

Abstract
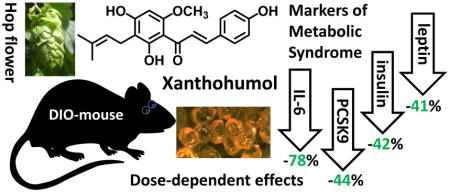
Xanthohumol (XN) is a prenylated flavonoid found in hops (Humulus lupulus) and beer. The dose-dependent effects of XN on glucose and lipid metabolism in a preclinical model of metabolic syndrome were the focus of our study. Forty-eight male C57BL/6J mice, 9 weeks of age, were randomly divided into three XN dose groups of 16 animals. The mice were fed a high-fat diet (60% kcal as fat) supplemented with XN at dose levels of 0, 30, or 60 mg/kg body weight/day, for 12 weeks. Dietary XN caused a dose-dependent decrease in body weight gain. Plasma levels of glucose, total triglycerides, total cholesterol, and MCP-1 were significantly decreased in mice on the 60 mg/kg/day treatment regimen. Treatment with XN at 60 mg/kg/day resulted in reduced plasma LDL-cholesterol (LDL-C), IL-6, insulin and leptin levels by 80%, 78%, 42%, and 41%, respectively, compared to the vehicle control group. Proprotein Convertase Subtilisin Kexin 9 (PCSK-9) levels were 44% lower in the 60 mg/kg dose group compared to the vehicle control group (p ≤ 0.05) which may account for the LDL-C lowering activity of XN. Our results show that oral administration of XN improves markers of systemic inflammation and metabolic syndrome in diet-induced obese C57BL/6J mice.
Keywords: metabolic syndrome, obesity, xanthohumol, C57BL/6J mice, lipidomics, PCSK9
Graphical abstract
1. Introduction
Metabolic syndrome is a condition defined by clinical diagnosis of three or more of these conditions: abdominal obesity, atherogenic dyslipidemia, insulin resistance and/or impaired glucose tolerance, hypertension, pro-inflammatory state, and prothrombotic state [1]. An estimated 25–34% of U.S. adults meet the criteria for metabolic syndrome which puts them at significantly increased risk for cardiovascular disease and type 2 diabetes [2]. Direct health care costs arising from obesity and/or related disorders account for ~7–10% of U.S. health care expenditures annually [3]. Researchers are currently investigating several complementary and alternative medicine-based therapies designed to target one or more features of metabolic syndrome [4]. There is currently no single agent effective in treating this disorder.
Studies published by us [5, 6] and others [7–9] suggest that it is both feasible to, and potentially practical to, effectively and safely treat metabolic syndrome with xanthohumol (XN, see structure in Table 1), a prenylated flavonoid found in hops. Based on animal and cell culture studies, XN could be beneficial in treating or mitigating obesity, dysregulation of glucose and lipid metabolism, atherosclerosis, and non-alcoholic fatty liver disease [10–13]. Rats fed a high-fat diet enriched with XN extract (1% w/w equivalent to a dose of 100 mg/kg body weight/day) gained less weight and had lower triacylglycerol levels in the plasma and in the liver compared to the no-treatment control group [11]. In a diet-induced animal model of non-alcoholic steatohepatitis, daily oral administration of XN at a dose level of approximately 1000 mg/kg body weight exhibited anti-inflammatory and anti-fibrogenic effects [9]. In ApoE−/− mice fed diets containing XN (300 mg/kg body weight/day) for 8 weeks, there was a decrease in hepatic triglyceride and cholesterol content accompanied by the activation of AMP-activated protein kinase, phosphorylation and inactivation of acetyl-CoA carboxylase, and reduced expression of hepatic sterol regulatory element-binding protein (SREBP) 1c mRNA [12]. In genetically obese KK-Ay mice, XN lowered fasting plasma glucose, plasma and hepatic triglyceride concentrations, and weights of white adipose tissue [14].
Table 1.
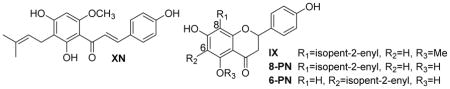
Concentrations of xanthohumol (XN) and its metabolites isoxanthohumol (IX), 8-prenylnaringenin (8-PN), and 6-prenylnaringenin (6-PN) in liver and plasma. 8-PN was only detected in the liver and then only in very small amounts, and no 6-PN was detected in either the low or high dose group.
| |||
|---|---|---|---|
| Plasma (nM) | Liver (nmol/g) | ||
| Low Dose (30 mg/kg BW/day) n=16 | XN | 84.4 ± 8.5 | 0.477 ± 0.119 |
| IX | 3.1 ± 1.2 | 0.279 ± 0.070 | |
| 8-PN | ND* | 0.010 ± 0.002 | |
| 6-PN | ND* | ND* | |
| Total | 87.5 ± 9.2 | 0.766 ± 0.191 | |
|
| |||
| High Dose (60 mg/kg BW/day) n=16 | XN | 95.1 ± 13.4 | 0.805 ± 0.201 |
| IX | 13.6 ± 2.1 | 0.746 ± 0.186 | |
| 8-PN | ND* | 0.006 ± 0.001 | |
| 6-PN | ND* | ND* | |
| Total | 109 ± 14 | 1.56 ± 0.39 | |
ND, not detectable.
In our previous study, we found that XN, orally administered to male Zucker fatty (fa) rats at a dose of 16.9 mg/kg body weight/day, produced a reduction in body weight gain and fasting plasma glucose levels compared to the control group [5]. Zucker (fa) rats lack physiological control of appetite due to dysfunctional leptin signaling. Leptin exerts its actions on appetite control via the cognate leptin receptor, Ob-R, but in Zucker (fa) rats, a missense A to C mutation in the Lepr gene on chromosome 5 (Leprfa) causes a Gln to Pro change in the Ob-R protein making the receptor non-functional [15]. In the present study, we selected a diet-induced obesity mouse model which more accurately reflects metabolic syndrome in humans [16]. The C57BL/6J mouse develops a metabolic syndrome-like phenotype when fed a high fat diet. They develop obesity, hyperglycemia, hyperinsulinemia, and hypertension on a high-fat diet but remain lean when fed a low-fat diet [17, 18].
The goal of this study was to determine the effects of XN on various endpoints of metabolic syndrome in male C57BL/6J mice fed a high fat diet. This study advances the knowledge on the in vivo effects of XN because it establishes a dose-effect relationship in a non-genetic, diet-induced rodent model of metabolic syndrome, it relates dose regimen to plasma and hepatic concentrations of XN and its main metabolites, and it provides a mechanism for the plasma LDLC-C lowering effect of XN through decreasing plasma levels of Proprotein Convertase Subtilisin Kexin 9 (PCSK9). PCSK9 is a negative regulator of plasma LDL-C clearance that acts by promoting the proteolytic degradation of the LDL receptor. In recent years, PCSK9 has received much attention as a pharmacological target to treat hypercholesterolemia. Induction of the expression of PCSK9 is now a recognized adverse effect of statin therapy [19]. In 2015, the U.S. Food & Drug Administration approved the first two monoclonal antibody inhibitors of PCSK9 for treating patients suffering from familial hypercholesterolemia [20]. To date, no small molecule regulators of plasma PCSK9 have reached the market despite significant efforts of the pharmaceutical industry.
2. Materials and methods
2.1. Reagents
Xanthohumol (purity 99+ %) was provided by Hopsteiner, Inc., New York. Oleic acid was purchased from TCI America, Portland, Oregon. Other chemicals were purchased from Sigma-Aldrich, St. Louis, MO.
2.2. Animals
All animal studies were conducted with the approval from the Institutional Animal Care and Use Committee of Oregon State University, Corvallis, Oregon, USA. Male C57BL/6J mice, 8 weeks of age, were purchased from The Jackson Laboratory, Bar Harbor, ME, USA, and maintained on a 12 h dark/light cycle and on a regular mouse chow diet. After 1 week of acclimation, the mice were randomly assigned into three groups of 16 animals each: Control, Low XN and High XN. All the animals were fed high-fat diets (5.12 kcal/g of diet; with 60 kcal% fat, 20 kcal% carbohydrate and 20% kcal protein), with or without (control group) XN. XN was first dissolved in a mixture of oleic acid:propylene glycol:Tween 80 (0.9:1:1 by weight) before it was mixed into the diet at a concentration of 0.033% (Low XN) or 0.066 % (High XN). The control diet contained an identical amount of the vehicle. The diets were given to the mice as pellets (Diets were prepared by Dyets, Inc., Bethlehem, PA) ad libitum. Food and water were supplied to the mice ad libitum. Food intake was measured three times a week and body weight was recorded weekly. After 12 weeks of feeding, the animals were sacrificed after an overnight fast and blood and liver were collected. Blood was centrifuged, and plasma was collected and frozen at −80° C for various biochemical analyses and ELISA measurements.
2.3. Measurement of plasma parameters
Plasma levels of glucose were measured by a test kit (Autokit Glucose) obtained from Wako Diagnostics (Mountain View, CA). Plasma triglycerides (TGs) and total cholesterol were analyzed by test kits purchased from Fisher-Scientific, Pittsburgh, PA. Plasma HDL cholesterol was determined using MaxDiscovery™HDL Cholesterol Assay Kit (Bioo Scientific Corporation, Austin, TX). Plasma LDL-C (in mg/dL) was estimated by the Friedewald Equation [21]: LDL-C = total cholesterol – HDL cholesterol – (total triglycerides/5). Plasma insulin was determined using a mouse insulin ELISA kit (Alpco Diagnostics, Salem, NH). Plasma Monocyte Chemoattractant Protein-1 (MCP-1 or CCL2) and Interleukin (IL)-6 were analyzed by ELISA kits from Life Technologies (Carlsbad, CA). Plasma leptin levels were estimated by an ELISA kit obtained from Crystal Chem, Inc., Downers Grove, IL. Mouse plasma Proprotein Convertase Subtilisin Kexin 9 (PCSK9) was measured using an ELISA kit purchased from R&D Systems (Minneapolis, MN).
2.4. XN and metabolite quantitation in liver and plasma
To determine the levels of XN and its metabolites isoxanthohumol (IX), 8-prenylnaringenin (8-PN), and 6-prenylnaringenin (6-PN), liver and plasma samples were analyzed using the paper strip extraction technique and LC-MS/MS quantification method as described by Legette et al. [22]. For plasma analyses, 50 μL of mouse plasma was combined with 430 μL of 0.1 M sodium acetate (pH 4.7), 10 μL of methanol, and 100 μL of Helix pomatia β-glucuronidase (Sigma, St. Louis, MO) enzyme mixture (600 Units/100 μL of 0.1 M sodium acetate buffer) in a 2 mL screw cap vial, for a total volume of 590 μL. Samples were then incubated in a water bath at 37 °C for two hours. Following incubation, 10 μL of 2 μM 4,2′- dihydroxychalcone (Indofine Chemical Company, Hillsborough, NJ) was added to each sample as an internal standard, and a small pointed strip (0.8 cm × 4.5 cm, W×L) of Whatman #1 filter paper was placed in each tube. The samples were then allowed to dry completely in a vacuum dessicator over Drierite™.
After all samples were completely dry, 500 μL of methanol containing 0.1% formic acid was added to each sample tube and then vortexed for 30 seconds. Sample tubes were then agitated on a microplate shaker (Scientific Industries, Bohemia, NY, USA, model no. SI-0400, speed 10) for 30 minutes. Following shaking, all samples were centrifuged at 15,700 ×g for 10 minutes, and the supernatant was pipetted into 1.5 mL glass autosampler vials (MicroSolve, Eatontown, NJ, Catalogue No. 9502S-1WCP). LC-MS/MS analysis was conducted using a Shimadzu SIL-HTC liquid autosampler, LC-10AD pumps, and separation was achieved with an Agilent 2×50 mm Zorbax 300SB-C8 (Agilent, Santa Clara, CA) column coupled to an Applied Biosystems Sciex 4000 QTRAP mass spectrometer (Sciex, Concord, ON, Canada).
Mouse liver samples were prepared by first homogenizing approximately 200 mg of liver tissue from each mouse in methanol:water (90:10; 500 μL/200 mg) using a bullet blender (Next Advance, Inc., Averill Park, NY) with 0.5 mm zirconium oxide beads (Next Advance, Inc., Averill Park, NY, product #ZrOB05). The homogenized tissue was then centrifuged at 15,700 ×g for 10 minutes, and 100 μL of supernatant was used per sample for extraction of XN and metabolites following the paper strip protocol described above.
2.5. Liver triglycerides by lipidomics analyses
C57BL/6J mice liver tissue (approximately 20 mg) was ground using a bullet blender (Next Advance, Inc., Averill Park, NY) with 0.5 mm zirconium oxide beads (Next Advance, Inc., Averill Park, NY, product #ZrOB05) in ice-cold methylene chloride:isopropanol:methanol (25:10:65; 800 μL/20 mg) after adding internal standard (2.0 μL of 100 μg/mL of tri(heptadecanoyl)glycerol (TG 51:0, Avanti Polar Lipids Inc. Alabaster, AL, USA). The homogenate was incubated at −20 °C for 1 h. The mixture was centrifuged at 15,700 ×g at 4 °C for 10 min, and an aliquot (3 μL) of the supernatant subjected to UPLC-QTOF analysis on a 5600 TripleTOF instrument (Sciex, Concord, ON, Canada) as described previously [23].
2.6. Statistical analyses
Statistical analyses were performed with the GraphPad Prism 5.0 software. Group differences were assessed by analysis of variance (ANOVA) followed by a post-hoc Dunnett’s test unless otherwise stated. Statistical significance was set at p ≤ 0.05. For processing of lipidomics data, PeakView (version 1.2, Sciex), LipidView (version 1.2, Sciex) and MetaboAnalyst 3.0 [24] were used.
3. Results
3.1. Mouse body weight and food intake
One week after feeding the test diets, body weights of mice fed low XN and high XN diets were significantly lower compared to the control mice (Fig. 1A). Differences in body weights between control mice and XN-fed mice continued to be significantly different at weekly intervals throughout the 12-week feeding period (Fig. 1A). Liver weights expressed as a percent of body weight (Fig. 1B) were significantly lower in mice fed the low or high XN diets versus the control diet. Food intake was not significantly different among the three treatment groups during the course of the study (data not shown).
Fig. 1.

Weekly body weights (A) and liver/body weight ratios (B) of male mice fed a high-fat diet with no XN (control), with 30 mg XN/kg body weight/day (low XN), or 60 mg XN/kg body weight/day (high XN) for 12 weeks. Values are expressed as mean ± SEM of 16 mice per group. Asterisks denote significantly different from the control group, p ≤ 0.05.
3.2. Plasma metabolic profiles
Mice given the high XN diet had significantly lower levels of plasma glucose (Fig. 2A), TGs (Fig. 2B), and total cholesterol (Fig. 2C) than the control mice. There was no significant effect of low XN diet on these plasma levels. Low and high XN treatment significantly increased plasma HDL-cholesterol (Fig. 2D) by 14.3% and 9.3%, respectively. Plasma LDL-C (Fig. 2E) was decreased 50% by low XN and 80% by high XN. The levels of the inflammatory marker, MCP-1 (Fig. 3A) were significantly reduced in mice fed the high XN diet but not those fed the low XN diet. In contrast, low XN and high XN significantly reduced plasma IL-6 levels (Fig. 3B) by 86% and 78%, respectively. Low and high XN significantly decreased plasma insulin by 33% and 42%, respectively (Fig. 4A). Leptin levels (Fig. 4B) were decreased 14% in the low XN and 41% in the high XN treatment group (p ≤ 0.05). Feeding mice low XN and high XN diets significantly reduced plasma PCSK9 by 22% and 44%, respectively (Fig. 4C).
Fig. 2.

Effects of dietary XN on plasma glucose (A), triglyceride (B), total cholesterol (C), HDL cholesterol (D), and LDL-C (E) levels of male mice fed a high-fat diet for 12 weeks. Values are expressed as mean ± SEM of 16 mice per group. Asterisks denote significantly different from the control group, p ≤ 0.05.
Fig 3.

Effects of dietary XN on plasma levels of MCP-1 (A) and IL-6 (B) in male mice fed a high-fat diet for 12 weeks. Values are expressed as mean ± SEM of 16 mice per group. Asterisks denote significantly different from the control group, p ≤ 0.05.
Fig 4.
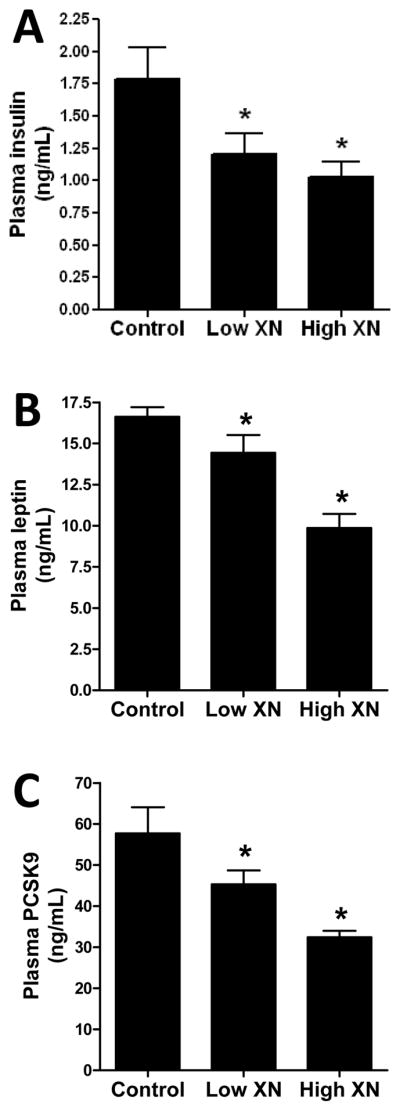
Effects of dietary XN on plasma insulin (A), plasma leptin (B), and plasma PCSK9 (C) levels of male mice fed a high-fat diet for 12 weeks. Values are expressed as mean ± SEM of 16 mice per group. Asterisks denote significantly different from the control group, p ≤ 0.05.
3.3. Triglycerides in liver
The XN-induced changes in liver triglycerides (TGs) are depicted as a heat map of the top 34 differentiating TGs, ranked by one-way ANOVA followed by Fisher’s LSD post-hoc analysis (p<0.01) (Fig. 5A). Detailed analysis of TG profiles revealed that XN treatment did not significantly affect the qualitative composition of TGs (data not shown). Total hepatic levels of TGs were significantly lower in mice fed low or high XN diets than in the control (Fig. 5B).
Fig. 5.

Lipid profiling and data processing using MetaboAnalyst 3.0. (A) Heat map revealing XN-induced changes in the relative levels of the top 34 triglyceride (TG) species ranked by one-way ANOVA followed by Fisher’s LSD post hoc analysis (p<0.01) to retain the most contrasting patterns. Data were normalized relative to the internal standard (TG51:0) and then Pareto-scaled. Color in the heat map reflects the relative TG abundance level with red being higher, and blue lower, than the mean value (the color-coded scale is derived from the Z-score). (B) The sum of chromatographic peak intensities of TGs relative to the peak intensity of the internal standard (Total TGs/TG51:0), averaged across liver samples within each treatment group (n=16/group, mean ± SEM). Hepatic TG levels in low and high XN groups are significantly different from controls (p<0.0001).
3.4. XN and metabolites in plasma and liver
The plasma concentrations of XN in the low XN and high XN groups were 84.4 nM and 95.1 nM, respectively (Table 1). In the liver of the low XN group, the XN concentration was 0.477 nmol/g whereas XN concentration in the high XN group was 0.805 nmol/g of liver tissue. IX was detected in plasma at concentrations of 3.1 nM and 13.6 nM for the low XN and high XN groups, respectively. The concentrations of IX in liver were 0.279 nmol/g and 0.746 nmol/g of liver tissue in low XN and high XN groups, respectively (Table 1). 6-PN and 8-PN were not detected in plasma of XN-treated mice. However, small quantities of 8-PN (close to 0.01 nmol/g of liver tissue) were detected in both low XN and high XN groups. There were no detectable levels of 6-PN in liver. The total concentrations of plasma XN (plus IX) in low XN and high XN groups were 87.5 and 109 nM, respectively (Table 1). Total concentrations of XN plus metabolites in liver of low XN and high XN mice were 0.766 and 1.56 nmol/g of liver tissue, respectively (Table 1).
4. Discussion
We selected diet-induced obese (DIO) male C56BL/6J mice as a model to examine the effects of XN on markers of metabolic syndrome. Obesity was induced by a high-fat diet (60% kcal as fat) to mimic the nutritional conditions which lead to metabolic syndrome in humans. This DIO mouse model has been well-studied for evidence of dysfunction of glucose and lipid metabolism and for changes in body weight gain as compared to the same strain of mice fed a normal or standard diet [25, 26]. Male C57BL/6J mice started on a high-fat diet (60% kcal as fat) at 4 weeks gained a mean of 22.4 g after 20 weeks whereas the mice on the control diet (10% kcal as fat) gained a mean of 12.8 g [26]. This difference in body weight gain between mice on a normal diet and mice on a high-fat diet was likewise observed as early as 12 weeks of age when feeding in both groups started at 5 weeks of age [25]. Statistically significant differences were also observed between mice fed a normal diet and high-fat fed mice in various biochemical parameters of carbohydrate and lipid metabolism such as glucose (10.2 mM in the high-fat diet group versus 7.7 mM in the normal diet group [25]), triglycerides, cholesterol, insulin, and leptin in serum [25, 26]. In our study, the DIO mice gained approximately 19 g over 12 weeks, at which point their fasting plasma glucose levels were 12.2 mM, indicating that the high-fat fed mice in our study developed obesity and elevated fasting plasma glucose levels.
In the present study, dietary XN produced a reduction in body weight gain induced by a high-fat diet. Based on a previous study [27], we used a food intake of 2.7 g for a 30-g mouse to select the amounts of XN as 0.033% and 0.066% for the low XN and high XN diets, respectively. At 0.033% and 0.066% in the diet, the dose of XN was estimated to be 30 mg/kg body weight/day and 60 mg/kg body weight/day, respectively. By using allometric interspecies scaling of dose [28], the tested mouse doses of 30 and 60 mg/kg/day correspond to human doses of 175 mg and 350 mg/day for a 70-kg person. Our doses of 30 and 60 mg XN/kg body weight/day were lower than those used in a study conducted by Miyata and co-workers [29], in which male C57BL/6J mice were given high-fat diets containing 0.2% XN and 0.4% XN, corresponding to 150 and 300 mg/kg/day, respectively. In Miyata’s study [29], 6-week old male mice were first fed a high-fat diet resembling our test diets (60% energy from fat) for 10 weeks and then fed the XN-supplemented diets for 50 days. With this protocol, the mice fed high doses of XN exhibited lower weight gain than those fed the high-fat diet alone. In our study, XN given at much lower doses during the 12-week feeding study mitigated the increase of body weight induced by the high-fat diet. A study published by Nozawa [14] demonstrated the anti-obesity effect of XN using genetically-altered (KK-Ay) mice which become obese even when fed a basal diet instead of a high-fat diet. In Nozawa’s study [14], the mice were fed basal diets supplemented with 0.3% or 1% XN, concentrations that are 10 times or greater compared to our XN diets. A feeding study with Wistar rats [11] showed the anti-obesity effect of a XN-rich hop extract (as 1% of the high-fat diet), but the hop extract also contained other flavonoids, so the actual effect of XN itself on obesity cannot be evaluated with certainty in this study.
We measured total XN (XN plus metabolites) concentrations in mouse plasma of 87.5 nM and 109 nM for the low XN and high XN treatment groups, respectively. In mouse liver, the concentrations of XN in low XN and high-XN groups were 0.766 nmol/g and 1.56 nmol/g, respectively (Table 1). These hepatic XN concentrations are proportionally similar to the levels reported by Zamzow and co-workers [27] who detected XN concentrations in liver of approximately 0.3 nmol/g in young mice and 0.5 nmol/g in old mice given an estimated daily dose of 20 mg/kg body weight as part of a normal diet. In our study, IX was detected in both mouse plasma and liver of mice treated with XN but the contribution of this XN metabolite to the anti-obesity effect of XN treatment is not known at this time.
Stevens and co-workers [30] conducted a single-dose human pharmacokinetics study with XN in men (n = 24) and women (n = 24) to determine dose-concentration relationships. Subjects received a single oral dose of 20, 60, or 180 mg XN. The maximum plasma XN concentrations (Cmax) increased linearly with dose and reached 0.37 μM following a single human dose of 180 mg (see Table 5 in [30]). Comparison of the human plasma Cmax levels with the (fasting) mouse plasma levels (Table 1) indicates that it is possible to reach human plasma levels resulting from a daily dose of 175 mg XN/day (equivalent to 30 mg/kg in mice) that exerts beneficial effects on several markers of metabolic syndrome in DIO mice given the equivalent dose. In animal studies, these dose levels have not resulted in adverse or toxic effects. Gerhauser and colleagues studied the safety of XN administered orally to Sprague-Dawley rats for four weeks [31]. At a daily dose of 1000 mg XN/kg body weight, equivalent to 11.7 g of XN per day for a 70 kg person, these authors reported a reduction of liver weight, but found no macroscopic or histopathologic changes of the liver, kidney, lung, heart, stomach and spleen. In a two-generation study of the teratogenic effects of XN, Gerhauser’s group found no differences in development of SD rats treated lifelong with a daily dose of 100 mg/kg body weight. Hellerbrand and coworkers [7] investigated the safety profile of XN in female BALB/c mice fed XN via a standard diet for three weeks. The daily dose was 1000 mg/kg bodyweight. They did not detect toxic effects of XN on liver, kidney, colon, lung, heart, spleen, and thymus by histopathological examination.
Several mechanisms have been proposed for the anti-obesity effect of XN in rat models and they include: inhibition of fat absorption from the intestine [11], reduced activity of enzymes involved in fatty acid synthesis [11], enhanced fatty acid oxidation resulting from mild mitochondrial uncoupling [6], and reduced SREBP-1c mRNA expression [11]. The anti-obesity effect of XN in mouse models might be explained by reduced SREBP1c mRNA expression [12], by decreased de novo lipogenesis through the inhibition of SREBP maturation in the mouse liver [29], and by reduced mRNA expression of hepatic genes involved in fatty acid synthesis and gluconeogenesis [14]. The decreased production of mature SREBPs was also demonstrated in cell culture studies using Huh-7 (a human hepatoma cell line) cells [29]. In 3T3-L1 adipocytes, XN treatment inhibited differentiation of preadipocytes and induced apoptosis in mature adipocytes [32]. Enhanced fatty acid oxidation and decreased de novo lipogenesis would explain our observation that XN treatment resulted in an overall reduction of hepatic TG content without altering the qualitative composition of TGs (Fig. 5).
Obesity is associated with systemic inflammation in which inflammatory cells are increased systemically and in adipose tissue of obese subjects [33]. There is an increased production of inflammatory cytokines not only in adipose tissue but also in liver, brain, pancreas and muscle tissue. These cytokines include TNF-α, IL-6, IL-1β, and MCP-1. MCP-1 increases the infiltration of macrophages into adipose tissues [34]. In our study, we showed for the first time that dietary XN caused a significant decrease in plasma IL-6 and plasma MCP-1 levels in C57BL/6J mice fed a high fat diet. This effect of XN has important implications in the prevention of inflammation associated with obesity and insulin resistance. Previously, the anti-inflammatory effect of XN has been demonstrated in a mouse model of warm ischemia/reperfusion injury of the liver [35], in western-type diet-fed apolipoprotein-E-deficient (ApoE−/−) mice [12], and in various cell culture models of inflammation [36–38]. XN may exert its anti-inflammatory activity through Nrf2-ARE signaling and up- regulation of downstream heme oxygenase-1 [38], or by suppressing LPS-induced TLR4 activation partly by interfering with LPS binding to the TLR4 co-receptor MD-2 in LPS-treated human monocytic THP-1 cells [36].
Inflammatory cytokines are implicated in insulin resistance in obesity by inhibiting insulin signaling [39]. An increase in the circulating levels of MCP-1 contributes to insulin resistance [40]. Obese diabetics are known to have higher levels of insulin compared with lean individuals. IL-6, secreted by adipose tissues, is elevated in plasma with increasing body fat content and also contributes to insulin resistance [41]. Hyperinsulinemia ensues in obesity and type 2 diabetes as a compensatory mechanism to remove glucose from the circulation [33]. In our study, we found that feeding high XN caused a significant 42% reduction in plasma insulin levels in mice on a high fat diet (Fig. 4A), suggesting that XN may be beneficial in preventing insulin resistance in obese subjects with type 2 diabetes.
Leptin, a hormone produced mainly in white adipose tissue, is an important factor linking obesity and metabolic syndrome. Leptin is a key regulator of body weight and food intake or energy balance by suppressing appetite [42, 43] and increased circulating leptin is a marker of leptin resistance and a common feature in obesity. Previous studies have shown that plasma leptin levels are elevated in C57BL/6J mice fed high fat diets [44]. These mice fed a normal diet had serum leptin levels below 3 ng/ml but those fed a high fat diet had serum levels of 6 ng/ml or higher. In our study, plasma leptin levels of mice fed a high fat diet were over 16 ng/ml as determined by ELISA (Fig. 4B) demonstrating a state of leptin resistance. Feeding low and high XN produced a significant decrease in plasma leptin levels. This finding is consistent with other studies showing that leptin resistance is independently associated with systemic inflammation and insulin resistance [45].
Hyperlipidemia, characterized by elevated levels of TGs in plasma or serum is one of the metabolic disorders induced by feeding a high-fat diet to C57BL/6J mice [46–48]. In our study, XN significantly decreased plasma TG levels in diet-induced obese C57BL/6J mice. The reduction in plasma TG levels by XN is consistent with the results obtained in rats fed a high-fat diet in which hepatic fatty acid synthesis is decreased possibly through the reduction of hepatic SREBP1c mRNA [11]. SREBPs stimulate the expression of enzymes involved in fatty acid biosynthesis and play a major role in the pathophysiology of metabolic syndrome [49]. Suppression of SREBP activation is one of the mechanisms by which XN reduces the de novo synthesis of fatty acid and cholesterol in diet-induced obese mice [29]. The reduction of liver TGs by XN further supports the anti-obesity effects of this compound which is in agreement with the findings of Kirkwood et al. [6] and Miyata et al. [29].
Elevated levels of circulating LDL-C are a well-known major risk factor of atherosclerotic cardiovascular disease which is the largest cause of premature death and morbidity worldwide [50]. LDL-C plays a major role in the atherogenic process in that its oxidation in macrophages in the arterial wall leads to inflammation and formation of foam cells eventually resulting in atherosclerotic plaque [51]. Reduction of LDL-C levels by cholesterol-lowering HMG-CoA reductase inhibitors (statins) has been shown to significantly reduce cardiovascular events in clinical trials [52]. The development of PCSK9 antibody inhibitors, e.g., evolocumab and alirocumab, offers a new therapeutic approach in the management of familial hypercholesterolemia. PCSK9 is a secreted protease that interacts with the epidermal growth factor-like repeat A (EGF-A) domain of low density lipoprotein receptor (LDL-R) to induce LDL-R degradation [53, 54]. Therefore, high levels of plasma PCSK9 will reduce the abundance of LDL-R on the surface of hepatocytes resulting in elevated levels of LDL-C. LDL-R clears LDL-C from the circulation by facilitating cellular uptake of LDL-C. Serum PCSK9 levels are directly associated with serum cholesterol levels [53, 54] and administration of PCSK9 neutralizing antibodies or agents that block or inhibit PCSK9 lower serum cholesterol levels [55, 56]. In our study, we found that XN is highly effective in lowering LDL-C and PCSK9 levels in plasma of mice fed a high-fat diet. In a previous study [29], XN (at dietary levels of 0.2% or 0.4%) was found to significantly decrease the levels of total cholesterol and LDL-C in C57BL/6J mice fed a high-fat diet for 50 days. In our study, XN at a much lower level (0.066% XN) in the diet produced a significant reduction in the plasma levels of total cholesterol and LDL-C in DIO mice, conceivably by lowering the production of PCSK9. Taken together, we have discovered PCSK9 as a new molecular target of XN which provides an explanation for its beneficial effects on LDL-C.
5. Conclusions
In conclusion, dietary XN reduced body weight gain and ameliorated hyperglycemia, dyslipidemia, insulin resistance and leptin resistance in DIO mice. Furthermore, dietary XN decreased the plasma levels of inflammatory cytokines which may contribute to the mitigation of obesity and insulin resistance in these mice. XN lowers total cholesterol and LDL-C in DIO mice which is consistent with the XN-related reduction of plasma PCSK9. Given the concentrations of XN found in beer (around 0.2 mg/L), it is unlikely that XN taken in the form of beer will provide beneficial effects in metabolic syndrome. By allometric interspecies scaling of dose, the two efficacious mouse doses administered daily in this study translate to human doses of 175 mg/day and 350 mg/day for a 70-kg person.
Acknowledgments
This work was supported by the Linus Pauling Institute, the OSU College of Pharmacy, Hopsteiner, Inc., New York, the OSU Foundation Buhler-Wang Research Fund, and the National Institutes of Health (Grants S10RR027878 and R01AT009168).
Abbreviations
- ARE
antioxidant response element
- DIO
diet-induced obesity
- IL-6
Interleukin-6
- IX
isoxanthohumol
- LDL-C
low-density lipoprotein cholesterol
- LDL-R
low-density lipoprotein receptor
- MCP-1
Monocyte Chemoattractant Protein-1
- Nrf2
nuclear erythroid 2-related factor 2
- PCSK9
Proprotein Convertase Subtilisin Kexin 9
- 6-PN
6-prenylnaringenin
- 8-PN
8-prenylnaringenin
- SREBP
sterol regulatory element-binding protein
- TG
triglyceride
- XN
xanthohumol
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Grundy SM, Brewer HB, Jr, Cleeman JI, Smith SC, Jr, Lenfant C. Circulation. 2004;109:433–438. doi: 10.1161/01.CIR.0000111245.75752.C6. [DOI] [PubMed] [Google Scholar]
- 2.Ervin RB. National Health Statistics Reports. US Center for Disease Control and Prevention; 2009. Prevalence of Metabolic Syndrome Among Adults 20 Years of Age and Over, by Sex, Age, Race and Ethnicity, and Body Mass Index: United States, 2003–2006. [PubMed] [Google Scholar]
- 3.Trogdon JG, Finkelstein EA, Feagan CW, Cohen JW. Obesity (Silver Spring) 2011 doi: 10.1038/oby.2011.169. [DOI] [PubMed] [Google Scholar]
- 4.Hollander JM, Mechanick JI. J Am Diet Assoc. 2008;108:495–509. doi: 10.1016/j.jada.2007.12.007. [DOI] [PubMed] [Google Scholar]
- 5.Legette LL, Luna AY, Reed RL, Miranda CL, Bobe G, Proteau RR, Stevens JF. Phytochemistry. 2013;91:236–241. doi: 10.1016/j.phytochem.2012.04.018. [DOI] [PubMed] [Google Scholar]
- 6.Kirkwood JS, Legette LL, Miranda CL, Jiang Y, Stevens JF. J Biol Chem. 2013;288:19000–19013. doi: 10.1074/jbc.M112.445452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dorn C, Bataille F, Gaebele E, Heilmann J, Hellerbrand C. Food Chem Toxicol. 2010;48:1890–1897. doi: 10.1016/j.fct.2010.04.030. [DOI] [PubMed] [Google Scholar]
- 8.Dorn C, Hellerbrand C. Z Gastroenterol. 2012;50:P1–09. Congress abstract. [Google Scholar]
- 9.Dorn C, Kraus B, Motyl M, Weiss TS, Gehrig M, Scholmerich J, Heilmann J, Hellerbrand C. Mol Nutr Food Res. 2010;54(Suppl 2):S205–213. doi: 10.1002/mnfr.200900314. [DOI] [PubMed] [Google Scholar]
- 10.Weiskirchen R, Mahli A, Weiskirchen S, Hellerbrand C. Front Physiol. 2015;6:140. doi: 10.3389/fphys.2015.00140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yui K, Kiyofuji A, Osada K. J Oleo Sci. 2014;63:159–168. doi: 10.5650/jos.ess13136. [DOI] [PubMed] [Google Scholar]
- 12.Doddapattar P, Radovic B, Patankar JV, Obrowsky S, Jandl K, Nusshold C, Kolb D, Vujic N, Doshi L, Chandak PG, Goeritzer M, Ahammer H, Hoefler G, Sattler W, Kratky D. Mol Nutr Food Res. 2013;57:1718–1728. doi: 10.1002/mnfr.201200794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liu M, Hansen PE, Wang G, Qiu L, Dong J, Yin H, Qian Z, Yang M, Miao J. Molecules. 2015;20:754–779. doi: 10.3390/molecules20010754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nozawa H. Biochem Biophys Res Commun. 2005;336:754–761. doi: 10.1016/j.bbrc.2005.08.159. [DOI] [PubMed] [Google Scholar]
- 15.Wang B, Chandrasekera PC, Pippin JJ. Curr Diabetes Rev. 2014;10:131–145. doi: 10.2174/1573399810666140508121012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fellmann L, Nascimento AR, Tibirica E, Bousquet P. Pharmacol Ther. 2013;137:331–340. doi: 10.1016/j.pharmthera.2012.11.004. [DOI] [PubMed] [Google Scholar]
- 17.Collins S, Martin TL, Surwit RS, Robidoux J. Physiol Behav. 2004;81:243–248. doi: 10.1016/j.physbeh.2004.02.006. [DOI] [PubMed] [Google Scholar]
- 18.Winzell MS, Ahren B. Diabetes. 2004;53(Suppl 3):S215–219. doi: 10.2337/diabetes.53.suppl_3.s215. [DOI] [PubMed] [Google Scholar]
- 19.Cariou B, Le May C, Costet P. Atherosclerosis. 2011;216:258–265. doi: 10.1016/j.atherosclerosis.2011.04.018. [DOI] [PubMed] [Google Scholar]
- 20.Shantha G, Robinson JG. Clin Pharmacol Ther. 2016;99:59–71. doi: 10.1002/cpt.281. [DOI] [PubMed] [Google Scholar]
- 21.Friedewald WT, Levy RI, Fredrickson DS. Clin Chem. 1972;18:499–502. [PubMed] [Google Scholar]
- 22.Legette LL, Reed RL, Murty L, Maier CS, Stevens JF. Spectroscopy (Springf) 2013;39:s18–s25. [PMC free article] [PubMed] [Google Scholar]
- 23.Choi J, Leonard SW, Kasper K, McDougall M, Stevens JF, Tanguay RL, Traber MG. J Lipid Res. 2015;56:1182–1190. doi: 10.1194/jlr.M058941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Xia J, Sinelnikov IV, Han B, Wishart DS. Nucleic Acids Res. 2015;43:W251–257. doi: 10.1093/nar/gkv380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hoffler U, Hobbie K, Wilson R, Bai R, Rahman A, Malarkey D, Travlos G, Ghanayem BI. Endocrine. 2009;36:311–325. doi: 10.1007/s12020-009-9224-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gallou-Kabani C, Vige A, Gross MS, Rabes JP, Boileau C, Larue-Achagiotis C, Tome D, Jais JP, Junien C. Obesity (Silver Spring) 2007;15:1996–2005. doi: 10.1038/oby.2007.238. [DOI] [PubMed] [Google Scholar]
- 27.Zamzow DR, Elias V, Legette LL, Choi J, Stevens JF, Magnusson KR. Behav Brain Res. 2014;275:1–10. doi: 10.1016/j.bbr.2014.08.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.FDA. Guidance for Industry: Estimating the Maximum Safe Starting Dose in Initial Clinical Trials for Therapeutics in Adult Healthy Volunteers. Food and Drug Administration; 2005. http://wwwfdagov/ohrms/dockets/98fr/02d-0492-gdl0002.pdf. [Google Scholar]
- 29.Miyata S, Inoue J, Shimizu M, Sato R. J Biol Chem. 2015;290:20565–20579. doi: 10.1074/jbc.M115.656975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Legette L, Karnpracha C, Reed RL, Choi J, Bobe G, Christensen JM, Proteau RR, Purnell J, Stevens JF. Mol Nutr Food Research. 2014;58:248–255. doi: 10.1002/mnfr.201300333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hussong R, Frank N, Knauft J, Ittrich C, Owen R, Becker H, Gerhauser C. Mol Nutr Food Res. 2005;49:861–867. doi: 10.1002/mnfr.200500089. [DOI] [PubMed] [Google Scholar]
- 32.Yang JY, Della-Fera MA, Rayalam S, Baile CA. Obesity (Silver Spring) 2008;16:1232–1238. doi: 10.1038/oby.2008.66. [DOI] [PubMed] [Google Scholar]
- 33.Gregor MF, Hotamisligil GS. Annu Rev Immunol. 2011;29:415–445. doi: 10.1146/annurev-immunol-031210-101322. [DOI] [PubMed] [Google Scholar]
- 34.Kanda H, Tateya S, Tamori Y, Kotani K, Hiasa K, Kitazawa R, Kitazawa S, Miyachi H, Maeda S, Egashira K, Kasuga M. J Clin Invest. 2006;116:1494–1505. doi: 10.1172/JCI26498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dorn C, Massinger S, Wuzik A, Heilmann J, Hellerbrand C. Exp Mol Pathol. 2013;94:10–16. doi: 10.1016/j.yexmp.2012.05.003. [DOI] [PubMed] [Google Scholar]
- 36.Peluso MR, Miranda CL, Hobbs DJ, Proteau RR, Stevens JF. Planta Med. 2010;76:1536–1543. doi: 10.1055/s-0029-1241013. [DOI] [PubMed] [Google Scholar]
- 37.Lupinacci E, Meijerink J, Vincken JP, Gabriele B, Gruppen H, Witkamp RF. J Agric Food Chem. 2009;57:7274–7281. doi: 10.1021/jf901244k. [DOI] [PubMed] [Google Scholar]
- 38.Lee IS, Lim J, Gal J, Kang JC, Kim HJ, Kang BY, Choi HJ. Neurochem Int. 2011;58:153–160. doi: 10.1016/j.neuint.2010.11.008. [DOI] [PubMed] [Google Scholar]
- 39.McArdle MA, Finucane OM, Connaughton RM, McMorrow AM, Roche HM. Front Endocrinol (Lausanne) 2013;4:52. doi: 10.3389/fendo.2013.00052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tateya S, Tamori Y, Kawaguchi T, Kanda H, Kasuga M. Endocrinology. 2010;151:971–979. doi: 10.1210/en.2009-0926. [DOI] [PubMed] [Google Scholar]
- 41.Hoene M, Weigert C. Obes Rev. 2008;9:20–29. doi: 10.1111/j.1467-789X.2007.00410.x. [DOI] [PubMed] [Google Scholar]
- 42.Park HK, Ahima RS. Metabolism. 2015;64:24–34. doi: 10.1016/j.metabol.2014.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Schwartz MW, Woods SC, Porte D, Jr, Seeley RJ, Baskin DG. Nature. 2000;404:661–671. doi: 10.1038/35007534. [DOI] [PubMed] [Google Scholar]
- 44.Munzberg H, Flier JS, Bjorbaek C. Endocrinology. 2004;145:4880–4889. doi: 10.1210/en.2004-0726. [DOI] [PubMed] [Google Scholar]
- 45.Martin SS, Qasim A, Reilly MP. J Am Coll Cardiol. 2008;52:1201–1210. doi: 10.1016/j.jacc.2008.05.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Liu X, Xu J, Xue Y, Gao Z, Li Z, Leng K, Wang J, Xue C, Wang Y. Food Funct. 2015;6:3428–3436. doi: 10.1039/c5fo00602c. [DOI] [PubMed] [Google Scholar]
- 47.Justo ML, Claro C, Zeyda M, Stulnig TM, Herrera MD, Rodriguez-Rodriguez R. Eur J Nutr. 2015 doi: 10.1007/s00394-015-1015-x. [DOI] [PubMed] [Google Scholar]
- 48.Kuo YH, Lin CH, Shih CC. J Agric Food Chem. 2015;63:2479–2489. doi: 10.1021/acs.jafc.5b00073. [DOI] [PubMed] [Google Scholar]
- 49.Soyal SM, Nofziger C, Dossena S, Paulmichl M, Patsch W. Trends Pharmacol Sci. 2015;36:406–416. doi: 10.1016/j.tips.2015.04.010. [DOI] [PubMed] [Google Scholar]
- 50.Ellulu MS, Patimah I, Khaza’ai H, Rahmat A, Abed Y, Ali F. Inflammopharmacology. 2016;24:1–10. doi: 10.1007/s10787-015-0255-y. [DOI] [PubMed] [Google Scholar]
- 51.Brown MS, Goldstein JL. Science. 1986;232:34–47. doi: 10.1126/science.3513311. [DOI] [PubMed] [Google Scholar]
- 52.C. Cholesterol Treatment Trialists. Fulcher J, O’Connell R, Voysey M, Emberson J, Blackwell L, Mihaylova B, Simes J, Collins R, Kirby A, Colhoun H, Braunwald E, La Rosa J, Pedersen TR, Tonkin A, Davis B, Sleight P, Franzosi MG, Baigent C, Keech A. Lancet. 2015;385:1397–1405. [Google Scholar]
- 53.Chan DC, Lambert G, Barrett PH, Rye KA, Ooi EM, Watts GF. Clin Chem. 2009;55:2049–2052. doi: 10.1373/clinchem.2009.128645. [DOI] [PubMed] [Google Scholar]
- 54.Lambert G, Ancellin N, Charlton F, Comas D, Pilot J, Keech A, Patel S, Sullivan DR, Cohn JS, Rye KA, Barter PJ. Clin Chem. 2008;54:1038–1045. doi: 10.1373/clinchem.2007.099747. [DOI] [PubMed] [Google Scholar]
- 55.Navarese EP, Kolodziejczak M, Schulze V, Gurbel PA, Tantry U, Lin Y, Brockmeyer M, Kandzari DE, Kubica JM, D’Agostino RB, Sr, Kubica J, Volpe M, Agewall S, Kereiakes DJ, Kelm M. Ann Intern Med. 2015;163:40–51. doi: 10.7326/M14-2957. [DOI] [PubMed] [Google Scholar]
- 56.Fitzgerald K, Frank-Kamenetsky M, Shulga-Morskaya S, Liebow A, Bettencourt BR, Sutherland JE, Hutabarat RM, Clausen VA, Karsten V, Cehelsky J, Nochur SV, Kotelianski V, Horton J, Mant T, Chiesa J, Ritter J, Munisamy M, Vaishnaw AK, Gollob JA, Simon A. Lancet. 2014;383:60–68. doi: 10.1016/S0140-6736(13)61914-5. [DOI] [PMC free article] [PubMed] [Google Scholar]