

Abstract
Background
Haemophagocytic lymphohistiocytosis (HLH) is a life-threatening disorder of immune regulation, and HLH patients with mutations in genes including PRF1, UNC13D, STX11, STXBP2, SH2D1A, XIAP, and ITK were reported to be primary HLH. Due to the different treatment options, the differentiation between primary and secondary HLH is critical. Our previous studies have showed that a Th1/Th2 cytokine profile is diagnostic for HLH, yet the cytokine profiles between primary and secondary HLH have not been compared. The aim of the study was to test whether the Th1/Th2 cytokine profile could be used as a tool to differentiate between primary and secondary HLH.
Methods
A total of 45 hospitalized Chinese children with HLH during the period of February 2010 through September 2012 were enrolled in the study. Fifty healthy children were enrolled as controls. Primary HLH related genes were sequenced using genomic DNA samples. The Th1/Th2 cytokine levels including interferon-γ (IFN-γ), tumor necrosis factor-alpha (TNF-α), interleukin (IL)-10, IL-6, IL-4 and IL-2 were quantitatively determined by cytometric bead assay techniques.
Results
Primary HLH group (n = 4) included one patient with biallelic heterozygous mutations in PRF1 gene, and three patients with hemizygous mutation in SH2D1A gene. Based on the available genetic data, the other 41 patients were classified into the secondary HLH group. When compared the cytokine levels between the two groups, IL-4 level in primary-HLH was significantly lower than that in secondary HLH (P = 0.025), while IFN-γ level in primary HLH had a tendency of statistically lower than that in secondary HLH (P = 0.051). Area under receiver operating characteristic (ROC) curves of IL-4 and IFN-γ, IL-10, TNF-α, IL-2, and IL-6 levels were 0.841, 0.799, 0.506, 0.494, 0.457, and 0.250, respectively. ROC curves showed that 1.7 pg/ml of IL-4 had sensitivity and specificity for differentiation between primary and secondary HLH as 70.7 and 100.0 %, while 433.9 pg/ml of IFN-γ had sensitivity and specificity as 51.2 and 100.0 %, respectively.
Conclusions
HLH patients with lower IL-4 and IFN-γ levels have higher possibility to be primary HLH. The cytokine profile may be used as an additional tool for the quick differential diagnosis between primary and secondary HLH.
Electronic supplementary material
The online version of this article (doi:10.1186/s13052-016-0262-7) contains supplementary material, which is available to authorized users.
Keywords: Haemophagocytic lymphohistiocytosis, Cytokines, Interleukin-4, Interferon-γ
Background
Haemophagocytic lymphohistiocytosis (HLH) is a life-threatening disorder of immune regulation, characterized by a highly stimulated, but ineffective immune response to antigens, which results in cytokine storm and inflammatory reaction [1]. HLH is not a single entity, but a clinical syndrome that can be encountered in association with various underlying diseases leading to similar characteristic clinical and laboratory presentations. Briefly, the diagnosis of HLH requires either a genetic diagnosis, or fulfillment of 5 out of 8 clinical criteria including fever, splenomegaly, bicytopenia, either hypofibrinoginemia or hypertriglyceridemia, hyperferritinemia, elevated soluble interleukin-2 receptor (sCD25 or sIL2R), impaired natural killer (NK) cell cytolytic function, or the observation of hemophagocytosis in bone marrow, spleen, or lymph nodes [2]. Based on the etiology, HLH can be classified into two types, primary HLH (with genetic mutations), and secondary HLH (no known mutations). Epstein–Barr virus (EBV) infections seem to be very common in HLH patients, especially in Asian countries [3–7]. Beside EBV infection, cytomegalovirus (CMV) infection is another common cause in HLH patients [8, 9]. Clinically, prompt and accurate diagnosis is critical to initiate definitive therapy. Unfortunately, the clinical features are identical in primary and secondary HLH, and both forms are often triggered by infections, so it is difficult to distinguish between these two types [8]. Furthermore, the diagnosis of HLH based on the current combination of clinical, laboratory and immunological criteria is challenging because all of the criteria are not specifically diagnostic for any HLH subtypes.
Primary HLH can be further divided into familial hemophagocytic lymphohistiocytosis (FHL) and immune deficiencies associated HLH [10]. Five genetic defects have been identified in FHL. A potential gene locus (FHL1) has been reported to be associated with HLH, but the specific gene involved has not yet been identified [11]. The first described FHL related gene is PRF1 gene (FHL2) [12], a gene encodes perforin protein. The next identified cause of FHL (FHL3) is UNC13D gene [13], which encodes Munc13-4 protein. Later, mutations of STX11 gene and STXBP2 gene are found to be the causes of FHL4 [14] and FHL5 [15, 16], respectively. Immune deficiencies associated HLH occur with significant frequency in X-linked lymphoproliferative syndrome type 1 (XLP1) [17], XLP type 2 (XLP2) [18], and IL-2–inducible T cell kinase deficiency-associated lymphoproliferation [19], which are characterized by mutations in SH2D1A, XIAP, and ITK genes, respectively.
Hypercytokinemia is a hallmark of HLH. Henter JI et al. suggested that hypercytokinemia may be caused by a genetic defect in cytokine regulation as FHL patients showed high cytokine levels [20]. Using the rapid cytometric bead array (CBA) technique, a specific cytokine profile (significant increase of IFN-γ and IL-10, combined with a slightly increased level of IL-6) for childhood HLH was described by our group in 2008 [21]. In our clinical practice, this cytokine profile was helpful for the early diagnosis of HLH and for the differentiation from other disease entities [22, 23]. However, one limitation of our previous cytokine assay was that we did not perform genetic sequencing of HLH involved genes simultaneously. In this study, we tried to sequence PRF1, UNC13D, STX11, STXBP2, SH2D1A, XIAP, and ITK genes, together with cytokine determination for HLH patients to test whether cytokine profile could be used as a tool to distinguish between primary HLH and secondary HLH patients.
Methods
Patients
The diagnoses of all our patients were made based on the HLH-2004 criteria [2]. A cohort of 45 consecutively hospitalized HLH patients were enrolled in the study, which included 27 males and 18 females with a male-to-female ratio of 1.5:1, and a mean age of 3.7 years with a range of 8 days through 12.3 years. Patients who had been treated by steroids before referral to our hospital were excluded. A total of 50 unrelated healthy individuals (30 males, and 20 females) matched for age, and race were recruited as healthy controls. All the patients and healthy controls were from our hospital between February 2010 and September 2012. No HLH patient showed signs of mucocutaneous albinism. The study protocol was reviewed and approved by the Ethics Committee at Children’s Hospital Zhejiang University School of Medicine and written informed consents were obtained from all participants’ parents or guardians before this study.
Genetic analysis
Genomic DNA was isolated from peripheral blood using QIAamp DNA Blood Mini Kit (Qiagen, Hilden, Germany). The coding regions and flanking intronic sequences of PRF1, UNC13D, STX11, STXBP2, SH2D1A, XIAP, and ITK genes, and the deep intronic sequences of intron 1 in UNC13D gene were amplified by a polymerase chain reaction (PCR) machine (BIO-RAD) in our laboratory, and the PCR products were sequenced by a DNA sequencer in Invitrogen Company (Shanghai, China). We analyzed PRF1, UNC13D, STX11, STXBP2, and ITK genes in all 45 patients and 50 controls, and all the male patients (n = 27) and male controls (n = 30) were sequenced in their SH2D1A and XIAP genes. The mutations were validated by re-sequencing an independent PCR-generated amplicon from the subjects. The variants were named according to the Human Genome Variation Society and journal requirements.
Database investigation and in silico prediction of mutations were checked, including the frequency of each variant in the general population investigated by Exome Aggregation Consortium (ExAC, Cambridge, MA, URL: http://exac.broadinstitute.org, March, 2016 accessed), previous reports of the same variants in the literature checked from Human Gene Mutation Database (HGMD, http://www.hgmd.cf.ac.uk) and Google Scholar (GS, https://scholar.google.com/), in silico prediction results by Polymorphism Phenotyping-2 (PolyPhen-2, http://genetics.bwh.harvard.edu/pph2/) and Sorting Intolerant From Tolerant (SIFT, http://sift.jcvi.org/). For PolyPhen-2, higher scores mean more deleterious. For SIFT, amino acids with probabilities < 0.05 were predicted to be deleterious.
Cytokine determination
All 45 HLH patients and 50 controls were tested for the six cytokines including IL-2, IL-4, IL-6, IL-10, TNF-α, and IFN-γ. Peripheral blood samples were collected, transferred to a serum separating tube and centrifuged at 1000 g at 20 °C for 20 min after clotting. The serum was carefully harvested, and the determination of the cytokines was performed immediately, or if the situation was not so urgent, the aliquot was temporarily stored at 2 °C to 8 °C until analysis (usually within 12 h). Concentrations of the six cytokines aforementioned were quantitatively determined using the CBA Human Th1/Th2 Cytokine Kit II (BD Biosciences, San Jose, California) as described previously [21]. The minimum and maximum limits of detection for all six cytokines were 1 and 5000 pg/ml, respectively.
Degranulation assays with flow cytometry
Degranulation assay were tested in 36 HLH patients and 43 controls. The assay was performed as previously described with modifications [24]. Peripheral blood mononuclear cells (PBMC) were isolated by Ficoll gradient centrifugation, rested in Iscove’s modified Dulbecco medium (IMDM, Invitrogen, Carlsbad, California, USA) supplemented with 10 % fetal bovine serum (FBS, Sijiqing, Hangzhou, China) at 37°C in a humidified atmosphere of 5 % CO2 for 2 h, and washed in 1 × PBS (phosphate buffer saline). K562 cells were obtained from the American Type Culture Collection (ATCC, Rockefeller, Maryland, USA) and cultured in RPMI1640 medium (Invitrogen, Carlsbad, California, USA) supplemented with 10 % FBS. All the antibodies including CD3-FITC, CD8-PerCP, CD56-APC, CD107a-PE (H4A3, IgG1), and isotype controls were purchased from Becton Dickinson (San Jose, CA, USA).
5 × 105 PBMCs were mixed with 5 × 104 sensitive target cells (K562) as stimulants in 12 flat bottom well plates and incubated for 2 h at 37°C. An equal volume of RPMI-1640 culture medium to replace the volume of K562 cells were used as negative control. Then the cells were stained with 10μl of CD3-FITC, 10μl of CD8-PerCP, 2.5μl of CD56-APC, and 10μl of CD107a-PE (isotype IgG1-PE as negative control) for 30 min at 4 °C in the dark. After two washes with 1 × PBS of the above samples, flow cytometric analyses were performed by a FACSCalibur cytometer (Becton-Dickinson, San Jose, CA, USA). CD3-CD56+ and CD3+CD8+ T cells were gated as NK cells and Cytotoxic T cells (CTLs), respectively. Data were acquired with CellQuest software (BD Bioscience).
Clinical data
Information at diagnosis of age, sex, with fever or not, hemoglobin levels, platelets numbers, white blood cell counts, percentage of neutrophils, total neutrophil counts, percentage of lymphocytes, total lymphocyte counts, triglyceride levels, fibrinogen levels, LDH levels, ferritin levels, sIL2R levels, and sIL2R/ferritin ratio were collected. Quantitative real-time PCR was used to detect EBV-DNA and CMV-DNA copies in sera.
Statistical analysis
Serum concentrations of individual cytokines, clinical data were compared between groups using the Mann-Whitney U test. A chi-square test was used to assess ratio differences between groups. Receiver operating characteristic (ROC) curves were derived from the cytokine levels for all HLH patients. In a ROC curve, the sensitivity and specificity of all six cytokines were calculated for the differentiation between primary and secondary HLH. All statistical analyses were performed using SPSS 17.0 software (SPSS Inc, Chicago, Illinois). A two-sided P-value <0.05 was considered to be statistically significant.
Results
Genetic results
The genetic findings of the 45 HLH patients were shown in Table 1, Additional file 1: Figure S1, Additional file 2: Figure S2, Additional file 3: Figure S3, Additional file 4: Figure S4, Additional file 5: Figure S5, and Additional file 6: Figure S6. All variants classified as pathogenic were not detected in the controls, while those classified as single nucleotide polymorphisms (SNPs) were found in both HLH patients and healthy controls. One patient with biallelic heterozygous mutations in PRF1 gene (P2), and three patients with hemizygous mutation in SH2D1A gene (P16, P17 and P26) were categorized into primary HLH group (n = 4). Based on the available genetic data, the other 41 patients were classified into the secondary HLH group, including nine HLH patients with single heterozygous mutation (mutations in patients P6, P10, and P45 were already reported in literature, while mutations in patients P1, P3, P4, P5, P7, and P11 were not reported before), three HLH patients with only SNPs (P8, P9, and P38), and the remaining 29 HLH patients without any SNPs.
Table 1.
Summary of genetic findings in 45 HLH patients
| Case | Gender | Age | Candidate gene | Exon/intron | Nucleotide change | Amino acid change | Genotype |
|---|---|---|---|---|---|---|---|
| P2 | F | 6Y3M | PRF1 | Exon3 | c.757G>A c.1061A>T |
p.Glu253Lys p.Asp354Val |
heterozygous heterozygous |
| P16 | M | 3Y3M | SH2D1A | Exon2 | c.191G>A | p.Trp64Ter | hemizygous |
| P17 | M | 11M24D | SH2D1A | Exon2 | c.162C>G | p.Tyr54Ter | hemizygous |
| P26 | M | 11M17D | SH2D1A | Exon2 | c.163C>T | p.Arg55 Ter | hemizygous |
| P1 | M | 1Y5M | PRF1 | Exon2 | c.385T>A | p.Trp129Arg | heterozygous |
| P3 | M | 4Y5M | UNC13D | Exon6 | c.478G>A | p.Val160 Met | heterozygous |
| P4 | M | 2Y8M | UNC13D | Exon6 | c.518C>T | p.Thr173Met | heterozygous |
| P5 | F | 1Y7M | UNC13D | Exon10 | c.760C>T | p.Arg254Cys | heterozygous |
| P6 | M | 8D | UNC13D | Exon23 | c.2296C>T | p.Glu766Ter | heterozygous |
| P7 | M | 25D | UNC13D | Exon32 | c.3259C>T | p.Arg1087Trp | heterozygous |
| XIAP | Exon5 | c.1268A>C | p.Gln423Proa | hemizygous | |||
| P8 | M | 7Y9M | STXBP2 | Exon7 | c.497C>T | p.Thr166Meta | heterozygous |
| P9 | M | 11Y11M | STXBP2 | Exon7 | c.497C>T | p.Thr166 Meta | heterozygous |
| P10 | F | 5Y8M | STXBP2 | Exon7 | c.575G>A | p.Arg192His | heterozygous |
| P11 | M | 1Y1M | STXBP2 | Exon9 | c.767T>C | p.Leu256Pro | heterozygous |
| P38 | M | 1Y1M | XIAP | Exon5 | c.1268A>C | p.Gln423Proa | hemizygous |
| P45 | M | 4Y | UNC13D | Intron1 | c.118-307G>A | Unknown | heterozygous |
Gender, M male, F female
Age, Y year, M month, D day
aSingle nucleotide polymorphism (SNP)
Ter Terminator, which would result in truncated protein
Mutations in PRF1, UNC13D, STX11, STXBP2, SH2D1A, XIAP, and ITK genes of all the 45 HLH cases accounted for 2/45 (4.4 %), 6/45 (13.3 %), 0/45 (0.0 %), 2/45 (4.4 %), 3/45 (6.7 %), 0/45 (0 %), and 0/45 (0.0 %), respectively. For the four primary HLH cases, mutations in PRF1 and SH2D1A genes accounted for 1/4 (25 %) and 3/4 (75 %), respectively. Three missense heterozygous variants in PRF1 gene were found in one male and one female (Additional file 1: Figure S1). One heterozygous c.385T>A (p.Trp129Arg) mutation was found in P1, a 1-year-5-month male, while compound heterozygous c.757G>A (p.Glu253Lys) and c.1061A>T (p.Asp354Val) in PRF1 gene were found in P2, a 6-year-3-month female, with c.757G>A inherited from her father and c.1061A>T from her mother. Five missense mutations of UNC13D gene (Additional file 2: Figure S2), c.478G>A (p.Val160 Met) in P3, c.518C>T (p.Thr173Met) in P4, c.760C>T (p.Arg254Cys) in P5, c.3259C>T (p.Arg1087Trp) in P7, and c.118-307G>A in P45, were found in four males and one female, respectively. Another heterozygous mutation c.2296C>T (p.Glu766Ter) in exon23 of UNC13D gene of an 8-day male patient (P6), with the cDNA study revealed that this mutation had a deleterious effect on splicing, c.2295_2298delGCAG (p.Glu765Aspfs*27) (Additional file 6: Figure S6), was already reported by our group as a special case [25]. Two heterozygous mutations of STXBP2 gene, c.575G>A (p.Arg192His), and c.767T>C (p.Leu256Pro), were identified in two patients (Additional file 3: Figure S3). Three hemizygous mutations of SH2D1A gene, c.162C>G (p.Tyr54Ter), c.163C>T (p.Arg55Ter), and c.191G>A (p.Trp64Ter), were identified in three male patients (Additional file 4: Figure S4). Two male patients (P8 and P9) with c.497C>T (p.Thr166Met, SNP rs181216956) of STXBP2 gene, and two male patients (P7 and P38) with c.1268A>C (p.Gln423Pro, SNP rs5956583) of XIAP gene, were identified in this cohort (Additional file 5: Figure S5). No mutation was detected from STX11 and ITK genes in all 45 HLH patients.
Database investigation and in silico prediction of mutations in HLH patients were shown in Table 2. The in silico prediction results between PolyPhen-2 and Sorting Intolerant From Tolerant (SIFT) were consistent with each other, except p.Arg1087Trp of UNC13D gene (PolyPhen-2 predicted this change was BENIGN with a score of 0.002, while SIFT indicated that this change would AFFECT PROTEIN FUNCTION with a score of 0.00).
Table 2.
Database investigation and in silico prediction of mutations in 45 HLH patients
| Case | Candidate gene | Nucleotide change | Amino acid change | ExAC allele frequency | HGMD/GS references | Polyphen-2 | SIFT | CD107a |
|---|---|---|---|---|---|---|---|---|
| P2 | PRF1 | c.757G>A c.1061A>T |
p.Glu253Lys p.Asp354Val |
0.00003296 Not reported |
[33] No reference |
PROBABLY DAMAGING with a score of 0.962 POSSIBLY DAMAGING with a score of 0.952 |
AFFECT PROTEIN FUNCTION with a score of 0.01 AFFECT PROTEIN FUNCTION with a score of 0.02 |
32.28 |
| P16 | SH2D1A | c.191G>A | p.Trp64Ter | Not reported | [34, 35] | Not Available | Not Available | 33.83 |
| P17 | SH2D1A | c.162C>G | p.Tyr54 Ter | Not reported | [36] | Not Available | Not Available | 0.72 |
| P26 | SH2D1A | c.163C>T | p.Arg55 Ter | Not reported | [36, 37] | Not Available | Not Available | No done |
| P1 | PRF1 | c.385T>A | p.Trp129Arg | Not reported | No reference | PROBABLY DAMAGING with a score of 1.000 | AFFECT PROTEIN FUNCTION with a score of 0.00 | 3.24 |
| P3 | UNC13D | c.478G>A | p.Val160 Met | Not reported | No reference | PROBABLY DAMAGING with a score of 0.999 | AFFECT PROTEIN FUNCTION with a score of 0.05 | 35.14 |
| P4 | UNC13D | c.518C>T | p.Thr173Met | 0.00001670 | No reference | PROBABLY DAMAGING with a score of 1.000 | AFFECT PROTEIN FUNCTION with a score of 0.01 | 0.43 |
| P5 | UNC13D | c.760C>T | p.Arg254Cys | 0.0003450 | No reference | PROBABLY DAMAGING with a score of 0.987 | AFFECT PROTEIN FUNCTION with a score of 0.02 | 5.69 |
| P6 | UNC13D | c.2296C>T | p.Glu766 Ter | Not reported | [25, 38, 39] | Not Available | Not Available | 0.51 |
| P7 | UNC13D | c.3259C>T | p.Arg1087Trp | 0.0001438 | No reference | BENIGN with a score of 0.002 | AFFECT PROTEIN FUNCTION with a score of 0.00 | 2.64 |
| XIAP | c.1268A>C | p.Gln423Pro | 0.3334 | [40–42] | BENIGN with a score of 0.002 | TOLERATED with a score of 0.30 | ||
| P8 | STXBP2 | c.497C>T | p.Thr166Met | 0.0002454 | No reference | BENIGN with a score of 0.022 | TOLERATED with a score of 0.10 | 3.51 |
| P9 | STXBP2 | c.497C>T | p.Thr166 Met | 0.0002454 | No reference | BENIGN with a score of 0.022 | TOLERATED with a score of 0.10 | 13.37 |
| P10 | STXBP2 | c.575G>A | p.Arg192His | Not reported | [15] | PROBABLY DAMAGING with a score of 1.000 | AFFECT PROTEIN FUNCTION with a score of 0.00 | 13.73 |
| P11 | STXBP2 | c.767T>C | p.Leu256Pro | Not reported | No reference | PROBABLY DAMAGING with a score of 1.000 | AFFECT PROTEIN FUNCTION with a score of 0.00 | 4.7 |
| P38 | XIAP | c.1268A>C | p.Gln423Pro | 0.3334 | [40–42] | BENIGN with a score of 0.002 | TOLERATED with a score of 0.30 | 0.07 |
| P45 | UNC13D | c.118-307G>A | Unknown | Not reported | [29, 39, 43] | Not Available | Not Available | 3.07 |
ExAC Exome Aggregation Consortitium, HGMD Human Gene Mutation Database, GS Google Scholar
PolyPhen-2 Polymorphism Phenotyping-2, SIFT Sorting Intolerant From Tolerant
Ter Termination, which would result to truncated protein
Cytokine levels
The mean (95 % Confidence Interval, CI) of serum IL-2, IL-4, IL-6, IL-10, TNF-α, and IFN-γ concentrations for healthy controls were 2.4 (2.2–2.7), 3.2 (2.9–3.5), 6.0 (4.0–7.9), 3.4 (3.0–3.9), 2.9 (2.5–3.4), 9.2 (8.3–10.0) pg/ml, respectively. As compared to control group, the levels of IL-4 in both primary and secondary HLH groups were significantly lower (P < 0.05) while those of IL-6, IL-10 and IFN-γ were significantly higher (P < 0.05) (Fig. 1). However, the levels of IL-2 and TNF-α were not statistically different among the three groups (all P > 0.05).
Fig. 1.
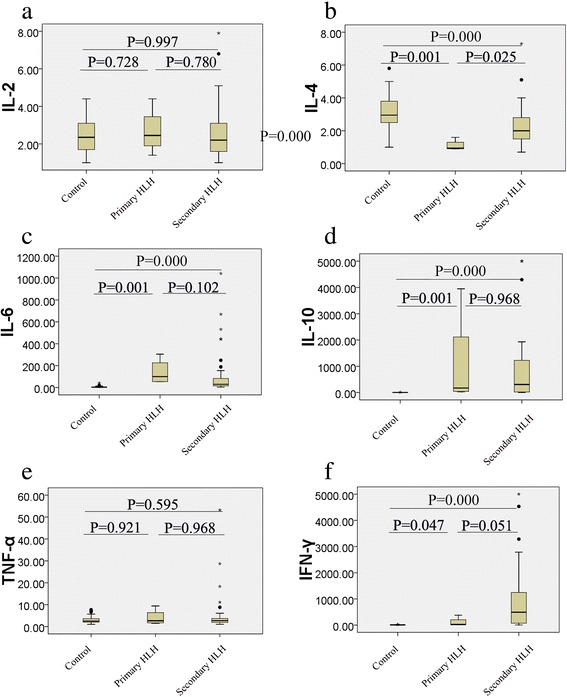
Comparisons of serum cytokine concentrations (pg/ml) among control, primary HLH, and secondary HLH. a IL-2; b IL-4; c IL-6; d IL-10; e TNF-α; f IFN-γ. The center horizontal line of the central box is the median (50th percentile), the bottom and top of the box are the 25th and 75th percentiles. The whiskers extend from each end of the box to the 5th and 95th percentiles of the values, respectively. Outliers are the data with values beyond the 5th and 95th percentiles
When we compared the levels of the cytokines between the primary and secondary HLH groups, the IL-4 level in primary-HLH was significantly lower than that in secondary HLH (P = 0.025), with mean (95 % CI) in primary and secondary HLH groups showed as 1.1 (0.6–1.6) and 2.3 (1.9–2.7), respectively. Additionally, IFN-γ level in primary HLH had a tendency of statistically lower than that in secondary HLH (P = 0.051), with mean (95 % CI) in primary and secondary HLH groups showed as 106.6 (minus-393.9) pg/ml and 905.7 (530.7–1280.6) pg/ml, respectively. The 95 % CI gap of IL-4 between primary and secondary HLH groups was 1.6–1.9 pg/ml, while the gap of IFN-γ between the two groups was 393.9–530.7 pg/ml. The levels of the remaining four cytokines including IL-2, IL-6, IL-10, and TNF-α were not significantly different between primary and secondary HLH groups (P = 0.78, P = 0.102, P = 0.968, and P = 0.968, respectively).
The area under the ROC curve, referred to as the AUC, is an appropriate measure for describing the overall accuracy of a diagnostic test, and higher AUC value mean better diagnostic value. AUCs of IL-4, IFN-γ, IL-10, TNF-α, IL-2, and IL-6 levels were calculated to be 0.841, 0.799, 0.506, 0.494, 0.457, and 0.250, respectively, indicating that levels of IL-4 and IFN-γ may be used as additional tools for the quick differential diagnosis between primary and secondary HLH (Fig. 2). ROC curves of IL-4 levels showed that 1.6 pg/ml, 1.7 pg/ml, 1.8 pg/ml, and 1.9 pg/ml had sensitivity and specificity as 73.2 and 75.0 %, 70.7 and 100.0 %, 68.3 and 100.0 %, 63.4 and 100 %, respectively. Furthermore, ROC curves of IFN-γ levels showed that 360.6 pg/ml, 433.9 pg/ml, and 539.7 pg/ml had sensitivity and specificity as 51.2 and 75.0 %, 51.2 and 100.0 %, 48.8 and 100 %, respectively. Taken the results of IL-4 and IFN-γ together, we proposed that HLH patients with IL-4 below 1.7 pg/ml and IFN-γ below 433.9 pg/ml had a higher chance to be primary HLH.
Fig. 2.
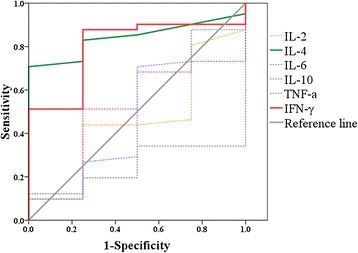
ROC curves of IL-2, IL-4, IL-6, IL-10, TNF-α, and IFN-γ between primary HLH and secondary HLH. The diagonal line is the reference line
Degranulation and clinical data
Information of surface CD107a level in resting NK cells, age at diagnosis, sex, with fever or not, hemoglobin levels, platelet counts, white blood cell counts, percentage of neutrophils, absolute neutrophil counts, percentage of lymphocytes, absolute lymphocyte counts, triglyceride levels, fibrinogen levels, LDH levels, ferritin levels, sIL2R levels, sIL2R/ferritin ratio, EBV-DNA, and CMV-DNA copies were shown in Table 3. The results showed that, except the percentage of neutrophils (P = 0.012) and the percentage of lymphocytes (P = 0.012), there was no significant difference between primary HLH and secondary HLH groups in any other factors.
Table 3.
Clinical information on 45 HLH patients
| Total (n = 45) | Primary (n = 4) | Secondary (n = 41) | P-value | |
|---|---|---|---|---|
| Age at diagnosis | ||||
| ≤12 months | 9/45 | 2/4 | 7/41 | 0.116 |
| >12 months | 36/45 | 2/4 | 34/41 | |
| Mean age (year, range) | 3.7 (0–12.3) | 2.9 (1.0–6.3) | 3.8 (0–12.3) | 0.646 |
| Sex (M/F) | 27/18 | 3/1 | 24/17 | 0.521 |
| Fever | 45/45 | 4/4 | 41/41 | |
| Hemoglobin (<90 g/L) | 16/45 | 1/4 | 15/41 | 0.644 |
| Mean (g/L, range) | 94.0 (46.0–135.0) | 93.8 (84.0–102.0) | 94.0 (46.0–135.0) | 0.905 |
| Platelets (<100x109/L) | 30/45 | 2/4 | 28/41 | 0.459 |
| Mean (100x109/L, range) | 82.5 (32.0–400.0) | 100.0 (5.0–215.0) | 80.8 (3.0–400.0) | 0.661 |
| White blood cells | ||||
| Mean (1x109/L, range) | 7.5 (0.3–41.3) | 6.5 (1.0–9.7) | 7.6 (0.3–41.3) | 0.842 |
| Percentage of neutrophils | ||||
| Mean (%, range) | 38.2 (2.0–81.5) | 12.8 (6.9–19.4) | 40.7 (2.0–81.5) | 0.012 |
| Total neutrophils (<1x109/L) | 20/45 | 2/4 | 18/41 | 0.815 |
| Mean (1x109/L, range) | 2.9 (0.1–22.1) | 0.9 (0.1–1.3) | 3.1 (0.1–22.1) | 0.413 |
| Percentage of lymphocytes | ||||
| Mean (%, range) | 50.4 (7.9–88) | 75.3 (71.8–77.7) | 48.0 (7.9–88.0) | 0.012 |
| Total lymphocytes | ||||
| Mean (1x109/L, range) | 3.6 (0.2–28.1) | 4.9 (0.7–7.5) | 3.4 (0.2–28.1) | 0.175 |
| Triglycerides (>3.0 mmol/L) | 15/45 | 3/4 | 12/41 | 0.064 |
| Mean (mmol/L, range) | 2.7 (0.8–8.9) | 3.0 (1.3–4.1) | 2.7 (0.8–8.9) | 0.425 |
| Fibrinogen (<1.5 g/L) | 30/45 | 4/4 | 26/41 | 0.138 |
| Mean (g/L, range) | 1.5 (0.2–4.9) | 0.9 (0.5–1.2) | 1.5 (0.2–4.9) | 0.14 |
| LDH (>500 IU/L) | 32/45 | 3/4 | 29/41 | 0.857 |
| Mean (IU/L, range) | 1018.0 (167.0–6565.0) | 814.0 (363.0–1644.0) | 1037.9 (167.0–6565.0) | 0.842 |
| Ferritin (>500 μg/L) | 45/45 | 4/4 | 41/41 | |
| Mean (μg/L, range) | 1477.7 (895.0–1500.0) | 1500.0 (1500.0–1500.0) | 1474.2 (895.0–1500.0) | 0.655 |
| sIL2R (≥2400 U/ml) | 26/27 | 4/4 | 22/23 | 0.671 |
| Mean (U/ml, range) | 34092.7 (155.6–85787.7) | 27198.1 (14250.9–38203.1) | 35239.6 (155.6–85787.7) | 0.922 |
| sIL2R/Ferritin | ||||
| Mean (U/pg, range) | 23.0 (0.1–57.2) | 18.4 (9.5–25.5) | 23.8 (0.1–57.2) | 0.811 |
| Degranulation (CD107a <5 %) | 16/36 | 1/3 | 15/33 | 0.795 |
| Mean (%, range) | 10.3 (0.1–35.1) | 22.3 (0.7–33.8) | 9.2 (0.1–35.1) | 0.407 |
| EBV-DNA (>1000 copies) | 26/45 | 2/4 | 24/41 | 0.741 |
| CMV-DNA (>1000 copies) | 1/45 | 0/4 | 1/41 | 0.752 |
P value: Comparisons between primary and secondary HLH groups
The degranulation assay results in healthy controls, primary HLH cases, and secondary HLH cases were shown in Fig. 3, with mean (95 % CI) in controls was 20.9 (18.19–23.6) %. We defined resting NK cell degranulation below 5 % as defective [26]. The results showed 0/1 (0 %) patient with FHL2, 1/3 (33.3 %) patient with XLP, and 15/33 (45.5 %) patients with a diagnosis of secondary HLH had resting NK degranulation below 5 % (Additional file 7: Table S1). There was a high possibility that patients with defective degranulation could carry undetected mutations.
Fig. 3.
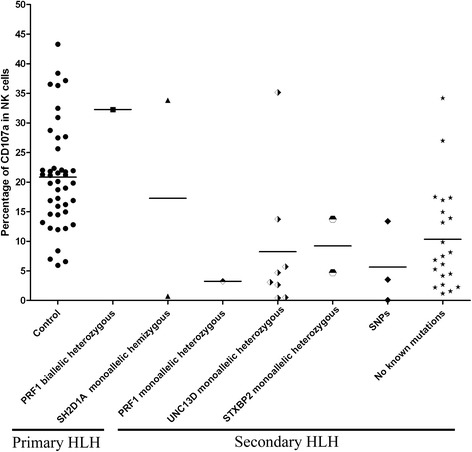
Degranulation results of healthy controls, primary HLH cases, and pecondary HLH cases. Shapes represent individual subjects, while horizontal bars show the mean in each group
Meanwhile, we re-grouped HLH patients based on their CD107a levels in resting NK cells: HLH with CD107a<5 % (n = 16), and HLH with CD107a>5 % (n = 20). Comparisons of serum cytokine concentrations (pg/ml) among Control, HLH with CD107a<5 %, and HLH with CD107a>5 % were shown in Additional file 8: Figure S7, indicated similar results to primary and secondary HLH grouping. Comparing to the healthy control group, both HLH with CD107a<5 % and CD107a>5 % had higher IL-6, IL-10 and IFN-γ levels, and lower IL-4 level (with all P < 0.05). Between HLH with CD107a<5 % and CD107a>5 % groups, all six cytokines showed no statistical significance.
Discussion
Inheritance of HLH is known to be autosomal recessive. Based on the data from published studies, primary HLH usually includes patients with homozygous, hemizygous, or compound heterozygous mutations, as mutations of these types affect protein function in a known way. In our HLH patients, no homozygous mutation was detected, and all were hemizygous or heterozygous variants. Similar to our results, Zhizhuo H et al. investigated 67 Chinese HLH patients and identified eight patients with variants in primary HLH related genes, included one patient with a hemizygous mutation of XIAP gene, three patients with compound heterozygous mutations, and four patients with single heterozygous mutation [27]. How does the single heterozygous mutation affect its coding protein’s function is complicated and remains to be elucidated. Based on the recent study of Spessott WA et al. in 2015 [28], some single heterozygous mutation in special site can cause primary HLH, yet the dominant negative effect can not be generalized, and people should do further study to prove the related protein function is damaged if they want to assign some HLH patients with monoallelic mutation as primary HLH. Another possibility is that the risk of having missed pathogenic variants is high, and possible additional variants, such as deep intronic variants similar to mutations in deep intron 1 of UNC13D gene [29–31], might have been missed for some cases.
In our previous study, we found that HLH patients presented a specific cytokine profile of highly increased levels of IFN-γ and IL-10, and a moderately increased level of IL-6 [23]. In this study, the levels of IL-6, IL-10, and IFN-γ in both primary and secondary HLH groups were significantly higher (P < 0.05) than those of controls, and no patient was overlapped with our previous study, which further confirmed the usefulness of this cytokine profile for the diagnosis of HLH.
When we compared the levels of the cytokines between the primary and secondary HLH groups, the IL-4 level in primary-HLH was significantly lower than that in secondary HLH (P = 0.025), with the gap of IL-4 between the two groups was 1.6–1.9 pg/ml, and IFN-γ level in primary HLH had a tendency of statistically lower than that in secondary HLH (P = 0.051), with the gap of IFN-γ between the two groups was 393.9–530.7 pg/ml. AUCs of six cytokines indicated that levels of IL-4 and IFN-γ may be used as additional tools for the quick differential diagnosis between primary and secondary HLH. Taken the results of IL-4 and IFN-γ together, we propose that HLH patients with IL-4 below 1.7 pg/ml and IFN-γ below 433.9 pg/ml have a higher possibility to be primary HLH.
There have been studies trying to find other discriminators to differentiate primary from secondary cases of HLH. According to the report by Bryceson et al. [26], degranulation assay has a high sensitivity and specificity rate for discrimination between two types of HLH (type1 includes FHL3, FHL4, and FHL5, while type2 includes FHL2, XLP1, XLP2, and secondary HLH cases). In this cohort, all primary HLH cases belong to type2 HLH including FHL2 and XLP1, and it is not surprising that we can not find difference of degranulation between primary HLH and secondary HLH groups. Yasumi et al. proposed that the percentage of total lymphocytes, serum levels of LDH, and the sIL2R/ferritin ratio could differentiate familial from secondary HLH [32]. In this study, when we compare to the secondary HLH patients, primary HLH patients have higher percentage of lymphocytes (mean level 75.3 % vs 48.0 %, P = 0.012), and lower percentage of neutrophils (12.8 % vs 40.7 %, P = 0.012), which is consistent with Yasumi et al’s findings.
Our study has some limitations. First, the risk of having missed pathogenic variants is high, and possible additional variants, such as deep intronic variants, regulatory variants, and complex structural variants, might have been missed for some cases, which would have great impact on the grouping strategy and then influence the interpretation of the results. Second, as this is a single-center study, and the results are based on data from small number of samples and incomplete genetic sequencing results, a multi-center study was required to be performed to validate the results.
Conclusions
Among 45 HLH patients, four HLH patients are identified as primary HLH with hemizygous or compound heterozygous mutations, and 41 HLH patients belong to secondary HLH. Variants in PRF1, UNC13D, STX11, STXBP2, SH2D1A, XIAP, and ITK genes account for 2/45 (4.4 %), 6/45 (13.3 %), 0/45 (0.0 %), 2/45 (4.4 %), 3/45 (6.7 %), 0/45 (0 %), and 0/45 (0.0 %), respectively. HLH patients with lower IL-4 level, lower IFN-γ level, higher percentage of lymphocytes, and lower percentage of neutrophils have higher possibility to be primary HLH. The cytokine profiles can be used as an additional tool for the quick differential diagnosis between primary and secondary HLH.
Acknowledgments
This work was supported in part by the grants from the National Natural Science Foundation of China (No. 81170502, 81470304), and the Zhejiang Provincial Natural Science Foundation of China (No. LZ12H08001). The authors would like to thank Ms. Baiqin Qian, Ms Ping Chen, and Mr. Hongqiang Shen for their excellent technical assistance. We thank all the anonymous reviewers for their helpful suggestions on the quality improvement of our paper.
Abbreviations
- 95 % CI
95 % Confidence Interval
- AUC
area under ROC curve
- CBA
cytometric bead assay
- CTLs
cytotoxic T cells
- ExAC
Exome Aggregation Consortitium
- FHL
familial hemophagocytic lymphohistiocytosis
- GS
Google Scholar
- HGMD
Human Gene Mutation Database
- HLH
hemophagocytic lymphohistiocytosis
- IFN-γ
interferon-γ
- IL
interleukin
- NK
natural killer
- PolyPhen-2
Polymorphism Phenotyping-2
- ROC
receiver operating characteristic
- SIFT
Sorting Intolerant From Tolerant
- sIL2R
soluble interleukin-2 receptor
- SNPs
Single Nucleotide Polymorphisms
- TNF-α
tumor necrosis factor-alpha
Additional files
Sequencing results of PRF1 gene. Genomic DNA sequencing results showed a 1-year-5-month male (P1) had heterozygous mutation c.385T>A (p.Trp129Arg), and a 6-year-3-month female (P2) had compound heterozygous c.757G>A (p.Glu253Lys) and c.1061A>T (p.Asp354Val) of PRF1 gene. (TIF 1391 kb)
Sequencing results of UNC13D gene. Five missense mutations of UNC13D gene, c.478G>A (p.Val160 Met) in P3, c.518C>T (p.Thr173Met) in P4, c.760C>T (p.Arg254Cys) in P5, c.3259C>T(p.Arg1087Trp) in P7, and c.118-307G>A in P45, were found in four males and one female, respectively. (TIF 171 kb)
Sequencing results of STXBP2 gene. Two mutations of STXBP2 gene, c.575G>A (p.Arg192His) in P10, and c.767T>C (p.Leu256Pro) in P11, were found in one male and one female, respectively. (TIF 985 kb)
Sequencing results of SH2D1A gene. Three hemizygous mutations of SH2D1A gene, c.191G> (p.Trp64Ter) in P16, c.162C>G(p.Tyr54Ter) in P17, and c.163C>T(p.Arg55Ter) in P26, were identified in three male patients, respectively. (TIF 136 kb)
SNPs of STXBP2 and XIAP genes. Two males patients, 7-year-9-month (P8) and 11-year-11-month (P9), showed c.497C>T (p.Thr166Met) in STXBP2 gene, numbered SNP rs181216956; of XIAP gene, two male patients (P7 and P38) were identified with c.1268A>C(p.Gln423Pro), numbered SNP rs5956583. (TIF 938 kb)
Sequencing results of UNC13D gene in a Chinese male neonate(P6) and his parents. In mRNA level of P6, the result manifested a heterozygous frameshift mutation c.2295_2298delGCAG (A), which was consistent with the heterozygous point mutation c.2296C>T in genomic DNA level (B). Sequencing results from cDNA and genomic DNA of P6 and his parents both showed that P6 inherited this mutation from his mother. (TIF 391 kb)
Percentage of CD107a expression in NK cells from 43 controls and 36 HLH patients. (DOCX 15 kb)
Comparisons of serum cytokine concentrations (pg/ml) among control, HLH with CD107a<5 %, and HLH with CD107a>5 %. A: IL-2; B: IL-4; C: IL-6; D: IL-10; E: TNF-α; F: IFN-γ. The center horizontal line of the central box is the median (50th percentile), the bottom and top of the box are the 25th and 75th percentiles. The whiskers extend from each end of the box to the 5th and 95th percentiles of the values, respectively. Outliers are the data with values beyond the 5th and 95th percentiles. (TIF 957 kb)
Footnotes
Competing interests
All authors declare that they have no competing interests.
Authors’ contributions
YYC, YMT designed the study; YYC, ZJW, ZBL, NZ, SLY made substantial contributions to acquisition and analysis of data; YYC drafted and rewrote the manuscript; YMT revised the manuscript. All authors read and approved the final manuscript.
References
- 1.Tang YM, Xu XJ. Advances in hemophagocytic lymphohistiocytosis: pathogenesis, early diagnosis/differential diagnosis, and treatment. ScientificWorldJournal. 2011;11:697–708. doi: 10.1100/tsw.2011.62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Henter JI, Horne A, Arico M, Egeler RM, Filipovich AH, Imashuku S, et al. HLH-2004: Diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer. 2007;48(2):124–31. doi: 10.1002/pbc.21039. [DOI] [PubMed] [Google Scholar]
- 3.Xiao L, Xian Y, Dai BT, Su YC, Xiao JW, Zheng QC, et al. Clinical features and outcome analysis of 83 childhood Epstein-Barr virus-associated hemophagocytic lymphohistiocytosis with HLH-2004 protocol. Zhonghua Xue Ye Xue Za Zhi. 2011;32(10):668–72. [PubMed] [Google Scholar]
- 4.Koh KN, Im HJ, Chung NG, Cho B, Kang HJ, Shin HY, et al. Clinical features, genetics, and outcome of pediatric patients with hemophagocytic lymphohistiocytosis in Korea: report of a nationwide survey from Korea Histiocytosis Working Party. Eur J Haematol. 2015;94(1):51–9. doi: 10.1111/ejh.12399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ishii E, Ohga S, Imashuku S, Yasukawa M, Tsuda H, Miura I, et al. Nationwide survey of hemophagocytic lymphohistiocytosis in Japan. Int J Hematol. 2007;86(1):58–65. doi: 10.1532/IJH97.07012. [DOI] [PubMed] [Google Scholar]
- 6.My LT, le Lien B, Hsieh WC, Imamura T, Anh TN, Anh PN, et al. Comprehensive analyses and characterization of haemophagocytic lymphohistiocytosis in Vietnamese children. Br J Haematol. 2010;148(2):301–10. doi: 10.1111/j.1365-2141.2009.07957.x. [DOI] [PubMed] [Google Scholar]
- 7.Ramachandran B, Balasubramanian S, Abhishek N, Ravikumar KG, Ramanan AV. Profile of hemophagocytic lymphohistiocytosis in children in a tertiary care hospital in India. Indian Pediatr. 2011;48(1):31–5. doi: 10.1007/s13312-011-0020-2. [DOI] [PubMed] [Google Scholar]
- 8.Janka GE. Familial and acquired hemophagocytic lymphohistiocytosis. Annu Rev Med. 2012;63:233–46. doi: 10.1146/annurev-med-041610-134208. [DOI] [PubMed] [Google Scholar]
- 9.Frederiksen JK, Ross CW. Cytomegalovirus-associated hemophagocytic lymphohistiocytosis in a patient with myasthenia gravis treated with azathioprine. Blood. 2014;123(15):2290. doi: 10.1182/blood-2014-01-548172. [DOI] [PubMed] [Google Scholar]
- 10.Janka GE, Lehmberg K. Hemophagocytic syndromes--an update. Blood Rev. 2014;28(4):135–42. doi: 10.1016/j.blre.2014.03.002. [DOI] [PubMed] [Google Scholar]
- 11.Ohadi M, Lalloz MR, Sham P, Zhao J, Dearlove AM, Shiach C, et al. Localization of a gene for familial hemophagocytic lymphohistiocytosis at chromosome 9q21.3-22 by homozygosity mapping. Am J Hum Genet. 1999;64(1):165–71. doi: 10.1086/302187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Stepp SE, Dufourcq-Lagelouse R, Le Deist F, Bhawan S, Certain S, Mathew PA, et al. Perforin gene defects in familial hemophagocytic lymphohistiocytosis. Science. 1999;286(5446):1957–9. doi: 10.1126/science.286.5446.1957. [DOI] [PubMed] [Google Scholar]
- 13.Feldmann J, Callebaut I, Raposo G, Certain S, Bacq D, Dumont C, et al. Munc13-4 is essential for cytolytic granules fusion and is mutated in a form of familial hemophagocytic lymphohistiocytosis (FHL3) Cell. 2003;115(4):461–73. doi: 10.1016/S0092-8674(03)00855-9. [DOI] [PubMed] [Google Scholar]
- 14.zur Stadt U, Schmidt S, Kasper B, Beutel K, Diler AS, Henter JI, et al. Linkage of familial hemophagocytic lymphohistiocytosis (FHL) type-4 to chromosome 6q24 and identification of mutations in syntaxin 11. Hum Mol Genet. 2005;14(6):827–34. doi: 10.1093/hmg/ddi076. [DOI] [PubMed] [Google Scholar]
- 15.zur Stadt U, Rohr J, Seifert W, Koch F, Grieve S, Pagel J, et al. Familial hemophagocytic lymphohistiocytosis type 5 (FHL-5) is caused by mutations in Munc18-2 and impaired binding to syntaxin 11. Am J Hum Genet. 2009;85(4):482–92. doi: 10.1016/j.ajhg.2009.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cote M, Menager MM, Burgess A, Mahlaoui N, Picard C, Schaffner C, et al. Munc18-2 deficiency causes familial hemophagocytic lymphohistiocytosis type 5 and impairs cytotoxic granule exocytosis in patient NK cells. J Clin Invest. 2009;119(12):3765–73. doi: 10.1172/JCI40732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schwartzberg PL, Mueller KL, Qi H, Cannons JL. SLAM receptors and SAP influence lymphocyte interactions, development and function. Nat Rev Immunol. 2009;9(1):39–46. doi: 10.1038/nri2456. [DOI] [PubMed] [Google Scholar]
- 18.Rigaud S, Fondaneche MC, Lambert N, Pasquier B, Mateo V, Soulas P, et al. XIAP deficiency in humans causes an X-linked lymphoproliferative syndrome. Nature. 2006;444(7115):110–4. doi: 10.1038/nature05257. [DOI] [PubMed] [Google Scholar]
- 19.Huck K, Feyen O, Niehues T, Ruschendorf F, Hubner N, Laws HJ, et al. Girls homozygous for an IL-2-inducible T cell kinase mutation that leads to protein deficiency develop fatal EBV-associated lymphoproliferation. J Clin Invest. 2009;119(5):1350–8. doi: 10.1172/JCI37901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Henter JI, Elinder G, Soder O, Hansson M, Andersson B, Andersson U. Hypercytokinemia in familial hemophagocytic lymphohistiocytosis. Blood. 1991;78(11):2918–22. [PubMed] [Google Scholar]
- 21.Tang Y, Xu X, Song H, Yang S, Shi S, Wei J, et al. Early diagnostic and prognostic significance of a specific Th1/Th2 cytokine pattern in children with haemophagocytic syndrome. Br J Haematol. 2008;143(1):84–91. doi: 10.1111/j.1365-2141.2008.07298.x. [DOI] [PubMed] [Google Scholar]
- 22.Xu XJ, Tang YM, Liao C, Song H, Yang SL, Xu WQ, et al. Inflammatory cytokine measurement quickly discriminates gram-negative from gram-positive bacteremia in pediatric hematology/oncology patients with septic shock. Intensive Care Med. 2013;39(2):319–26. doi: 10.1007/s00134-012-2752-4. [DOI] [PubMed] [Google Scholar]
- 23.Xu XJ, Tang YM, Song H, Yang SL, Xu WQ, Zhao N, et al. Diagnostic accuracy of a specific cytokine pattern in hemophagocytic lymphohistiocytosis in children. J Pediatr. 2012;160(6):984–90. doi: 10.1016/j.jpeds.2011.11.046. [DOI] [PubMed] [Google Scholar]
- 24.Marcenaro S, Gallo F, Martini S, Santoro A, Griffiths GM, Arico M, et al. Analysis of natural killer-cell function in familial hemophagocytic lymphohistiocytosis (FHL): defective CD107a surface expression heralds Munc13-4 defect and discriminates between genetic subtypes of the disease. Blood. 2006;108(7):2316–23. doi: 10.1182/blood-2006-04-015693. [DOI] [PubMed] [Google Scholar]
- 25.Chen Y, Wang Z, Cheng Y, Tang Y. Novel mutations in the UNC13D gene carried by a Chinese neonate with hemophagocytic lymphohistiocytosis. Yonsei Med J. 2013;54(4):1053–7. doi: 10.3349/ymj.2013.54.4.1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bryceson YT, Pende D, Maul-Pavicic A, Gilmour KC, Ufheil H, Vraetz T, et al. A prospective evaluation of degranulation assays in the rapid diagnosis of familial hemophagocytic syndromes. Blood. 2012;119(12):2754–63. doi: 10.1182/blood-2011-08-374199. [DOI] [PubMed] [Google Scholar]
- 27.Zhizhuo H, Junmei X, Yuelin S, Qiang Q, Chunyan L, Zhengde X, et al. Screening the PRF1, UNC13D, STX11, SH2D1A, XIAP, and ITK gene mutations in Chinese children with Epstein-Barr virus-associated hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer. 2012;58(3):410–4. doi: 10.1002/pbc.23216. [DOI] [PubMed] [Google Scholar]
- 28.Spessott WA, Sanmillan ML, McCormick ME, Patel N, Villanueva J, Zhang K, et al. Hemophagocytic lymphohistiocytosis caused by dominant-negative mutations in STXBP2 that inhibit SNARE-mediated membrane fusion. Blood. 2015;125(10):1566–77. doi: 10.1182/blood-2014-11-610816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Entesarian M, Chiang SC, Schlums H, Meeths M, Chan MY, Mya SN, et al. Novel deep intronic and missense UNC13D mutations in familial haemophagocytic lymphohistiocytosis type 3. Br J Haematol. 2013;162(3):415–8. doi: 10.1111/bjh.12371. [DOI] [PubMed] [Google Scholar]
- 30.Seo JY, Song JS, Lee KO, Won HH, Kim JW, Kim SH, et al. Founder effects in two predominant intronic mutations of UNC13D, c.118-308C>T and c.754-1G>C underlie the unusual predominance of type 3 familial hemophagocytic lymphohistiocytosis (FHL3) in Korea. Ann Hematol. 2013;92(3):357–64. doi: 10.1007/s00277-012-1628-6. [DOI] [PubMed] [Google Scholar]
- 31.Meeths M, Chiang SC, Wood SM, Entesarian M, Schlums H, Bang B, et al. Familial hemophagocytic lymphohistiocytosis type 3 (FHL3) caused by deep intronic mutation and inversion in UNC13D. Blood. 2011;118(22):5783–93. doi: 10.1182/blood-2011-07-369090. [DOI] [PubMed] [Google Scholar]
- 32.Yasumi T, Hori M, Hiejima E, Shibata H, Izawa K, Oda H, et al. Laboratory parameters identify familial haemophagocytic lymphohistiocytosis from other forms of paediatric haemophagocytosis. Br J Haematol. 2015;170(4):532–8. doi: 10.1111/bjh.13461. [DOI] [PubMed] [Google Scholar]
- 33.Kobayashi Y, Salih HM, Kajiume T, Nakamura K, Miyagawa S, Sato T, et al. Successful treatment with liposteroid followed by reduced intensity stem cell transplantation in an infant with perforin deficiency presenting with hemophagocytic lymphohistiocytosis. J Pediatr Hematol Oncol. 2007;29(3):178–82. doi: 10.1097/MPH.0b013e3180335030. [DOI] [PubMed] [Google Scholar]
- 34.Overwater E, Smulders Y, van der Burg M, Lombardi MP, Meijers-Heijboer HE, Kuijpers TW, et al. The value of DNA storage and pedigree analysis in rare diseases: a 17-year-old boy with X-linked lymphoproliferative disease (XLP) caused by a de novo SH2D1A mutation. Eur J Pediatr. 2014;173(12):1695–8. doi: 10.1007/s00431-014-2313-7. [DOI] [PubMed] [Google Scholar]
- 35.Sumegi J, Huang D, Lanyi A, Davis JD, Seemayer TA, Maeda A, et al. Correlation of mutations of the SH2D1A gene and epstein-barr virus infection with clinical phenotype and outcome in X-linked lymphoproliferative disease. Blood. 2000;96(9):3118–25. [PubMed] [Google Scholar]
- 36.Meazza R, Tuberosa C, Cetica V, Falco M, Parolini S, Grieve S, et al. Diagnosing XLP1 in patients with hemophagocytic lymphohistiocytosis. J Allergy Clin Immunol. 2014;134(6):1381–7. doi: 10.1016/j.jaci.2014.04.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Coffey AJ, Brooksbank RA, Brandau O, Oohashi T, Howell GR, Bye JM, et al. Host response to EBV infection in X-linked lymphoproliferative disease results from mutations in an SH2-domain encoding gene. Nat Genet. 1998;20(2):129–35. doi: 10.1038/2424. [DOI] [PubMed] [Google Scholar]
- 38.Mougiakakos D, Machaczka M, Jitschin R, Klimkowska M, Entesarian M, Bryceson YT, et al. Treatment of familial hemophagocytic lymphohistiocytosis with third-party mesenchymal stromal cells. Stem Cells Dev. 2012;21(17):3147–51. doi: 10.1089/scd.2012.0214. [DOI] [PubMed] [Google Scholar]
- 39.Qian Y, Johnson JA, Connor JA, Valencia CA, Barasa N, Schubert J, et al. The 253-kb inversion and deep intronic mutations in UNC13D are present in North American patients with familial hemophagocytic lymphohistiocytosis 3. Pediatr Blood Cancer. 2014;61(6):1034–40. doi: 10.1002/pbc.24955. [DOI] [PubMed] [Google Scholar]
- 40.Ameratunga R, Woon ST. Customised molecular diagnosis of primary immune deficiency disorders in New Zealand: an efficient strategy for a small developed country. N Z Med J. 2009;122(1304):46–53. [PubMed] [Google Scholar]
- 41.Weiss KH, Runz H, Noe B, Gotthardt DN, Merle U, Ferenci P, et al. Genetic analysis of BIRC4/XIAP as a putative modifier gene of Wilson disease. J Inherit Metab Dis. 2010;33(Suppl 3):S233–40. doi: 10.1007/s10545-010-9123-5. [DOI] [PubMed] [Google Scholar]
- 42.Ferretti M, Gattorno M, Chiocchetti A, Mesturini R, Orilieri E, Bensi T, et al. The 423Q polymorphism of the X-linked inhibitor of apoptosis gene influences monocyte function and is associated with periodic fever. Arthritis Rheum. 2009;60(11):3476–84. doi: 10.1002/art.24905. [DOI] [PubMed] [Google Scholar]
- 43.Li W, Gao C, Cui L, Liu S, Zhao X, Zhang R, et al. DNMT3A mutations and prognostic significance in childhood acute lymphoblastic leukemia. Leuk Lymphoma. 2015;56(4):1066–71. doi: 10.3109/10428194.2014.947607. [DOI] [PubMed] [Google Scholar]