subclinical, pro-inflammatory phenotype, which has been previously linked to an increased risk for CVD events [39].
Potential limitations
We conducted a cross-sectional study, in which several limitations need to be considered. First, as aforementioned, confounding by indication may have contributed to our findings, which we tried to minimize regarding RA-related features. However, we cannot account completely for disease severity with the measures provided in this study. Moreover, the design precludes conclusions on disease course and influences of previous medication use. Combined with the small study samples, we thus cannot draw causal conclusions on the observations made in our study. Traditional cardiovascular risk factors are also increased in RA subjects [40], and may be influenced by factors such as disease severity or treatment [11, 41]. In our study, except for systolic blood pressure, which was 20 mmHg higher in RA patients using anti-TNF therapy, traditional CVD risk factors were equally distributed between both RA groups. In line with previous studies [15, 42], we found no correlation between blood pressure and arterial wall inflammation, suggesting that the contribution of systolic blood pressure to arterial wall inflammation in RA subjects is limited. The use of 18F-FDG PET/CT for BM and splenic activity has not been fully established and thus interpretation should be done with caution. Particularly concerning BM, distinction between direct involvement in RA and metabolic activity leading to increased production of inflammatory cells cannot be made, as we did not assess synovial activity of the joint affected in RA in the present study. Yet, the vertebrae of the spinal column were used to assess BM activity, an organ not known to be affected in RA.
Conclusions
We present, for the first time, a study addressing functional arterial wall changes in RA subjects in prolonged stable clinical remission. Overall, subjects in long-term remission had levels of arterial wall inflammation compared to age- and sex-matched controls. However, by using a unique combination of cellular and imaging data, we show that the subset of patients requiring continuation of anti-TNF therapy to maintain remission of their joint disease had increased arterial wall and cellular inflammation compared to those using DMARDs only. In these subjects, RA disease duration was the most important contributor to the level of arterial wall inflammation. This enhanced inflammatory state implies an increased cardiovascular risk in these patients, notwithstanding prolonged clinical remission. In view of this increased CV risk, stringent risk management may be necessary in this group, with perhaps a role for lower treatment thresholds. Finally, the increased hematopoietic activity in these patients also holds a promise for novel therapeutic strategies aimed at attenuating the inflammatory state by targeting immune cell mobilization from the bone marrow.
Acknowledgements
We sincerely thank Miranda Versloot and Alinda W. Schimmel for assistance in the monocyte studies; Dorothea A. Greven for assistance in patient recruitment; and Lianne Kraal en Marian Turkenberg-Hoogeveen for assessing disease activity scores.
Funding
This work was supported by a grant from the Netherlands Heart Foundation (CVON 2011/B019: Generating the best evidence-based pharmaceutical targets for atherosclerosis (GENIUS).
Abbreviations
- 18F-FDG
18F-fluorodeoxyglucose
- ACS
acute coronary syndrome
- BM
bone marrow
- BMI
body mass index
- CCP
cyclic citrullinated peptide
- CRP
C-reactive protein
- CT
computed tomography
- CVD
cardiovascular disease
- DAS
disease activity score
- DBP
diastolic blood pressure
- DMARD
disease-modifying antirheumatic drug
- ESR
erythrocyte sedimentation rate
- HDL
high-density lipoprotein cholesterol
- IMT
intima media thickness
- LDL
low-density lipoprotein cholesterol
- MDS
most diseased segment
- MFI
median fluorescence intensity
- MTX
methotrexate
- PET
positron emission tomography
- RA
Rheumatoid arthritis
- RF
rheumatoid factor
- ROI
regions of interest
- SBP
systolic blood pressure
- SUV
standardized uptake values
- TBR
target-to-background ratio
- TEM
transendothelial migration
- TNF
tumor necrosis factor
Additional file
Antibodies used for flow cytometry. Table S2. Univariate linear regression analysis in RA subjects. Table S3. Baseline – ex vivo experiments. Table S4. Ex vivo baseline – RA-specific characteristics. (PDF 45 kb)
Footnotes
Competing interests
ESS has received lecturing fees from Merck, Novartis, ISIS, Amgen, Sanofi-Aventis and Chiesi, none of which are related to the contents of this manuscript. MTN has received lecturing and (ad hoc) consulting fees from Abbvie, Pfizer, BMS, Roche and Janssen, none of which are related to the contents of this manuscript. All other authors declare that they have no conflict of interest and no relationships with any industry relevant to this study.
Authors’ contributions
SBM, FV, and ESS designed the study. SBM, FV, AS, LS, and JK performed the research. EK and MTN assisted in patient recruitment. JB and HV provided analytic tools. SBM, FV, and ESS drafted the manuscript; all authors critically reviewed the manuscript. All authors read and approved the final manuscript.
References
- 1.Peters MJL, van Halm VP, Voskuyl AE, Smulders YM, Boers M, Lems WF, et al. Does rheumatoid arthritis equal diabetes mellitus as an independent risk factor for cardiovascular disease? A prospective study. Arthritis Rheum. 2009;61:1571–9. doi: 10.1002/art.24836. [DOI] [PubMed] [Google Scholar]
- 2.Del Rincón ID, Williams K, Stern MP, Freeman GL, Escalante A. High incidence of cardiovascular events in a rheumatoid arthritis cohort not explained by traditional cardiac risk factors. Arthritis Rheum. 2001;44:2737–45. doi: 10.1002/1529-0131(200112)44:12<2737::AID-ART460>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- 3.Van der Heijde D, Klareskog L, Boers M, Landewé R, Codreanu C, Bolosiu HD, et al. Comparison of different definitions to classify remission and sustained remission: 1 year TEMPO results. Ann Rheum Dis. 2005;64:1582–7. doi: 10.1136/ard.2004.034371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Micha R, Imamura F, Wyler von Ballmoos M, Solomon DH, Hernán MA, Ridker PM, et al. Systematic review and meta-analysis of methotrexate use and risk of cardiovascular disease. Am J Cardiol. 2011;108:1362–70. doi: 10.1016/j.amjcard.2011.06.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Westlake SL, Colebatch AN, Baird J, Kiely P, Quinn M, Choy E, et al. The effect of methotrexate on cardiovascular disease in patients with rheumatoid arthritis: A systematic literature review. Rheumatology. 2010;49:295–307. doi: 10.1093/rheumatology/kep366. [DOI] [PubMed] [Google Scholar]
- 6.Dadoun S, Zeboulon-Ktorza N, Combescure C, Elhai M, Rozenberg S, Gossec L, et al. Mortality in rheumatoid arthritis over the last fifty years: systematic review and meta-analysis. Joint Bone Spine. 2013;80:29–33. doi: 10.1016/j.jbspin.2012.02.005. [DOI] [PubMed] [Google Scholar]
- 7.Gabriel SE. Why do people with rheumatoid arthritis still die prematurely? Ann Rheum Dis. 2008;67(3):iii30–4. doi: 10.1136/ard.2008.098038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Grigor C, Capell H, Stirling A, McMahon AD, Lock P, Vallance R, et al. Effect of a treatment strategy of tight control for rheumatoid arthritis (the TICORA study): a single-blind randomised controlled trial. Lancet. 2004;364:263–9. doi: 10.1016/S0140-6736(04)16676-2. [DOI] [PubMed] [Google Scholar]
- 9.Schipper LG, Vermeer M, Kuper HH, Hoekstra MO, Haagsma CJ, Den Broeder AA, et al. A tight control treatment strategy aiming for remission in early rheumatoid arthritis is more effective than usual care treatment in daily clinical practice: a study of two cohorts in the Dutch Rheumatoid Arthritis Monitoring registry. Ann Rheum Dis. 2012;71:845–50. doi: 10.1136/annrheumdis-2011-200274. [DOI] [PubMed] [Google Scholar]
- 10.McKellar GE, McCarey DW, Sattar N, McInnes IB. Role for TNF in atherosclerosis? Lessons from autoimmune disease. Nat Rev Cardiol. 2009;6:410–7. doi: 10.1038/nrcardio.2009.57. [DOI] [PubMed] [Google Scholar]
- 11.Popa C, Netea MG, Radstake T, Van der Meer JWM, Stalenhoef AFH, van Riel PLCM, et al. Influence of anti-tumour necrosis factor therapy on cardiovascular risk factors in patients with active rheumatoid arthritis. Ann Rheum Dis. 2005;64:303–5. doi: 10.1136/ard.2004.023119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Barnabe C, Martin B-J, Ghali WA. Systematic review and meta-analysis: anti-tumor necrosis factor α therapy and cardiovascular events in rheumatoid arthritis. Arthritis Care Res (Hoboken) 2011;63:522–9. doi: 10.1002/acr.20371. [DOI] [PubMed] [Google Scholar]
- 13.Roubille C, Richer V, Starnino T, McCourt C, McFarlane A, Fleming P, et al. The effects of tumour necrosis factor inhibitors, methotrexate, non-steroidal anti-inflammatory drugs and corticosteroids on cardiovascular events in rheumatoid arthritis, psoriasis and psoriatic arthritis: a systematic review and meta-analysis. Ann Rheum Dis. 2015;74:480–9. doi: 10.1136/annrheumdis-2014-206624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tam LS, Kitas GD, González-Gay MA. Can suppression of inflammation by anti-TNF prevent progression of subclinical atherosclerosis in inflammatory arthritis? Rheumatology (Oxford) 2014;53:1108–19. doi: 10.1093/rheumatology/ket454. [DOI] [PubMed] [Google Scholar]
- 15.Mäki-Petäjä KM, Elkhawad M, Cheriyan J, Joshi FR, Ostör AJK, Hall FC, et al. Anti-tumor necrosis factor-α therapy reduces aortic inflammation and stiffness in patients with rheumatoid arthritis. Circulation. 2012;126:2473–80. doi: 10.1161/CIRCULATIONAHA.112.120410. [DOI] [PubMed] [Google Scholar]
- 16.Lioté F, Boval-Boizard B, Weill D, Kuntz D, Wautier JL. Blood monocyte activation in rheumatoid arthritis: increased monocyte adhesiveness, integrin expression, and cytokine release. Clin Exp Immunol. 1996;106:13–9. doi: 10.1046/j.1365-2249.1996.d01-820.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ghattas A, Griffiths HR, Devitt A, Lip GYH, Shantsila E. Monocytes in coronary artery disease and atherosclerosis: where are we now? J Am Coll Cardiol. 2013;62:1541–51. doi: 10.1016/j.jacc.2013.07.043. [DOI] [PubMed] [Google Scholar]
- 18.Dutta P, Courties G, Wei Y, Leuschner F, Gorbatov R, Robbins CS, et al. Myocardial infarction accelerates atherosclerosis. Nature. 2012;487:325–9. doi: 10.1038/nature11260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Aletaha D, Neogi T, Silman AJ, Funovits J, Felson DT, Bingham CO, et al. 2010 Rheumatoid arthritis classification criteria: An American College of Rheumatology/European League Against Rheumatism collaborative initiative. Arthritis Rheum. 2010;62:2569–81. doi: 10.1002/art.27584. [DOI] [PubMed] [Google Scholar]
- 20.Felson DT, Smolen JS, Wells G, Zhang B, van Tuyl LHD, Funovits J, et al. American College of Rheumatology/European League Against Rheumatism provisional definition of remission in rheumatoid arthritis for clinical trials. Arthritis Rheum. 2011;63:573–86. doi: 10.1002/art.30129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Prevoo MLL, Van’T Hof MA, Kuper HH, Van Leeuwen MA, Van De Putte LBA, Van Riel PLCM. Modified disease activity scores that include twenty-eight-joint counts development and validation in a prospective longitudinal study of patients with rheumatoid arthritis. Arthritis Rheum. 1995;38:44–8. doi: 10.1002/art.1780380107. [DOI] [PubMed] [Google Scholar]
- 22.Rudd JHF, Myers KS, Bansilal S, Machac J, Pinto CA, Tong C, et al. Atherosclerosis inflammation imaging with 18F-FDG PET: carotid, iliac, and femoral uptake reproducibility, quantification methods, and recommendations. J Nucl Med. 2008;49:871–8. doi: 10.2967/jnumed.107.050294. [DOI] [PubMed] [Google Scholar]
- 23.Emami H, Singh P, MacNabb M, Vucic E, Lavender Z, Rudd JHF, et al. Splenic metabolic activity predicts risk of future cardiovascular events. JACC Cardiovasc Imaging. 2015;8:121–30. doi: 10.1016/j.jcmg.2014.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.van Der Valk FM, Kroon J, Potters WV, Thurlings RM, Bennink RJ, Verberne HJ, et al. In vivo imaging of enhanced leukocyte accumulation in atherosclerotic lesions in humans. J Am Coll Cardiol. 2014;64:1019–29. doi: 10.1016/j.jacc.2014.06.1171. [DOI] [PubMed] [Google Scholar]
- 25.Figueroa AL, Abdelbaky A, Truong QA, Corsini E, MacNabb MH, Lavender ZR, et al. Measurement of arterial activity on routine FDG PET/CT images improves prediction of risk of future CV events. JACC Cardiovasc Imaging. 2013;6:1250–9. doi: 10.1016/j.jcmg.2013.08.006. [DOI] [PubMed] [Google Scholar]
- 26.Saleem B, Keen H, Goeb V, Parmar R, Nizam S, Hensor EM, et al. Patients with RA in remission on TNF blockers: when and in whom can TNF blocker therapy be stopped? Ann Rheum Dis. 2010;69:1636–42. doi: 10.1136/ard.2009.117341. [DOI] [PubMed] [Google Scholar]
- 27.Masuda H, Miyazaki T, Shimada K, Tamura N, Matsudaira R, Yoshihara T, et al. Disease duration and severity impacts on long-term cardiovascular events in Japanese patients with rheumatoid arthritis. J. Cardiol. 2014;64:366–70. doi: 10.1016/j.jjcc.2014.02.018. [DOI] [PubMed] [Google Scholar]
- 28.Gabriel SE, Crowson CS, Kremers HM, Doran MF, Turesson C, O’Fallon WM, et al. Survival in rheumatoid arthritis: A population-based analysis of trends over 40 years. Arthritis Rheum. 2003;48:54–8. doi: 10.1002/art.10705. [DOI] [PubMed] [Google Scholar]
- 29.Sattar N, McCarey DW, Capell H, McInnes IB. Explaining how “high-grade” systemic inflammation accelerates vascular risk in rheumatoid arthritis. Circulation. 2003;108:2957–63. doi: 10.1161/01.CIR.0000099844.31524.05. [DOI] [PubMed] [Google Scholar]
- 30.Solomon DH, Reed GW, Kremer JM, Curtis JR, Farkouh ME, Harrold LR, et al. Disease activity in rheumatoid arthritis and the risk of cardiovascular events. Arthritis Rheumatol. 2015;67:1449–55. doi: 10.1002/art.39098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Brown AK, Quinn MA, Karim Z, Conaghan PG, Peterfy CG, Hensor E, et al. Presence of significant synovitis in rheumatoid arthritis patients with disease-modifying antirheumatic drug-induced clinical remission: evidence from an imaging study may explain structural progression. Arthritis Rheum. 2006;54:3761–73. doi: 10.1002/art.22190. [DOI] [PubMed] [Google Scholar]
- 32.van Vollenhoven RF, Nagy G, Tak PP. Early start and stop of biologics: has the time come? BMC Med. 2014;12:25. doi: 10.1186/1741-7015-12-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rogacev KS, Cremers B, Zawada AM, Seiler S, Binder N, Ege P, et al. CD14++CD16+ monocytes independently predict cardiovascular events: A cohort study of 951 patients referred for elective coronary angiography. J Am Coll Cardiol. 2012;60:1512–20. doi: 10.1016/j.jacc.2012.07.019. [DOI] [PubMed] [Google Scholar]
- 34.Han KH, Tangirala RK, Green SR, Quehenberger O. Chemokine receptor CCR2 expression and monocyte chemoattractant protein-1-mediated chemotaxis in human monocytes. A regulatory role for plasma LDL. Arterioscler Thromb Vasc Biol. 1998;18:1983–91. doi: 10.1161/01.ATV.18.12.1983. [DOI] [PubMed] [Google Scholar]
- 35.Boring L, Gosling J, Cleary M, Charo IF. Decreased lesion formation in CCR2−/− mice reveals a role for chemokines in the initiation of atherosclerosis. Nature. 1998;394:894–7. doi: 10.1038/29788. [DOI] [PubMed] [Google Scholar]
- 36.Gower RM, Wu H, Foster GA, Devaraj S, Jialal I, Ballantyne CM, et al. CD11c/CD18 expression is upregulated on blood monocytes during hypertriglyceridemia and enhances adhesion to vascular cell adhesion molecule-1. Arterioscler Thromb Vasc Biol. 2011;31:160–6. doi: 10.1161/ATVBAHA.110.215434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tacke F, Randolph GJ. Migratory fate and differentiation of blood monocyte subsets. Immunobiology. 2006;211:609–18. doi: 10.1016/j.imbio.2006.05.025. [DOI] [PubMed] [Google Scholar]
- 38.Emami H, Singh P, MacNabb M, Figueroa A, Abdelbaky A, Nasir K, et al. Increased bone barrow metabolic activity is associated with increased risk of future cardiovascular events. J Am Coll Cardiol. 2014;63:A976. doi: 10.1016/S0735-1097(14)60976-1. [DOI] [Google Scholar]
- 39.Nahrendorf M, Frantz S, Swirski FK, Mulder WJM, Randolph G, Ertl G, et al. Imaging systemic inflammatory networks in ischemic heart disease. J Am Coll Cardiol. 2015;65:1583–91. doi: 10.1016/j.jacc.2015.02.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Skeoch S, Bruce IN. Atherosclerosis in rheumatoid arthritis: is it all about inflammation? Nat Rev. Rheumatol. 2015;11:390–400. doi: 10.1038/nrrheum.2015.40. [DOI] [PubMed] [Google Scholar]
- 41.Zhang J, Chen L, Delzell E, Muntner P, Hillegass WB, Safford MM, et al. The association between inflammatory markers, serum lipids and the risk of cardiovascular events in patients with rheumatoid arthritis. Ann Rheum Dis. 2014;73:1301–8. doi: 10.1136/annrheumdis-2013-204715. [DOI] [PubMed] [Google Scholar]
- 42.Emami H, Vijayakumar J, Subramanian S, Vucic E, Singh P, MacNabb MH, et al. Arterial 18F-FDG uptake in rheumatoid arthritis correlates with synovial activity. JACC Cardiovasc Imaging. 2014;7:959–60. doi: 10.1016/j.jcmg.2014.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]