

Abstract
Background
Copy number variation (CNV) represents an important source of genetic divergence that can produce drastic phenotypic differences and may therefore be subject to selection during domestication and environmental adaptation. To investigate the evolutionary dynamics of CNV in the yak genome, we used a read depth approach to detect CNV based on genome resequencing data from 14 wild and 65 domestic yaks and determined CNV regions related to domestication and adaptations to high-altitude.
Results
We identified 2,634 CNV regions (CNVRs) comprising a total of 153 megabases (5.7 % of the yak genome) and 3,879 overlapping annotated genes. Comparison between domestic and wild yak populations identified 121 potentially selected CNVRs, harboring genes related to neuronal development, reproduction, nutrition and energy metabolism. In addition, we found 85 CNVRs that are significantly different between domestic yak living in high- and low-altitude areas, including three genes related to hypoxia response and six related to immune defense. This analysis shows that genic CNVs may play an important role in phenotypic changes during yak domestication and adaptation to life at high-altitude.
Conclusions
We present the first refined CNV map for yak along with comprehensive genomic analysis of yak CNV. Our results provide new insights into the genetic basis of yak domestication and adaptation to living in a high-altitude environment, as well as a valuable genetic resource that will facilitate future CNV association studies of important traits in yak and other bovid species.
Electronic supplementary material
The online version of this article (doi:10.1186/s12864-016-2702-6) contains supplementary material, which is available to authorized users.
Keywords: Yak, Copy number variation, Domestication, High altitude adaptation
Background
Copy number variations (CNVs), a form of genomic structural variation, are defined as duplications or deletions of DNA fragments that range in size from at least 50 base-pairs (bp) to more than several megabase-pairs (Mb), causing a different copy number of specific genomic regions among individuals of a species [1–4]. CNV represents an important source of genetic variation complementary to SNP data, but which affects a higher percentage of genomic sequences and has potentially stronger effects on phenotypic diversity and evolutionary adaptation, through changing gene dosage and transcript structure, and regulating gene expression and function [5].
As a common feature of vertebrate genomes, the foundational studies of CNV have been conducted in humans. Around 2,057,368 CNVs that correspond to over 24,032 CNV regions have been identified in humans and these may account for >9.5 % of the human genome, with over 12 % of gene sequences involved in CNV regions [6]. CNVs that underlie complex traits such as obesity, diabetes, Alzheimer’s disease and Autism spectrum disorders have been detected in human patients [5]. In recent years, advances in genomic technologies have made it increasingly feasible to screen comprehensively for CNVs, and so interest in CNV detection has extended to livestock species, with considerable advances being made [7–9]. CNV maps have been constructed for cattle [10], horse [3], goat [11], sheep [12], pig [13], dog [14] and chicken [15], providing a very valuable resource for evolution and genetic improvement research in livestock. In addition, there is growing evidence for CNVs associated with phenotypic variation, disease susceptibility, environmental adaptation and production traits in livestock species. For example, the copy number variation of the AMY2B gene probably allowed dogs to thrive on a relatively starch-rich diet during their domestication [16]. The ASIP CNV allele is almost entirely associated with different coat colors in different goat breeds [17] and in Tibetan sheep [18]. The most recognizable chicken pea-comb phenotype is attributed to a duplication near the SOX5 gene [19]. A CNV involving the AKR1C gene is considered possibly responsible for testicular androgen synthesis and sexual development in horse [3]. The DNA dosage and EST expression of CNVs overlapping with the Ntn1 gene may influence meat quality in pigs [20]. In bovid species, several important genes related to clear phenotypic changes and breed differences have been modified by CNVs: the copy number variations of the HSFY and ZNF280BY genes are associated with male reproductive traits in Holstein bulls [21]; PLA2G2D located in a CNV region is associated with aspects of body size in Chinese bulls such as heart girth and hucklebone width [22]; hereditary myopathy of diaphragmatic muscles in Holstein-Friesian cattle has been linked to deletion of the HSPA1B gene [23]; and the APOL3 gene involved in lipid transport is highly duplicated in beef breeds [24]. These studies reveal that many beneficial CNVs may have been artificially selected in livestock during domestication and could be associated with or affect important traits of economic interest. However, only a few studies provide a comprehensive characterization of the evolutionary impact of CNVs comparing a wild and domestic population.
The yak (Bos grunniens) is the only major livestock that can survive the extremely cold, harsh and oxygen-poor conditions and take full advantage of the limited grassland resources on the Qinghai-Tibetan Plateau (QTP) [25]. Yak were domesticated by the early nomadic people from wild yak more than 7,300 years ago [26]. Nowadays, there are more than 14 million domestic yaks, providing necessities for Tibetans and other nomadic pastoralists in high-altitude environments; in addition, there are still 15–20 thousand wild yaks roaming the northwestern QTP [27]. It should be noted that the domestic yak is the only large animal that still coexists with its wild ancestors in a similar environment, as wild progenitors of other domestic livestock are now extinct or geographically dispersed [25, 28]. Therefore, the wild and domestic yak provide a good framework for studying effects of CNV in large livestock domestication [29]. Indeed, a more comprehensive understanding of CNVs at the whole-genome level could provide additional evidence for unraveling the genetic basis of yak domestication. However, as far as we know, there is only one published dataset reporting only 161 CNV regions based on just two yak individuals using the cattle-specific Nimblegen3x720K CGH array [22].
Although comparative genomic hybridization (CGH) and SNP arrays are routinely used for CNV identification, the performance of these methods are heavily depending on the marker density and the specially designed non-polymorphic probes [30–32]. The advent of next-generation sequencing (NGS) technologies and complementary analysis has provided innovative approaches to systematically screen CNVs at a whole-genome level, especially in non-model organisms [3, 30, 33]. Among different analysis programs to detect CNVs using sequence data, the read depth (RD) method have become a major approach due to its stronger ability to estimate the exact copy numbers and identify CNVs in complex genomic regions [33, 34]. Thus, the recent completion of the yak genome and population genome studies now allow for a comprehensive screening of CNV [26, 35, 36].
Here we describe the first genome-wide and systematic analysis of CNVs in yak using NGS genome re-sequencing data from wild and domestic yak. This CNV map was constructed for three main reasons. First, to develop and enrich new genomic variations in order to facilitate further research on yak and other bovid species and make it available to the scientific community. Second, to determine genes affected by CNV that differ between wild and domestic yak, and evaluate whether extensive CNV is related to domestication. Third, to compare the CNVs in individuals from different altitudes and assess the evolutionary impact of CNVs in adaptations to life at high altitude.
Results and discussion
CNV discovery and data set statistics
We used whole genome re-sequenced data from our previous study, based on 14 wild and 65 domestic yaks from widely spaced locations across the QTP (Fig. 1), representing three highly diverged mitochondrial lineages and broad genetic diversity [28]. A total of 1.56 Tb of sequences with an average depth of 6.7× were used (Additional file 1) [26]. Mapping these reads to the yak reference genome [35] revealed that at least 93 % of the genome was covered by reads from a single individual, and, on average, 98 % of the reference genome was covered, indicating that the data are sufficient and of high enough quality for CNV detection [37].
Fig. 1.
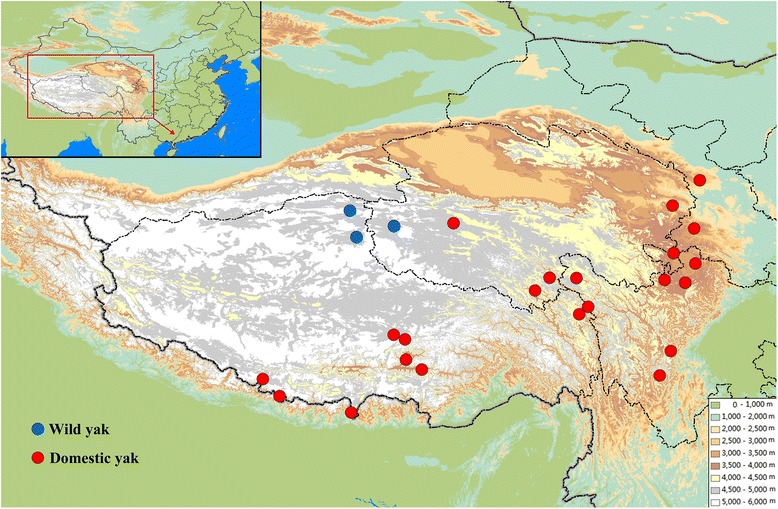
Geographical distribution of the yak samples used in this study. The sampling locations and altitudes for wild (blue) and domestic (red) yak were mapped using ArcGIS software. Geographic (ESRI: www.esri.com) and altitude (WorldClim: www.worldclim.org) parameters were obtained from freely accessible data layers
Using the CNVnator software based on the RD method [37], we detected a total of 98,441 CNV events from the 79 individuals (Fig. 2 and Additional file 2). The average number of CNVs per individual was 1,246 (ranging from 954 to 2,952) with an average of 291 gain and 955 loss events. The size of the CNVs identified varied from 1.5 kb to 1,226.0 kb with an average size of 15.6 kb and a median size of 8.0 kb. Specifically, only 1.8 % of all CNV calls were present in a single individual (Additional file 3). The results of CNVs identified and the location information for each individual are listed in Additional file 2.
Fig. 2.
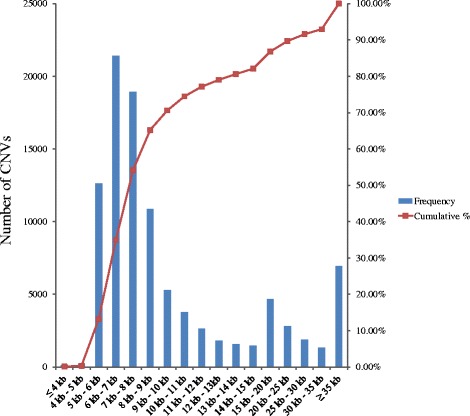
CNV size interval distribution. Average CNV size is 15.6 kb and the median size is 8.0 kb
We further defined 2,634 regions with copy number variations (termed CNVRs) by merging all overlapping calls across multiple individuals into unique regions and filtering the ones that were present in fewer than four individuals. These CNVRs occupied a total of 153 Mb or 5.7 % of the yak genome, substantially more than the recent study in two yak individuals based on a CGH array approach (33 Mb, 1.25 %) [38]. The length of the CNVRs varied from 3 kb to 1,309 kb with an average of 58 kb. Among all CNVRs, 234 (8.9 %) were found to be common to all 79 individuals (Fig. 3 and Additional file 4). According to the type of CNVRs, they were divided into three categories, including 785 gain, 1,575 loss and 274 both (gain and loss within the same regions from different individuals) events (Fig. 3). Although it is difficult to compare the CNVRs in different studies due to the different diversity of individuals and the technology used for detection, our study based on next-generation sequencing, using 79 different wild as well as domestic yaks resulted in better resolution and higher confidence in calling CNVRs than past research [22]. Thus, most of the CNVRs discovered in this study are novel compared to previous studies and they represent the largest catalog of yak specific CNVRs to date.
Fig. 3.
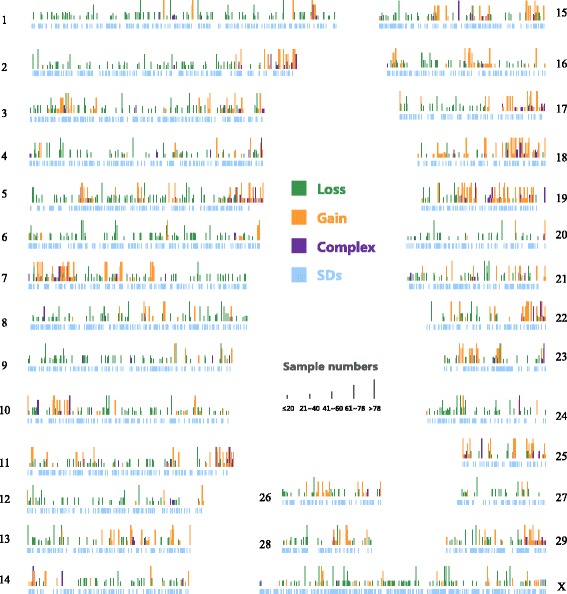
Genomic landscape of yak copy number variation regions and segmental duplications. The SDs are plotted as blue bars. The CNVRs are illustrated above the SDs in green (loss), orange (gain) and purple (complex of gains and losses). The chromosome numbers (1–29, X) are shown nearby correspond CNV landscape, and the bar height represents different sample numbers (≤20, 21–40, 41–60, 61–78, > 78 )
We evaluated the accuracy of individual copy number variations predicted by performing a series of real time-polymerase chain reactions (qPCR). The experimental validation showed that 88 % of the CNVs (56/64) had an accurate copy number and 100 % were in agreement with the predicted patterns (Additional file 5). It should be emphasized that not all true CNVs could be detected by qPCR, especially some low-copy duplications with lower sequence similarities. Thus, a 12 % false positive rate is a conservative estimate in our CNV analysis, representing a relatively low false positive rate in our CNV calling and CNVR definitions [24, 39, 40]. We further estimated the accuracy using 6 deep-coverage (30×) yak genomes, including three domestic and three wild individuals, revealing that 85 % of our original CNV calls were supported by higher coverage genome data. Moreover, considering that the quality of the assembled reference as well as the annotated repeats is crucial to discovering CNVs using the RD method, additional experimental validation, such as qPCR from more individuals, array comparative genomic hybridization (aCGH) and fluorescent in situ hybridization (FISH), will be required to obtain more accurate information about the CNVs and to exclude false positives [3, 24].
SD detection and comparison with CNVRs
Segmental duplications (SDs) were shown to be one of the catalysts and hotspots for CNV formation in many species [3, 24, 41], and we tested whether the non-random association between CNVs and SDs was preserved in yak. Based on whole-genome assembly comparison (WGAC) [42] and whole-genome shotgun sequence detection (WSSD) [43] methods, we first identified 47,740 and 5,381 putative segment duplication events, respectively. By merging these two data sets, we finally predicted 50,800 segments, spanning 113.8 Mb of DNA sequence and comprising 4.3 % of the yak genome (Additional file 6), a similar level to that of the bovine genome (3.11 %) [44]. We subsequently compared the overlap between the SDs identified above and the CNVRs and found that 56 % of large CNVRs (>15 kb) directly overlapped with SDs. Random simulations further confirmed significant relationships between 5 Mb flanking regions of CNVRs and their overlap with SDs (Additional file 7), consistent with the previously studied CNVs that were enriched with SDs [10].
Gene content of CNV regions
Among the 2,634 CNVRs described above, 961 (36.5 %) harbored a total of 3,879 protein-coding genes according to the current yak genome (Additional file 8). These represent a valuable resource for future studies on the relationship between CNV genes and phenotypic variation. Gene ontology (GO) enrichment analysis indicated that CNVRs harbored genes that were mainly involved in sensory perception and olfactory receptor activity (Additional file 9), such as the GO categories “olfactory receptor activity”, “sensory perception”, “sensory perception of chemical stimulus” and “sensory perception of smell”, which was consistent with the observation that olfactory gene families are large and associated with CNVs [10, 13, 39]. In addition, gene families involved in sensory perception are usually fast evolving due to their importance in the organism responding to rapid changes in the environment and they have been repeatedly detected in CNV regions of several livestock genomes. Our previous comparative genome study also found that gene families related to sensory perception were substantially expanded in yak compared to other mammals [35].
Population-differentiated CNVRs between wild and domestic yak
To reveal the potential contributions of CNVs in the process of yak domestication, we carried out a comparative study to find the CNVRs with significantly different copy number between domestic and wild yak. We identified 121 CNVRs potentially selected during domestication, containing 100 annotated protein coding genes (Additional file 10).
Strong selection on reducing aggressive behavior and neurological traits is often involved in the initial step of animal domestication [45], and our previous domestication selective sweep analysis based on SNP data identified 38 genes related to these functions [26]. In this study, we found eight more genes with high CN differentiation between domestic and wild yak that are involved in neuronal development and are associated with behavior, further indicating that genes related to brain and nervous system development might underlie the processes that led to the successful domestication of the yak. For example, GRIN2D is related to normal brain development and associated with learning, working memory and behavioral attention [46]; FTL encodes the ferritin protein and mutations in this gene could lead to behavioral abnormalities and cognitive impairment [47]; NTN5 is highly expressed in neurogenic regions of the adult brain and controls neurogenesis [48]; SHANK3 plays a role in synapse formation and dendritic spine maturation [49]; KCNJ14 has a function in controlling the excitability of motor neurons [50]; CA11 is abundantly expressed in the brain and has a general role in the central nervous system [51]; NTF4 encodes a neurotrophic factor which controls neuron differentiation [52]; and ARSA is related to many neurodegenerative diseases [53].
Another important consequence of livestock domestication is a change in their reproductive efficiency, such as age of puberty, sperm production, ovulation rate and embryonic mortality [54]. We found seven CNV-associated genes involved in reproductive performance traits. TSEG2, AKR1C3 and IZUMO1 are sperm-specific expressed genes and have a role in spermatogenic cell development and fertility [55–57]. LHB is expressed in the pituitary gland and promotes spermatogenesis and ovulation; mutations in this gene are associated with polycystic ovary syndrome in women [58]. SPACA4 encoded protein is retained on the inner acrosomal membrane of spermatozoa and plays a role in sperm–egg binding and fertilization [59]. FGF21 encodes fibroblast growth factor protein, which is involved in embryonic development [60]. DMBT1 encoding lactoprotein as a scavenger receptor protein in milk, which can suppress various bacterial infections in vitro and plays an important role in the innate immunity of breast-fed infants [61].
Previous studies have revealed that domestication leads to relaxation of selective constraints in the yak mitochondrial genome, because the domestic yak and wild yak have different energy demands and metabolic efficiency [62]. We found four genes, ND4L [63], SCO2 [64], CO1 [65] and ND1 [66], related to mitochondrial oxidative phosphorylation (OXPHOS) that exhibit marked CN variation between wild and domestic yak, indicating that CNV may affect genes involved in energy metabolism during yak domestication.
The progress of domestication of livestock would also affect nutritional uptake, absorption and metabolism [67]. We found that seven interesting CNVRs harbored genes related to nutrition metabolism and there were significant differences in copy number between domestic and wild yak. For instance, MOGAT2 [68], GYS1 [66] and DHDH [69] are involved in the metabolism of sugars (Fig. 4); HSD17B14 [70] and CPT1B [71] are involved in lipid metabolism; BCAT2 is involved in amino acid metabolism [72]; and MIOX is involved in carbohydrate metabolism [73]. In addition, we identified four genes (CHRM3 [74], KLF6 [75], GPC1 [76] and CHKB [77]) related to meat production and quality, which is also an economically significant trait that has been extensively considered during the artificial selection process.
Fig. 4.
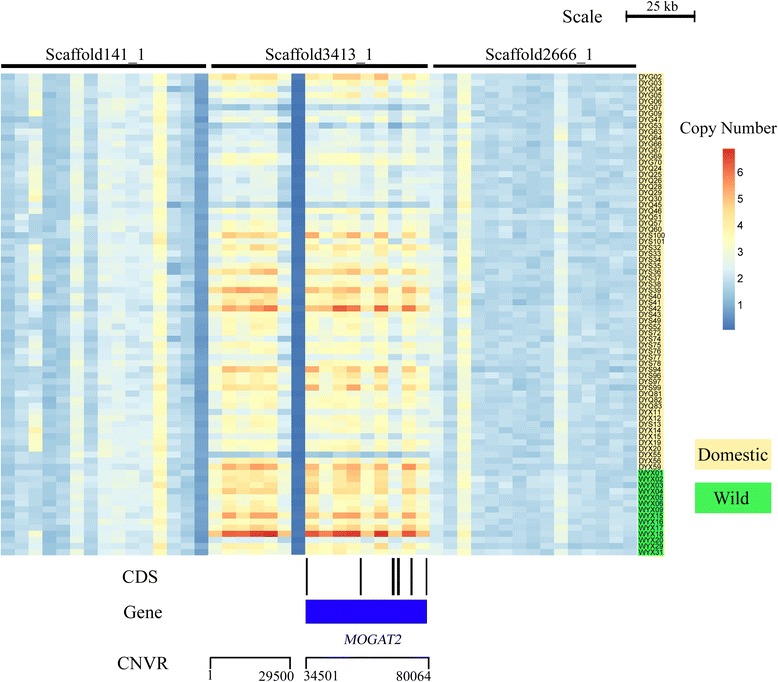
Heatmap of CNVR with MOGAT2 gene located in scaffold3413_1. Average read depths were plotted every 5 kb of the genome. The CNVR with the MOGAT2 gene represents different average normalized read depths of a specific region. The domestic yak (yellow) exhibited a lower copy number than the wild ones (green). CDS and gene are shown at the bottom (black rectangle, coding sequence; blue box, whole gene)
Taken together, our results suggest that some CNVRs may have been under selection pressure during yak domestication and these are associated with behavior, physical characteristics and economically significant traits. Consequently, additional functional experiments are needed to verify the contributions of the identified genes located in CNVRs to domestication.
Population-differentiated CNVRs between yak living at high and low altitudes
In order to understand better the evolutionary impact of CNVs in adaptations to high altitude, we compared the CNVs between domestic yak living in high-altitude (>4,000 m) and low-altitude (<2,500 m) areas. We found 85 CNVRs that are significantly different between these two groups: 29 of them harbor 41 protein-coding genes (Additional file 11). First, compared to the low-altitude population, yak living at high-altitudes must overcome the severe environmental challenges of hypoxia and an increased risk of high-altitude illness, such as pulmonary arterial hypertension and emphysema. We identified three genes involved in the response to hypoxia – MRP4 [78], DCC [79] and DEXI [80] – for which the copy number differed between high and low-altitude domestic yak. The MRP4 gene encodes ATP-binding cassette (ABC) transporters, regulating intracellular levels of cAMP and cGMP in arterial smooth muscle cells; inhibition of MRP4 expression can prevent and reverse hypoxia-induced pulmonary arterial hypertension [78]. The DCC gene encodes the Netrin 1 receptor, which plays an important mediating function in protecting against hypoxia-induced mitochondrial apoptosis [79]. DEXI expression was increased in emphysema tissues and is associated with disease progression [80]. Second, we found six CNV genes related to the immune system that may be involved in high altitude adaptation. ULBP17 encodes the immune system-activating receptor on T-cells and is related to pathogen- and parasite-resistance [24]. CIITA is a positive regulator of class II major histocompatibility and mutations in this gene are associated with diseases of the immune system [81]. CATHL1 encodes cathelicidin, which is important in innate immune defense against bacterial infection (Fig. 5) [82], BoLA-DQA2, BoLA-DQA3 and BoLA-DQB are key leukocyte antigen genes associated with the immune response. These CNVRs that span potential genes influencing the immune system may reflect the substantially different diseases triggered by the parasites and arbovirus at high and low altitudes [83–85]. These results suggest that CNV is an important type of genetic variation that may play a role in yak adaptation to high-altitude environments.
Fig. 5.
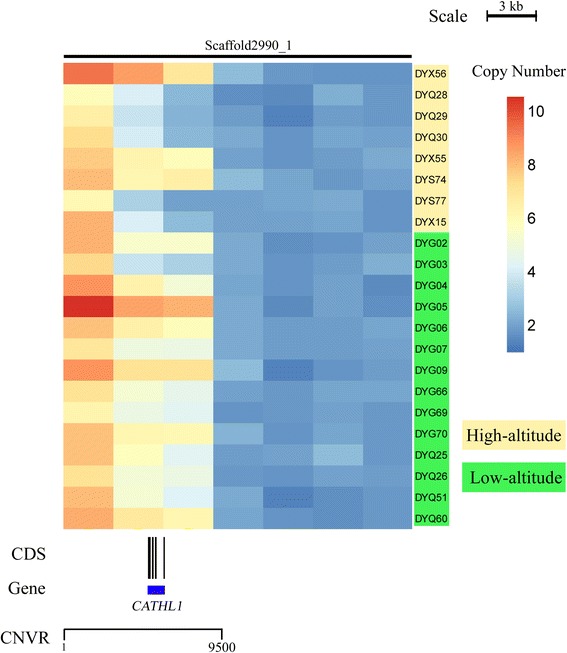
Heatmap of CNVR with CATHL1 gene located in scaffold2990_1. Average read depths were plotted every 3 kb of the genome. The CNVRs with the CATHL1 gene represents different average normalized read depths of a specific region. The yaks living in low-altitude (yellow) exhibited a lower copy number than the ones living in high-altitude (green). CDS and gene are shown at the bottom (black rectangle, coding sequence; blue box, whole gene)
Conclusions
We performed genome-wide CNV detection based on whole genome sequencing data for 14 wild and 65 domestic yaks. A total of 2,634 CNVRs comprising 153 megabases of the yak genome were identified; this represents the largest source of CNVs identified and the highest-resolution individualized CNV map constructed for the yak genome to date. The inferred CNVRs contain 3,879 functionally annotated genes and further functional analyses reveal CNVRs enriched for genes related to sensory perception and responses to stimuli. Comparing the different CNVs in wild and domestic yak suggests that many genes overlapping with CNVRs play a role in behavior, physical characteristics and economically significant traits. In addition, some novel genes, including DEXI, DCC, and MRP4, were found to be covered by CNVRs related to adaptation to high-altitude environments. Our study represents a comprehensive genomic analysis of CNVs in yak and provides valuable insights into the evolutionary dynamics of copy number variation, in the context of domestication and high-altitude adaptation. The database presented in this study will provide an important genetic resource for future work on phenotypic variation and breeding in both yak and other bovid species.
Methods
Data sets
The whole genome sequencing data were obtained from our previously study (Additional file 1, which were deposited in the European Molecular Biology Laboratory (EMBL/EBI) Nucleotide Sequence Database under the accession number PRJNA285834). Briefly, the muscle samples were obtained from 14 wild yaks and 65 domestic yaks of diverse breeds from widely spaced locations across the QTP. DNA was extracted using a standard phenol/chloroform extraction method; paired-end sequencing libraries with an insert size of 500 bp were sequenced using an Illumina HiSeq 2000 platform.
Sequence quality checking and read alignment
Using a custom Perl script, we filtered out the low quality reads based on the following criteria: (i) reads with ≥10 % unidentified nucleotides (N); (ii) reads for which more than 65 % of the read length had a Phred quality value ≤7; (iii) reads with more than 10 bp aligned to the adapter, allowing 2 bp mismatches; and (iv) duplicate reads. Reads were also trimmed if they had three consecutive base pairs with a Phred quality value of 13 or below, and discarded if they were shorter than 45 bp. The pair-end sequence reads were mapped to the B. grunniens reference genome using BWA-MEM (0.7.10-r789) with default parameters. SAMTOOLS [86] was used to sort and index the resulting Binary Alignment Map (BAM) format files. In order to reduce the inaccuracy alignment, the Genome Analysis Toolkit [87] was used to realign reads located in regions around indels [36] after assigning read group information pertaining to library, lane and sample identity in the Picard software (http://broadinstitute.github.io/picard/, version 1.92).
CNV detection and CNVR definition
CNVs were detected through read-depth analysis, in which the number of copies presented is inferred from sequence depth of whole genome sequencing data [13]. By combining the established mean-shift approach, multiple-bandwidth partitioning and GC correction, CNVnator was used to detect CNVs. It has been suggested that this approach is superior to other methods with respect to accuracy of the copy number estimate and the precision of sensitivity and specificity [34]. For each individual, the realigned BAM file was processed in CNVnator. Following the authors’ recommendations, we choose the optimal parameters for different individuals, and trimmed the boundaries of CNVs on the basis of 500-bp bins to avoid bias caused by different coverage. The fraction of reads that can map to two or more locations was denoted q0. All CNV calls with q0 >50 % were filtered out, followed by any remaining CNV events whose RD significantly differed from the average genome RD (t-test corrected for multiple hypotheses testing; p < 0.05). Previous studies have shown that RD based analysis have reduced power and reliability to detect small CNVs events [37, 88]. To reduce the false positive discovery rate and avoid misinterpreting the results, we only retained the CNVs longer than 1.5 kb for further analysis [13, 39]. In consideration of the fact that the yak is diploid, to reflect homozygous and heterozygous deletions, we normalized the copy number to 2.
The copy number variation region (CNVR) is defined as a combined region of overlapping CNVs on the genome. CNVRs are merged from different samples with any amount of overlap by extending the boundaries of the overlapping CNVs [89]. Here, all the CNVRs were defined using a custom Perl script. To reduce the false positive discovery rate further, only the CNVRs present in more than four samples were used for functional and comparative analysis, thus minimizing the bias caused by uniformity of sequencing coverage.
qPCR and high depth resequencing validation
To evaluate the accuracy of our copy number assignments, we randomly selected eight gain and eight loss genic CNVRs. Thirty-two different individuals were randomly selected to validate these CNVRs by quantitative PCR (qPCR). A segment of bovine basic transcription factor 3 (BTF3) gene was chosen as a reference location for all qPCR experiments, because neither CNVs nor SDs overlapping this gene were found in our dataset nor in any previous studies of bovid species [24, 90]. Primers for qPCR validation were designed using the Primer3 webtool (http://frodo.wi.mit.edu/). The amplicon length was set to 100–200 bp, and the regions with GC percentage were between 30 and 60 % (Additional file 5).
Genomic DNA was extracted and purified from tissue using the standard phenol–chloroform method [91]. Using the CFX96 Real-Time PCR System (BIO-RAD), we performed the qPCR experiments in a total reaction volume of 20 μl, which contained 50 ng of template DNA, 10 mM primers and the reagents in the SYBR Green PCR kit (Takara). The copy number differences were determined using a standard ΔΔCt method and compared to the diploid reference gene BTF3 as previously described [24].
We also used a high-depth (30×) re-sequencing dataset based on three domestic and three wild yaks to estimate the accuracy of our original identified CNV events. We calculated the coefficient of variation (cv%) between the high-depth-sequencing predicted and our original predicted copy numbers. The difference was considered to be acceptable if cv % was <0.25.
Segmental duplication detection and association with CNV distribution
Using the yak_1.1 yak genome assembly, we applied two well-established computational approaches, whole-genome shotgun sequence detection (WSSD) and whole-genome assembly comparison (WGAC) to detect putative segmental duplications. Briefly, WGAC identifies paralogous sequences >1 kb in length with >90 % sequence identity, and WSSD identifies genomic regions that exhibit significant depth of coverage by aligning whole-genome shotgun sequencing reads to the reference genome sequence. The final SD dataset was obtained by merging these two results. Association between CNVs and SDs was tested by random simulations using a custom Perl script as previously published [10].
Gene content and gene ontology
By searching for the coordinate of each CNVR in the yak genome assembly and gene annotation, we assessed the gene contents of each CNVR. Only the genes that were primarily comprised of CNVRs (>50 %) were retained. Functional classification of GO categories was performed using the Blast2GO program [92]. Enrichment analysis was performed and the hypergeometric test was used to calculate the statistical significance of enrichment. The P values were adjusted by FDR and the adjusted P value cut-off was 0.05. GO terms associated with the CNVRs and whole genome background were plotted by WEGO (http://wego.genomics.org.cn/cgi-bin/wego/index.pl).
Comparison between different populations
We carried out two comparative studies to identify CNV genes with high differentiation between populations. One was domestication related, including 65 domestic and 14 wild samples; the other was high-altitude adaptation related, only including domestic yak living at high (>4,000 m, eight samples) and low-altitude (<2,500 m, 14 samples) areas. With the aim of identifying population-differentiated CNVRs more accurately, two statistical measures were used during comparisons: a metric analogous to FST (the fixation index) named VST [93] and the student’s t-test. For each locus, the VST was calculated using the formula VST = (VT–VS)/VT as previously published. Here, VT is the total variance in CN between the two populations and VS is the average of the variance within each population, weighted for its sample size [39]. The distributions of CNs between different groups were also compared using the student’s t-test. Only loci exhibiting significantly different CNs based on these two statistics (top 5 % of all the VST values and P-value < 0.05) were considered to be population-differentiated CNVs, representing a rigorous criteria to identify differential CNVRs between populations. All these statistics were calculated using the R package.
Heatmap analysis
We performed heatmap analyses based on the depth of each sample. For each individual, the depth of every base was computed using the “depth” command in SAMTOOLS [86]. As the estimated copy number, the ratio of the average depth of each window to the effective depth of each individual was then calculated. The pheatmap R package was used to plot these effective copy number values for all individuals.
Ethics statement
All animal specimens were collected legally. Animal collection and utility protocols were approved by the Ethics Committee of Animal Experiments at Lanzhou University and in accordance with guidelines from the China Council on Animal Care.
Consent for publication
Not applicable.
Availability of data and material
The datasets supporting the conclusions of this article are available in the European Molecular Biology Laboratory (EMBL/EBI) Nucleotide Sequence Database repository (accession code PRJNA285834). Other supporting data are included as Additional files: 1, 2, 3, 4, 5, 6, 7, 8, 9, 10 and 11.
Acknowledgments
This study was supported by grants from the National Natural Science Foundation of China (31322052), the National High Technology Research and Development Program of China (2013AA102505 3–2), the Fok Ying Tung Education Foundation (151105), and the State Scholarship Fund of China Scholarship Council (201406185015 and 201306185026).
Abbreviations
- ABC
ATP-binding cassette
- CGH
comparative genomic hybridization
- CN
copy number
- CNV
copy number variation
- CNVR
region of copy number variations
- GO
gene ontology
- MHC
major histocompatibility complex
- NGS
next generation sequencing
- OXPHOS
oxidative phosphorylation
- qPCR
real time-polymerase chain reactions
- QTP
Qinghai-Tibetan Plateau
- RD
read depth
- SD
segmental duplication
- WGAC
whole-genome assembly comparison
- WSSD
whole-genome shotgun sequence detection
Additional files
Basic information relating to yak used in this study, their types, geographical distributions and resequencing data used in the CNV analysis. (XLS 45 kb)
List of all 98,441 CNV events in this study. (XLSX 2719 kb)
Summary of CNV events in different individuals. (XLS 35 kb)
List of all CNVRs in the yak genome. (XLSX 784 kb)
Primer sequences and confirmation results for qPCR. (XLS 39 kb)
Statistics for yak segmental duplication. (XLS 28 kb)
{kind=link}
Relationships between flanking distances and numbers of yak CNVRs overlapping with SDs. The observed overlaps between CNVRs and SDs are plotted as little rings; the results of every 5000× random simulations are shown as filled dots with error bar. (PNG 152 kb)
{kind=link}
Gene ontology annotations for genes covered by whole genome and CNVRs. The left y-axis indicates the percentage of a specific category of genes in a main category, while the right axis indicates the number of genes in it. (PNG 1006 kb)
Over-representation / under-representation of molecular function, cellular component and biological process terms. (XLS 30 kb)
Population-differentiated CNVRs with annotated protein coding genes when comparing wild and domestic yak. (XLS 40 kb)
Population-differentiated CNVRs with annotated protein coding genes when comparing yak living at high and low altitudes. (XLS 35 kb)
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
DW and QQ conceived and designed the experiments. XZ, KW, LW, YY, ZN and XX analyzed the data. XZ, KW, XS and JH performed the experiments. XZ and QQ wrote the paper. All authors read and approved the final manuscript.
Contributor Information
Dongshi Wan, Email: wandsh@lzu.edu.cn.
Qiang Qiu, Email: qiuqiang@lzu.edu.cn.
References
- 1.Weischenfeldt J, Symmons O, Spitz F, Korbel JO. Phenotypic impact of genomic structural variation: insights from and for human disease. Nat Rev Genet. 2013;14(2):125–138. doi: 10.1038/nrg3373. [DOI] [PubMed] [Google Scholar]
- 2.Conrad DF, Pinto D, Redon R, Feuk L, Gokcumen O, Zhang Y, Aerts J, Andrews TD, Barnes C, Campbell P, et al. Origins and functional impact of copy number variation in the human genome. Nature. 2010;464(7289):704–712. doi: 10.1038/nature08516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ghosh S, Qu Z, Das PJ, Fang E, Juras R, Cothran EG, McDonell S, Kenney DG, Lear TL, Adelson DL, et al. Copy number variation in the horse genome. Plos Genet. 2014;10(10):e1004712. doi: 10.1371/journal.pgen.1004712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mills RE, Walter K, Stewart C, Handsaker RE, Chen K, Alkan C, Abyzov A, Yoon SC, Ye K, Cheetham RK, et al. Mapping copy number variation by population-scale genome sequencing. Nature. 2011;470(7332):59–65. doi: 10.1038/nature09708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhang F, Gu W, Hurles ME, Lupski JR. Copy number variation in human health, disease, and evolution. Annu Rev Genomics Hum Genet. 2009;10:451–481. doi: 10.1146/annurev.genom.9.081307.164217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zarrei M, MacDonald JR, Merico D, Scherer SW. A copy number variation map of the human genome. Nat Rev Genet. 2015;16(3):172–183. doi: 10.1038/nrg3871. [DOI] [PubMed] [Google Scholar]
- 7.Liu GE, Xu L, Huang KS. Recent advances in studying of copy number variation and gene expression. Gene Expression to Genetical Genomics. 2014;7:1. doi: 10.4137/GGG.S14286. [DOI] [Google Scholar]
- 8.Clop A, Vidal O, Amills M. Copy number variation in the genomes of domestic animals. Anim Genet. 2012;43(5):503–517. doi: 10.1111/j.1365-2052.2012.02317.x. [DOI] [PubMed] [Google Scholar]
- 9.Bickhart DM, Liu GE. The challenges and importance of structural variation detection in livestock. Front Genet. 2014;5:37. doi: 10.3389/fgene.2014.00037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liu GE, Hou YL, Zhu B, Cardone MF, Jiang L, Cellamare A, Mitra A, Alexander LJ, Coutinho LL, Dell’Aquila ME, et al. Analysis of copy number variations among diverse cattle breeds. Genome Res. 2010;20(5):693–703. doi: 10.1101/gr.105403.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fontanesi L, Martelli PL, Beretti F, Riggio V, Dall’Olio S, Colombo M, Casadio R, Russo V, Portolano B. An initial comparative map of copy number variations in the goat (Capra hircus) genome. BMC Genomics. 2010;11:639. doi: 10.1186/1471-2164-11-639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liu J, Zhang L, Xu L, Ren H, Lu J, Zhang X, Zhang S, Zhou X, Wei C, Zhao F, et al. Analysis of copy number variations in the sheep genome using 50 K SNP BeadChip array. BMC Genomics. 2013;14:229. doi: 10.1186/1471-2164-14-229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Paudel Y, Madsen O, Megens HJ, Frantz LA, Bosse M, Bastiaansen JW, Crooijmans RP, Groenen MA. Evolutionary dynamics of copy number variation in pig genomes in the context of adaptation and domestication. BMC Genomics. 2013;14:449. doi: 10.1186/1471-2164-14-449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Alvarez CE, Akey JM. Copy number variation in the domestic dog. Mamm Genome. 2012;23(1–2):144–163. doi: 10.1007/s00335-011-9369-8. [DOI] [PubMed] [Google Scholar]
- 15.Zhang H, Du ZQ, Dong JQ, Wang HX, Shi HY, Wang N, Wang SZ, Li H. Detection of genome-wide copy number variations in two chicken lines divergently selected for abdominal fat content. BMC Genomics. 2014;15:517. doi: 10.1186/1471-2164-15-517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Axelsson E, Ratnakumar A, Arendt ML, Maqbool K, Webster MT, Perloski M, Liberg O, Arnemo JM, Hedhammar A, Lindblad-Toh K. The genomic signature of dog domestication reveals adaptation to a starch-rich diet. Nature. 2013;495(7441):360–364. doi: 10.1038/nature11837. [DOI] [PubMed] [Google Scholar]
- 17.Fontanesi L, Beretti F, Riggio V, Gomez Gonzalez E, Dall’Olio S, Davoli R, Russo V, Portolano B. Copy number variation and missense mutations of the agouti signaling protein (ASIP) gene in goat breeds with different coat colors. Cytogenet Genome Res. 2009;126(4):333–347. doi: 10.1159/000268089. [DOI] [PubMed] [Google Scholar]
- 18.Han JL, Yang M, Yue YJ, Guo TT, Liu JB, Niu CE, Yang BH. Analysis of agouti signaling protein (ASIP) gene polymorphisms and association with coat color in Tibetan sheep (Ovis aries) Genet Mol Res. 2015;14(1):1200–1209. doi: 10.4238/2015.February.6.22. [DOI] [PubMed] [Google Scholar]
- 19.Wright D, Boije H, Meadows JR, Bed’hom B, Gourichon D, Vieaud A, Tixier-Boichard M, Rubin CJ, Imsland F, Hallbook F, et al. Copy number variation in intron 1 of SOX5 causes the Pea-comb phenotype in chickens. Plos Genet. 2009;5(6):e1000512. doi: 10.1371/journal.pgen.1000512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang L, Xu L, Liu X, Zhang T, Li N, el Hay H, Zhang Y, Yan H, Zhao K, Liu GE, et al. Copy number variation-based genome wide association study reveals additional variants contributing to meat quality in Swine. Sci Rep. 2015;5:12535. doi: 10.1038/srep12535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yue XP, Dechow C, Chang TC, DeJarnette JM, Marshall CE, Lei CZ, Liu WS. Copy number variations of the extensively amplified Y-linked genes, HSFY and ZNF280BY, in cattle and their association with male reproductive traits in Holstein bulls. BMC Genomics. 2014;15:113. doi: 10.1186/1471-2164-15-113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang L, Jia S, Yang M, Xu Y, Li C, Sun J, Huang Y, Lan X, Lei C, Zhou Y, et al. Detection of copy number variations and their effects in Chinese bulls. BMC Genomics. 2014;15:480. doi: 10.1186/1471-2164-15-480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sugimoto M, Furuoka H, Sugimoto Y. Deletion of one of the duplicated Hsp70 genes causes hereditary myopathy of diaphragmatic muscles in Holstein-Friesian cattle. Anim Genet. 2003;34(3):191–197. doi: 10.1046/j.1365-2052.2003.00990.x. [DOI] [PubMed] [Google Scholar]
- 24.Bickhart DM, Hou YL, Schroeder SG, Alkan C, Cardone MF, Matukumalli LK, Song JZ, Schnabe RD, Ventura M, Taylor JF, et al. Copy number variation of individual cattle genomes using next-generation sequencing. Genome Res. 2012;22(4):778–790. doi: 10.1101/gr.133967.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wiener G, Han JL, Long RJ. The Yak. 2. Bangkok: Regional Office for Asia and the Pacific Food and Agriculture Organization of the United Nations; 2003. [Google Scholar]
- 26.Qiu Q, Wang L, Wang K, Yang Y, Ma T, Wang Z, Zhang X, Ni Z, Hou F, Long R, et al. Yak whole-genome resequencing reveals domestication signatures and prehistoric population expansions. Nat Commun. 2015;6:10283. doi: 10.1038/ncomms10283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Qian Y. The origin of domesticated animal: biohistory. Beijing: China Science Press; 1979. [Google Scholar]
- 28.Wang ZF, Shen X, Liu B, Su JP, Yonezawa T, Yu Y, Guo SC, Ho SYW, Vila C, Hasegawa M, et al. Phylogeographical analyses of domestic and wild yaks based on mitochondrial DNA: new data and reappraisal. J Biogeogr. 2010;37(12):2332–2344. doi: 10.1111/j.1365-2699.2010.02379.x. [DOI] [Google Scholar]
- 29.Rubin CJ, Megens HJ, Martinez Barrio A, Maqbool K, Sayyab S, Schwochow D, Wang C, Carlborg O, Jern P, Jorgensen CB, et al. Strong signatures of selection in the domestic pig genome. Proc Natl Acad Sci U S A. 2012;109(48):19529–19536. doi: 10.1073/pnas.1217149109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Alkan C, Coe BP, Eichler EE. Genome structural variation discovery and genotyping. Nat Rev Genet. 2011;12(5):363–376. doi: 10.1038/nrg2958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.McCarroll SA, Kuruvilla FG, Korn JM, Cawley S, Nemesh J, Wysoker A, Shapero MH, de Bakker PI, Maller JB, Kirby A, et al. Integrated detection and population-genetic analysis of SNPs and copy number variation. Nat Genet. 2008;40(10):1166–1174. doi: 10.1038/ng.238. [DOI] [PubMed] [Google Scholar]
- 32.Ramos AM, Crooijmans RPMA, Affara NA, Amaral AJ, Archibald AL, Beever JE, Bendixen C, Churcher C, Clark R, Dehais P, et al. Design of a High Density SNP Genotyping Assay in the Pig Using SNPs Identified and Characterized by Next Generation Sequencing Technology. PLoS One. 2009;4(8):e6524. doi: 10.1371/journal.pone.0006524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhao M, Wang Q, Jia P, Zhao Z. Computational tools for copy number variation (CNV) detection using next-generation sequencing data: features and perspectives. BMC Bioinformatics. 2013;14(Suppl 11):S1. doi: 10.1186/1471-2105-14-S11-S1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Duan J, Zhang JG, Deng HW, Wang YP. Comparative studies of copy number variation detection methods for next-generation sequencing technologies. PLoS One. 2013;8(3):e59128. doi: 10.1371/journal.pone.0059128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Qiu Q, Zhang G, Ma T, Qian W, Wang J, Ye Z, Cao C, Hu Q, Kim J, Larkin DM, et al. The yak genome and adaptation to life at high altitude. Nat Genet. 2012;44(8):946–949. doi: 10.1038/ng.2343. [DOI] [PubMed] [Google Scholar]
- 36.Wang K, Hu Q, Ma H, Wang L, Yang Y, Luo W, Qiu Q. Genome-wide variation within and between wild and domestic yak. Mol Ecol Resour. 2014;14(4):794–801. doi: 10.1111/1755-0998.12226. [DOI] [PubMed] [Google Scholar]
- 37.Abyzov A, Urban AE, Snyder M, Gerstein M. CNVnator: an approach to discover, genotype, and characterize typical and atypical CNVs from family and population genome sequencing. Genome Res. 2011;21(6):974–984. doi: 10.1101/gr.114876.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhang L, Jia S, Plath M, Huang Y, Li C, Lei C, Zhao X, Chen H. Impact of Parental Bos taurus and Bos indicus Origins on Copy Number Variation in Traditional Chinese Cattle Breeds. Genome Biol Evol. 2015;7(8):2352–2361. doi: 10.1093/gbe/evv151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pezer Z, Harr B, Teschke M, Babiker H, Tautz D. Divergence patterns of genic copy number variation in natural populations of the house mouse (Mus musculus domesticus) reveal three conserved genes with major population-specific expansions. Genome Res. 2015;25(8):1114–1124. doi: 10.1101/gr.187187.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhao Q, Han MJ, Sun W, Zhang Z. Copy number variations among silkworms. BMC Genomics. 2014;15:251. doi: 10.1186/1471-2164-15-251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nicholas TJ, Cheng Z, Ventura M, Mealey K, Eichler EE, Akey JM. The genomic architecture of segmental duplications and associated copy number variants in dogs. Genome Res. 2009;19(3):491–499. doi: 10.1101/gr.084715.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bailey JA, Yavor AM, Massa HF, Trask BJ, Eichler EE. Segmental duplications: organization and impact within the current human genome project assembly. Genome Res. 2001;11(6):1005–1017. doi: 10.1101/gr.GR-1871R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bailey JA, Gu Z, Clark RA, Reinert K, Samonte RV, Schwartz S, Adams MD, Myers EW, Li PW, Eichler EE. Recent segmental duplications in the human genome. Science. 2002;297(5583):1003–1007. doi: 10.1126/science.1072047. [DOI] [PubMed] [Google Scholar]
- 44.Liu GE, Ventura M, Cellamare A, Chen L, Cheng Z, Zhu B, Li C, Song J, Eichler EE. Analysis of recent segmental duplications in the bovine genome. BMC Genomics. 2009;10:571. doi: 10.1186/1471-2164-10-571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Carneiro M, Rubin CJ, Di Palma F, Albert FW, Alfoldi J, Barrio AM, Pielberg G, Rafati N, Sayyab S, Turner-Maier J, et al. Rabbit genome analysis reveals a polygenic basis for phenotypic change during domestication. Science. 2014;345(6200):1074–1079. doi: 10.1126/science.1253714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kalsi G, Whiting P, Bourdelles BL, Callen D, Barnard EA, Gurling H. Localization of the human NMDAR2D receptor subunit gene (GRIN2D) to 19q13.1-qter, the NMDAR2A subunit gene to 16p13.2 (GRIN2A), and the NMDAR2C subunit gene (GRIN2C) to 17q24-q25 using somatic cell hybrid and radiation hybrid mapping panels. Genomics. 1998;47(3):423–425. doi: 10.1006/geno.1997.5132. [DOI] [PubMed] [Google Scholar]
- 47.Wild EJ, Mudanohwo EE, Sweeney MG, Schneider SA, Beck J, Bhatia KP, Rossor MN, Davis MB, Tabrizi SJ. Huntington’s disease phenocopies are clinically and genetically heterogeneous. Mov Disord. 2008;23(5):716–720. doi: 10.1002/mds.21915. [DOI] [PubMed] [Google Scholar]
- 48.Yamagishi S, Yamada K, Sawada M, Nakano S, Mori N, Sawamoto K, Sato K. Netrin-5 is highly expressed in neurogenic regions of the adult brain. Front Cell Neurosci. 2015;9:146. doi: 10.3389/fncel.2015.00146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Durand CM, Perroy J, Loll F, Perrais D, Fagni L, Bourgeron T, Montcouquiol M, Sans N. SHANK3 mutations identified in autism lead to modification of dendritic spine morphology via an actin-dependent mechanism. Mol Psychiatry. 2012;17(1):71–84. doi: 10.1038/mp.2011.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Topert C, Doring F, Derst C, Daut J, Grzeschik KH, Karschin A. Cloning, structure and assignment to chromosome 19q13 of the human Kir2.4 inwardly rectifying potassium channel gene (KCNJ14) Mamm Genome. 2000;11(3):247–249. doi: 10.1007/s003350010047. [DOI] [PubMed] [Google Scholar]
- 51.Fujikawa-Adachi K, Nishimori I, Taguchi T, Yuri K, Onishi S. cDNA sequence, mRNA expression, and chromosomal localization of human carbonic anhydrase-related protein, CA-RP XI. Biochim Biophys Acta. 1999;1431(2):518–524. doi: 10.1016/S0167-4838(99)00067-9. [DOI] [PubMed] [Google Scholar]
- 52.Shen Y, Inoue N, Heese K. Neurotrophin-4 (ntf4) mediates neurogenesis in mouse embryonic neural stem cells through the inhibition of the signal transducer and activator of transcription-3 (stat3) and the modulation of the activity of protein kinase B. Cell Mol Neurobiol. 2010;30(6):909–916. doi: 10.1007/s10571-010-9520-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Biffi A, Montini E, Lorioli L, Cesani M, Fumagalli F, Plati T, Baldoli C, Martino S, Calabria A, Canale S, et al. Lentiviral hematopoietic stem cell gene therapy benefits metachromatic leukodystrophy. Science. 2013;341(6148):1233158. doi: 10.1126/science.1233158. [DOI] [PubMed] [Google Scholar]
- 54.Setchell BP. Domestication and Reproduction. Anim Reprod Sci. 1992;28(1–4):195–202. doi: 10.1016/0378-4320(92)90106-N. [DOI] [Google Scholar]
- 55.Hu T, Wang Z, Zeng F, Chen X, Gu Z, Zheng L, Tong Q. Expression pattern of testis-specific expressed gene 2 in cryptorchidism model and its role in apoptosis of spermatogenic cells. J Huazhong Univ Sci Technolog Med Sci. 2010;30(2):193–197. doi: 10.1007/s11596-010-0212-3. [DOI] [PubMed] [Google Scholar]
- 56.Baba T. Current concept of sperm/egg fusion in mouse: From Fertilin to Izumo1. Biol Reprod. 2008;78(1 Supplement):276. [Google Scholar]
- 57.Ashley RA, Yu Z, Fung KM, Frimberger D, Kropp BP, Penning TM, Lin HK. Developmental evaluation of aldo-keto reductase 1C3 expression in the cryptorchid testis. Urology. 2010;76(1):67–72. doi: 10.1016/j.urology.2009.09.046. [DOI] [PubMed] [Google Scholar]
- 58.Man PS, Wells T, Carter DA. Cellular distribution of Egr1 transcription in the male rat pituitary gland. J Mol Endocrinol. 2014;53(2):271–280. doi: 10.1530/JME-14-0158. [DOI] [PubMed] [Google Scholar]
- 59.Shetty J, Wolkowicz MJ, Digilio LC, Klotz KL, Jayes FL, Diekman AB, Westbrook VA, Farris EM, Hao Z, Coonrod SA, et al. SAMP14, a novel, acrosomal membrane-associated, glycosylphosphatidylinositol-anchored member of the Ly-6/urokinase-type plasminogen activator receptor superfamily with a role in sperm-egg interaction. J Biol Chem. 2003;278(33):30506–30515. doi: 10.1074/jbc.M301713200. [DOI] [PubMed] [Google Scholar]
- 60.Itoh N. The Fgf families in humans, mice, and zebrafish: their evolutional processes and roles in development, metabolism, and disease. Biol Pharm Bull. 2007;30(10):1819–1825. doi: 10.1248/bpb.30.1819. [DOI] [PubMed] [Google Scholar]
- 61.Ronellenfitsch S, Weiss C, Frommhold D, Koch L, Mollenhauer J, Poeschl J, Muller H. High DMBT1 concentrations in breast milk correlate with increased risk of infection in preterm and term neonates. BMC Pediatr. 2012;12:157. doi: 10.1186/1471-2431-12-157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wang Z, Yonezawa T, Liu B, Ma T, Shen X, Su J, Guo S, Hasegawa M, Liu J. Domestication relaxed selective constraints on the yak mitochondrial genome. Mol Biol Evol. 2011;28(5):1553–1556. doi: 10.1093/molbev/msq336. [DOI] [PubMed] [Google Scholar]
- 63.Flaquer A, Baumbach C, Kriebel J, Meitinger T, Peters A, Waldenberger M, Grallert H, Strauch K. Mitochondrial genetic variants identified to be associated with BMI in adults. PLoS One. 2014;9(8):e105116. doi: 10.1371/journal.pone.0105116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Papadopoulou LC, Sue CM, Davidson MM, Tanji K, Nishino I, Sadlock JE, Krishna S, Walker W, Selby J, Glerum DM, et al. Fatal infantile cardioencephalomyopathy with COX deficiency and mutations in SCO2, a COX assembly gene. Nat Genet. 1999;23(3):333–337. doi: 10.1038/15513. [DOI] [PubMed] [Google Scholar]
- 65.Davis RE, Miller S, Herrnstadt C, Ghosh SS, Fahy E, Shinobu LA, Galasko D, Thal LJ, Beal MF, Howell N, et al. Mutations in mitochondrial cytochrome c oxidase genes segregate with late-onset Alzheimer disease. Proc Natl Acad Sci U S A. 1997;94(9):4526–4531. doi: 10.1073/pnas.94.9.4526. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 66.Kirby DM, McFarland R, Ohtake A, Dunning C, Ryan MT, Wilson C, Ketteridge D, Turnbull DM, Thorburn DR, Taylor RW. Mutations of the mitochondrial ND1 gene as a cause of MELAS. J Med Genet. 2004;41(10):784–789. doi: 10.1136/jmg.2004.020537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kohane MJ, Parsons PA. Evolutionary Biology: Volume 23. Boston, MA: Springer US; 1988. Domestication; pp. 31–48. [Google Scholar]
- 68.Nelson DW, Gao Y, Spencer NM, Banh T, Yen CL. Deficiency of MGAT2 increases energy expenditure without high-fat feeding and protects genetically obese mice from excessive weight gain. J Lipid Res. 2011;52(9):1723–1732. doi: 10.1194/jlr.M016840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Arimitsu E, Aoki S, Ishikura S, Nakanishi K, Matsuura K, Hara A. Cloning and sequencing of the cDNA species for mammalian dimeric dihydrodiol dehydrogenases. Biochem J. 1999;342(Pt 3):721–728. doi: 10.1042/bj3420721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lukacik P, Keller B, Bunkoczi G, Kavanagh KL, Lee WH, Adamski J, Oppermann U. Structural and biochemical characterization of human orphan DHRS10 reveals a novel cytosolic enzyme with steroid dehydrogenase activity. Biochem J. 2007;402(3):419–427. doi: 10.1042/BJ20061319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Miljkovic I, Yerges LM, Li H, Gordon CL, Goodpaster BH, Kuller LH, Nestlerode CS, Bunker CH, Patrick AL, Wheeler VW, et al. Association of the CPT1B gene with skeletal muscle fat infiltration in Afro-Caribbean men. Obesity (Silver Spring) 2009;17(7):1396–1401. doi: 10.1038/oby.2008.677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Lackey DE, Lynch CJ, Olson KC, Mostaedi R, Ali M, Smith WH, Karpe F, Humphreys S, Bedinger DH, Dunn TN, et al. Regulation of adipose branched-chain amino acid catabolism enzyme expression and cross-adipose amino acid flux in human obesity. Am J Physiol Endocrinol Metab. 2013;304(11):E1175–1187. doi: 10.1152/ajpendo.00630.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Tominaga T, Dutta RK, Joladarashi D, Doi T, Reddy JK, Kanwar YS. Transcriptional and Translational Modulation of myo-Inositol Oxygenase (Miox) by Fatty Acids: Implications in renal tubular injury induced in obesity and diabetes. J Biol Chem. 2016;291(3):1348–1367. doi: 10.1074/jbc.M115.698191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Yamada M, Miyakawa T, Duttaroy A, Yamanaka A, Moriguchi T, Makita R, Ogawa M, Chou CJ, Xia B, Crawley JN, et al. Mice lacking the M3 muscarinic acetylcholine receptor are hypophagic and lean. Nature. 2001;410(6825):207–212. doi: 10.1038/35065604. [DOI] [PubMed] [Google Scholar]
- 75.Serao NV, Gonzalez-Pena D, Beever JE, Bollero GA, Southey BR, Faulkner DB, Rodriguez-Zas SL. Bivariate genome-wide association analysis of the growth and intake components of feed efficiency. PLoS One. 2013;8(10):e78530. doi: 10.1371/journal.pone.0078530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Velleman SG. Meat Science and Muscle Biology Symposium: extracellular matrix regulation of skeletal muscle formation. J Anim Sci. 2012;90(3):936–941. doi: 10.2527/jas.2011-4497. [DOI] [PubMed] [Google Scholar]
- 77.Sherriff JL, O’Sullivan TA, Properzi C, Oddo JL, Adams LA. Choline, Its Potential Role in Nonalcoholic Fatty Liver Disease, and the Case for Human and Bacterial Genes. Adv Nutr. 2016;7(1):5–13. doi: 10.3945/an.114.007955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Hara Y, Sassi Y, Guibert C, Gambaryan N, Dorfmuller P, Eddahibi S, Lompre AM, Humbert M, Hulot JS. Inhibition of MRP4 prevents and reverses pulmonary hypertension in mice. J Clin Invest. 2011;121(7):2888–2897. doi: 10.1172/JCI45023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Son TW, Yun SP, Yong MS, Seo BN, Ryu JM, Youn HY, Oh YM, Han HJ. Netrin-1 protects hypoxia-induced mitochondrial apoptosis through HSP27 expression via DCC- and integrin alpha6beta4-dependent Akt, GSK-3beta, and HSF-1 in mesenchymal stem cells. Cell Death Dis. 2013;4:e563. doi: 10.1038/cddis.2013.94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Edgar AJ, Birks EJ, Yacoub MH, Polak JM. Cloning of dexamethasone-induced transcript: a novel glucocorticoid-induced gene that is upregulated in emphysema. Am J Respir Cell Mol Biol. 2001;25(1):119–124. doi: 10.1165/ajrcmb.25.1.4417. [DOI] [PubMed] [Google Scholar]
- 81.Chang CH, Guerder S, Hong SC, van Ewijk W, Flavell RA. Mice lacking the MHC class II transactivator (CIITA) show tissue-specific impairment of MHC class II expression. Immunity. 1996;4(2):167–178. doi: 10.1016/S1074-7613(00)80681-0. [DOI] [PubMed] [Google Scholar]
- 82.Storici P, Tossi A, Lenarcic B, Romeo D. Purification and structural characterization of bovine cathelicidins, precursors of antimicrobial peptides. Eur J Biochem. 1996;238(3):769–776. doi: 10.1111/j.1432-1033.1996.0769w.x. [DOI] [PubMed] [Google Scholar]
- 83.M-y Y, D-h Z, J-z L, J-z C, X-q Z. Prevalence of yak main parasitic diseases in China and strategies for its control. China Animal Hus Vet Med. 2014;05:227–230. [Google Scholar]
- 84.Liu A, Guan G, Liu Z, Liu J, Leblanc N, Li Y, Gao J, Ma M, Niu Q, Ren Q, et al. Detecting and differentiating Theileria sergenti and Theileria sinensis in cattle and yaks by PCR based on major piroplasm surface protein (MPSP) Exp Parasitol. 2010;126(4):476–481. doi: 10.1016/j.exppara.2010.05.024. [DOI] [PubMed] [Google Scholar]
- 85.Gao S, Qiu C, Zhou J, Zhang Y. Serologic Monitoring of Bovine Viral Diarrhea/Mucosal Disease in Yellow Cattle and Yaks in Partial regions of the South-western and North-western Five Provinces. Chin J Vet Sci Technol. 1999;07:17–18. [Google Scholar]
- 86.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25(16):2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20(9):1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Marques-Bonet T, Kidd JM, Ventura M, Graves TA, Cheng Z, Hillier LW, Jiang Z, Baker C, Malfavon-Borja R, Fulton LA, et al. A burst of segmental duplications in the genome of the African great ape ancestor. Nature. 2009;457(7231):877–881. doi: 10.1038/nature07744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Lou H, Li S, Jin W, Fu R, Lu D, Pan X, Zhou H, Ping Y, Jin L, Xu S. Copy number variations and genetic admixtures in three Xinjiang ethnic minority groups. Eur J Hum Genet. 2015;23(4):536–542. doi: 10.1038/ejhg.2014.134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Xu Y, Shi T, Cai H, Zhou Y, Lan X, Zhang C, Lei C, Qi X, Chen H. Associations of MYH3 gene copy number variations with transcriptional expression and growth traits in Chinese cattle. Gene. 2014;535(2):106–111. doi: 10.1016/j.gene.2013.11.057. [DOI] [PubMed] [Google Scholar]
- 91.Sambrook JRD. Molecular Cloning: A Laboratory Manual. 3. New York: Cold Spring Harbor Laborator; 2001. pp. 1.31–1.43. [Google Scholar]
- 92.Conesa A, Gotz S. Blast2GO: A comprehensive suite for functional analysis in plant genomics. Int J Plant Genomics. 2008;2008:619832. doi: 10.1155/2008/619832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Redon R, Ishikawa S, Fitch KR, Feuk L, Perry GH, Andrews TD, Fiegler H, Shapero MH, Carson AR, Chen W, et al. Global variation in copy number in the human genome. Nature. 2006;444(7118):444–454. doi: 10.1038/nature05329. [DOI] [PMC free article] [PubMed] [Google Scholar]