

Abstract
Background
Next-generation sequencing now allows for total RNA extracts to be sequenced in non-model organisms such as bamboos, an economically and ecologically important group of grasses. Bamboos are divided into three lineages, two of which are woody perennials with bisexual flowers, which undergo gregarious monocarpy. The third lineage, which are herbaceous perennials, possesses unisexual flowers that undergo annual flowering events.
Results
Transcriptomes were assembled using both reference-based and de novo methods. These two methods were tested by characterizing transcriptome content using sequence alignment to previously characterized reference proteomes and by identifying Pfam domains. Because of the striking differences in floral morphology and phenology between the herbaceous and woody bamboo lineages, MADS-box genes, transcription factors that control floral development and timing, were characterized and analyzed in this study. Transcripts were identified using phylogenetic methods and categorized as A, B, C, D or E-class genes, which control floral development, or SOC or SVP-like genes, which control the timing of flowering events. Putative nuclear orthologues were also identified in bamboos to use as phylogenetic markers.
Conclusions
Instances of gene copies exhibiting topological patterns that correspond to shared phenotypes were observed in several gene families including floral development and timing genes. Alignments and phylogenetic trees were generated for 3,878 genes and for all genes in a concatenated analysis. Both the concatenated analysis and those of 2,412 separate gene trees supported monophyly among the woody bamboos, which is incongruent with previous phylogenetic studies using plastid markers.
Electronic supplementary material
The online version of this article (doi:10.1186/s12864-016-2707-1) contains supplementary material, which is available to authorized users.
Keywords: Transcriptome, Bambusoideae, Woody bamboos, RNA-Seq, MADS-box
Background
The Bambusoideae are a subfamily of perennial forest grasses endemic to every continent except Europe and Antarctica and comprise approximately 1,450 species [1, 2]. Bamboos exhibit a combination of characters that uniquely distinguish the subfamily within the larger evolutionary radiation of Poaceae. Bambusoideae is divided phylogenetically into three well-supported lineages: temperate woody (Arundinarieae), tropical woody (Bambuseae) and herbaceous (Olyreae) bamboos [2, 3]. Both lineages of woody bamboos are characterized by complex rhizome systems, a tree-like habit with highly lignified and usually hollow culms, well-differentiated culm leaves, well developed aerial branching, foliage leaf blades with outer ligules, and bisexual spikelets. Woody bamboos typically exhibit gregarious flowering cycles followed by death of the parent plants (monocarpy) [4]. They also serve as an economically important resource as they produce timber, fiber, food and other products. Herbaceous bamboos lack well differentiated culm leaves and outer ligules combined with relatively weakly lignified culms, restricted vegetative branching, unisexual spikelets and seasonal flowering [4].
Despite the uncertainty of their phylogenetic relationships [2, 3, 5–9], the two woody bamboo lineages share aspects of phenology and sexual systems suggestive of common ancestry. Their phenological patterns can be especially striking as they can exhibit extremely long intervals between flowering periods (3—120 years), which may be synchronized between disjunct populations [10]. The subsequent die-off following a flowering event can result in sudden ecological consequences such as lower shade levels in former bamboo forests.
Floral characteristics of Olyreae contrasts with those of woody bamboos in both phenology and sexual systems as the herbaceous species flower annually and possess unisexual spikelets, which are either segregated into different inflorescences or found together in a mixed inflorescence, in both cases on monoecious plants. Phenological differences between herbaceous and woody bamboos impact phylogenetic studies. Members of Olyreae generally exhibit elevated mutation rates compared to those of the woody bamboos, which are correlated with shorter generation times and longer branch lengths in phylogenetic trees [2, 11–13].
Floral development in angiosperms has been found to be largely controlled by MADS-box genes. Named after four of their homologues (MCM1, AGAMOUS, DEFICIENS, SRF), these transcription factors control the development of each of the four floral whorls (sepal, petal, stamen, and carpel). The function of MADS box genes in floral development has been extensively studied using another grass, Oryza sativa, as well as the eudicots Antirrhinum majus and Arabidopsis thaliana. The mechanism of development can be generally described using the classical ‘ABC’ model [14]. In this model, sepals develop with the expression of A-class genes, petals develop with the expression of A and B-class genes, stamens develop with the expression of B and C-class genes, and carpels develop with the expression of C-class genes. Subsequent studies have added D and E-class genes to the model in which expression of E-class genes is required for B and C-class function [15] and D-class gene expression is required for ovule development [16, 17] A further refinement on this understanding of floral developmental genetics is the quartet model, which suggests that MADS-box proteins work in groups of four to initiate transcription [18].
In addition to development of floral structures, the expression of some MADS box genes can affect the timing of flowering. The SUPRRESSION OF OVER-EXPRESSION OF CONSTANS 1 (SOC1) gene in A. thaliana as well as its homolog OsMADS56 in O. sativa have been characterized as being involved in several steps in the process of inducing floral development [19, 20]. The SHORT VEGETATIVE PHASE (SVP) in A. thaliana and OsMADS22 + 55 in O. sativa have been also shown to control flowering and can act as antagonists of the SOC1 genes [21, 22]. These genes are of particular interest in bamboos because of the aforementioned phenological characteristics. Next-generation sequencing (NGS) has allowed gene expression to be examined at a large scale for non-model organisms using the RNA-Seq method [23]. RNA-Seq typically uses mRNA selected by the poly-A tails to filter only for eukaryotic protein-coding transcripts. The RNA is reverse-transcribed into cDNA, which is fragmented and sequenced on a NGS platform such as Illumina. The RNA-Seq method had first been used in a bamboo species by Zhang et al. [24] to characterize the floral transcriptome of Dendrocalamus latiflorus (Bambuseae) with a follow up study on seed, leaf, stem, shoot and root tissue of the same species by Liu et al. [25]. Changes in transcript abundance in shoots of Phyllostachys edulis (Arundinarieae) during development were examined by Peng et al. [26] and a similar study on flowers was conducted by Gao et al. [27].
In this study, floral transcriptomes are characterized from four species representing three major bamboo lineages: Guadua inermis and Otatea acuminata (tropical woody Bambuseae), Phyllostachys aurea (temperate woody Arundinarieae) and Lithachne pauciflora (herbaceous Olyreae). The floral structure of these species vary as L. pauciflora has unisexual florets while the three woody bamboos have hermaphroditic florets. P. aurea and O. acuminata have three stamens [28, 29] while G. inermis has six. G. inermis, O. acuminata, and L. pauciflora have two stigmas [30, 31] while P. aurea has three [29].
These transcriptomes were analyzed to meet three complementary objectives. First, the content of each transcriptome was characterized by transcript identification and categorization. Transcripts conserved across the grass family and other plants are identified as well as bamboo-specific transcripts. Two specific methods of assembly were compared. In the past two years, the first draft nuclear genome for a bamboo (Phyllostachys heterocycla) was sequenced and published [32]. Our sampling scheme includes transcriptome data from the congeneric P. aurea, which allows for a reference-based assembly to be tested and compared to a de novo assembly.
Second, the evolutionary histories of the genes important to floral timing and development in grasses were explored. Because both bisexual and unisexual flowers and two very distinct phenological patterns are present within the taxa examined here, these includes the developmental A, B, C, D and E-class MADS-box genes as well as the SVP and SOC1 gene families, which are involved in reproductive timing.
Lastly, the evolutionary history of the bamboo species examined here was examined by alignment and analysis of putative nuclear orthologues. Genes were selected based on their single-copy status in two reference taxa from the Bambusoideae-Oryzoideae-Pooideae (BOP) clade (Brachypodium distachyon and O. sativa). The goal of this portion of the study was to find markers from transcriptomic data that were available for phylogenetic analysis and to recover the overall phylogenetic signal from them. All analyses were performed on a gene-by-gene basis and also in one large concatenated alignment.
Methods
Mature spikelets from Guadua inermis, Lithachne pauciflora, Otatea acuminata, and Phyllostachys aurea were collected from tropical seasonal forests in the state of Veracruz, Mexico during the mid-dry season when florets were at full anthesis (Fig. 1). Harvested material was immediately placed in RNA-later solution (Qiagen, Valencia, CA, USA). Herbarium vouchers were collected for each species and taxonomic identities were verified (Table 1). Samples were stored at −20 °C prior to RNA extraction. Spikelets were homogenized in liquid nitrogen and a full RNA extraction and on-column DNase digestion was performed using the RNeasy extraction kit (Qiagen, Valecia, CA, USA) following the manufacturer’s protocol. Extractions were quantified with a Nanodrop 1000 (ThermoFisher Scientific, Wilmington, DE, USA) to verify a minimum concentration of 50 ng/ul. Extractions from male and female spikelets of L. pauciflora were pooled to represent transcripts from both genders. All other species had bisexual flowers. Library preparation and sequencing were performed at the Carver Biotechnology Center (University of Illinois, Urbana-Champaign, IL, USA). Four samples were sequenced paired-end on the HiSeq 2000 platform (Illumina, San Diego, CA, USA). Sequence areas that showed a significantly low quality score (p < 0.05) were deleted from each set of reads using the DynamicTrim function of SolexaQA [33]. Adapter sequences were trimmed from each read using CutAdapt v 1.7 [34].
Fig. 1.

a Guadua inermis pseudospikelets. b Otatea acuminata, spikelets. c Phyllostachys aurea, pseudospikelets. d Lithachne pauciflora, male and female spikelets (floral structures of the other three species are bisexual). Note that photos (a, b and c) represent the actual specimens collected for this study. Photo (d) represents a separate individual of the same species. Photos by E. Ruiz-Sanchez
Table 1.
Species used in this study, with collection sites, collectors, collector numbers, the herbarium where the specimen vouchers are deposited and the number of paired-end reads generated for each specimen
| Taxon | Country/state | Collectors/number | Herbarium | Number of read pairs |
|---|---|---|---|---|
| Guadua inermis Rupr. ex E. Fourn | Mexico, Veracruz | E. Ruiz-Sanchez & W. Wysocki, 466 | IEB | 54,488,684 |
| Otatea acuminata (Munro) Calderón & Soderstr. | Mexico, Veracruz | E. Ruiz-Sanchez & W. Wysocki, 469 | IEB | 55,890,954 |
| Phyllostachys aurea Carrière ex Rivière & C. Rivière | Mexico, Veracruz | E. Ruiz-Sanchez & W. Wysocki, 470 | IEB | 46,405,022 |
| Lithachne pauciflora (Sw.) P. Beauv. | Mexico, Veracruz | E. Ruiz-Sanchez & W. Wysocki, 470a | IEB | 58,017,553 |
Transcript assembly
Two transcriptome assemblies were performed for each taxon. 1) Reads were assembled de novo into contiguous sequences (contigs) using Trinity v. r20140717 [35]. Contigs were clustered by sequence similarity using the Chrysalis function of Trinity with default settings to reduce redundancy. 2) A reference-guided assembly was performed using Tophat v. 2.0.13 [36] and Bowtie2 v. 0.12.7 [37] to map reads to the previously sequenced Phyllostachys heterocycla nuclear scaffolds. The parameters of Bowtie2 were optimized to account for differences between the target and reference genomes by using the ‘very-sensitive’ setting in end-to-end mode and increasing the allowed number of mismatches in each read alignment to ten. Mapped reads were then assembled using Cufflinks v. 2.2.1 [36] and the gffread function of Cufflinks was used to extract transcripts from the P. heterocycla genome and to identify exon boundaries. Reads were mapped to these sets of transcripts using the same parameters and the consensus sequence of each was extracted using the mpileup function of samtools [38]. Transcripts were then clustered by 100 % sequence similarity using CD-HIT v. 4.6 [39]. All subsequent analyses were performed on both sets of assembled transcripts.
TransDecoder (http://transdecoder.github.io) was used to predict putative reading frames, provide translations for each reading frame and further cluster transcripts. Putative reading frames were then screened for coding potential using CPAT v. 1.2.1 [40]. Known coding and non-coding RNA transcripts from three well-annotated plant genomes (A. thaliana, O. sativa and Hordeum vulgare) were used as training data to generate a logit model for coding potential assessment. Putative reading frames were screened for a coding potential probability over 98 %. Putative reading frames that fulfilled these coding potential criteria are henceforth referred to as putative coding transcripts (PCTs). PCTs were used as the basis of most transcriptomic quantitation in this study as they confidently reflect the protein coding assemblage and serve as a computationally efficient method of transcriptome annotation. TransDecoder was also used in combination with HMMER 3.0 [41] to predict functional domains in the PCTs using hidden Markov models (HMMs) and the Pfam database [42].
Transcriptome content analysis
Both sets of PCTs were queried against the rice (O. sativa) proteome using BLASTX from the BLAST software package v. 2.2.25 [43] to identify putative function. Rice was chosen as a reference because of its phylogenetic proximity to the bamboos and high level of annotation. The BLAST query was repeated twice to screen for matches to the A. thaliana and P. heterocycla proteomes. A. thaliana was chosen as it has the most thoroughly explored and annotated plant genome and P. heterocycla was chosen to identify any bamboo-specific sequences. All PCTs that matched to at least one of these proteomes will be referred to as plant-PCTs (pPCTs).
A BLASTN query of pPCTs from the de novo assembly against those produced by the reference-based assembly was performed for each taxon with a threshold e-value of 10−5 and an identity cutoff of 95 % to determine which pPCTs were assembled by both methods and which were specific to their assembly method. All contigs were queried against the Guadua weberbaueri plastid genome (plastome) (GenBank: KP793062) using BLASTN with an e-value threshold of 10−5 to test for plastid contamination within the Illumina libraries.
MADS box identification and evolutionary analysis
MADS box homologues were identified by querying those from B. distachyon against our respective transcript sets using BLASTP. Homologues from B. distachyon were used because a thorough and recent survey of the full genome was performed to identify MADS box genes [44]. Hits were filtered for redundancy and for sequences over 100 amino acids (aa) in length. Bamboo proteins were aligned conspecifically to identify copies that were assembled by both methods but differed because of assembly artifacts. Because the two most similar copies of MADS-box genes from B. distachyon are 93 % identical, copies from the same species that shared over 95 % identity were either reduced to one copy or merged to encompass regions of the protein that were determined using both assemblies. Bamboo sequences and previously identified homologues from B. distachyon and O. sativa were aligned using CLUSTALW [45]. Previously identified MADS-box genes from the genetically well-characterized A. thaliana were added to the alignment to aid in gene copy identification.
Geneious Pro v.8.1.7 (Biomatters, Auckland, New Zealand) was used to generate a neighbor-joining (NJ) tree, with 1,000 bootstrap pseudoreplicates, for all gene copies used in this study. Sequences were assigned to clades identified in Wei et al. [44] based on the presence of non-bamboo reference genes and were assigned names based on the analysis performed in Wei et al. [44]. Note that the copies from B. distachyon are labeled by the numbering system in Genbank rather than the numbering system in the tree generated by Wei et al. [44]. When the gene families of interest were identified, each set of protein sequences that corresponded to each family were grouped separately and outgroup OTUs were assigned based on the original NJ tree. A CLUSTALW alignment and NJ analysis was performed on each gene family separately to generate the best trees. All subsequent tree annotations were performed using the ETE Python package [46].
Nuclear orthologue phylogenetics
Only the transcripts assembled de novo were used because they were less likely to be overrepresented based on preliminary results. Single-copy syntenic orthologous coding sequences, determined by Schnable et al. [47], were extracted from the O. sativa and B. distachyon genomes. BLASTN was used to query coding sequences from O. sativa against pPCTS from the four bamboo species that were assembled de novo and combined with all identified coding sequences from B. distachyon and P. heterocycla. Blast hits were filtered for a maximum e-value of 10−5, a minimum alignment length of 100 bp and a minimum sequence identity of 70 % following methods from Zhang et al. [48]. BLASTN results were queried for instances where one copy from O. sativa exhibited exactly one hit to a copy from B. distachyon and vice versa. The best hit from each bamboo species to each copy from O. sativa was located based on the highest bit-score. The homologous portions of each gene were extracted into clusters with the corresponding O. sativa transcript. Only groups that contained all bamboo species were used in subsequent steps. Each cluster, that included five bamboo sequences and a reference sequence from O. sativa, was then aligned using the LINSI algorithm of MAFFT [49], which is optimized to align nucleotide sequences accurately.
A maximum-likelihood (ML) phylogeny was then estimated using RAxML [50] for each alignment. The GTRGAMMA-I model was used for all trees. A majority consensus tree was produced from all ML trees using the Consense function in the PHYLIP software package [51]. An additional matrix was produced by concatenating all transcript alignments and removing all nucleotide positions that contained at least one gap to reduce ambiguity in downstream analyses. A ML analysis was also performed with RAxML using this matrix. The GTRGAMMA-I model was used and 1,000 ML bootstrap pseudoreplicates were performed with the concatenated alignment to assess topological support.
Results
A total of 214,802,213 pairs of reads were sequenced for the four taxa. A total of 505,939 unique transcripts were produced from the de novo assembly and 613,319 were produced from the reference-based assembly. Open reading frame (ORF) detection followed by a coding potential filtering step in the de novo assembly yielded over 78,000 PCTs in O. acuminata and between 37,000 and 48,000 PCTs in the three remaining species. ORF detection in the reference-based assembly yielded between 43,000 and 105,000 PCTs. The number of reads is reported (Table 1). The number of contigs and PCTs produced for each species and method is also reported (Table 2).
Table 2.
The number of contigs, PCTs and pPCTs generated for each taxon. Results from both assemblies are reported here
| Taxon | Contigs | PCTS | pPCTs | |
|---|---|---|---|---|
| De novo | G. inermis | 108438 | 44252 | 19254 |
| O. acuminata | 111831 | 78443 | 31498 | |
| P. aurea | 88256 | 48001 | 21033 | |
| L. pauciflora | 197414 | 37848 | 17212 | |
| Reference | G. inermis | 96762 | 43226 | 42788 |
| O. acuminata | 180171 | 97094 | 95665 | |
| P. aurea | 197627 | 104413 | 103165 | |
| L. pauciflora | 144759 | 77433 | 76278 |
A BLASTX query of PCTs assembled de novo against the O. sativa, A. thaliana, and P. heterocycla proteomes (pPCTs) produced hits that covered 40–45 % of PCTs in the four bamboo species. The same query of the reference-based assembly covered > 98 % of PCTs in all four bamboo species (Table 2). The de novo assembly produced the highest number of unique pPCTs from the BLASTX search to the O. sativa proteome while the reference based assembly produced the highest number of unique pPCTs from the P. heterocycla proteome. All three genomes were able to recover 85.8 % of the pPCTs assembled de novo and 90.7 % of the pPCTs assembled using a reference genome. The numbers of pPCTs that were recovered using all reference proteomes are reported (Fig. 2).
Fig. 2.
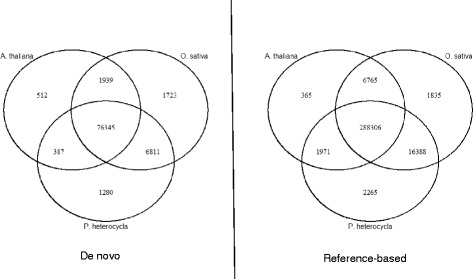
Venn diagrams reporting the number of pPCT hits that are unique to each reference proteome and shared by them. Diagrams are separated by assembly type. Venn diagrams were generated using the VennDiagram R-package
The BLASTN comparison between the pPCTs assembled de novo and using a reference genome revealed that in G. inermis, O. acuminata and P. aurea 90–98 % of pPCTs were represented in the de novo assembly by at least 95 % sequence similarity. In L. pauciflora 53 % of pPCTs generated using a reference genome were represented in the de novo assembly. In all taxa, 43–76 % of pPCTS assembled de novo were represented in the reference-based assemblies (Fig. 3) with the P. aurea assembly containing the largest portion.
Fig. 3.
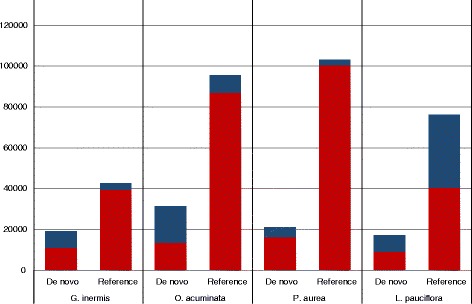
Bar graph indicating the number of pPCTs (putative transcripts that show a close plant homolog) for each taxon for both assemblies. The red portion of each bar indicates the number of redundant transcripts that exhibit at least 95 % nucleotide sequence identity to the other assembly. The blue portion represents transcripts that do not reach this criterion and are unique to their respective assembly
The HMM search on the Pfam database revealed 129,695 Pfam domains in 77,429 of the pPCTs assembled de novo (1.67 domains/pPCT) and 379,169 domains in 248,140 of the pPCTs assembled using a reference genome (1.53 domains/pPCT). A total of 13,650 unique domain types were represented in both species. The most numerous type of domain in both assemblies was the ‘protein kinase domain.’ Between 62 and 69 % of all represented Pfam domains were present in both assemblies while 7–22 % were found only in the de novo assembly and 13–25 % were only found in the reference-based assembly (Fig. 4).
Fig. 4.
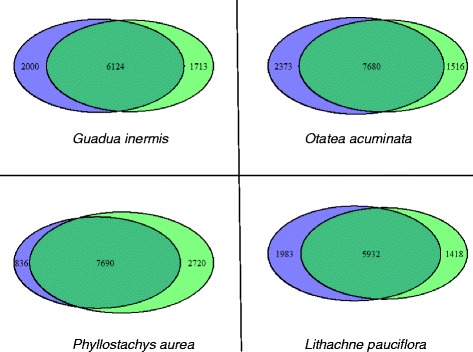
Venn diagrams reporting the number of Pfam domains that are unique to each assembly and shared by them both. Diagrams are separated by taxon. Venn diagrams are proportional to their values and were generated using the VennDiagram R-package
Querying the contig sets for similarities to the G. weberbaueri full plastome produced 94 matches from transcripts assembled de novo and 101 from transcripts assembled using a reference genome. The transcripts assembled de novo ranged from 1,286–47,612 bp (see Discussion section for explanation) and those assembled using a reference ranged from 352 – 6,151 bp.
Exploration of expressed floral genes
In the following, the terms “genes” and “gene copies” refer to those that were expressed and assembled from our transcriptomic data set. After elimination of redundant copies from the transcript sets, a total of 72 MADS-box genes were identified within the six gene families of interest: A, B, C/D, E-class, SOC-like and SVP-like MADS-box genes. The fewest number of copies were identified in the SVP-like gene family (4) and the most were identified in the B-class gene family (17). The fewest number of identified copies were expressed in G. inermis (13), while the other three taxa expressed comparable numbers (19–20). The total number of copies expressed by each taxon for all gene families is reported (Table 3). Bamboo gene copies that exhibit putative orthology to copies from O. sativa are listed (Additional file 1: Table S1).
Table 3.
A total of 72 MADS-box genes were identified in this study. Numbers from each gene class and taxon are reported
| Class | G. inermis | O. acuminata | P. aurea | L. pauciflora | Total |
|---|---|---|---|---|---|
| A | 2 | 2 | 3 | 3 | 10 |
| B | 3 | 4 | 3 | 7 | 17 |
| C/D | 2 | 4 | 5 | 3 | 14 |
| E | 3 | 5 | 4 | 3 | 15 |
| SOC-Like | 2 | 3 | 4 | 3 | 12 |
| SVP-Like | 1 | 1 | 1 | 1 | 4 |
| Total | 13 | 19 | 20 | 20 | 72 |
Six neighbor-joining gene trees were generated using peptides from each of the six gene families. The gene tree topologies did not uniformly reflect previously recovered taxonomic relationships among species, but did exhibit notable patterns. A sister relationship to other bamboo species was found in 25 % of all copies from L. pauciflora, with the other 75 % associating either with copies from reference taxa or with clades composed of both reference and bamboo copies. A total of 55 % of copies from P. aurea, 69.2 % from G. inermis, and 73.7 % from O. acuminata exhibited a sister relationship to other bamboo species.
The relationships between copies from bamboos and the reference taxa from other grass subfamilies were noted when subtrees contained representative copies from Bambusoideae, Oryzoideae (O. sativa) and Pooideae (B. distachyon) and the copies from Bambusoideae exhibited monophyly. These subtrees could be classified into three distinct categories: 1) Bamboo copies exhibit a sister relationship to copies from O. sativa + B. distachyon, 2) the copy from B. distachyon exhibits monophyly with bamboos and this clade exhibits a sister relationship to copies from O. sativa, 3) the copy from O. sativa exhibits monophyly with bamboos and this clade exhibits a sister relationship to copies from B. distachyon (see clades marked with asterisks in Figs. 5 and 6). A total of 18 distinct instances could be classified into these categories. Eight instances fell under category 1, while five instances could be classified under each of categories 2 and 3.
Fig. 5.
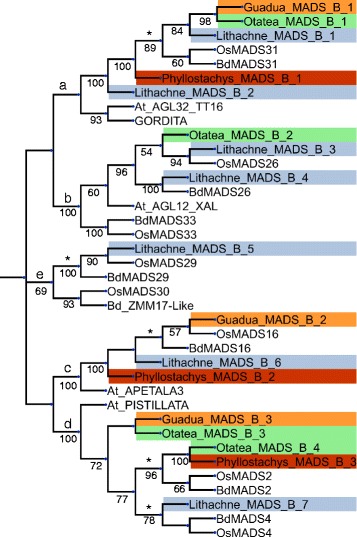
Neighbor-joining gene tree representing the B-class MADS box genes. Gene copies assembled in this study are labeled by genus, colored according to taxa (orange: G. inermis, green: O. acuminata, dark red: P. aurea, blue: L. pauciflora) and numbered redundantly to distinguish copies. Reference gene copies are not colored, are abbreviated by binomial (At: Arabidopsis thaliana, Bd: Brachypodium distachyon, Os: Oryza sativa) and are numbered according to their labeling in Genbank. Nodes that were supported at over 50 % bootstrap are indicated
Fig. 6.
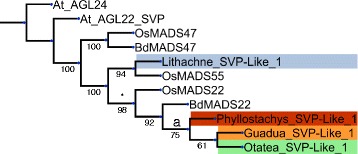
Neighbor-joining gene tree representing the SVP-like MADS box genes. Gene copies assembled in this study are labeled by genus, colored according to taxa (orange: G. inermis, green: O. acuminata, dark red: P. aurea, blue: L. pauciflora) and numbered redundantly to distinguish copies. Reference gene copies are not colored, are abbreviated by binomial (At: Arabidopsis thaliana, Bd: Brachypodium distachyon, Os: Oryza sativa) and are numbered according to their labeling in Genbank. Nodes that were supported at over 50 % bootstrap are indicated
The B-class genes (Fig. 5) formed four clades that associate with copies of MADS-box genes from A. thaliana and a fifth grass-specific clade. Clade a, which associated with AGL32 and GORDITA from A. thaliana, united a bamboo specific subclade with one copy each from O. sativa and B. distachyon. One additional gene copy from both P. aurea and L. pauciflora are also included in clade a diverging in intermediate positions. Clade b, which associated with AGL12/XAL from A. thaliana, comprised one copy from O. acuminata and two copies from L. pauciflora. Each of the copies from L. pauciflora in clade b formed a sister relationship to a copy from O. sativa or B. distachyon. Clade c associated with APETALA1 from A. thaliana and contained one copy each from G. inermis, L. pauciflora and P. aurea. Clade d associated with PISTILLATA from A. thaliana and contained two copies from O. acuminata, and one from each of the other three bamboo species. Clade d also possessed two copies each from O. sativa and B. distachyon. The grass-specific clade (e) comprised one copy from L. pauciflora, which associated with copies OsMADS29 and BdMADS29.
The A-class genes formed two distinct clades that included only grasses and one additional large clade that included grasses and A. thaliana (Additional file 2: Figure S1; A). Clade a associates with OsMADS18 and includes one copy from L. pauciflora, O. acuminata and P. aurea, as well as a gene copy from B. distachyon. The second grass-specific clade (b) consisted of genes from only two species of grasses (OsMADS20 + BdMADS20) and did not include any bamboo copies. Another grass clade associated with the FRUITFULL, CAULIFLOWER, and APETALA1 genes from A. thaliana (c). Clade c included two gene copies from all bamboo species except for O. acuminata, which was represented by one copy.
The C/D class genes are paraphyletic and were combined into one tree (Additional file 2: Figure S1; C/D). The D-class genes from grasses form clade a, while another clade (b) includes C-class genes from grasses (subclades c and d), C-class genes from A. thaliana(e) and one D-class gene from A. thaliana (SEEDSTICK). The four copies from A. thaliana formed a clade with one copy from O. acuminata and no other grasses (e + f). The remainder of the clades (a, c, d) were grass specific. Clade a-1, which associates with OsMADS21 contains two copies from P. aurea, one copy from G. inermis and one gene copy from L. pauciflora. The other exclusively C-class clade (a-2) associates with OsMADS13 and contains one gene copy from all bamboo species except for G. inermis. The next grass-specific clade of D-class genes (c) associated with OsMADS3 and BdMADS3, contained one gene copy from all four bamboo species. The other grass-specific D-class clade (d) contained one gene copy from O. acuminata, one from P. aurea, two copies from O. sativa and one from B. distachyon.
The E-class gene tree (Additional file 3: Figure S2; E) formed three clades that associated with MADS-box gene copies from A. thaliana(a, b, c) and one grass specific clade (d). One gene copy from O. acuminata and one from P. aurea associated with RSB from A. thaliana in clade a along with OsMADS6, BdMADS6, and OsMADS17. Clade b contained SEPALLATA3 from A. thaliana, which formed a sister relationship to two subclades of grasses that each associated with a gene copy from O. sativa. The first subclade (b-1) contained one gene copy from G. inermis, one copy from L. pauciflora, and OsMADS8. The second subclade (b-2) contained one gene copy from O. acuminata, one copy from P. aurea, OsMADS7 and BdMADS7. Clade c contained one gene copy from L. pauciflora and one from O. acuminata, which formed a sister relationship to SEPALLATA1 + SEPALLATA2 from A. thaliana. The distribution of gene copies from O. sativa and B. distachyon suggests that the grass specific clade (d) diverged into three subclades. The first subclade (d-1) contained one gene copy from P. aurea and associates with OsMADS34 and BdMADS34. The second subclade (d-2) contained one gene copy from all species but L. pauciflora and associates with OsMADS55 and BdMADS55. The third subclade (d-3) contains gene copies from all species except for P. aurea and associates with OsMADS1 and BdMADS1.
The SVP-like gene tree (Fig. 2) contains only one copy of each bamboo species with the woody bamboos forming clade a. The copy from L. pauciflora, which is separated from the woody bamboo clade, formed a sister relationship to a copy from O. sativa. The SOC-Like gene tree (Additional file 3: Figure S2; SOC) formed a clade of grass specific genes (a) separate from five A. thaliana copies. One subclade of grasses (b) contained copies from all four species of bamboos, but also formed a sister relationship to a second gene copy from L. pauciflora. Subclade b also associated with OsMADS56, BdMADS56 and BdMADS50. This subclade (c) contained three copies from P. aurea, two from O. acuminata and one each from G. inermis and L. pauciflora. The second subclade (c) is sparsely populated with bamboo copies as four copies from B. distachyon (BdMADS56, BdMADS50, BdMADS7, BdMADS22) are present along with two copies from O. sativa (OsMADS56, OsMADS37), but only seven bamboo copies, from four species, are present.
Nuclear orthologue phylogenetics
A total of 3,878 clusters were produced that met the criteria set for this study. The length of each alignment ranged from 219 to 10,755 bp. The concatenated alignment had a length of 5,736,540 bp, which was reduced to 2,698,410 bp after gapped positions were removed.
The concatenated analysis produced a fully-resolved ML tree and was rooted at O. sativa based on previous phylogenetic results (Fig. 7). In this case, B. distachyon formed a sister relationship with the rest of the taxa (bamboos). Within Bambusoideae, L. pauciflora formed a sister relationship with the rest of the bamboos, which were all woody. The tropical woody bamboos, G. inermis + O. acuminata formed a sister relationship to the temperate woody bamboos, P. aurea + P. heterocycla. All relationships were supported at 100 % ML bootstrap.
Fig. 7.
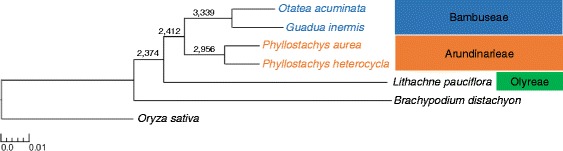
A maximum-likelihood tree generated using 3,878 concatenated gene alignments (2,698,410 bp). Branch lengths reflect number of substitution mutations. The tropical woody (Bambuseae), temperate woody (Arundinarieae) and herbaceous (Olyreae) bamboo tribes are indicated. Numbered nodes denote the number of the 3,878 best trees from each separate gene that support each node. All nodes were supported at 100 % ML bootstrap
Out of the 3,878 best gene trees, 2,374 trees (61.21 %) produced a monophyletic Bambusoideae when rooted at O. sativa. Monophyly in the woody bamboos was recovered in 2,412 trees (62.19 %). A P. aurea + P. heterocycla sister relationship was recovered in 2,956 trees (76.22 %) and a G. inermis + O. acuminata sister relationship was recovered in 3,339 trees (86.10 %). A relationship consistent with the chloroplast phylogeny was recovered in 215 trees (5.54 %).
Discussion
Comparison of de novo and reference-based transcriptome assemblies
While both de novo and reference-based assemblies have been used to describe full transcriptomes, these two methods, which were performed here on identical sets of reads, exhibited strikingly different transcriptomic results. The reference-based assembly produced PCT sets that were consistently shown to have higher percentages of pPCTS. This clearly reflects the nature of reference-based methods in which only reads that met a sequence similarity threshold to a previously-sequenced plant genome were assembled. All species used here exhibited either higher or comparable levels of pPCT abundance in the reference-based assembly.
The pPCTs recovered using reference proteomes from A. thaliana, O. sativa and P. heterocycla, could be placed into general overlapping subgroups. Those pPCTs recovered using A. thaliana were representative of the transcripts that could be generally found in most angiosperms given the phylogenetic distance of the eudicots to the bamboos. Those recovered using O. sativa were representative of genes that could be found in grasses, and those recovered using P. heterocycla represented genes specific to bamboos. While these taxonomic levels are not precisely defined (i.e., some hits to the O. sativa proteome may be indicative of BOP-clade specific transcripts), the overlap exhibits predictable patterns. In both assemblies, pPCTs that were uniquely recovered using A. thaliana form the smallest group while the largest group uniquely recovered in the reference assembly is from P. heterocycla. However, more pPCTs were uniquely recovered from O. sativa in the de novo assembly. This could be indicative of a weakness in the de novo assembly in which transcripts are present, but not at high enough abundances to assemble them without the aid of a reference genome. It also could be an artifact of the reference-based assembly and reliance on the P. heterocycla genome. The lower representation level in the de novo assembly may be due to its incompleteness.
When a 95 % sequence identity threshold was used to assess redundancy, a much larger portion of the pPCTs assembled de novo is unique to each assembly, except those from L. pauciflora. For the three woody species, this would indicate that the de novo assembly produced a set of unique transcripts while the reference based assembly produced transcripts that were largely represented in the de novo assembly. This is likely indicative of under-assembly in the reference-based trancripts. While this could be an indication that a reference-based assembly is largely unnecessary and could artificially inflate the number of transcripts produced, the number of uniquely represented transcripts in the reference-based L. pauciflora assembly may suggest otherwise.
The reference-based assembly likely recovered a large number of transcripts in L. pauciflora with coverage too low to be assembled de novo. This does not reflect any inherent property of the L. pauciflora reads (lower number, quality, etc.) but may reflect a difference in genomic properties. While exact ploidy levels are not known for the species analyzed in this study, herbaceous bamboos are known to be primarily diploid, while woody bamboos tend to exhibit higher levels of ploidy likely resulting from hybridizations [9, 52, 53]. A lower ploidy level in L. pauciflora could have been conducive to a larger spread of transcriptomic representation due to higher coverage levels for each gene.
Further support for performing both types of assembly comes from the Pfam analysis in the pPCTs. Only 62-69 % of all represented Pfam domains were present in both assemblies even though both shared a considerable proportion of unique protein domains (Fig. 4). The reference-based assembly for P. aurea produced more than three times the number of unique Pfam domains than the de novo assembly. While the other three produced fewer, this is most likely due to the phylogenetic proximity of P. aurea to the congeneric reference genome [54].
Presence of plastid genome sequences
Although the Illumina libraries were sequenced after selection for transcripts containing poly-A tails, sequences exhibiting plastome homology were likely assembled due to the high number of plastids present in each plant cell. The presence of plastid sequences may have been retained during the poly-A selection step due to the AT richness of the plastome. A large number of these transcripts, which were assembled de novo, were much larger than mRNA transcripts that were observed in eukaryotic plastomes > 10 kbp and contained more than one coding sequence. One explanation for these unusually large transcripts is that plastid genomes are typically very compact and contain relatively small intergenic regions. If overlapping UTRs were present in the sequenced transcripts, it could be an artifact of the de novo assembly method, which takes no reference genome into account and produces assemblies based solely on sequence identity.
Floral gene analysis
Because the field-harvested sources of our RNA extracts were spikelets with fully developed and emergent florets, not all MADS-box genes (A-E class) were expected to be expressed. This is apparent as significantly fewer MADS box genes were found in each bamboo transcriptome (13–20) than the 57 that were found in the fully sequenced B. distachyon genome [44]. Between two and five genes were identified in all classes for each taxon except for L. pauciflora, which expressed seven distinct B-class genes. Because B-class genes are important in lodicule and stamen development in grasses [55], they are required in the development of both types of unisexual florets that are produced by herbaceous bamboos. We hypothesize that duplications of two B class genes may have allowed separate copies to be expressed differentially in either male (lodicule and stamen development) or female (lodicule only development) florets. Though O. sativa and B. distachyon have hermaphroditic florets and are both represented with seven copies, their data is genomic and the lower number of transcripts in the three woody bamboos is likely a result of differential expression. The first of these, involving gene copies B1 and B2, are both found in clade a (Fig. 5). Gene copy B1 clusters with (OsMADS31 + BdMADS31). Gene copy B2 is sister to all of the grass-specific members of this clade, rather than to any specific gene copy, although copy B1 of P. aurea is the next copy in the clade to diverge. In the second case there are two copies, B3 and B4, which are found in clade b (Fig. 5) and cluster with OsMADS26 and BdMADS26, respectively. Unexpectedly copy B5 from L. pauciflora does not cluster with any other copies from Bambusoideae, so its origin is obscure. This hypothesis could be verified by sequencing B-class genes from separate staminate and carpellate florets.
In several cases, correlations between gene copy number and differences in floral phenotypes can be hypothesized. The PISTILLATA homologues from O. sativa, OsMADS2 and OsMADS4 were shown to have complementary importance in lodicule development and about equal importance in stamen development [56]. One copy each from O. acuminata and P. aurea associate with OsMADS2 (Fig. 5; clade d), which may be indicative of phenotype as both have three stamens compared to G. inermis, which has six stamens [57]. However, sister to the OsMADS2-4 subclade within clade d are one copy each from G. inermis and O. acuminata which may be indicative of a duplication resulting in a novel, bamboo-specific, B-class gene. Another potentially bamboo-specific gene can be found in one copy from L. pauciflora and O. acuminata to the E-class SEPALLATA1-2 genes from A. thaliana (Additional file 3 Figure S2; E-clade c). The placement of these copies seems to be indicative of a gene duplication in bamboos or a copy deletion in non-bamboo grasses.
Within the C/D-class clade, which are paraphyletic in regard to function as previously noted in broad studies of fruit development genes [58], OsMADS3 and OsMADS58 are known to be C-class genes. OsMADS3 has been shown to be important in carpel diversification [59] and clusters with copies from G. inermis, O. acuminata, and L. pauciflora (Additional file 2: Figure S1; C/D-clade c). These three genes also distantly associate to a copy from P. aurea. This could potentially be indicative of the phenotypic difference in carpels between the clade formed by copies from G. inermis, O. acuminata. L. pauciflora, O. sativa, and B. distachyon, which have two stigmas, and P. aurea, which has three stigmas and forms a sister relationship with the two-stigma clade.
Another potential connection to phenotype can be found in the gene tree for SVP-like genes, which has been tied to flowering time. One copy from each species of bamboo was present in this tree (Fig. 6). The copy from L. pauciflora is the immediate sister to a copy from O. sativa, which also flowers annually, rather than with the other three bamboo species, the latter of which formed clade a and exhibited 92.6 % sequence similarity. The copy from L. pauciflora exhibited between 69 and 82 % sequence similarity to the other bamboo copies. This may be explained by the different phenological patterns found among these species; L. pauciflora is a perennial that flowers approximately annually while the other three taxa flower at very long intervals [4].
These phenotypic connections to expressed gene copy number and evolutionary history are interesting, and could be confirmed with subsequent testing. In situ hybridization and transcriptome sampling at different stages of floral development could be performed to verify these hypotheses. The bioinformatics-based survey performed in this study is a foundation to further elucidating the complete flowering mechanisms of these, and other, bamboo species.
The presence of sister orthologues of O. sativa and B. distachyon that do not associate with bamboo copies is observed at least three times (Fig. 5, clade b: OsMADS33-BdMADS33; clade e: OsMADS30-Bd_ZMM17-Like; Additional file 2: Figure S1, clade b: OsMADS20-BdMADS20). The most probably explanation for this pattern is that orthologues (or close homologues) of these genes were not expressed in the four tissues that were used in this study. This is especially likely as some A/B-class genes are known to be expressed earlier in floral development and the florets harvested for this study were fully developed. The second possibility is that there was a deletion of these copies in the Bambusoideae. The possibility of gene duplication in the B. distachyon and O. sativa lineage(s) is very unlikely as previous phylogenetic studies have placed the Bambusoideae either sister to B. distachyon or sister to O. sativa (see below).
One important caveat to any of the transcriptome comparisons made within this study is that inconsistencies may arise from the method of tissue retrieval and the study system used. While the floral tissue was harvested from spikelets of approximately equal maturity, the stresses and conditions endured by each plant (i.e., soil type, climate, herbivory) may have been significantly different. This method of collecting floral tissue is necessary when using bamboos as a study system since their flowering cycles are typically very long and unpredictable, and they are difficult to cultivate as flowering specimens under greenhouse conditions with few exceptions [60]. Genome sequencing followed by an extensive survey for functional genes would allow us to more confidently confirm the presence or absence of specific gene copies.
Floral genes and phylogeny
The repeated instances of gene copies from L. pauciflora being isolated from copies from other bamboos in our gene trees (Figs. 5 and 6) could be explained by the differences between Olyreae and the two woody bamboo tribes (Arundinarieae and Bambuseae) in phenology, sexual systems, floral development and structure. This could be an indication that Olyreae are evolutionarily separate from the woody bamboo lineages, but contradicts previous studies that have placed Olyreae in a sister relationship to Bambuseae [2, 13, 61]. This potential indication of shared ancestry between Arundinarieae and Bambuseae is compatible with the results of Triplett et al. [9], who proposed a monophyletic woody bamboo clade (Bambuseae + Arundinarieae) on the basis of phylogenetic analysis of single-copy nuclear markers. However, complete genomic sequencing and annotation would be required to rule out the possibility that these topologies are a result of unexpressed, and therefore missing, copies.
Most of the subfamilial grass phylogeny that could be inferred from these trees showed a topology in which bamboos are more closely related to O. sativa than to B. distachyon. This contrasts markedly with previously published phylogenetic studies that use plastid markers [2, 13, 61], which place the Oryzoideae (O. sativa) in a sister relationship to the Bambusoideae + Pooideae (B. distachyon) clade. However, some studies that used plastid markers have placed Bambusoideae in a sister relationship to Oryzoideae [7, 62].
The use of nuclear transcripts as phylogenetic markers has been controversial because of the shorter length of single transcripts, selective effects on coding mRNAs and the ambiguity in differentiating orthologues from paralogues (or other homologues). The aforementioned speculations based on MADS box gene tree topological patterns can be tested extensively, using a large variety of nuclear markers, to draw robust conclusions about the evolutionary relationships among these taxa.
Nuclear orthologue phylogenetics
The recovery of two conflicting tribal topologies in the Bambusoideae brings two main questions into consideration: 1) Which topology reflects the actual species tree and evolutionary history of Bambusoideae? 2) Which evolutionary events would cause these conflicts to emerge? The trees produced using concatenated nuclear genes strongly supported the hypothesis of a woody bamboo clade (Fig. 7). A monophyletic relationship among the woody bamboos is also supported in the majority of separate gene trees. We will refer to this topology, ((Arundinarieae + Bambuseae), Olyreae), as the ‘nuclear hypothesis’ of bamboo evolution. Phylogenetic trees that use complete sets of coding and noncoding regions from plastid genomes have recovered a well-supported paraphyly in the woody bamboos, ((Olyreae + Bambuseae), Arundinarieae), which we will refer to as the ‘plastid hypothesis.’
Based purely on the quantity of data, the nuclear hypothesis is supported by over an order of magnitude more than the plastid hypothesis. The nuclear hypothesis is supported by the phylogenetic signal given by over 3,700 loci, while the complete plastid genome is inherited cytoplasmically and theoretically gives the phylogenetic signal equivalent to one locus. However, the single-locus attribute of the plastid genome gives it a much higher degree of certainty in its use as a phylogenetic marker between plant species. Although care was taken to minimize our level of uncertainty, nuclear genes can have multiple paralogous copies. Their use as phylogenetic markers can also be complicated by allopolyploidy, which many grasses have been shown to exhibit [63].
Although the use of morphological characteristics in the determination of phylogenetic relatedness is controversial, the morphological and phenological characteristics of the three bamboo tribes are notable. Arundinarieae and Bambuseae share a suite of characteristics with highly lignified shoots, bisexual flowers and intermittent flowering events followed by a die-off. Olyreae have shoots with significantly less lignification, unisexual florets and annual flowering. The nuclear hypothesis suggests a single origin of these characteristics and the plastid hypothesis suggests two origins or one origin followed by a loss of these characteristics in Olyreae. The duplicate origin of similar characteristics seems unlikely, but a loss of certain characters could be biologically feasible.
A hypothesis for the validity of both phylogenetic signals involves an ancient hybridization. With the species tree following the nuclear hypothesis, the Bambuseae and Arundinarieae would exhibit a sister relationship to Olyreae. If a progenitor species of Bambuseae had hybridized with a sympatric progenitor species of Olyreae, followed by a back-cross in the paternal species, the maternally inherited plastid signal would place Bambuseae phylogenetically sister to Olyreae rather than Arundinarieae. An alternative explanation for the incongruence of phylogenetic signal could be selection in which either tropical bamboos (Bambuseae and Olyreae) or woody bamboos (Bambuseae and Arundinarieae) accumulated homoplasious mutations. However, it would be very unlikely that over 60 % of the genes sampled in our analyses skewed the phylogenetic signal identically, placing Bambuseae sister to Arundinarieae, due to selection. Long-branch attraction can be eliminated as a possibility because both Bambuseae and Arundinarieae produce very short branches within each tribe [13].
Triplett et al. [9] also recovered monophyly in the woody bamboos using three low copy nuclear genes. However, each gene copy was classified into respective ancient genomes based on the hypothesis of woody bamboos being a product of allopolyploidization. Our study assumed orthology based on the highest sequence similarity and the presence of one orthologous copy per species. While the single-orthologue approach does not account for multiple orthologous copies from allopolyploids, the overall phylogenetic signal from the concatenated alignment and trees is robust. If the putative orthologues from each taxon originated from different progenitor woody bamboo genomes, we might expect the support for nodes within the woody bamboo clade to reflect this.
Conclusions
This marks the first study that compared transcriptomes across Bambusoideae and the first transcriptome generated from an herbaceous bamboo. Methodologically it was demonstrated that transcriptome assembly can be performed using de novo or reference-based methods. De novo methods may have over-assembled the reads (in that more transcripts in vivo were represented by fewer transcripts in silico) while reference-based methods may have under-assembled (in which more transcripts were produced in silico that represented fewer in vivo). This study also identified expressed MADS-box genes in the bamboo species represented here. Although these genes do not represent an exhaustive genomic survey, they can be used in futures studies to thoroughly examine bamboo floral development. The lack of phylogenetic information in MADS-box genes was also clarified, at least in bamboos. The phylogenetic utility of a full transcriptome was demonstrated by identifying putative orthologues in each transcriptome and performing a maximum likelihood analysis. The origin of woody bamboos was supported as monophyletic, which contrasted with many studies that used plastid markers. Sequencing of full nuclear genomes would be required to confirm orthology although it is unlikely that most gene analyses identically produced erroneous results.
Availability of data and materials
All reads were deposited into the SRA database at the NCBI and can be accessed under the experimental accession SRX1553102.
Acknowledgements
We thank R. Macias for access to his collection at the El Riscal bamboo plantation and A. Hernandez for aid in preparing samples for RNA sequencing. Sequencing and library preparation were supported by the Northern Illinois University Department of Biological Sciences and the Plant Molecular Biology Center. This project was also funded by NSF/NASA grant DEB1342782 to MRD. Any opinions, findings, and conclusions or recommendations expressed in this material are those of the authors and do not necessarily reflect the views of the National Science Foundation.
Additional files
MADS-box gene copies from bamboos matched to corresponding orthologs from Oryza sativa. MADS-box gene classes are noted. Gene copies in parentheses denote orthologs that were inferred ambiguously based on topological relationships. (DOC 58 kb)
Neighbor-joining gene tree representing the A and C/D-class MADS box genes. Gene copies assembled in this study are labeled by genus, colored according to taxa (orange: G. inermis, green: O. acuminata, dark red: P. aurea, blue: L. pauciflora) and numbered redundantly to distinguish copies. Reference gene copies are not colored, are abbreviated by binomial (At: Arabidopsis thaliana, Bd: Brachypodium distachyon, Os: Oryza sativa) and are numbered according to their labeling in Genbank. Nodes that were supported at over 50 % bootstrap are indicated. (PDF 331 kb)
Neighbor-joining gene tree representing the SOC-like and E-class MADS box genes. Gene copies assembled in this study are labeled by genus, colored according to taxa (orange: G. inermis, green: O. acuminata, dark red: P. aurea, blue: L. pauciflora) and numbered redundantly to distinguish copies. Reference gene copies are not colored, are abbreviated by binomial (At: Arabidopsis thaliana, Bd: Brachypodium distachyon, Os: Oryza sativa) and are numbered according to their labeling in Genbank. Nodes that were supported at over 50 % bootstrap are indicated. (PDF 465 kb)
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
WPW collected floral tissue, extracted RNA and carried out transcriptome assembly, dowstream analyses, and drafted the manuscript. ERS helped locate and identify bamboo populations in Veracruz and expertise on floral anatomy. YY provided expertise in bioinformatics analyses. MRD aided in drafting the manuscript and in RNA extraction. All authors read and contributed written sections of the final manuscript. All authors read and approved the final manuscript.
References
- 1.Bamboo Phylogeny Group [BPG]. An updated tribal and subtribal classification of the bamboos (Poaceae: Bambusoideae). In: Gielis J, Potters G, editors. P 9th World Bamboo Congr. American Bamboo Society. Antwerp; 2012. p. 3–27.
- 2.Kelchner SA, Bamboo Phylogeny Group Higher level phylogenetic relationships within the bamboos (Poaceae: Bambusoideae) based on five plastid markers. Mol Phylogenet Evol. 2013;67:404–13. doi: 10.1016/j.ympev.2013.02.005. [DOI] [PubMed] [Google Scholar]
- 3.Sungkaew S, Stapleton CMA, Salamin N, Hodkinson TR. Non-monophyly of the woody bamboos (Bambuseae; Poaceae): a multi-gene region phylogenetic analysis of Bambusoideae s.s. J Plant Res. 2009;122:95–108. doi: 10.1007/s10265-008-0192-6. [DOI] [PubMed] [Google Scholar]
- 4.Clark LG, Londoño X, Ruiz-Sanchez E. Bamboo taxonomy and habitat. In: Laslo P, Köehl M, (eds). Bamboo. Series: Tropical Forestry Handbook. Springer. 2015. (In press)
- 5.Zhang WP. Phylogeny of the Grass Family (Poaceae) from rpl16 Intron Sequence Data. Mol Phylogenet Evol. 2000;15:135–46. doi: 10.1006/mpev.1999.0729. [DOI] [PubMed] [Google Scholar]
- 6.Zhang WP, Clark LG. Phylogeny and classification of the Bambusoideae (Poaceae) In: Jacobs SWL, Everett J, editors. Grass Syst Evol. Melbourne: CSIRO; 2000. pp. 35–42. [Google Scholar]
- 7.Grass Phylogeny Working Group [GPWG] Phylogeny and subfamilial classification of the grasses (Poaceae) Ann Missouri Bot Gard. 2001;88:373–457. doi: 10.2307/3298585. [DOI] [Google Scholar]
- 8.Bouchenak-Khelladi Y, Salamin N, Savolainen V, Forest F, van der Bank M, et al. Large multi-gene phylogenetic trees of the grasses (Poaceae): progress towards complete tribal and generic level sampling. Mol Phylogenet Evol. 2008;47:488–505. doi: 10.1016/j.ympev.2008.01.035. [DOI] [PubMed] [Google Scholar]
- 9.Triplett JK, Clark LG, Fisher AE, Wen J. Independent allopolyploidization events preceded speciation in the temperate and tropical woody bamboos. New Phytol. 2014;204:66–73. doi: 10.1111/nph.12988. [DOI] [PubMed] [Google Scholar]
- 10.Janzen DH. Why bamboos wait so long to flower. Ann Rev Ecol Syst. 1976;7:347–91. doi: 10.1146/annurev.es.07.110176.002023. [DOI] [Google Scholar]
- 11.Gaut BS, Clark LG, Wendel JF, Muse SV. Comparisons of the molecular evolutionary process at rbcL and ndhF in the grass family (Poaceae) Mol Biol Evol. 1997;14:769–77. doi: 10.1093/oxfordjournals.molbev.a025817. [DOI] [PubMed] [Google Scholar]
- 12.Oliveira RP, Clark LG, Schnadelbach AS, Monteiro SH, Borba EL, et al. A molecular phylogeny of Raddia and its allies within the tribe Olyreae (Poaceae, Bambusoideae) based on noncoding plastid and nuclear spacers. Mol Phylogenet Evol. 2014;78:105–17. doi: 10.1016/j.ympev.2014.04.012. [DOI] [PubMed] [Google Scholar]
- 13.Wysocki WP, Clark LG, Attigala L, Ruiz-Sanchez E, Duvall MR. Evolution of the bamboos (Bambusoideae; Poaceae): a full plastome phylogenomic analysis. BMC Evol Biol. 2015;15(1):50. doi: 10.1186/s12862-015-0321-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Coen ES, Meyerowitz EM. The war of the whorls: genetic interactions controlling flower development. Nature. 1991;353(6339):31–7. doi: 10.1038/353031a0. [DOI] [PubMed] [Google Scholar]
- 15.Pelaz S, Ditta GS, Baumann E, Wisman E, Yanofsky MF. B and C floral organ identity functions require SEPALLATA MADS-box genes. Nature. 2000;405(6783):200–3. [DOI] [PubMed]
- 16.Skinner DJ, Hill TA, Gasser CS. Regulation of ovule development. Plant Cell. 2004;16:S32–45. doi: 10.1105/tpc.015933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dreni L, Jacchia S, Fornara F, Fornari M, Ouwerkerk PB, et al. The D‐lineage MADS‐box gene OsMADS13 controls ovule identity in rice. Plant J. 2007;52(4):690–9. doi: 10.1111/j.1365-313X.2007.03272.x. [DOI] [PubMed] [Google Scholar]
- 18.Theißen G, Saedler H. Plant biology: floral quartets. Nature. 2001;409(6819):469–71. doi: 10.1038/35054172. [DOI] [PubMed] [Google Scholar]
- 19.Ryu CH, Lee S, Cho LH, Kim SL, Lee YS, et al. OsMADS50 and OsMADS56 function antagonistically in regulating long day (LD)‐dependent flowering in rice. Plant Cell Environ. 2009;32(10):1412–27. doi: 10.1111/j.1365-3040.2009.02008.x. [DOI] [PubMed] [Google Scholar]
- 20.Lee J, Lee I. Regulation and function of SOC1, a flowering pathway integrator. J Exp Bot. 2010;61(9):2247–54. doi: 10.1093/jxb/erq098. [DOI] [PubMed] [Google Scholar]
- 21.Hartmann U, Höhmann S, Nettesheim K, Wisman E, Saedler H, Huijser P. Molecular cloning of SVP: a negative regulator of the floral transition in Arabidopsis. Plant J. 2000;21(4):351–60. doi: 10.1046/j.1365-313x.2000.00682.x. [DOI] [PubMed] [Google Scholar]
- 22.Lee JH, Park SH, Ahn JH. Functional conservation and diversification between rice OsMADS22/OsMADS55 and Arabidopsis SVP proteins. Plant Sci. 2012;185:97–104. doi: 10.1016/j.plantsci.2011.09.003. [DOI] [PubMed] [Google Scholar]
- 23.Wang Z, Gerstein M, Snyder M. RNA-Seq: a revolutionary tool for transcriptomics. Nature Rev Genet. 2009;10(1):57–63. doi: 10.1038/nrg2484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang XM, Zhao L, Larson-Rabin Z, Li DZ, Guo ZH. De novo sequencing and characterization of the floral transcriptome of Dendrocalamus latiflorus (Poaceae: Bambusoideae) PLoS One. 2012;7(8):e42082. doi: 10.1371/journal.pone.0042082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liu M, Qiao G, Jiang J, Yang H, Xie L, et al. Transcriptome sequencing and de novo analysis for ma bamboo (Dendrocalamus latiflorus Munro) using the Illumina platform. PLoS One. 2012;7(10):e46766. doi: 10.1371/journal.pone.0046766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Peng Z, Zhang C, Zhang Y, Hu T, Mu S, et al. Transcriptome sequencing and analysis of the fast growing shoots of Moso bamboo (Phyllostachys edulis) PloS One. 2013;8(11):e78944. doi: 10.1371/journal.pone.0078944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gao J, Zhang Y, Zhang C, Qi F, Li X, et al. Characterization of the Floral Transcriptome of Moso Bamboo (Phyllostachys edulis) at Different Flowering Developmental Stages by Transcriptome Sequencing and RNA-Seq Analysis. PLoS One. 2014;9(6):e98910. doi: 10.1371/journal.pone.0098910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ruiz-Sanchez E, Sosa V, Mejía-Saules MT, Londoño X, Clark LG. A taxonomic revision of Otatea (Poaceae: Bambusoideae: Bambuseae) including four new species. Syst Bot. 2011;36:314–36. doi: 10.1600/036364411X569516. [DOI] [Google Scholar]
- 29.Kellogg EA. V. Subfamily Bambusoideae Luerss (1893). In: Flowering Plants. Monocots. Switzerland: Springer International Publishing; 2015. p. 151–98.
- 30.Kellogg EA. IV. Subfamily Ehrhartoideae Link (1827). In: Flowering Plants. Monocots. Switzerland: Springer International Publishing; 2015. p. 143–50.
- 31.Kellogg EA. VI. Subfamily Pooideae Benth (1861). In: Flowering Plants. Monocots. Switzerland: Springer International Publishing; 2015. p. 199–265.
- 32.Peng Z, Lu Y, Li L, Zhao Q, Feng Q, et al. The draft genome of the fast-growing non-timber forest species moso bamboo (P. heterocycla) Nature Genet. 2013;45(4):456–461. doi: 10.1038/ng.2569. [DOI] [PubMed] [Google Scholar]
- 33.Cox MP, Peterson DA, Biggs PJ. SolexaQA: At-a-glance quality assessment of Illumina second-generation sequencing data. BMC Bioinformatics. 2010;11(1):485. doi: 10.1186/1471-2105-11-485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 2011;17(1):10. doi: 10.14806/ej.17.1.200. [DOI] [Google Scholar]
- 35.Grabherr MG, Haas BJ, Yassour M, Levin JZ, Thompson DA, et al. Full-length transcriptome assembly from RNA-seq data without a reference genome. Nat Biotech. 2011;29(7):644–52. doi: 10.1038/nbt.1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Trapnell C, Pachter L, Salzberg SL. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 2009;25(9):1105–11. doi: 10.1093/bioinformatics/btp120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9(4):357–9. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, et al. The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25(16):2078–9. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li W, Godzik A. Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics. 2006;22(13):1658–9. doi: 10.1093/bioinformatics/btl158. [DOI] [PubMed] [Google Scholar]
- 40.Wang L, Park HJ, Dasari S, Wang S, Kocher JP, et al. CPAT: Coding-Potential Assessment Tool using an alignment-free logistic regression model. Nucleic Acids Res. 2013;41(6):e74. doi: 10.1093/nar/gkt006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Finn RD, Clements J, Eddy SR. HMMER web server: interactive sequence similarity searching. Nucleic Acids Res. 2011;39(Web Server issue):W29-–37. doi: 10.1093/nar/gkr367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bateman A, Coin L, Durbin R, Finn RD, Hollich V, et al. The Pfam protein families database. Nucleic Acids Res. 2004;32(suppl 1):D138–41. doi: 10.1093/nar/gkh121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25(17):3389–402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wei B, Zhang RZ, Guo JJ, Liu DM, Li AL, et al. Genome-wide analysis of the MADS-box gene family in Brachypodium distachyon. PLoS One. 2014;9(1):e84781. doi: 10.1371/journal.pone.0084781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Thompson JD, Higgins DG, Gibson TJ. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22(22):4673–80. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Huerta-Cepas J, Dopazo J, Gabaldón T. ETE: a python Environment for Tree Exploration. BMC Bioinformatics. 2010;11(1):24. doi: 10.1186/1471-2105-11-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Schnable JC, Freeling M, Lyons E. Genome-wide analysis of syntenic gene deletion in the grasses. Genome Biol Evol. 2012;4(3):265–77. doi: 10.1093/gbe/evs009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhang LN, Zhang XZ, Zhang YX, Zeng CX, Ma PF, Zhao L, Guo ZH, Li DZ. Identification of putative orthologous genes for the phylogenetic reconstruction of temperate woody bamboos (Poaceae: Bambusoideae) Mol Ecol Resources. 2014;14(5):988–99. doi: 10.1111/1755-0998.12248. [DOI] [PubMed] [Google Scholar]
- 49.Katoh K, Standley DM. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 2013;30(4):772–80. doi: 10.1093/molbev/mst010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Stamatakis A. RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics. 2006;22(21):2688–90. doi: 10.1093/bioinformatics/btl446. [DOI] [PubMed] [Google Scholar]
- 51.Felsenstein J. PHYLIP Seattle: Department of Genome Science. Seattle, WA: University of Washington; 2005. p. 3.
- 52.Hunziker JH, Wulff AF, Soderstrom TR. Chromosome studies on the Bambusoideae (Gramineae) Brittonia. 1982;34(1):30–5. doi: 10.2307/2806397. [DOI] [Google Scholar]
- 53.Chokthaweepanich H. Phylogenetics and evolution of the paleotropical woody bamboos (Poaceae: Bambusoideae: Bambuseae). Dissertation. Ames, IA: Iowa State University; 2014.
- 54.Triplett JK, Clark LG. Phylogeny of the Temperate Bamboos (Poaceae: Bambusoideae: Bambuseae) with an emphasis on Arundinaria and Allies. Syst Bot. 2010;35:102–120.X. doi: 10.1600/036364410790862678. [DOI] [Google Scholar]
- 55.Whipple CJ, Ciceri P, Padilla CM, Ambrose BA, Bandong SL, Schmidt RJ. Conservation of B-class floral homeotic gene function between maize and Arabidopsis. Development. 2004;131(24):6083–91. doi: 10.1242/dev.01523. [DOI] [PubMed] [Google Scholar]
- 56.Yao SG, Ohmori S, Kimizu M, Yoshida H. Unequal genetic redundancy of rice PISTILLATA orthologs, OsMADS2 and OsMADS4, in lodicule and stamen development. Plant Cell Physiol. 2008;49(5):853–7. doi: 10.1093/pcp/pcn050. [DOI] [PubMed] [Google Scholar]
- 57.Judziewicz EJ, Clark LG, Londoño X, Stern MJ. American bamboos. Washington D.C.: Smithsonian Institution Press; 1999.
- 58.Pabón-Mora N, Wong GS, Ambrose BA. Evolution of fruit development genes in flowering plants. Frontiers in plant science. 2014;5:300. doi: 10.3389/fpls.2014.00300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Yamaguchi T, Lee DY, Miyao A, Hirochika H, An G, Hirano HY. Functional diversification of the two C-class MADS box genes OSMADS3 and OSMADS58 in Oryza sativa. Plant Cell. 2006;18(1):15–28. doi: 10.1105/tpc.105.037200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lin CS, Lin CC, Chang WC. Shoot regeneration, re-flowering and post flowering survival in bamboo inflorescence culture. Plant Cell Tiss Org Cult. 2005;82(3):243–9. doi: 10.1007/s11240-005-0883-9. [DOI] [Google Scholar]
- 61.Wu ZQ, Ge S. The phylogeny of the BEP clade in grasses revisited: evidence from the whole-genome sequences of chloroplasts. Mol Phylogenet Evol. 2012;62(1):573–8. doi: 10.1016/j.ympev.2011.10.019. [DOI] [PubMed] [Google Scholar]
- 62.Clark LG, Zhang W, Wendel JF. A phylogeny of the grass family (Poaceae) based on ndhF sequence data. Syst Bot. 1995;20:436–60. doi: 10.2307/2419803. [DOI] [Google Scholar]
- 63.Levy AA, Feldman M. The impact of polyploidy on grass genome evolution. Plant Physiol. 2002;130(4):1587–93. doi: 10.1104/pp.015727. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All reads were deposited into the SRA database at the NCBI and can be accessed under the experimental accession SRX1553102.