

Abstract
Introduction
The fixed-dose, long-acting bronchodilator combination of umeclidinium/vilanterol (UMEC/VI) has not previously been compared with a combination of a long-acting muscarinic antagonist and long-acting β2-agonist in patients with chronic obstructive pulmonary disease (COPD).
Methods
This 12-week, randomized, blinded, triple-dummy, parallel-group, non-inferiority study compared once-daily UMEC/VI 62.5/25 mcg with once-daily tiotropium (TIO) 18 mcg + indacaterol (IND) 150 mcg in patients with moderate-to-very-severe COPD. The primary endpoint was the trough forced expiratory volume in 1 s (FEV1) on day 85 (predefined non-inferiority margin −50 mL), and the secondary endpoint was the 0- to 6-h weighted mean (WM) FEV1 on day 84. Other efficacy endpoints [including rescue medication use, the Transition Dyspnea Index (TDI) focal score, and the St. George’s Respiratory Questionnaire (SGRQ) score] and safety endpoints [adverse events (AEs), vital signs, and COPD exacerbations] were also assessed.
Results
Trough FEV1 improvements were comparable between treatment groups [least squares (LS) mean changes from baseline to day 85: UMEC/VI 172 mL; TIO + IND 171 mL; treatment difference 1 mL; 95 % confidence interval (CI) −29 to 30 mL], demonstrating non-inferiority between UMEC/VI and TIO + IND. The treatments produced similar improvements in the trough FEV1 at other study visits and the 0- to 6-h WM FEV1 (LS mean changes at day 84: UMEC/VI 235 mL; TIO + IND 258 mL; treatment difference −23 mL; 95 % CI −54 to 8 mL). The results for patient-reported measures (rescue medication use, TDI focal score, and SGRQ score) were comparable; both treatments produced clinically meaningful improvements in TDI and SGRQ scores. The incidence of AEs and COPD exacerbations, and changes in vital signs were similar for the two treatments.
Conclusion
UMEC/VI and TIO + IND, given once daily, provided similar improvements in lung function and patient-reported outcomes over 12 weeks in patients with COPD, with comparable tolerability and safety profiles.
Trial numbers
ClinicalTrials.gov study ID NCT02257385; GSK study no. 116961.
Electronic supplementary material
The online version of this article (doi:10.1007/s40268-016-0131-2) contains supplementary material, which is available to authorized users.
Key Points
| The combination of umeclidinium/vilanterol was considered non-inferior to tiotropium + indacaterol in terms of the primary lung function endpoint (the trough forced expiratory volume in 1 s at day 85) on the basis of a predefined non-inferiority margin. |
| The fixed-dose umeclidinium/vilanterol combination and the free combination of tiotropium + indacaterol, given once daily, provided similar improvements in lung function and patient-reported outcomes over 12 weeks in patients with chronic obstructive pulmonary disease. |
| The treatments had similar tolerability and safety profiles. |
Introduction
Long-acting bronchodilators are central to the pharmacological management of chronic obstructive pulmonary disease (COPD) [1]. However, many patients remain symptomatic despite the availability of effective long-acting monotherapies. In a real-world study, Dransfield et al. [2] reported that patients receiving tiotropium (TIO), formoterol, or salmeterol as long-acting bronchodilator maintenance therapy continued to report dyspnea and high levels of supplemental rescue medication use, irrespective of the level of airflow limitation.
Dual long-acting bronchodilator therapy represents an alternative to long-acting bronchodilator monotherapy. The fixed combination of the long-acting muscarinic antagonist (LAMA) umeclidinium (UMEC) and the long-acting β2-agonist (LABA) vilanterol (VI), at a dose of 62.5/25 mcg once daily, is an approved maintenance treatment for COPD in the USA, Canada, the EU, and several other countries [3–5]. Dual bronchodilation with UMEC/VI has been shown to provide greater improvements in lung function than UMEC, VI, or TIO alone, with similar or greater improvements in measures of dyspnea, rescue medication use, and health-related quality of life (HRQoL) [6–8]. However, a direct comparison between UMEC/VI and another once-daily inhaled LAMA/LABA combination has not been performed.
This is the first study to compare UMEC/VI 62.5/25 mcg with a combination of a LAMA (TIO 18 mcg) and a LABA [indacaterol (IND) 150 mcg] in patients with moderate-to-very-severe COPD. TIO and IND are both approved as single therapies for the maintenance treatment of COPD [9–12]. TIO is a widely used and well-characterized LAMA bronchodilator for the treatment of COPD [1, 13], and once-daily IND at a dose of 150 mcg has been shown to provide greater improvements in lung function, symptoms, and HRQoL than placebo [14, 15]. IND 150 mcg is the approved dose in all countries apart from the USA, where the recommended dose is 75 mcg [11, 12].
Methods
Study Design
This was a multicenter, randomized, blinded, triple-dummy, parallel-group study (GSK study no. 116961; Clinicaltrials.gov study ID NCT02257385), conducted between October 2014 and May 2015 at 86 centers across Argentina, Chile, Estonia, France, Germany, Hungary, Italy, Peru, Poland, Romania, the Russian Federation, and Slovakia.
Patients meeting eligibility criteria at screening (visit 1) entered a 5- to 7-day run-in period prior to randomization at visit 2. The use of short-acting muscarinic antagonists, inhaled corticosteroids (ICS), and albuterol were permitted during the run-in period. Eligible patients were then randomized to one of two treatments (UMEC/VI or TIO + IND) over a 12-week period. Clinic visits took place on days 2, 14, 28, 56, 84, and 85, with a follow-up period of approximately 7 days. The total duration of the study was approximately 14 weeks.
The study was approved by a national, regional, or investigational center ethics committee/institutional review board in each country and was performed in accordance with the International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use Good Clinical Practice (ICH-GCP) guidelines [16], all applicable subject privacy requirements, and the ethical principles outlined in the Declaration of Helsinki, 2013 [17]. Written, informed consent was obtained from all patients who participated in the study.
Patients
Eligible patients were male or female; were ≥40 years of age; had an established clinical history of COPD (in accordance with the American Thoracic Society/European Respiratory Society criteria [18]); were current or former cigarette smokers with a history of cigarette smoking of ≥10 pack-years; had pre- and post-bronchodilator forced expiratory volume in 1 s (FEV1) values of ≤70 % predicted normal; had pre- and post-bronchodilator FEV1/forced vital capacity (FVC) ratios of <0.70; had a score of ≥2 on the modified Medical Research Council Dyspnea Scale; and had a corrected QT (QTc) interval (corrected for the heart rate, according to Fridericia’s formula) of <450 or <480 ms for patients with bundle branch block.
Patients were excluded from the study if they were of childbearing potential (unless they were practicing acceptable birth control methods); had a current diagnosis of asthma; had alpha-1 antitrypsin deficiency, an active lung infection (such as tuberculosis), lung cancer, or another clinically significant disease/abnormality; had an abnormal or significant electrocardiogram finding; had a history of allergy or hypersensitivity to any anticholinergic, muscarinic receptor antagonist, β2-agonist, lactose/milk protein, or magnesium stearate; had been hospitalized for COPD or pneumonia within 12 weeks prior to visit 1; had undergone lung volume reduction surgery within 12 months prior to visit 1; were receiving long-term oxygen therapy; had received any of the prohibited medications listed in Table S1 in the Electronic Supplementary Material; or were in the acute phase of a pulmonary rehabilitation program. The use of ICS/LABAs, phosphodiesterase-4 inhibitors, theophyllines, oral β2-agonists, LAMAs, LABAs, and LAMA/LABA combinations (other than those under study) was not allowed.
Patients were randomized to treatment only if they had not experienced COPD exacerbation between visits 1 and 2, and if they had not used any prohibited medication during the run-in period or at visit 2.
Treatments
Patients were randomized in accordance with a centralized randomization schedule, using a randomization code generated by a validated computerized system (RandAll Version NG, GSK). Patients were randomized using an interactive voice recognition system.
Patients meeting the eligibility criteria were randomized 1:1 to receive UMEC/VI 62.5/25 mcg once daily via an ELLIPTA™ dry powder inhaler + placebo once daily via a HandiHaler® + placebo once daily via a Breezhaler®, or TIO 18 mcg once daily via a HandiHaler® + IND 150 mcg once daily via a Breezhaler® + placebo once daily via an ELLIPTA™.
Patients were instructed to take one dose each morning from the ELLIPTA™, the HandiHaler®, and the Breezhaler®. All patients had albuterol provided for as-needed use.
A triple-dummy study design was used to achieve study blinding, whereby patients were given three inhalers containing either an active drug or placebo. All patients and investigators were blinded to the assigned treatment during the study. However, exact physical placebo matches for the TIO and IND capsules and for the IND blister packs were not available, although they were closely matched in color. As the study was of a parallel-group design, the capsule type was consistent for each patient for the duration of the study. Both the TIO and IND blister packages containing the capsules were covered with opaque over-labels. The HandiHaler® and Breezhaler® devices were covered with labels to mask identifying markings. The study medication was prepared and provided to the patient by study personnel who were independent of safety and efficacy procedures.
Outcomes and Assessments
Efficacy Endpoints
The primary objective of the study was to determine whether the efficacy of UMEC/VI was non-inferior to that of TIO + IND as assessed by the trough FEV1 at day 85 (the primary endpoint was the trough FEV1 at day 85, defined as the mean of the FEV1 values obtained at 23 and 24 h after dosing on day 84). The secondary endpoint of the study was the weighted mean (WM) FEV1 over 0–6 h postdose at day 84, calculated from the predose FEV1 values (obtained 30 and 5 min before dosing) and the postdose FEV1 measurements at 1, 3, and 6 h.
Other efficacy endpoints included the trough FEV1 at days 28 and 56; 0- to 6-h WM FEV1 at day 1; 0- to 6-h serial FEV1 at days 1 and 84; serial and trough FVC; mean number of puffs of rescue medication per day over days 1–84; percentage of rescue-free days over days 1–84; Transition Dyspnea Index (TDI) focal score and proportion of TDI responders at days 28, 56, and 84 (defined as patients with a TDI focal score of ≥1 unit [19]); and St. George’s Respiratory Questionnaire (SGRQ) score and proportion of responders at days 28, 56, and 84 (defined as having a total SGRQ score ≥4 units below baseline [20]).
TDI and SGRQ assessments were performed prior to spirometry testing. Patients used a paper diary to record their rescue medication use in the previous 24 h.
Safety assessments took into account the incidence rates of adverse events (AEs), vital signs, and COPD exacerbations. AEs and COPD exacerbations (defined as worsening of COPD symptoms, requiring use of additional treatment other than the prescribed bronchodilator, such as antibiotics, systemic corticosteroids, and/or emergency treatment or hospitalization) were assessed throughout the study. Vital signs (the pulse rate and systolic and diastolic blood pressure) were assessed at screening and either at day 85 or at an early withdrawal visit.
Statistical Analysis
The sample size calculations used a one-sided 2.5 % significance level and an estimated residual standard deviation for the trough FEV1 of 220 mL, based on a mixed-model repeated measures (MMRM) analysis of previous studies in patients with COPD (GSK; data on file). A study with 760 evaluable patients for the primary analysis would have 88 % power to detect non-inferiority of UMEC/VI to TIO + IND on the trough FEV1, when the margin of non-inferiority was −50 mL. If the lower limit of the 95 % confidence interval (CI) fell above −50 mL but below 0, then UMEC/VI could be considered statistically non-inferior to TIO + IND.
The intent-to-treat (ITT) population comprised all patients randomized to treatment who received at least one dose of randomized study medication, while the per-protocol (PP) population comprised all patients in the ITT population who were not identified as full protocol deviators.
The PP population was used for treatment comparisons of the primary endpoint of the trough FEV1 at day 85, as use of the ITT population (and inclusion of data from protocol deviators) would tend to bias the results toward equivalence. This could potentially make an inferior treatment appear non-inferior [21]. The approach used in the present study has also been used in other non-inferiority COPD studies [22, 23] and was intended to maximize true differences between treatments. The ITT population was used for comparisons of the secondary and other efficacy endpoints, as well as safety endpoints. This approach was employed because the study was not designed to detect non-inferiority on the other endpoints; therefore, the ITT population was used because it followed the randomization procedure [24, 25].
It was estimated that approximately 10 % of patients providing an assessment at day 84 would be excluded from the PP population, giving 844 evaluable patients for the ITT analysis, with >90 % power. With allowance for a 12 % withdrawal rate, it was planned that a total of 960 patients would be randomized.
The efficacy parameters were assessed using MMRM analysis, with the exception of the proportions of TDI and SGRQ responders (assessed using a logistic regression model), and the percentage of rescue-free days over weeks 1–12 (assessed using a non-parametric analysis).
Results
Patients
Of the 1190 patients enrolled, 967 were randomly assigned to treatment (distribution by country is shown in Table S2 in the Electronic Supplementary Material). The ITT population comprised 961 patients, as six patients were randomized in error and did not receive study medication. The majority of these patients (96 %) were included in the PP population, which comprised 918 patients (UMEC/VI, n = 463; TIO + IND, n = 455). In total, 917 patients (95 %) completed the study. The most common reason for study withdrawal was AEs, which accounted for a similar proportion of patients withdrawing from each treatment group [UMEC/VI, n = 12 (2 %); TIO + IND, n = 8 (2 %)] (Fig. 1).
Fig. 1.
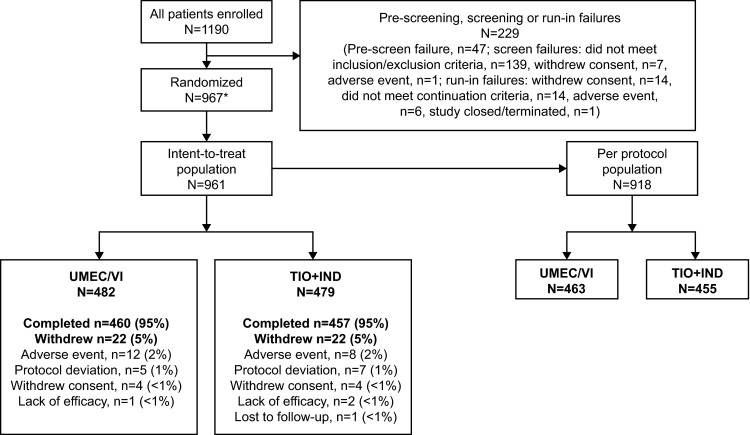
Summary of patient disposition. Asterisk six patients were randomized in error and are included in the screening failure and run-in failure rates. IND indacaterol, TIO tiotropium, UMEC umeclidinium, VI vilanterol
Patients in the ITT population had moderate-to-very-severe COPD [Global initiative for chronic Obstructive Lung Disease (GOLD) stage 2–4, primarily GOLD grades B and D], and the majority of patients were male and white (Table 1). The baseline demographics and clinical characteristics of the ITT and PP populations were similar.
Table 1.
Patient demographics and baseline characteristics (intent-to-treat population)
| UMEC/VI, N = 482 |
TIO + IND, N = 479 |
|
|---|---|---|
| Mean age [years (SD)] | 64 (7.75) | 64 (8.44) |
| Male [n (%)] | 355 (74) | 341 (71) |
| Race [n (%)] | ||
| White | 453 (94) | 450 (94) |
| American Indian or Alaska native | 24 (5) | 27 (6) |
| Asian | 5 (1) | 2 (<1) |
| Current smoker at screening [n (%)] | 198 (41) | 218 (46) |
| Mean smoking pack-years (SD)a | 43.17 (22.71) | 42.30 (23.12) |
| ICS use at screening [n (%)] | 269 (56) | 243 (51) |
| Post-albuterol FEV1 [L; mean (SD)]b | 1.369 (0.46) | 1.357 (0.48) |
| Post-albuterol FEV1/FVC [mean (SD)]b | 45.70 (11.09) | 45.55 (11.12) |
| Reversible with albuterol [n (%)]c,d | 126 (26) | 125 (26) |
| % Reversibility with albuterol [mean (SD)]c,d | 12.2 (13.06) | 12.5 (12.87) |
| GOLD category [n (%)]b | ||
| II | 209 (44) | 200 (42) |
| III | 222 (46) | 220 (46) |
| IV | 48 (10) | 57 (12) |
| GOLD grade according to CAT [n (%)]e | ||
| A: low risk, less symptoms | 26 (5) | 19 (4) |
| B: low risk, more symptoms | 139 (29) | 146 (31) |
| C: high risk, less symptoms | 22 (5) | 25 (5) |
| D: high risk, more symptoms | 293 (61) | 287 (60) |
| Mean BDI focal score (SD) | 5.9 (1.89) | 6.0 (1.67) |
BDI Baseline Dyspnea Index, CAT COPD assessment test, COPD chronic obstructive pulmonary disease, FEV 1 forced expiratory volume in 1 s, FVC forced vital capacity, GOLD Global initiative for chronic Obstructive Lung Disease, ICS inhaled corticosteroid, IND indacaterol, SD standard deviation, TIO tiotropium, UMEC umeclidinium, VI vilanterol
aSmoking pack-years = (number of cigarettes smoked per day/20) × number of years of smoking
bUMEC/VI, n = 479; TIO + IND, n = 477
cReversibility was defined as an increase in the FEV1 of ≥12 % and ≥200 mL following administration of albuterol
dUMEC/VI, n = 477; TIO + IND, n = 477
eUMEC/VI, n = 480; TIO + IND, n = 477
Efficacy
Lung Function
In the two treatment groups, similar improvements from baseline were observed for the primary endpoint of the trough FEV1 at day 85, with a difference of +1 mL for UMEC/VI versus TIO + IND (95 % CI −29 to 30 mL; PP population) (Table 2). The treatment difference was above the prespecified non-inferiority margin of −50 mL, demonstrating non-inferiority of the two treatments. Similar improvements from baseline in the trough FEV1 were observed at all time points with UMEC/VI and TIO + IND (Fig. 2). Similar results for the trough FEV1 at day 85 were observed in the ITT population [treatment difference for UMEC/VI versus TIO + IND: 7 mL (95 % CI −22 to 35 mL)].
Table 2.
Summary of lung function endpoints
| UMEC/VI, PP population, N = 463 |
TIO + IND, PP population, N = 455 |
||
|---|---|---|---|
| Trough FEV1 [mL; PP population] | |||
| Day 85 | n = 392 | n = 392 | |
| LS mean change from baseline (SE) | 172 (11) | 171 (11) | |
| Difference (95 % CI) | 1 (−29 to 30) | ||
| 0- to 6-h WM FEV1 [L; ITT population] | |||
| Day 1 | n = 478 | n = 471 | |
| LS mean change from baseline (SE) | 184 (7) | 185 (7) | |
| Difference (95 % CI) | −1 (−20 to 18) | ||
| Day 84 | n = 455 | n = 452 | |
| LS mean change from baseline (SE) | 235 (11) | 258 (11) | |
| Difference (95 % CI) | −23 (−54 to 8) | ||
| Trough FVC [mL; ITT population] | |||
| Day 85 | n = 455 | n = 455 | |
| LS mean change from baseline (SE) | 239 (18) | 220 (18) | |
| Difference (95 % CI) | 20 (−29 to 68) | ||
CI confidence interval, FEV 1 forced expiratory volume in 1 s, FVC forced vital capacity, IND indacaterol, ITT intent-to-treat, LS least squares, PP per-protocol, SE standard error, TIO tiotropium, UMEC umeclidinium, VI vilanterol, WM weighted mean
Fig. 2.

Least squares (LS) mean [95 % confidence interval (CI)] changes from baseline in the trough forced expiratory volume in 1 s (FEV1) over days 2–85 in the per-protocol (PP) population. IND indacaterol, TIO tiotropium, UMEC umeclidinium, VI vilanterol
The mean changes from baseline in the 0- to 6-h WM FEV1 at day 84 (the secondary endpoint) also showed similar improvements with UMEC/VI versus TIO + IND treatment [day 84 difference −23 mL (95 % CI −54 to 8 mL); ITT population] (Table 2).
The mean serial FEV1 values showed a consistent pattern of improvement versus baseline in both treatment groups at days 1 and 84 (see Fig. S1a, b in the Electronic Supplementary Material).
The improvement from baseline in the mean trough FVC values was comparable in the UMEC/VI and TIO + IND treatment groups at all time points from day 2 through to day 85 (Fig. 3; Table 2). Serial FVC measurements also showed a consistent pattern of improvement versus baseline in both treatment groups at days 1 and 84 (see Fig. S2a, b in the Electronic Supplementary Material).
Fig. 3.

Least squares (LS) mean [95 % confidence interval (CI)] changes from baseline in the trough forced vital capacity (FVC) in the intent-to-treat (ITT) population. IND indacaterol, TIO tiotropium, UMEC umeclidinium, VI vilanterol
Rescue Medication Use
The least squares (LS) mean changes from baseline in rescue medication use were similar in the UMEC/VI and TIO + IND treatment groups over weeks 1–12 [difference 0.1 puffs (95 % CI −0.1 to 0.3)] (Table 3), and no difference was detected in the percentages of rescue-free days over weeks 1–12 in the two treatment groups [median difference 0 days (95 % CI 0 to 2.6); Hodges Lehmann estimate and Wilcoxon rank sum test].
Table 3.
Summary of other efficacy endpoints (intent-to-treat population)
| UMEC/VI, N = 482 |
TIO + IND, N = 479 |
||
|---|---|---|---|
| Rescue use | |||
| Days 1–84 | n = 474 | n = 472 | |
| LS mean change in mean puffs per day from baseline (SE) | −1.5 (0.06) | −1.4 (0.06) | |
| Difference (95 % CI) | 0.1 (−0.1 to 0.3) | ||
| TDI focal score | |||
| Day 28 | n = 470 | n = 461 | |
| LS mean change from baseline (SE) | 2.07 (0.12) | 2.02 (0.12) | |
| Difference (95 % CI) | 0.05 (−0.29 to 0.39) | ||
| Day 56 | n = 462 | n = 459 | |
| LS mean change from baseline (SE) | 2.08 (0.12) | 2.14 (0.12) | |
| Difference (95 % CI) | −0.05 (−0.38 to 0.28) | ||
| Day 84 | n = 459 | n = 455 | |
| LS mean change from baseline (SE) | 2.32 (0.13) | 2.62 (0.13) | |
| Difference (95 % CI) | −0.30 (−0.65 to 0.05) | ||
| Proportion of responders according to TDI scorea | |||
| Day 28 | n = 478 | n = 471 | |
| Responders [n (%)] | 302 (63) | 295 (63) | |
| Odds ratio (95 % CI) | 1.01 (0.77 to 1.33) | ||
| Day 56 | n = 477 | n = 474 | |
| Responders [n (%)] | 287 (60) | 289 (61) | |
| Odds ratio (95 % CI) | 0.95 (0.73 to 1.25) | ||
| Day 84 | n = 478 | n = 476 | |
| Responders [n (%)] | 298 (62) | 309 (65) | |
| Odds ratio (95 % CI) | 0.88 (0.67 to 1.16) | ||
| SGRQ total score | |||
| Day 28 | n = 461 | n = 456 | |
| LS mean change from baseline (SE) | −4.41 (0.50) | −3.28 (0.50) | |
| Difference (95 % CI) | −1.12 (−2.50 to 0.26) | ||
| Day 56 | n = 448 | n = 458 | |
| LS mean change from baseline (SE) | −5.26 (0.54) | −4.89 (0.53) | |
| Difference (95 % CI) | −0.37 (−1.86 to 1.12) | ||
| Day 84 | n = 450 | n = 453 | |
| LS mean change from baseline (SE) | −4.93 (0.58) | −5.01 (0.57) | |
| Difference (95 % CI) | 0.08 (−1.52 to 1.67) | ||
| Proportion of responders according to SGRQ total scoreb | |||
| Day 28 | n = 470 | n = 466 | |
| Responders [n (%)] | 210 (45) | 185 (40) | |
| Odds ratio (95 % CI) | 1.19 (0.92 to 1.55) | ||
| Day 56 | n = 464 | n = 472 | |
| Responders [n (%)] | 229 (49) | 224 (47) | |
| Odds ratio (95 % CI) | 1.04 (0.80 to 1.35) | ||
| Day 84 | n = 471 | n = 476 | |
| Responders [n (%)] | 223 (47) | 216 (45) | |
| Odds ratio (95 % CI) | 1.05 (0.81 to 1.37) | ||
CI confidence interval, IND indacaterol, LS least squares, SE standard error, SGRQ St. George’s Respiratory Questionnaire, TDI Transition Dyspnea Index, TIO tiotropium, UMEC umeclidinium, VI vilanterol
aTDI focal score of ≥1 unit
bProportion of responders is defined by a difference of −4 units or lower from the visit score to the baseline score
Symptoms and Health-Related Quality of Life
The mean TDI focal scores and the proportions of responders based on the TDI score were similar with the two treatments throughout the study (Table 3). The LS mean TDI scores at day 84 were 2.32 and 2.62 in patients receiving UMEC/VI and TIO + IND, respectively (Table 3). Additionally, UMEC/VI and TIO + IND resulted in comparable improvements from baseline in SGRQ scores and the proportion of responders (having a total SGRQ score ≥4 units below baseline) at all assessments (Table 3). UMEC/VI and TIO + IND resulted in LS mean changes from the baseline SGRQ scores of −4.93 and −5.01 points, respectively (Table 3).
Safety
The overall incidence of on-treatment AEs during the study was generally similar in the two treatment groups, with headache and nasopharyngitis reported most frequently. There were low incidence rates of non-fatal serious AEs and AEs leading to study withdrawal in both the UMEC/VI and TIO + IND treatment groups (Table 4).
Table 4.
Summary of adverse events (AEs) and chronic obstructive pulmonary disease (COPD) exacerbations (intent-to-treat population)
| UMEC/VI, N = 482 |
TIO + IND, N = 479 |
|
|---|---|---|
| On-treatment AE [n (%)] | 202 (42) | 186 (39) |
| On-treatment drug-related AE [n (%)] | 30 (6) | 37 (8) |
| Any AE leading to study withdrawal/discontinuation of medication [n (%)] | 12 (2) | 8 (2) |
| On-treatment non-fatal serious AE [n (%)] | 17 (4) | 15 (3) |
| On-treatment fatal serious AE [n (%)] | 4 (<1) | 1 (<1) |
| AEs reported in ≥3 % of patients [n (%)] | ||
| Headache | 36 (7) | 28 (6) |
| Nasopharyngitis | 32 (7) | 27 (6) |
| Cough | 16 (3) | 17 (4) |
| Patients experiencing COPD exacerbation [n (%)] | 48 (10) | 49 (10) |
| Cardiovascular events of special interest [n (%)]a | 11 (2) | 9 (2) |
AEs with an onset during the follow-up period were considered on-treatment and were assigned to the treatment previously received
IND indacaterol, TIO tiotropium, UMEC umeclidinium, VI vilanterol
aStandardized Medical Dictionary for Regulatory Activities (MedDRA) terms included cardiac arrhythmias, cardiac failure, ischemic heart disease, central nervous system hemorrhages, and cerebrovascular conditions
Five deaths occurred on-treatment during the study (UMEC/VI: n = 4, <1 %; TIO + IND: n = 1, <1 %), including one fatal event of pneumonia and respiratory failure in the UMEC/VI treatment group (onset at day 77) and one fatal event of pneumonia in the TIO + IND treatment group (onset at day 72). The three other deaths in the UMEC/VI treatment group were related to individual events of ventricular fibrillation (onset at day 76), circulatory collapse (onset at day 7), and cardiac arrest (onset at day 41).
The three deaths relating to ventricular fibrillation, cardiac arrest, and pneumonia in the UMEC/VI treatment group occurred ≥14 days after the last dose of study medication, as recorded in the electronic case report forms. The death relating to cardiac arrest was reported as a sudden event, occurring with exertion 41 days after the first dose. The cardiac arrest was reported by the investigator as being related to the study medication. No other deaths were identified by the study investigators as being related to the study medication.
The incidence rates of COPD exacerbations were low and the same in both groups (10 %; Table 4). The mean changes in vital signs were small and similar in the two treatments groups (data not shown).
Discussion
Previous studies have shown that UMEC/VI provides greater improvements in lung function than UMEC, VI, or TIO alone, with similar or greater improvements in measures of dyspnea, rescue medication use, and HRQoL [6–8]. This is the first clinical trial to evaluate UMEC/VI in comparison with another combination of a LAMA and LABA—specifically, co-administration of TIO and IND, both of which are well-characterized, once-daily, long-acting bronchodilators indicated for COPD [13–15]. As both treatments comprised a LAMA and LABA, it was anticipated that the combinations would have comparable beneficial effects on the trough FEV1. The study was therefore designed to test for non-inferiority for the primary efficacy measure of the trough FEV1.
UMEC/VI was shown to be non-inferior to TIO + IND by the prespecified non-inferiority margin (−50 mL) for the trough FEV1 at day 85, with similar magnitudes of improvement from baseline being shown with the two treatments. The LS mean treatment differences between the two treatment groups were also similarly small at all other evaluations of the trough FEV1. The trough FEV1 results, obtained at the end of the once-daily dosing interval, were supported by similar findings in the two treatment groups for evaluations of the initial bronchodilator effect, as measured by serial FEV1 assessments obtained over the first 6 h postdose and the 0- to 6-h WM FEV1, indicating similar peak effects and onsets of action.
The improvements in lung function measures were paralleled by comparable improvements in patient-reported measures of dyspnea, HRQoL, and rescue medication use. For the assessment of dyspnea, both treatments produced a TDI score that exceeded 1 unit [the minimum clinically important difference (MCID) [19] at all assessments]. Improvements in HRQoL were indicated by a total SGRQ score ≥4 units below baseline (the established MCID for the SGRQ [20]) from day 28 onwards for UMEC/VI, and from day 56 onward for TIO + IND. These findings support the results of other randomized controlled trials, in which UMEC/VI treatment led to clinically meaningful improvements in TDI and SGRQ scores from baseline [6–8].
TIO and IND were chosen as open dual comparators in the present study as they were the only two once-daily LAMA and LABA medications approved for COPD at the time of the study initiation. TIO is also a well-characterized LAMA bronchodilator for the treatment of COPD [1, 13], and IND 150 mcg once daily has been shown to improve lung function, symptoms, and HRQoL in comparison with placebo [14, 15]. Although TIO administered via a HandiHaler® is approved at a single dose of 18 mcg once daily, IND is approved at multiple doses; an IND dose of 150 mcg was selected in the present study as this is the approved dose in all countries apart from the USA, where the recommended dose is 75 mcg [11, 12]. As additional LABA and LAMA monotherapies and LAMA/LABA fixed-dose combinations are now available for the treatment of COPD [1], further head-to-head studies will be required to evaluate the comparative safety and efficacy of other combinations.
Previous studies have shown that UMEC/VI have tolerability and safety profiles similar to those of UMEC, VI, and TIO alone, and placebo [6–8]. In this study, both UMEC/VI and TIO + IND were generally well tolerated, with AEs consistent with the patient population under study. The incidence of dry mouth was low (1 % in each treatment group), and cough was reported in 3 and 4 % of UMEC/VI and TIO + IND patients, respectively. The incidence rates of cardiovascular events of special interest—which included AE terms for cardiac arrhythmias, cardiac failure, ischemic heart disease, and hemorrhages—were low and the same in the two groups (2 % of patients). The incidence rates of death were similar in the two groups (<1 %), although there was a numerical imbalance in the number of deaths reported in the UMEC/VI group (n = 4) compared with the TIO + IND group (n = 1). Overall, the nature of the events was consistent with the population under study. Interpretation of the imbalance is limited because of the small number of fatal events overall and the confounding of three events in the UMEC/VI group, for which the event date was reported as ≥14 days after the last dose of the study medication.
A limitation of this study was the imperfect blinding to the study medications, as exact physical matches for the TIO and IND capsules were not available. However, safeguards were in place to prevent the unblinding of study personnel, and study blinding coordinators independent of other clinical trial procedures were involved in the preparation and administration of treatment to patients.
Conclusion
This study demonstrated that treatment over 12 weeks with UMEC/VI 62.5/25 mcg once daily resulted in improvements in measures of lung function, symptoms, and health status similar to those achieved with the free combination of TIO 18 mcg + IND 150 mcg once daily in patients with moderate-to-very-severe COPD, with comparable safety profiles.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Acknowledgments
The authors acknowledge Chiara Gussetti for her invaluable assistance with the conduct of the study.
Compliance with Ethical Standards
Ethics Approval and Consent to Participate
All procedures in this study were performed in accordance with the ethical standards of the responsible institutional and national committees on human experimentation and with the Helsinki Declaration of 1964, as revised in 2013 [17]. Informed consent was obtained from all patients included in the study.
Authorship
All authors participated in the data analysis and interpretation. Amy Newlands, Chris Kalberg, Dmitry Galkin, and Dianne O’Dell were involved in the conception and design of the study. All authors contributed to the writing and reviewing of the manuscript. All named authors meet the International Committee of Medical Journal Editors (ICMJE) criteria for authorship of this manuscript, take responsibility for the integrity of the work as a whole, and have given final approval of the version to be published.
Funding
The work presented here—including the conduct of the study and the data analysis and interpretation—was supported by GSK (GSK study no. 116961; ClinicalTrials.gov study ID NCT02257385).
Editorial Assistance
Editorial assistance in the preparation of the manuscript (in the form of writing assistance, assembling tables and figures, collating author comments, grammatical editing, and referencing) was provided by Natasha Thomas, PhD, of Fishawack Indicia Ltd (Knutsford, UK). Support for this assistance was funded by GSK.
Conflict of interest
Chris Kalberg, Dmitry Galkin, Amy Newlands, and William A. Fahy are employees of GSK and hold stocks/shares in GSK. At the time of the study, Dianne O’Dell was an employee of GSK and held stocks/shares in GSK; at the time of publication, she was an employee of Pearl Therapeutics, Inc. (Durham, NC, USA).
References
- 1.Global Initiative for Chronic Obstructive Lung Disease [GOLD]. Global strategy for the diagnosis, management and prevention of chronic obstructive pulmonary disease, 2015. Available at http://www.goldcopd.com. Accessed on 03 Dec 2015.
- 2.Dransfield MT, Bailey W, Crater G, Emmett A, O’Dell DM, Yawn B. Disease severity and symptoms among patients receiving monotherapy for COPD. Prim Care Respir J. 2011;20:46–53. doi: 10.4104/pcrj.2010.00059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Blair HA, Deeks ED. Umeclidinium/vilanterol: a review of its use as maintenance therapy in adults with chronic obstructive pulmonary disease. Drugs. 2015;75:61–74. doi: 10.1007/s40265-014-0326-1. [DOI] [PubMed] [Google Scholar]
- 4.GSK. Anoro™ ELLIPTA™ prescribing information, 2014. Available at https://www.gsksource.com/gskprm/htdocs/documents/ANORO-ELLIPTA-PI-MG.PDF. Accessed on 03 Dec 2015.
- 5.GSK. Anoro™ ELLIPTA™ summary of product characteristics, 2014. Available at http://www.medicines.org.uk/emc/medicine/28949#INDICATIONS. Accessed on 03 Dec 2015.
- 6.Donohue JF, Maleki-Yazdi MR, Kilbride S, Mehta R, Kalberg C, Church A. Efficacy and safety of once-daily umeclidinium/vilanterol 62.5/25 mcg in COPD. Respir Med. 2013;107:1538–1546. doi: 10.1016/j.rmed.2013.06.001. [DOI] [PubMed] [Google Scholar]
- 7.Decramer M, Anzueto A, Kerwin E, et al. Efficacy and safety of umeclidinium plus vilanterol versus tiotropium, vilanterol, or umeclidinium monotherapies over 24 weeks in patients with chronic obstructive pulmonary disease: results from two multicentre, blinded, randomised controlled trials. Lancet Respir Med. 2014;2:472–486. doi: 10.1016/S2213-2600(14)70065-7. [DOI] [PubMed] [Google Scholar]
- 8.Maleki-Yazdi MR, Kaelin T, Richard N, Zvarich M, Church A. Efficacy and safety of umeclidinium/vilanterol 62.5/25 mcg and tiotropium 18 mcg in chronic obstructive pulmonary disease: results of a 24-week, randomized, controlled trial. Respir Med. 2014;108:1752–1760. doi: 10.1016/j.rmed.2014.10.002. [DOI] [PubMed] [Google Scholar]
- 9.Boehringer Ingelheim. Spiriva® HandiHaler® prescribing information, 2014. Available at http://docs.boehringer-ingelheim.com/Prescribing%20Information/PIs/Spiriva/Spiriva.pdf. Accessed on 03 Dec 2015.
- 10.Boehringer Ingelheim. Spiriva® HandiHaler® summary of product characteristics, 2015. Available at https://www.medicines.org.uk/emc/medicine/10039. Accessed on 03 Dec 2015.
- 11.Novartis. Arcapta™ Neohaler™ prescribing information, 2011. Available at https://www.pharma.us.novartis.com/product/pi/pdf/arcapta.pdf. Accessed on 03 Dec 2015.
- 12.Novartis. Onbrez® Breezhaler® summary of product characteristics, 2014. Available at https://www.medicines.org.uk/EMC/medicine/23260/SPC/Onbrez+Breezhaler+150+and+300+microgram+inhalation+powder,+hard+capsules/. Accessed on 03 Dec 2015.
- 13.Tashkin DP, Celli B, Senn S, et al. A 4-year trial of tiotropium in chronic obstructive pulmonary disease. N Engl J Med. 2008;359:1543–1554. doi: 10.1056/NEJMoa0805800. [DOI] [PubMed] [Google Scholar]
- 14.Donohue JF, Fogarty C, Lötvall J, et al. Once-daily bronchodilators for chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2010;182:155–162. doi: 10.1164/rccm.200910-1500OC. [DOI] [PubMed] [Google Scholar]
- 15.Feldman G, Siler T, Prasad N, et al. Efficacy and safety of indacaterol 150 μg once-daily in COPD: a double-blind, randomised, 12-week study. BMC Pulm Med. 2010;10:11. doi: 10.1186/1471-2466-10-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use [ICH]. ICH harmonised tripartite guideline: guideline for good clinical practice E6(1), 1996 (cited 2013 May). Available at http://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Efficacy/E6_R1/Step4/E6_R1__Guideline.pdf. Accessed on 03 Dec 2015.
- 17.World Medical Association [WMA]. WMA Declaration of Helsinki—ethical principles for medical research involving human subjects, 2013 [cited 2013 May]. Available at http://www.wma.net/en/30publications/10policies/b3/index.html. Accessed on 03 Dec 2015.
- 18.Celli BR. MacNee W; ATS/ERS Task Force. Standards for the diagnosis and treatment of patients with COPD: a summary of the ATS/ERS position paper. Eur Respir J. 2004;23:932–946. doi: 10.1183/09031936.04.00014304. [DOI] [PubMed] [Google Scholar]
- 19.Mahler DA, Witek TJ., Jr The MCID of the Transition Dyspnea Index is a total score of one unit. COPD. 2005;2:99–103. doi: 10.1081/COPD-200050666. [DOI] [PubMed] [Google Scholar]
- 20.Jones PW. St. George’s Respiratory Questionnaire: MCID. COPD. 2005;2:75–79. doi: 10.1081/COPD-200050513. [DOI] [PubMed] [Google Scholar]
- 21.Snapinn SM. Noninferiority trials. Curr Control Trials Cardiovasc Med. 2000;1:19–21. doi: 10.1186/CVM-1-1-019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chapman KR, Beeh KM, Beier J, et al. A blinded evaluation of the efficacy and safety of glycopyrronium, a once-daily long-acting muscarinic antagonist, versus tiotropium, in patients with COPD: the GLOW5 study. BMC Pulm Med. 2014;14:4. doi: 10.1186/1471-2466-14-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vogelmeier C, Ramos-Barbon D, Jack D, et al. Indacaterol provides 24-hour bronchodilation in COPD: a placebo-controlled blinded comparison with tiotropium. Respir Res. 2010;11:135. doi: 10.1186/1465-9921-11-135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lewis JA, Machin D. Intention to treat—who should use ITT? Br J Cancer. 1993;68:647–650. doi: 10.1038/bjc.1993.402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use [ICH]. ICH harmonised tripartite guideline: statistical principles for clinical trials E9, 1998. Available at http://www.ich.org/products/guidelines/efficacy/efficacy-single/article/statistical-principles-for-clinical-trials.html. Accessed on 03 Dec 2015.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.