

Abstract
Background
Lytic polysaccharide monooxygenases (LPMOs) belong to the “auxiliary activities (AA)” enzyme class of the CAZy database. They are known to strongly improve the saccharification process and boost soluble sugar yields from lignocellulosic biomass, which is a key step in the efficient production of sustainable economic biofuels. To date, most LPMOs have been characterized from terrestrial fungi, but novel fungal LPMOs isolated from more extreme environments such as an estuary mangrove ecosystem could offer enzymes with unique properties in terms of salt tolerance and higher stability under harsh condition.
Results
Two LPMOs secreted by the mangrove-associated fungus Pestalotiopsis sp. NCi6 (PsLPMOA and PsLPMOB) were expressed in the yeast Pichia pastoris and produced in a bioreactor with >85 mg L−1 for PsLPMOA and >260 mg L−1 for PsLPMOB. Structure-guided homology modeling of the PsLPMOs showed a high abundance of negative surface charges, enabling enhanced protein stability and activity in the presence of sea salt. Both PsLPMOs were activated by a cellobiose dehydrogenase (CDH) from Neurospora crassa, with an apparent optimum of interaction at pH 5.5. Investigation into their regioselective mode of action revealed that PsLPMOA released C1- and C4-oxidized cello-oligosaccharide products, while PsLPMOB released only C4-oxidized products. PsLPMOA was found to cleave polymeric cellulose in the presence of up to 6 % sea salt, which emphasizes the use of sea water in the industrial saccharification process with improved ecological footprints.
Conclusions
Two new LPMOs from the mangrove fungus Pestalotiopsis sp. NCi6 were found to be fully reactive against cellulose. The combined hydrolytic activities of these salt-responsive LPMOs could therefore facilitate the saccharification process using sea water as a reaction medium for large-scale biorefineries.
Electronic supplementary material
The online version of this article (doi:10.1186/s13068-016-0520-3) contains supplementary material, which is available to authorized users.
Keywords: AA9, LPMO, Cellobiose dehydrogenase, Cellulose, Biorefinery
Background
Pessimistic forecasts of fossil-fuel reserves and the environmental impacts of oil production and use have spurred extensive worldwide research into the conversion of different local lignocellulosic biomasses as alternative sustainable sources for biofuel production [1–4]. Lignocellulosic plant cell walls are very resistant to degradation, as cellulose and hemicellulose are embedded into a lignin matrix which limits the ability of cellulases, the major hydrolytic enzymes in the saccharification process, to access their sites of action. The term “cellulases” covers a cocktail of enzymes that are exploited for the deconstruction or modification of plant biomass within the biorefinery concept [4–6]. However, cost-effective use of such enzyme cocktails is a limiting step for the production of biofuels and commodity chemicals, making the discovery of efficient and more robust enzymes from biodiversity a necessary challenge.
Copper-dependent lytic polysaccharide monooxygenases (LPMOs) are emerging as important components of the cellulase cocktail, [7–9], where they have even been described as “cellulase boosters” [10]. Recent intensive efforts have started to unravel their function in the oxidative degradation of cellulose [11–14] and other plant polysaccharides such as hemicellulose [9] and starch [15, 16]. LPMOs are currently classified as “auxiliary activities (AA)” within 4 families of the Carbohydrate Active Enzyme (CAZy) database [17, 18]: the fungal AA9, formerly known as glycosyl hydrolases 61; the bacterial AA10, formerly known as carbohydrate-binding modules 33 [17]; the newly characterized AA11 [19]; and the starch-oxidative LPMOs of AA13 [16]. Although the CAZy database contains more than 200 AA9 proteins, less than 20 have been enzymatically characterized and shown to oxidatively cleave the β-(1→4) glycosidic bonds of cellulose [20, 21]. AA9 LPMOs share a common structural fold with a flat substrate-binding surface, a conserved N-terminal histidine residue involved in the coordination of an essential copper ion, and the dependency of the activity on the presence of an electron donor. The reaction mechanism of the majority of the characterized LPMOs involves oxidation of the C1 carbon of a glucose molecule, leading to the formation of an aldonic acid and a break in the cellulose chain, whereas other members of the AA9 family have been shown to generate oxidation at C4 [20, 22]. AA9 LPMOs can be split into three classes on the basis of preferred site of oxidation, i.e., type 1 oxidizing at C1, type 2 oxidizing at C4, and type 3 oxidizing at both the C1 and C4 carbon atoms of glucose [15]. The oxidative cleavage performed by LPMOs occurs in the presence of small redox-active molecules such as ascorbic acid, reduced glutathione, or gallate [8, 11, 23]. The peculiarity of fungal AA9 LPMOs is their action in concert with cellobiose dehydrogenases (CDHs) that results in redox-mediated glycosidic bond cleavage in cellulose and points to a key role of this oxidative system in fungi [21, 24, 25].
Many LPMOs have been successfully produced heterologously from fungi, but few reports have detailed their biochemical characterization. One reason is the lack of a fast direct activity test. Kittl et al. [26] found that the Amplex Red assay can be modeled for biochemical characterization of LPMOs based on the generation of hydrogen peroxide as a side-reaction of the enzyme which is dependent on the availability of a suitable reductant for the LPMO type-2 copper center.
For the sustainable success of emerging biorefineries, apart from the need of novel biocatalytic systems another important aspect to consider is the large water consumption that lignocellulose biomass-processing plants will require at large-scale operations [27]. The assessment of non-potable water resources as reaction media for such biorefineries appears to be a promising field of research, which can result in integrative systems [28]. From these viewpoints, novel bio-catalysts should demonstrate its reactivity in crude aqueous media such as seawater. The marine fungi genera Pestalotiopsis sp. have lignocellulolytic enzymes of interest in terms of high salt tolerance and higher stability under harsh conditions [29]. Hence, we selected two putative lpmo genes from Pestalotiopsis sp. NCi6 (Ps) [30] for heterologous expression in the yeast Pichia pastoris. Their regioselectivity was tested on cellulose in the presence and the absence of sea salt for the possible application in the degradative processes of plant cell wall in saline/non-saline environment.
Results and discussion
Gene expression and batch-scale production of LPMOs
Our previous transcriptome analysis of the mangrove fungus Pestalotiopsis sp. NCi6 identified five putative lpmo sequences, two of which were strictly secreted under saline growth conditions. Three other candidates, one having a CBM, were found in both saline and non-saline growth conditions [30]. Among them, lpmoa (only secreted in saline conditions) and lpmob (secreted in saline/non-saline conditions) were chosen for heterologous production in P. pastoris. The two genes were selected based on the following criteria: (1) the corresponding proteins were secreted by Pestalotiopsis sp. NCi6 when grown on mangrove wood [30]; (2) one protein was strictly induced in saline conditions (PsLPMOA), while the second protein was present in both saline and non-saline growth conditions (PsLPMOB) [30]; (3) the amino acid sequences are diverse among each other and compared to previously characterized LPMOs (Additional file 1: Figure S1). The selected PsLPMOs have a sequence identity of only 40 % and belong to two separate phylogenetic branches, although they share a strictly conserved copper-binding site which consists of two histidines (one at N-terminal position) and one tyrosine. The codon-optimized genes encoding PsLPMOA and PsLPMOB were inserted into the pPICZαA expression vector for subsequent expression in the yeast P. pastoris. The best transformants that showed high protein secretion and the presence of masses corresponding to the expected molecular mass of PsLPMOA and PsLPMOB (as estimated by SDS-PAGE) were selected for production in 500-mL submerged cultures under agitation. A 2-L bioreactor was chosen for a scale-up protein production. Fermentation of the PsLPMOA-producing transformant was performed with 1.5 % sea salt in the batch cultivation medium to get active enzyme, whereas PsLPMOB was cultivated without sea salt. These cultivations were monitored for wet biomass, extracellular protein concentration (Fig. 1). LPMO secretion was verified by SDS-PAGE (data not shown). After the batch and glycerol fed-batch phases for biomass build-up, wet biomass concentrations in the fermentations of PsLPMOA and PsLPMOB reached 200 and 130 g L−1, respectively. Both bioreactors were harvested after around 118 h with protein levels of 0.4 and 1.0 g L−1 for PsLPMOA and PsLPMOB, respectively. These protein yields are comparable with other previously characterized LPMOs from terrestrial fungi [21, 25].
Fig. 1.
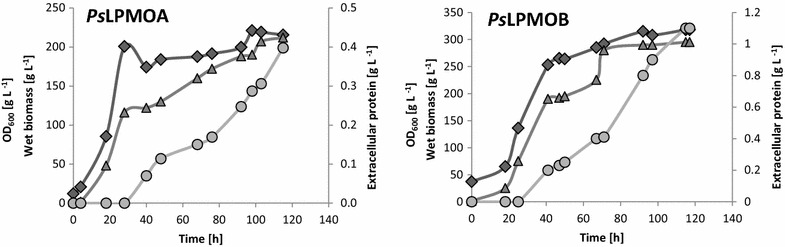
Production of PsLPMOs in P. pastoris. Squares wet biomass; triangles optical density (OD); circles extracellular protein concentration
The culture supernatants were harvested and purified by single-step affinity chromatography (Table 1). PsLPMOB was purified to homogeneity, whereas PsLPMOA showed two bands on SDS-PAGE (Additional file 1: Figure S2), suggesting that two isoforms were co-produced. This could be a result of varying protein processings, such as a difference in glycosylation. SDS-PAGE analysis showed that both proteins are heavily glycosylated resulting in diffuse bands of considerably larger sizes than the masses expected from the amino acid sequences. Deglycosylation with PNGase F under denaturing conditions was performed, but no visible effect on the molecular mass or appearance of the proteins on SDS-PAGE was observed for PsLPMOB (Additional file 1: Figure S2). This is consistent with the fact that no N-glycosylation sites were predicted for PsLPMOB. Conversely, PsLPMOA possesses two N-glycosylation sequences, and deglycosylation with PNGase F showed a clear shift yielding a unique band, the upper band corresponding to the PNGase F. The terminal sequence of the PsLPMOA, as determined following an Edman degradation on the purified protein, corresponded to HYTFPD, indicating that this protein was correctly matured in P. pastoris. On the other hand, the N-terminal sequence of PsLPMOB showed a mix of two sequences, EAEAHTI (60 % abundance) and HTI (40 %), revealing that the main portion of the protein was not correctly processed. The presence of the N-terminal histidine is essential because it coordinates the active-site copper ion, which is involved in oxygen activation to cleave glycosidic bonds in cellulose. An additional amino acid at the N-terminus, as was observed for PsLPMOB (Glu, Ala, Glu, Ala) would most likely render the protein inactive [31]. Immunodetection of the PsLPMOs was performed using antibodies raised against the poly-HIS tag. Western blotting showed bands corresponding to the mass of proteins, demonstrating that PsLPMOA and PsLPMOB recombinant proteins were not truncated at the C-terminus.
Table 1.
Purification steps using immobilized metal affinity chromatography (IMAC) of recombinant LPMOs
| Purification | Volume (mL) | Total activity (U) | Protein (mg) | Specific activity (U g−1) | Activity yield | Purification factor (fold) | |
|---|---|---|---|---|---|---|---|
| PsLPMOA | Culture medium | 1500 | 1.2 | 495 | 2.42 | 100 | 1.0 |
| IMAC | 2.7 | 0.51 | 130 | 3.92 | 42.5 | 1.6 | |
| PsLPMOB | Culture medium | 1060 | 0.25 | 1160 | 0.22 | 100 | 1.0 |
| IMAC | 1.9 | 0.62 | 285 | 2.18 | 248.0 | 9.9 |
Structural basis of salt tolerance
Salt-adapted hydrolases typically contain a high number of negative surface charges that enable enhanced protein stability and activity in extreme osmolytic habitats [32, 33]. To test whether this feature also applies to the PsLPMOs, structure-guided homology models were generated and their surface charge distribution was compared to LPMOs from other fungal origins. The overall topologies of the homology models bear the hallmarks of cellulose-active LPMOs including a central β-sandwich fold and a partially solvent-exposed active-site copper chelated by a histidine brace (Additional file 1: Figure S3). The “flat face” of LPMOs, which is oriented toward the cellulose substrate during catalysis, seems to contain a fairly balanced number of positive and negative surface charges in all investigated structures (Fig. 2). The residual solvent-exposed surfaces of PsLPMOA and PsLPMOB, however, showed a high abundance of negative charges relative to LPMO9F from Neurosporacrassa [34] and Hypocrea jecorina Cel61B [35]. PsLPMOA and PsLPMOB showed a 4.8-times and 4.3-times higher abundance of negative amino acids (D + E) over positively charged amino acids (R + K). None of the 21 LPMO sequences analyzed along with the PsLPMOs showed an equally high ratio of (D + E)/(R + K), although the sequences of the thermophilic fungi Thermoascus aurantiacus [12] (ratio 2.7) and Myriococcum thermophilum MYCTH112089 [34] (ratio 2.0) as well as LPMO GH61D from the mesophilic basidiomycete Phanerochaete chrysosporium [36] (ratio 2.4; Fig. 2) showed a significantly higher number of Asp and Glu residues than other non-halophilic LPMOs (Additional file 1: Table S1). These examples illustrate that dominance of negative surface charges is a strong indication for adaption to saline conditions, but is sometimes also observed in LPMOs from non-halophilic organisms. PsLPMOs tend to have lower isoelectric points (pI) than other fungal LPMOs. The experimentally determined pIs of PsLPMOA and PsLPMOB were between pH 4.2 and 4.7, which is in good agreement with the calculated pIs based on the amino acid composition.
Fig. 2.
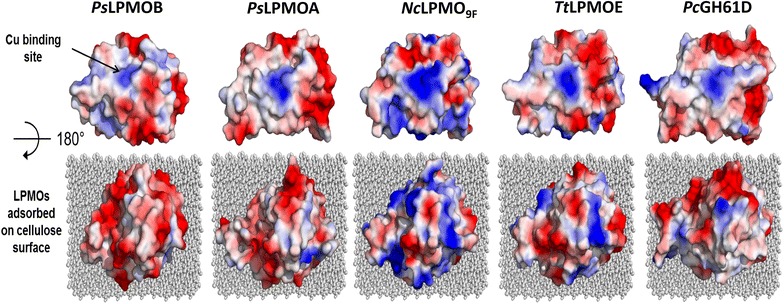
Surface charge plots of PsLPMOA and PsLPMOB, N. crassa LPMO9F (UniProt Q1K4Q1 [34]), H. jecorina CEL61B (UniProt: Q7Z9M7 [35]), and P. chrysosporium GH61D (UniProt: BAL43430 [36]). Top panels illustrate the copper-binding “histidine brace” on the flat cellulose-binding patch (indicated by arrows). LPMOs adsorbed on cellulose (gray sheets) are shown in the lower panels. Surface charge distribution is shown in color code (red negative charges; blue positive charges). The surface potentials were calculated using the vacuum electrostatics function of the PyMOL molecular graphics system (Schrödinger, New York, NY)
Activity determination, pH, and temperature stabilities
The high abundance of negative surface charges of the PsLPMOs relative to other LPMOs had no apparent effect on the interaction with the CDHs from N. crassa. Initial characterization of the recombinant PsLPMOs was carried out by measuring their capacity to produce H2O2 in the absence of a cellulosic substrate. We found that the two PsLPMOs were readily reduced by both N. crassa CDHs (NcCDHIIA, UniProt: Q7RXM0 and NcCDHIIB, UniProt: Q7S0Y1), indicating productive interactions. In addition, the reducing agent ascorbic acid also gave a concentration-dependent signal with the PsLPMOs. Oxygen turnover by PsLPMOs was routinely measured in combination with NcCDHIIA using its native substrate cellobiose. Specific oxygen-reducing activities of 3.9 and 2.2 U g−1 were determined for PsLPMOA and PsLPMOB, respectively. These values are in the same range as previously published LPMOs from terrestrial fungi [21, 26]. The lower specific activity of PsLPMOB could be related to the fact that only 40 % of the enzyme is correctly processed. The effects of pH and temperature on enzyme stabilities were determined within a pH range from 4.0–8.0. The pH-dependent activities for both PsLPMOs showed bell-shaped curves with optima at pH 5.5 (Fig. 3a). Both enzymes retained more than 50 % residual activity between pH 5.0 and pH 6.5. This is in good agreement with the pH-dependence of NcCDHIIA for artificial one- and two-electron acceptors [24]. At more acidic pH-values below 4.5, no activity could be detected. This could be related to the low pIs of both PsLPMOs. The high density of negative charges on their surfaces could result in substantial charge repulsion at the CDH/LPMO interfaces at low pH and potentially prevent effective electron exchange, as was shown for the internal cofactor-to-cofactor electron transfer of a CDH from M. thermophilum [34]. The effect of sea salt (3.5 % w/v) on the thermal stability of the PsLPMOs was examined at 50 °C. As shown in Additional file 1: Figure S4, sea salt had no noticeable effect on PsLPMOA, which showed similar half-lives under all experimental conditions, but a strong negative effect on PsLPMOB, which showed half-lives shortened below 5 min. Temperature stabilities of PsLPMOA and PsLPMOB measured in the absence of sea salt are shown in Fig. 3b-1, b-2, respectively. Both enzymes were stable at 30 and 40 °C, retaining 100 % activity for 150 min and approximately 80 % after 1000 min of incubation. PsLPMOB was considerably stable at 50 and 60 °C, whereas PsLPMOA showed fast degradation at 60 °C with an estimated half-life of 2 min. At 50 °C, PsLPMOA had a ten-fold shorter half-life than PsLPMOB. To the best of our knowledge, pH-dependent activities and temperature-profiles of LPMOs have not been reported so far. The thermal stability of four N. crassa LPMOs was evaluated in a previous study using differential scanning calorimetry, which showed similar transition midpoint temperatures ranging from 63.0–68.9 °C for all enzymes [26].
Fig. 3.
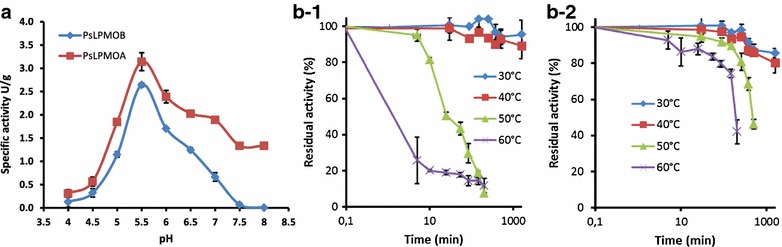
Amplex Red-based PsLPMOs characterization. a pH profiles of PsLPMOs (red line PsLPMOA; blue line PsLPMOB). b-1 Temperature stability of PsLPMOA and b-2 temperature stability of PsLPMOB (diamonds 30 °C; squares 40 °C; triangles 50 °C; stars 60 °C)
Cellulose degradation capacity and oxidative regioselectivity of LPMOs
To investigate the regioselectivity of the PsLPMOs, activity assays were performed on phosphoric acid swollen cellulose (PASC) in the presence of ascorbic acid as electron donor. Product profiles were resolved by high-performance anion-exchange chromatography (HPAEC) as described in previous works [21, 37]. Figure 4a shows HPAEC elution pattern of a released mixture of non-oxidized and oxidized soluble oligosaccharides for PsLPMOA. The degrees of polymerization (DP) of the products ranged from DP2 to DP5 for both non-oxidized and oxidized oligosaccharides. C1-oxidized oligosaccharides with DPs from two to five were observed in the assay on PsLPMOA, indicating that its action on cellulose yields C1-oxidized oligosaccharides. Generated lactones spontaneously hydrolyze to aldonic acids in bulk water. In 24-h reactions, we also observed a small peak corresponding to C1–C4 oxidation at around 42–44 min in the chromatogram for PsLPMOA, while PsLPMOB showed no oxidized oligosaccharide products at similar reaction time. Prolonging the reaction time from 24 to 72 h to clarify the regioselectivity of LPMOs resulted in C1-oxidized oligosaccharides and other peaks which eluted at 27, 37, and 40 min for PsLPMOA (zoom panel, Fig. 4a), while only the three later peaks were observed for PsLPMOB, as shown in Fig. 4b. These latter peaks correspond to the C4-oxidized species as was confirmed by Bennati-Granier et al. [21] using Mass Spectrometry analysis. The soluble fraction of the 72 h reaction from PsLPMOs was further treated with NcCDHIIA to oxidize the reducing (C1) ends of the C4-oxidized cello-oligosaccharides. Analysis showed a loss of peaks probably corresponding to C4-oxidized species and an increase in peaks at later retention times (Fig. 4). For both PsLPMOA and PsLPMOB, addition of NcCDHIIA led to an increase of C1-oxidized oligosaccharides and a clear change of products eluting after 25 min. Indeed, oxidized species at 27, 37, and 40 min (C4-oxidized species) vanished with the concomitant appearance of new peak at around 42–44 min, which based on the analysis [21, 22] may correspond to a C1/C4-double-oxidized DP2 and longer double-oxidized products, respectively. In conclusion, these experiments confirm that oxidative cleavage of cellulose occurs at both C1 and C4 for PsLPMOA but only at C4 for PsLPMOB. Comparison with results from similar experiments [21, 22, 25] shows that the purified proteins are active LPMOs and emphasizes the importance of CDH as electron donor. It has been suggested that the presence of native cellodextrins could originate from oxidative cleavages near the reducing ends releasing an intact non-reducing moiety [7, 11]. PsLPMOB produced mainly peaks corresponding to C4-oxidized cello-oligosaccharides, whereas PsLPMOA produced doubly oxidized compounds corresponding to C4 oxidation (4-keto or gem-diol sugar) and C1 oxidation (aldonic acid). Furthermore, we showed that PsLPMOB falls into the class of type-2 LPMOs (C4 oxidation), which fits with the regioselectivity pattern previously predicted based on phylogenetic analysis [15]. However, ionic chromatography evidenced that PsLPMOA behaves as a type-3 LPMO (oxidation at the C1 and C4 positions), although it is predicted to be a type-1 LPMO (oxidation at C1 position only); suggesting that this classification based on sequence alignments may be unable to depict the full scope of action in LPMOs. It is therefore essential to note that AA9s show low overall sequence identities across the entire family [20, 21, 38].
Fig. 4.
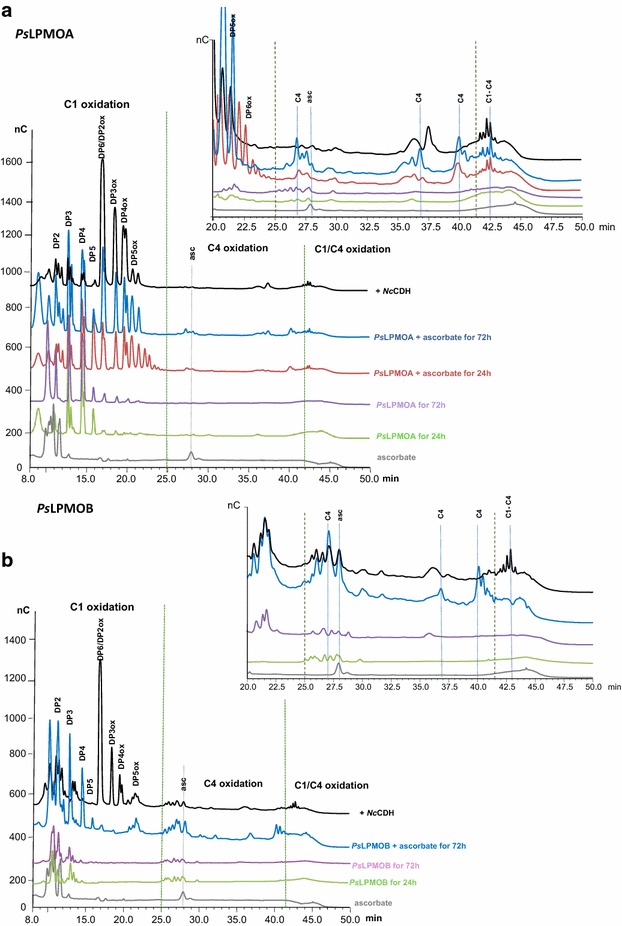
Analysis of oxidized products generated from cellulose by action of PsLPMOs. a HPAEC chromatograms of the oligosaccharides products released by action of PsLPMOA. b HPAEC chromatograms of the oligosaccharides products released by action of PsLPMOB. The peak annotations are based on comparison with oligosaccharide standards oxidized at the C1 position (DP2ox-DP5ox). Peaks eluting at 27, 37, and 41 min are annotated with dotted lines. Controls ascorbate (gray line), PsLPMO with PASC for 24 h (green line) or 72 h (purple line). Reaction samples PASC (0.1 %) with 4.4 µM PsLPMOs in the presence of 1 mM ascorbate, at 50 °C for 24 h (blue line) or 72 h (red line), and same reaction followed by the incubation with NcCDHIIA at 50 °C for 16 h (black line)
Cellulose degradation in the presence of sea salt
As PsLPMOA was produced under saline conditions, we used a cellulose cleavage assay to test the effect of sea salt on the performance of this enzyme. Activity on PASC with ascorbate as the electron donor was measured by HPAEC in presence of different concentrations of sea salt. Figure 5 shows that both PsLPMOs were fully active in the presence of up to 3.5 % (w/v) of sea salt. Both the HPAEC chromatograms showed slight shifts of oxidized product peak retention times due to the presence of sea salt in the samples. Nevertheless, significant amounts of C1- and C4-oxidized oligosaccharides were detected in the presence and the absence of sea salt for PsLPMOA, whereas (only) C4-oxidized oligosaccharides were detected for PsLPMOB. Furthermore, PsLPMOA remained active even at 6.0 % (w/v) sea salt (Additional file 1: Figure S5), whereas PsLPMOB showed no activity at this sea salt concentration. These results are contrasted by the peroxide-detecting Amplex Red assays (Additional file 1: Figure S3), which showed fast inactivation of PsLPMOB in the presence of 3.5 % (w/v) sea salt (Additional file 1: Figure S3). These assays, however, were performed in the absence of a suitable substrate for LPMO, and may therefore indicate that binding of LPMO to polysaccharide surfaces contributes to the overall stability of the enzyme.
Fig. 5.
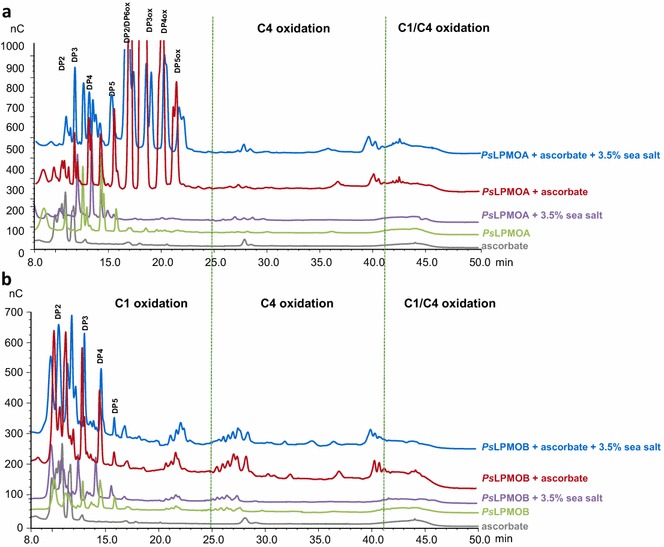
HPAEC chromatogram showing products released in the presence and absence of sea salt by action of PsLPMOs. a Cellulose degradation products by action of PsLPMOA. b Cellulose degradation products by action of PsLPMOB. The peak annotations are based on comparison with oligosaccharide standards oxidized at the C1 position (DP2ox-DP5ox). Controls ascorbate (gray line), PsLPMO with PASC without sea salt for 72 h (green line), or with 3.5 % sea salt (purple line). Reaction samples PASC (0.1 %) with 4.4 µM PsLPMOs with 1 mM ascorbate in the presence of 3.5 % sea salt, at 50 °C for 72 h (blue line) and same reaction without sea salt (red line)
Conclusions
Pestalotiopsis sp. NCi6 contains a complete arsenal of cellulolytic enzymes and represents an interesting model organism for studying the oxidative deconstruction of lignocellulose in extreme halophilic habitats. Here, we successfully produced two PsLPMOs (PsLPMOA and PsLPMOB) and characterized them in depth. The Amplex Red assay was found to be an easy and fast assay to determine biochemical properties and behavior patterns in the heterologously produced LPMOs. Furthermore, both enzymes show a highly reactive and more stable activity at 50 °C for cellulose degradation. PsLPMOA gave doubly oxidized compounds corresponding to C4 oxidation (4-keto or gem-diol sugar) and C1 oxidation (aldonic acid), while PsLPMOB showed C4-oxidized cello-oligosaccharides products. This study sheds light on the molecular and structural basis for halotolerance in enzymes, raising prospects for engineering this characteristic to suit industrial biofuel production needs. Using salt-responsive PsLPMOA could allow the use of sea water in biorefineries.
Methods
Chemicals, gene sequence, and microorganisms
All chemicals were of the highest purity grade available and were purchased from Sigma-Aldrich (Saint-Quentin-Fallavier, France) unless stated otherwise. Amplex Red (10-acetyl-3,7-dihydroxyphenoxazine) was purchased from VWR (Fontenay-sous-Bois, France). Restriction endonucleases and T4 DNA ligase were obtained from Fermentas-Thermo Fisher Scientific (Illkirch, France) and used as recommended by the manufacturer. Putative lpmo nucleotide sequences were identified in a transcriptome analysis of the mangrove fungus Pestalotiopsis sp. NCi6 [30]. Two of these sequences, pslpmoa (Genebank ID KR825270) and pslpmob (Genebank ID KR825269), were selected for expression. The cDNAs of pslpmoa and pslpmob were artificially synthesized with their native signal sequences and were codon-optimized for expression in P. pastoris. N. crassa CDH (IIA and IIB) was produced and purified as previously reported [24].
Cloning and protein production of Pestalotiopsis sp. NCi6 LPMOs
The synthetic pslpmoa and pslpmob genes were digested with the restriction enzymes XhoI and XbaI and ligated into the equally treated vector pPICZα A (Invitrogen, Cergy-Pontoise, France) using the Rapid DNA Ligation Kit (Fermentas). The procedures resulted in plasmids carrying genes encoding proteins with their native signal sequences cloned under the control of the methanol-inducible AOX1 promoter. C-terminal 6× histidine tags for purification were included. The plasmids were linearized with the restriction enzyme SacI and transformed into electro-competent P. pastoris cells. Transformants were grown on YPD plates (10 g L−1 yeast extract, 20 g L−1 peptone, 10 g L−1 glucose, 15 g L−1 agar) containing 100 mg L−1 Zeocin. Zeocin-resistant P. pastoris transformants were then screened for protein production by the platform method [39]. The best-producing transformant was grown in 1 L of BMGY containing 1 mL L−1 of PTM4 salts (2 g L−1 CuSO4·5H2O; 3 g L−1 MnSO4·H2O; 0.2 g L−1 Na2MoO4·2H2O; 0.02 g L−1 H3BO3; 0.5 g L−1 CaSO4·2H2O; 0.5 g L−1 CaCl2; 12.5 g L−1 ZnSO4·7H2O; 22 g L−1 FeSO4·7H2O; biotin 0.2 g L−1; conc. H2SO4 1 mL L−1) in shaken flasks at 30 °C in an orbital shaker (200 rpm) for 16 h to an OD600 of 2–6. Gene expression was induced by transferring cells into 200 mL BMMY containing 1 mL L−1 of PTM4 salts at 25 °C in an orbital shaker (200 rpm) for another 3 days. The medium was supplemented with fresh 3 % (v/v) methanol every day.
Recombinant production of LPMOs in a bioreactor and protein purification
The batch phase was performed in a 2-L bioreactor (MBR Electronics, Wald, Switzerland) with a starting volume of 1.2 L at 30 °C with the agitation set to 600 rpm. pH was kept constant at 5.0 using ammonium hydroxide (28 % v/v). Total O2 flow was between 0.1 and 0.2 vvm throughout the batch phase. The fermentations were started by adding 100 mL preculture grown overnight on YPD medium in 1-L baffled shaking flasks at 150 rpm and 30 °C. The cultivations were performed according to Invitrogen’s Pichia fermentation process guidelines with some alterations. After depletion of glycerol in the batch medium, the fed-batch phase was started with a constant feed of 36 mL h−1 50 % glycerol containing 12 mL L−1 PTM1 trace salts overnight to increase biomass. For induction, the feed was switched to 100 % methanol containing 12 mL L−1 PTM1 trace salts at an initial feed rate of 12 mL h−1 until the culture was fully adapted to methanol. After induction, cultivation temperature was reduced to 25 °C. Dissolved oxygen in the bioreactor was held at 20 %. Samples were taken twice a day at 6 h intervals to measure wet biomass, protein concentration, and OD600.
The fermentation broths were centrifuged at 6000×g for 30 min and 4 °C, and sodium hydroxide (2 M) was used to adjust the pH to 7.8. An additional centrifugation step was performed before loading the clear supernatant onto a 64-mL Nickel-chelated ion metal affinity column (GE Healthcare Life Sciences, Marlborough, MA) equilibrated with 50 mM Tris–HCl, pH 7.8 containing 150 mM NaCl and 10 mM imidazole. (His)6-tagged enzymes were eluted within a linear gradient from 0 to 50 % of 500 mM imidazole within 3 column volumes, and 10-mL fractions were collected. Fractions containing recombinant enzymes were pooled, concentrated, and dialyzed against 50 mM potassium phosphate buffer pH 6.0 and stored at 4 °C. Bulk fractions were concentrated by diafiltration using a Vivaflow 50 cross-flow module (Sartorius, Göttingen, Germany) with a polyether-sulfone membrane and a cut-off of 10 kDa. Subsequently, fractions were further concentrated by Amicon centrifugation tubes (Millipore, Guyancourt, France) (cut-off 10 kDa, 3200×g for 15 min at 4 °C).
Protein analysis and Western blotting
Protein contents of crude preparations or partially purified fractions were determined by the Bradford dye-binding method using a pre-fabricated assay (Biorad Laboratories, Marnes-la-Coquette, France) and BSA as calibration standard. Protein concentrations of purified enzymes were measured based on their molar absorption coefficients calculated from the mature amino acid sequence using ProtParam (http://www.web.expasy.org/protparam/). The molar absorption coefficients of PsLPMOA and PsLPMOB at 280 nm are 30.62 and 33.14 mM−1 cm−1, respectively. Concentrations of NcCDHIIA and NcCDHIIB were determined at 420 nm (103 and 87 mM−1 cm−1, respectively). Protein purification steps and fermentation progress were tracked by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) (Bio-Rad Laboratories). Protein bands were visualized with Coomassie Brilliant Blue R-250. The molecular mass under denaturing conditions was determined with reference standard proteins (PageRuler Prestained Protein Ladder, Thermo Fisher Scientific). All procedures followed the manufacturer’s recommendations (Bio-Rad Laboratories). Electrophoresed proteins were electroblotted onto polyvinylidene difluoride membranes following the manufacturer’s procedure (iBlot, Life Technologies, Saint-Aubin, France) [40].
LPMO activity based on H2O2 analysis
The futile oxygen-reducing activity of LPMOs in the absence of a cellulosic substrate was used as proxy for enzyme activity. The assay is based on the peroxidase-dependent conversion of Amplex Red to the fluorescent resorufin and allows the time-resolved quantification of peroxide. The reaction stoichiometry (H2O2:Resorufin) is 1:1. [26]. Assays were performed in 96-well black polystyrene plates (total volume 200 µL) using a Perkin Elmer EnSpire Multimode plate reader (Perkin Elmer, Waltham, MA). An experimentally determined excitation wavelength of 569 nm and an emission wavelength of 585 nm were used. All reactions were performed in 100 mM sodium phosphate buffer, pH 5.5, containing 50 μM Amplex Red, 7.1 U mL−1 horseradish peroxidase with either 30 µM ascorbate, or 0.4 µM NcCDH at 22 °C. We used 500 µM of cellobiose as an electron donor for NcCDH. In reference experiments without LPMO, the background signal (H2O2 production by NcCDH) was measured and subtracted from the assays. To determine the pH profile for PsLPMOs, the signal intensity of resorufin fluorescence at different pH-values (pH 4.0–8.0) was determined by adding assorted concentrations of H2O2 (0.1–3.0 µM) to the assay for 20 min. A linear relation of fluorescence signals and H2O2 concentrations was found at all pH-values. Slopes were used to calculate the pH-dependent enzyme factors. LPMO activity was defined as one µmol H2O2 generated per minute under the defined assay conditions. LPMOs alone or in combination with cellobiose showed no signal that exceeded the signal of Amplex Red alone over the investigated timespan. Time-dependent degradation curves for the PsLPMOs were measured by incubating sample aliquots of 70 µL for up to 1500 min at varying temperatures. Samples were sequentially placed in a thermomixer (±1 °C) and harvested at the same time to minimize measurement errors. Furthermore, sea salt effects on the activity of PsLPMOs were tested using the standard Amplex Red assay and addition of 3.5 % (w/v) sea salt to the buffer. All measurements were performed at least in triplicate.
N-terminal amino acid sequence determination and protein deglycosylation
The N-terminal amino acid sequences of purified PsLPMOA and PsLPMOB were determined according to Edman degradation. Samples were taken from PsLPMOs electroblotted onto a polyvinylidene difluoride membrane (iBlot, Life Technologies). Analyses were carried out on an Applied Biosystems 470A by the proteomics platform at the Institut de Microbiologie de la Méditerranée, CNRS-Aix-Marsille University, Marseille, France. Recombinant PsLPMOs were deglycosylated using N-glycosidase F (PNGase F) as per manufacturer’s procedure (New England Biolabs, Évry, France) under denaturing conditions. Glycosylation sequences were predicted using NetNGlyc 4.0 (http://www.cbs.dtu.dk/services/NetNGlyc/) and NetOGlyc 3.1 (http://www.cbs.dtu.dk/services/NetOGlyc/).
Cellulolytic activity and regioselectivity of PsLPMOs
Phosphoric acid swollen cellulose (PASC, prepared from Avicel as described in [41]) was incubated with PsLPMOs and analyzed for cellulose-oligosaccharides (Megazyme, Wicklow, Ireland). All cleavage assays (300 μL liquid volume) contained 5 μM PsLPMOs, 1 mM ascorbic acid and 0.1 % (w/v) PASC in 50 mM sodium acetate buffer, pH 5.5. Control experiments were performed without ascorbic acid in the presence of PsLPMOs. All experiments were performed in 2-mL tubes incubated in a thermomixer (Eppendorf, Montesson, France) at 50 °C and 850 rpm for 24 or 72 h. All samples were boiled at 100 °C for 10 min to stop the enzymatic reaction and then centrifuged at 16,000×g for 15 min at 4 °C to separate the soluble fraction from the remaining insoluble fraction before product analysis. NcCDHIIA was used in the subsequent reactions to determine the regioselectivity of PsLPMOs. NcCDHIIA was used at a concentration of 1.2 U mL−1 along with 400 µM cellobiose as electron donor and incubated for 22 h with the soluble fraction obtained after action of PsLPMOs. The reactions were carried out as described above. To determine effect of sea salt on activity of PsLPMOs, experiments were performed as described above with addition of 0.0–6.0 % (w/v) of sea salt into reaction mixtures. Blank controls were performed under the same conditions without ascorbic acid or NcCDHIIA, and PsLPMOs. All assays were performed as triplicate-independent experiments.
Analysis of oxidized and non-oxidized cello-oligosaccharides
Mono- and oligosaccharides as well as their corresponding aldonic acid forms generated after PASC cleavage were analyzed by HPAEC as described previously [21, 37]. Non-oxidized cello-oligosaccharides were used as standards. Corresponding oxidized standards were produced from non-oxidized cello-oligosaccharides by CDH treatment as described in [21].
Sequence analysis and homology models of PsLPMOs
A sequence alignment of PsLPMOs with biochemically characterized LPMOs from other species was performed using the Clustal Omega multiple sequence alignment program (http://www.ebi.ac.uk/Tools/msa/clustalo/). Structural homology models for all PsLPMOs were generated using the SWISS-MODEL server (http://www.swissmodel.expasy.org/) [42]. Quality assessments including Ramachandran plots for all models were performed with MolProbity (http://www.molprobity.biochem.duke.edu/). Structures were visualized using the PyMOL molecular graphics system, version 1.4 (Schrödinger, New York, NY, USA).
Authors’ contributions
IP and ER designed and coordinated the study. IP, DK, SM, SG, and MH performed the laboratory experiments and analyzed the results. IP wrote the manuscript. CF, JGB, and RL advised on experimental set-up and reviewed the manuscript. All authors read and approved the final manuscript.
Acknowledgements
The authors thank the French National Institute for Agricultural Research (INRA) for its generous support. This work was performed within INDOX, a project under the EU’s 7th Framework Program; Grant Number KBBE-2013-7-613549. The authors thank Y. Mathieu for his kind help in gene cloning, G. Anasontzis for help in preparing the phylogenetic tree, and C. Bennati-Granier for preparing the PASC. Finally, we would like to thank the reviewers for all the comments that helped us to improve the manuscript.
Availability of supporting data
The datasets supporting the conclusions of this article are included within the article and its additional file.
pslpmoa (Genebank ID KR825270, http://www.ncbi.nlm.nih.gov/nuccore/1023155790/?report=genbank).
pslpmob (Genebank ID KR825269, http://www.ncbi.nlm.nih.gov/nuccore/1023155788/?report=genbank).
pslpmoc (Genebank ID KR825268, http://www.ncbi.nlm.nih.gov/nuccore/1023155786/?report=genbank).
pslpmod (Genebank ID KR825271, http://www.ncbi.nlm.nih.gov/nuccore/1023155792/?report=genbank).
pslpmoe (Genebank ID KR825272, http://www.ncbi.nlm.nih.gov/nuccore/1023155794/?report=genbank).
Competing interests
The authors declare that they have no competing interests.
Funding
This work was performed within INDOX, a project under the EU’s 7th Framework Program; Grant Number KBBE-2013-7-613549. There is no role of the funding body in the design of the study, analysis, interpretation of data, and in writing the manuscript.
Abbreviations
- LPMOs
lytic polysaccharide monooxygenases
- AA
auxiliary activities
- CDH
cellobiose dehydrogenase
- PASC
phosphoric acid swollen cellulose
- HPAEC
high-performance anion-exchange chromatography
Additional file
10.1186/s13068-016-0520-3 Production, purification and characterization of lytic polysaccharide monooxygenases secreted by Pestalotiopsis sp. NCi6.
Contributor Information
Ilabahen Patel, Email: ila.patel@univ-amu.fr.
Daniel Kracher, Email: daniel.kracher@boku.ac.at.
Su Ma, Email: su.ma@boku.ac.at.
Sona Garajova, Email: sona.garajova@univ-amu.fr.
Mireille Haon, Email: mireille.haon@univ-amu.fr.
Craig B. Faulds, Email: craig.faulds@univ-amu.fr
Jean-Guy Berrin, Email: jean-guy.berrin@univ-amu.fr.
Roland Ludwig, Email: roland.ludwig@boku.ac.at.
Eric Record, Email: eric.record@univ-amu.fr.
References
- 1.Meng X, Ragauskas AJ. Recent advances in understanding the role of cellulose accessibility in enzymatic hydrolysis of lignocellulosic substrates. Curr Opin Biotechnol. 2014;27:150–158. doi: 10.1016/j.copbio.2014.01.014. [DOI] [PubMed] [Google Scholar]
- 2.Prasad S, Dhanya MS, Gupta N, Kumar A. Biofuels from biomass: a sustainable alternative to energy and environment. Biochem Cell Arch. 2012;12:255–260. [Google Scholar]
- 3.Lynd LR, Laser MS, Bransby D, Dale BE, Davison B, Hamilton R, et al. How biotech can transform biofuels. Nat Biotechnol. 2008;26:169–172. doi: 10.1038/nbt0208-169. [DOI] [PubMed] [Google Scholar]
- 4.Ragauskas AJ, Williams CK, Davison BH, Britovsek G, Cairney J, Eckert CA, et al. The path forward for biofuels and biomaterials. Science. 2006;311(5760):484–489. doi: 10.1126/science.1114736. [DOI] [PubMed] [Google Scholar]
- 5.Mba Medie F, Davies GJ, Drancourt M, Henrissat B. Genome analyses highlight the different biological roles of cellulases. Nat Rev Microbiol. 2012;10(3):227–234. doi: 10.1038/nrmicro2729. [DOI] [PubMed] [Google Scholar]
- 6.Henrissat B, Driguez H, Viet C, Schülein M. Synergism of cellulases from Trichoderma reesei in the degradation of cellulose. Nat Biotechnol. 1985;3:722–726. doi: 10.1038/nbt0885-722. [DOI] [Google Scholar]
- 7.Phillips CM, Beeson WT, Cate JH, Marletta MA. Cellobiose dehydrogenase and a copper-dependent polysaccharide monooxygenase potentiate cellulose degradation by Neurospora crassa. ACS Chem Biol. 2011;6:1399–1406. doi: 10.1021/cb200351y. [DOI] [PubMed] [Google Scholar]
- 8.Vaaje-Kolstad G, Westereng B, Horn SJ, Liu Z, Zhai H, Sørlie M, et al. An oxidative enzyme boosting the enzymatic conversion of recalcitrant polysaccharides. Science. 2010;330:219–222. doi: 10.1126/science.1192231. [DOI] [PubMed] [Google Scholar]
- 9.Agger JW, Isaksen T, Várnai A, Vidal-Melgosa S, Willats WGT, Ludwig R, et al. Discovery of LPMO activity on hemicelluloses shows the importance of oxidative processes in plant cell wall degradation. Proc Natl Acad Sci USA. 2014;111:6287–6292. doi: 10.1073/pnas.1323629111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Harris PV, Welner D, McFarland KC, Re E, Navarro Poulsen JC, Brown K, et al. Stimulation of lignocellulosic biomass hydrolysis by proteins of glycoside hydrolase family 61: structure and function of a large, enigmatic family. Biochemistry. 2010;49(15):3305–3316. doi: 10.1021/bi100009p. [DOI] [PubMed] [Google Scholar]
- 11.Westereng B, Ishida T, Vaaje-Kolstad G, Wu M, Eijsink VGH, Igarashi K, et al. The putative endoglucanase PcGH61D from Phanerochaete chrysosporium is a metal-dependent oxidative enzyme that cleaves cellulose. PLoS ONE. 2011;6:e27807. doi: 10.1371/journal.pone.0027807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Langston JA, Shaghasi T, Abbate E, Xu F, Vlasenko E, Sweeney MD. Oxidoreductive cellulose depolymerization by the enzymes cellobiose dehydrogenase and glycoside hydrolase 61. Appl Environ Microbiol. 2011;77:7007–7015. doi: 10.1128/AEM.05815-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Quinlan RJ, Sweeney MD, Lo Leggio L, Otten H, Poulsen J-CN, Johansen KS, et al. Insights into the oxidative degradation of cellulose by a copper metalloenzyme that exploits biomass components. Proc Natl Acad Sci USA. 2011;108:15079–15084. doi: 10.1073/pnas.1105776108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Beeson WT, Phillips CM, Cate JHD, Marletta MA. Oxidative cleavage of cellulose by fungal copper-dependent polysaccharide monooxygenases. J Am Chem Soc. 2012;134:890–892. doi: 10.1021/ja210657t. [DOI] [PubMed] [Google Scholar]
- 15.Vu VV, Beeson WT, Span EA, Farquhar ER, Marletta MA. A family of starch-active polysaccharide monooxygenases. Proc Natl Acad Sci USA. 2014;111:13822–13827. doi: 10.1073/pnas.1408090111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lo Leggio L, Simmons TJ, Poulsen J-CN, Frandsen KEH, et al. Structure and boosting activity of a starch-degrading lytic polysaccharide monooxygenase. Nat Commun. 2015;6:5961. doi: 10.1038/ncomms6961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lombard V, Golaconda Ramulu H, Drula E, Coutinho PM, Henrissat B. The carbohydrate-active enzymes database (CAZy) in 2013. Nucleic Acids Res. 2014;42((Database issue)):D490–D495. doi: 10.1093/nar/gkt1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Levasseur A, Drula E, Lombard V, Coutinho PM, Henrissat B. Expansion of the enzymatic repertoire of the CAZy database to integrate auxiliary redox enzymes. Biotechnol Biofuels. 2013;6:41. doi: 10.1186/1754-6834-6-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hemsworth GR, Henrissat B, Davies GJ, Walton PH. Discovery and characterization of a new family of lytic polysaccharide monooxygenases. Nat Chem Biol. 2014;10(2):122–126. doi: 10.1038/nchembio.1417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hemsworth GR, Johnston EM, Davies GJ, Walton PH. Lytic polysaccharide monooxygenases in biomass conversion. Trends Biotechnol. 2015;33:747–761. doi: 10.1016/j.tibtech.2015.09.006. [DOI] [PubMed] [Google Scholar]
- 21.Bennati-Granier C, Garajova S, Champion C, Sacha Grisel, Haon M, Zhou S, et al. Substrate specificity and regioselectivity of fungal AA9 lytic polysaccharide monooxygenases secreted by Podospora anserina. Biotechnol Biofuels. 2015;8:90. doi: 10.1186/s13068-015-0274-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Isaksen T, Westereng B, Aachmann FL, Agger JW, Kracher D, Kittl R, et al. A C4-oxidizing lytic polysaccharide monooxygenase cleaving both cellulose and cello-oligosaccharides. J Biol Chem. 2014;289(5):2632–2642. doi: 10.1074/jbc.M113.530196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Forsberg Z, Vaaje-Kolstad G, Westereng B, Bunæs AC, Stenstrøm Y, MacKenzie A, et al. Cleavage of cellulose by a CBM33 protein. Protein Sci. 2011;20:1479–1483. doi: 10.1002/pro.689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sygmund C, Kracher D, Scheiblbrandner S, Zahma K, Felice AKG, Harreither W, et al. Characterization of the two Neurospora crassa cellobiose dehydrogenases and their connection to oxidative cellulose degradation. Appl Environ Microbiol. 2012;78:6161–6171. doi: 10.1128/AEM.01503-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bey M, Zhou S, Poidevin L, Henrissat B, Coutinho PM, Berrin JG, et al. Cello-oligosaccharide oxidation reveals differences between two lytic polysaccharide monooxygenases (family GH61) from Podospora anserina. Appl Environ Microbiol. 2013;79:488–496. doi: 10.1128/AEM.02942-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kittl R, Kracher D, Burgstaller D, Haltrich D, Ludwig R. Production of four Neurospora crassa lytic polysaccharide monooxygenases in Pichia pastoris monitored by a fluorimetric assay. Biotechnol Biofuels. 2012;5:79. doi: 10.1186/1754-6834-5-79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sibilla F, Domínguez de María P. Integrating white biotechnology in lignocellulosic biomass transformations: from enzyme-catalysis to metabolic engineering. In: Triantafyllidis K, Lappas A, Stöcker M, editors. The role of catalysis for the sustainable production of bio-fuels and bio-chemicals. Amsterdam: Elsevier; 2012. pp. 445–466. [Google Scholar]
- 28.Domínguez de María P. On the use of seawater as reaction media for large-scale applications in biorefineries. ChemCatChem. 2013;5:1643–1648. doi: 10.1002/cctc.201200877. [DOI] [Google Scholar]
- 29.Chen HY, Xue DS, Feng XY, Yao SJ. Screening and production of ligninolytic enzyme by a marine-derived fungal Pestalotiopsis sp. J63. Appl Biochem Biotechnol. 2011;165:1754–1769. doi: 10.1007/s12010-011-9392-y. [DOI] [PubMed] [Google Scholar]
- 30.Arfi Y, Chevret D, Henrissat B, Berrin J-G, Levasseur A, Record E. Characterization of salt-adapted secreted lignocellulolytic enzymes from the mangrove fungus Pestalotiopsis sp. Nat Commun. 2013;4:1810. doi: 10.1038/ncomms2850. [DOI] [PubMed] [Google Scholar]
- 31.Lin X, Beeson WT, Phillips CM, Marletta MA, Cate JHD. Structural basis for substrate targeting and catalysis by fungal polysaccharide monooxygenases. Structure. 2012;20:1051–1061. doi: 10.1016/j.str.2012.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lanyi JK. Salt-dependent properties of proteins from extremely halophilic bacteria. Bacteriol Rev. 1974;38:272–290. doi: 10.1128/br.38.3.272-290.1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kern M, McGeehan JE, Streeter SD, Martin RNA, Besser K, Elis L, et al. Structural characterization of a unique marine animal family 7 cellobiohydrolase suggests a mechanism of cellulase salt tolerance. PNAS. 2013;110:10189–109194. doi: 10.1073/pnas.1301502110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tan T-C, Kracher D, Gandini R, Sygmund C, Kittl R, Haltrich D, et al. Structural basis for cellobiose dehydrogenase action during oxidative cellulose degradation. Nat Commun. 2015;6:7542. doi: 10.1038/ncomms8542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Karkehabadi S, Hansson H, Kim S, Piens K, Mitchinson C, Sandgren M. The first structure of a glycoside hydrolase family 61 member, Cel61B from Hypocrea jecorina, at 1.6 A resolution. J Mol Biol. 2008;31:144–154. doi: 10.1016/j.jmb.2008.08.016. [DOI] [PubMed] [Google Scholar]
- 36.Wu M, Beckham GT, Larsson AM, Ishida T, Kim S, et al. Crystal structure and computational characterization of the lytic polysaccharide monooxygenase GH61D from the Basidiomycota fungus Phanerochaete chrysosporium. J Biol Chem. 2013;288:12828–12839. doi: 10.1074/jbc.M113.459396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Westereng B, Agger JW, Horn SJ, Vaaje-Kolstad G, Aachmann FL, Stenstrøm YH, et al. Efficient separation of oxidized cello-oligosaccharides generated by cellulose degrading lytic polysaccharide monooxygenases. J Chromatogr A. 2013;1271:144–152. doi: 10.1016/j.chroma.2012.11.048. [DOI] [PubMed] [Google Scholar]
- 38.Busk PK, Lange L. Classification of fungal and bacterial lytic polysaccharide monooxygenases. BMC Gemomics. 2015;16:368. doi: 10.1186/s12864-015-1601-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Haon M, Grisel S, Navarro D, Gruet A, Berrin J-G, Bignon C. Recombinant protein production facility for fungal biomass-degrading enzymes using the yeast Pichia pastoris. Front Microbiol. 2015;6:1002. doi: 10.3389/fmicb.2015.01002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Piumi F, Levasser A, Navarro D, Zhou S, Mathieu Y, Ropartz D, Ludwig R, Faulds CB, Record E. A novel glucose dehydrogenase from the white-rot fungus Pycnoporus cinnabarinus: production in Aspergillus niger and physicochemical characterization of the recombinant enzyme. Appl Microbiol Biotechnol. 2014;98:10105–10118. doi: 10.1007/s00253-014-5891-4. [DOI] [PubMed] [Google Scholar]
- 41.Wood TM. Preparation of crystalline, amorphous, and dyed cellulase substrates. Methods Enzym. 1988;160:19–25. doi: 10.1016/0076-6879(88)60103-0. [DOI] [Google Scholar]
- 42.Arnold K, Bordoli L, Kopp J, Schwede T. The SWISS-MODEL workspace: a web-based environment for protein structure homology modelling. Bioinformatics. 2006;22:195–201. doi: 10.1093/bioinformatics/bti770. [DOI] [PubMed] [Google Scholar]