

Summary
Live cell imaging is a valuable technique that allows the characterization of the dynamic processes of the HIV-1 life-cycle. Here, we present a method of production and imaging of dual-labeled HIV viral particles that allows the visualization of two events. Varying release of the intravirion fluid phase marker reveals virion fusion and the loss of the integrity of HIV viral cores with the use of live wide-field fluorescent microscopy.
Keywords: Live cell imaging, HIV uncoating, HIV fusion, wide-field microscopy, HIV early-steps of infection, HIV viral core
1. Introduction
The two steps that define retroviral infections are the reverse transcription of the positive sense viral RNA genome into double stranded DNA, and the integration of this reverse transcribed DNA into the host genome. Our understanding of these earliest aspects of the HIV replication cycle remain incomplete even though there have been significant recent advances in our knowledge of the viral and cellular determinants in the pathway of infection. Interestingly, determinants in the viral capsid can have a large influence on events that happen in the nucleus including the pathway of nuclear import and integration site selection. During virion maturation, the capsid (CA) protein assembles into a conical shell that contains the viral genome known as the “viral core”. This structure is able to assemble through the interaction of CA monomers that form hexameric rings, which arrange themselves into a conical structure that shields and contains several viral proteins such as RT, NC, IN, PR, Vpr, the viral RNA as well as several host cell proteins such as Cyclophilin A.
During the progression of reverse transcription, the conical capsid is lost. The timing of the disassembly of the conical core structure, a process known as uncoating, remains controversial and appears to be dynamic. For example, multiple lines of evidence have revealed a relationship between reverse transcription and the disassembly of the viral core. However, details of the changes in the conical capsid structure are poorly defined relating to the spatial, temporal and mechanistic specifics of HIV-1 uncoating. Single particle imaging of HIV has revealed important details about the viral life cycle informing our understanding of trafficking and the step-progressive and sequential steps of uncoating from in situ imaging of fixed cells (1–6). However, an in vivo fluorescent assay needs to be developed to provide insights into the dynamics and kinetics of the uncoating process. Here, we present a detailed method for live cell imaging of HIV-1 allowing changes in the integrity of the conical capsid to be detected in real time. These changes in the conical capsid are considered to be the earliest steps in the process of uncoating.
It has been previously reported that Vpr remains attached to HIV-1 reverse transcribing complexes (RTC) after the uncoating process has taken place as revealed by the loss of readily detectable CA. (3, 4, 7). Based on these observations, HIV-1 particles can be tracked by fusing fluorescent proteins to Vpr, such as GFP, mCherry or tdTomato (3). Recently, a new system has been developed that labels HIV particles using a fluid phase marker. This is accomplished by the insertion of a fluorescent protein (GFP or mCherry) between the matrix (MA) and CA cleavage sites of Gag. This insertion allows the selected fluorophore to be released from Gag by proteolytic cleavage and trapped inside the virion upon budding (8). The fluid phase fluorescent protein is located both within the assembled core and in the virion space between the core and the viral membrane. Based on calculation of a volume estimate for a virion, only a minority of the fluid phase marker (eGFP or mCherry) will be located in the intact conical core. We have previously reported that the fluid phase marker can be retained within intact cores as shown by membrane-stripping ultracentrifugations and with a TRIM5 capture assay in the presence of MG132 (9). With the knowledge of these data it is possible to perform live cell time-lapse imaging using dual labeled HIV-1 particles. Therefore, by using this system during infection we anticipate two changes in the levels of intravirion fluid phase markers that co-localize with signal from tagged Vpr. The first loss of fluid phase marker will take place upon fusion, as has been previously reported (10). However, the subsequent complete loss of the fluid phase marker reveals changes in the integrity of the conical capsid structure allowing the fluorescent protein to escape the reverse transcribing complex, a change consistent with the initiation of uncoating. This system can be utilized to provide important new insights into the process of uncoating and the behavior of the HIV viral particles and the genome moving towards its ultimate goal of integrating into the target cell genome. Here, we present a method to produce and image the infection of HIV-iGFP + Cherry-Vpr or HIV-iCherry + GFP-Vpr viruses.
2. Materials
2.1 Cell culture materials
HEK-293T cells (ATCC). Keep in culture with DMEM supplemented with 10%FBS and 2mM L-Glutamine.
CHO-pgsA-745 cells (ATCC Catalog Number CRL-2242). Keep in culture with DMEM supplemented with 10%FBS, 2mM L-Glutamine and MEM-NEAA (see Note 1).
Dulbecco's Modified Eagle's Medium (DMEM).
Fetal Bovine Serum (FBS.
FluoroBrite DMEM Media (Life technologies).
L-Glutamine
MEM Non-Essential Amino Acids Solution (MEM-NEAA).
Polybrene (Hexadimethrine bromide).
10cm diameter culture Dishes.
0.45µm filters.
20ml Syringes.
Cryovials.
2.2 Virus production
PEI 1mg/ml in water.
HIV-Gag-iGFPΔEnv (AidsReagent) or HIV-Gag-iCherryΔEnv plasmids. These molecular clones of HIV (pNL4-3 derived) had GFP or Cherry inserted in gag. The fluorophores are coded between MA and CA. After HIV-1 protease cleavage of MA and CA the fluorophore is trapped inside the virion (8, 10).
The vector pCMV-VSV-G that codes for the G protein of Vesicular Stomatitis Virus. This protein is used to pseudotype the viral particles with an Envelope protein capable of infecting a wide range of cells through the interaction with LDL-Receptor therefore mimicking VSV tropism (11).
pGFPVpr/pCherryVpr - HIV-1 Vpr protein fusions can be used to track HIV-1 RTCs. These plasmids provide a fusion protein between either GFP or Cherry to Vpr (1, 3).
(OPTIONAL) pSPAX2 or pCMV-dR8.2 dVpr. 2nd or 3rd generation lentiviral packaging plasmid (gag-pol).
2.3 Wide-field Imaging components
The protocol below was optimized for a DeltaVision Wide-field microscope equipped with an EMCCD camera, a SSI light path and a 100× Olympus Lens. The system must be located within the appropriate bio-containment environment to maintain safe experimental conditions.
Delta T Culture Dish Controller (Bioptechs).
Delta TPG Culture Dishes 0.17mm thick (Bioptechs).
Lens heater system: Objective Heater Controller (Bioptechs)and an Objective Heater adapted to the diameter of the objective (Bioptechs).
CO2 system with BloodGas Mixture tank (5% CO2, 20% Oxygen).
Light-Duty Tissue Wipers.
(OPTIONAL) Oxyfluor (OXYRASE) at a 1:200 dilution supplemented with 15mM Sodium DL-latate.
(OPTIONAL) DRAQ5 for nuclear staining.
2.4 Data analysis software
Many data analysis software are available (Imaris, ImageJ/Fiji and IDL or python libraries for Crocker and Grier algorithms). The analysis described below was performed with the TrackMate Plugin for ImageJ according to the author’s description (12).
3. Methods
Carry out all procedures that involve cell lines at 37°C – 5% CO2 unless otherwise specified.
3.1 Dual labeled HIV production
Day -1 : Plate 1.5×106 HEK-293T cells into 10cm diameter cell culture plates.
-
Day 0: Transfect cells:
In a 1.5ml Eppendorf tube, mix:-
a)6µg of either HIV-Gag-iGFPΔEnv or HIV-Gag-iCherryΔEnv plasmid (for hybrid viruses containing WT gag with higher viral titer mix 3µg of pSPAX2 with 3µg of HIV-Gag-iGFPΔEnv/HIV-Gag-iCherryΔEnvplasmid).
-
b)4µg of pCMV-VSVg (to pseudotype the virions with G-protein from Vesicular Stomatitis virus).
-
c)1.5µg of pCMV-CherryVpr/pCMV-GFPVpr (Vpr accompanies the virus through the process of fusion, uncoating and reverse transcription (4)). If using iGFP, complement with pCMV-CherryVpr (or vice-versa for fluorescent protein tag).
-
d)1000 µl Serum free DMEM/OptiMEM.
-
e)Add 40µl of PEI.
-
f)Shake and incubate for 15minutes (Do not vortex since it might cause DNA shearing).
-
g)Apply the solution drop-by-drop to the 10cm HEK-293T cell culture plate.
-
a)
Day 1 (14–16h post-transfection): Change the Medium with pre-warmed DMEM.
- Day 2 (36 to 48h post-transfection):
-
h)Recover the supernatant with an appropriate sized syringe and filter through a 0.45um filter.
-
i)Aliquot into Cryovials and store at −80°C. Titer the viruses to determine the infectivity of your stocks. A virus with a titer higher than 1×104 TU/ml (transducing units /ml) is highly recommended for an efficient experimentation (see Note 2).
-
h)
3.2 Preparing CHO-pgsA-745 cells for imaging
Day -2: Plate 40 × 103 CHO-pgsA-745 cells on a DeltaT dish diluted with appropriate DMEM medium (DMEM supplemented with 10%FBS, NEAA, and L-glutamine) up to a volume of 1.5ml.
- Day 0:
- Change the DMEM medium with 1.5ml of pre-warmed 20% FBS- FluoroBrite DMEM Media (or phenol red-free DMEM) complemented with MEM-NEAA and L-glutamine.
- Add polybrene to a final concentration of 5µg/µl.
If nuclear staining is required for cell or nuclear identification (see Note 3).
If photobleaching and phototoxicity occur (see Note 4).
3.2 Setting-up the imaging system
Install an appropriate DeltaT plate holder into the microscope.
Turn on the microscope heating system several hours prior to the experiment (overnight if possible), allowing the system temperature to stabilize at 37°C. Turn on both the environmental chamber and the lens warmer. Both temperature regulating systems are necessary for temperature stability.
Carefully, apply a drop of imaging oil to the objective.
In case of the use of a non-sealed lid: minutes prior to the imaging process and without compromising the microscope integrity, humidify the area around the DeltaT holder in order to avoid evaporation to the fullest, optionally, place tissue wipers soaked with deionized water around the culture plate.
Set-up the blood gas mixture with a cover on top of the DeltaT dish holder in order to provide CO2 to stabilize the pH level. Start diffusing the system several minutes before imaging.
Place the DeltaT dish containing the cells in the appropriate DeltaT dish holder.
Cover the plate with the included lid to avoid evaporation to the fullest. Preferably, utilize a transparent lid with the DeltaT, so that manual eye focus is possible. In the case of the requirement of Differential Interference Contrast (DIC, white light), the use of a clear DeltaT lid is absolutely required (see Note 5).
(OPTIONAL) Focus at the bottom of the plate in order to optimize the acquisition to the whole cell. Configure the microscope so that the software recognizes the bottom of the plate as the set-up point.
Add multiple points to the visit list (see Note 6 for details on multiple visits points).
Utilize the ultimate focus system (or similar focus maintaining system) to maintain focus throughout the whole acquisition (see Note 7 for details).
Set-up the acquisition thickness according to the cell type (number of z’s), in order to image the whole cell from top to bottom. CHO-pgSA-745 cells usually have a thickness from 6 to 9µm. Set-up the Z-stacking spacing between 0.4µm and 0.6µm.
Set-up the time-lapse settings: the time between acquisitions per cell should depend on the study at hand. A quick process with multiple steps, several intensity changes, and a high number of particles and/or high movement of viral particles requires a higher rate of acquired frames/minute. A slow process with particles that have a low displacement rate, require a lower rate of acquired frames/minute. For example, the settings for a good measurement of HIV-VSVg membrane fusion would be 1 minute per z-stack acquisition for a total duration of 45–60mins.
Add viruses to the medium in order to have approximately 10–15 particles per field of view (empirical assessment is required; a typical volume for the production referred above can vary from 10 to 50 µl).
(OPTIONAL) – Check if the selected “visit points” maintained focus, if not, re-set the correct focus of the previously selected visit points. Maintaining focus over extended periods is necessary for a successful time-lapse experiment. If problems arise, return to step h and begin again.
3.3 Setting-up the system – Choosing the right amount of light to correctly detect dual labeled HIV-1 particles
Set-up transmission and exposure for every required wavelength so that the particles have a signal-to-noise ratio that will allow post-acquisition analyses. An increase on the amount of light transmission increases photo-bleaching while a lower transmission and higher exposure time might result in spatial disparities to the particles between acquisition wavelengths, causing a “tail-chase” effect (see Note 8). Optimize according to your system. The optimal amount of light is highly dependent on the particular microscope, therefore it should be empirically assessed according to the signal to noise ratio needed for detection and analysis.
Start acquisition.
3.4 Signal to noise ratio optimization
-
a)
Deconvolve the image files in order to increase signal-to-noise ratio.
-
b)
Z project the files using “Max Intensity” method (If 2D analysis is preferred).
-
c)
Assure yourself that you are able to perform the required analysis with the present signal-to-noise ratio with the software of your choice. If it is not possible to correctly analyze the expected phenotype with the current settings, a higher exposure time or transmission percentage settings might be required.
An example of acquired time-lapses and analysis of deconvolved Z projections is shown in Figure 1.
-
d)
Be sure that you are able to detect all the desired particles. For instance, while employing the iGFP/iCherry system there is a first drop of intensity at the moment of membrane fusion; if the level of light and exposure are not properly set, a perception of full loss of signal might occur despite the remaining of fluorophore inside a given number of particles.
(For an example of how to set-up the exposure/transmission values see Note 9)
-
e)
Repeat the setting-up process with cells that have not been infected with fluorescent viral particles. For this comparison with infected cells, the same transmission and exposure conditions have to be used. This control is crucial to ascertain that auto-fluorescence events are not being confused with real particle events.
Figure 1.
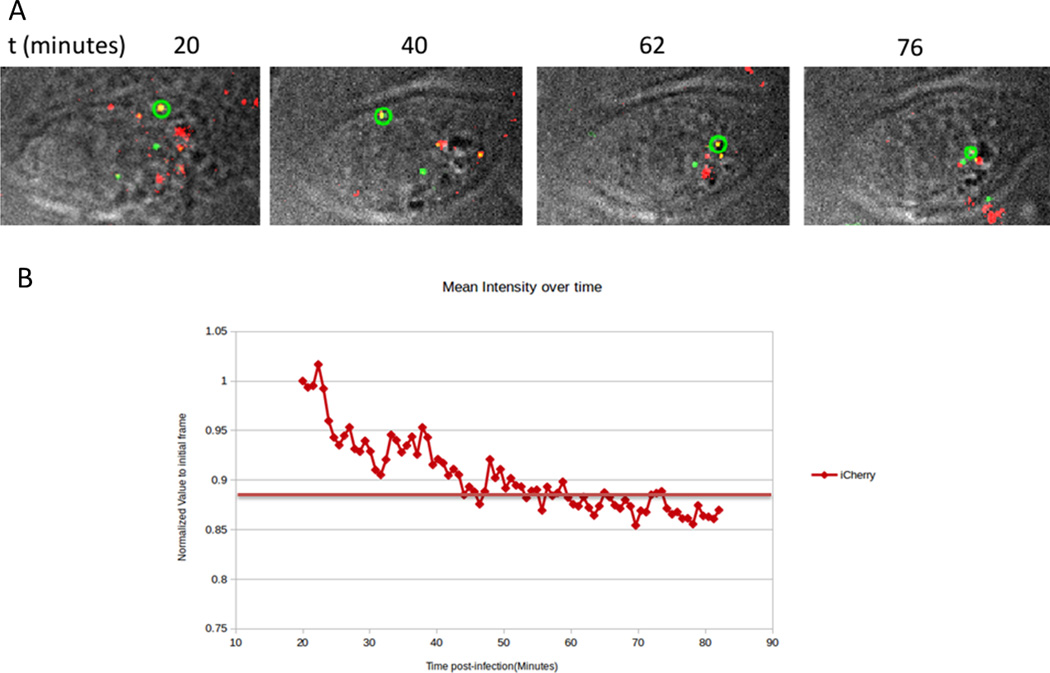
Time-lapse wide-field imaging of dual labeled iCherry/GFP Vpr HIV-1 particles. A. CHO-pgsA-745 cells were imaged for 60 minutes (starting 20 minutes post infection) the snapshots represent different time-points. GFP tagged Vpr (emission at 528nm) is represented in green; iCherry is represented in red (emission at 608nm) and DIC acquisition is represented in gray. The images were Deconvolved and Z-projected operating Softworks software from DeltaVision. The tracked particle of interest is encircled in green B. GFP-Vpr particles were tracked with TrackMate plugin for ImageJ and the mean intensities for the acquisition at 608nm (Cherry) of the particle of interest are represented. An initial fusion event is observed around 22minutes post-infection and a second drop of signal is observed which is believed to be uncoating. Horizontal bar represent the limit of detection to the given wavelength.
3.5 Setting-up the system – Reducing photobleaching and phototoxicity
Two of the biggest challenges to live cell imaging are photobleaching and phototoxicity. While performing live cell imaging experiments, there is a need of fast acquisition of multiple xy “snapshots” to produce a z-stack that encompasses the whole cell. For a single time point it is not uncommon to take 30 to 50 snapshots to a given cell. The amount of light that is applied to the sample might have the undesired effects of photobleaching and phototoxicity. Even though these processes are interconnected and the steps that are taken to prevent them are frequently the same, these are two distinct phenomena. Photobleaching is the process of the irreversible destruction of a fluorophore or dye upon light exposure, while phototoxicity is the process where continuous or repetitive imaging leads to a deregulation of the cell (or other biological processes sensitive to light), resulting in cell death or the incapacity to image the desired phenotype. For an extended review and further study, read Diaspro A. et al. (2006) and Tinevez J-Y et al. (2012) (13, 14). Here we will report several procedures that might be taken to reduce problems arising due to both of these phenomena.
The measures that are taken to reduce photobleaching are generally effective in reducing phototoxicity, and vice-versa, since both optimizations rely on both the reduction of the light that is applied to the sample and the reduction of toxic species in the sample.
a) Reducing the amount of acquisitions
While performing live imaging experiments, the experimenter has to adapt their settings to the specifics of the experiment at hand. For example, if the phenotype that is meant to be observed is in the order of minutes with particles that are relatively static in the field of view, it might be futile to acquire an image every 200ms. If a longer time interval between acquisitions is possible this will reduce the amount of light that is applied to the sample, therefore reducing both photobleaching and phototoxicity.
b) Binning
Binning is a process that sums the result of pixel signals at the level of the camera.
Augmenting the binning from 1×1 to 2×2, or higher, results in an increase of signal-to-noise ratio while sacrificing resolution. Image binning sums the values of pixel intensities (4 pixels in a 2×2 setting), while leaving the noise relatively the same as the value of one pixel, thereby increasing the signal-to-noise ratio (up to 8:1 in a 2×2 setting). This higher signal-to-noise ratio will allow us to decrease the amount of light that is applied to the sample and therefore protect the fluorophores.
c) Z-stack spacing
Yet another optimization that can diminish the total amount of light applied to a cell is to increase the spacing between z’s (z-stack spacing). Again, this will always depend on the experiment at hand. Regarding the experiment portrayed in this text, the major goal is to measure the timing that viral particles fuse or uncoat, therefore resolution in z is not an absolute priority. Throughout optimization experiments it was observed that a stacking of 0.55 µm would be enough for the study at hand. Again, reducing the spacing between z’s will reduce the number of acquisitions and exposures to the sample and consequently reduce the amount of light applied to the sample.
d) Oxygen scavengers
The repetitive and intense amount of light that is used to detect fluorescence, results in the production of oxygen free radicals. Production of these species results in a reduction of fluorescence duration and fluorescence intensity. Moreover, oxygen free radicals might also lead to a toxic environment to both the cell and the viral particles that often results in the loss of the biological function under study (phototoxicity). Oxyrase/Oxyfluor (www.oxyrase.com) is an enzyme system that removes dissolved oxygen from the cell environment, therefore protecting the fluorophores from photobleaching and cells from oxygen free radicals.
Footnotes
This cell line was derived from CHO-K1 cells. It was screened for deficiency in proteoglycan synthesis resulting in a lower level of particles that are lost due to deleterious endocytosis (15).
Add DRAQ5 if nuclear staining is desired. DRAQ5 is preferred over live-Hoescht since the presence of UV light results in significant photobleaching. Other dyes, might be added for cell tracing, such as the Lipophilic Tracers, Dil, DiO, DiD, DiA, and DiR.
In case of a high level of photobleaching and phototoxicity, add an oxygen scavenger, such as Oxyfluor at 100× dilution complemented with 15mM of Sodium DL-Lactate. This will decrease photobleaching and photoxicity.
If differential interference contrast (DIC) will be used during acquisition, for instance, to detect cell position, clear dishes are required. If only fluorescence based acquisition is required, the black version will allow a lower interference from the environmental light in the room.
In order to acquire as much data as possible, multiple visit points should be selected. The areas of acquisition should be as distant as possible to each other to avoid photobleaching/phototoxicity effects. The chosen amount of points should take into account the time it takes to acquire one cell in every wavelength and the full Z-stack and the desired frame-rate per cell.
Regarding the example in Figure 1, the Ultimate Focus settings that used were: move threshold = 300nm, number of iterations =2, performed in every time point = 2. Note that performing higher numbers of iterations in between time points will increase the acquisition time.
In order to minimize “tail chase” spatial effects to the acquired particles, set-up the microscope to acquire every wavelength first and then Z, possibly sacrificing some acquisition speed per cell.
In order to access the correct amount of light that is needed to detect HIV particles, an accumulation of fused viral cores has to be performed. It is possible to accumulate HIV-1 viral cores containing GFP/Cherry using cells expressing TRIM5 proteins in the presence of MG132 (9). With this system, a certain amount of fused particles will accumulate in TRIM5 bodies since a high number of intact viral cones contain GFP/mCherry proteins.
References
- 1.Fassati A, Goff SP. Characterization of intracellular reverse transcription complexes of human immunodeficiency virus type 1. J Virol. 2001;75:3626–3635. doi: 10.1128/JVI.75.8.3626-3635.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Goff SP. Intracellular trafficking of retroviral genomes during the early phase of infection: viral exploitation of cellular pathways. J Gene Med. 2001;3:517–528. doi: 10.1002/1521-2254(200111)3:6<517::AID-JGM234>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- 3.McDonald D, Vodicka MA, Lucero G, Svitkina TM, Borisy GG, Emerman M, Hope TJ. Visualization of the intracellular behavior of HIV in living cells. J Cell Biol. 2002;159:441–452. doi: 10.1083/jcb.200203150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hulme AE, Perez O, Hope TJ. Complementary assays reveal a relationship between HIV-1 uncoating and reverse transcription. Proc Natl Acad Sci. 2011;108:9975–9980. doi: 10.1073/pnas.1014522108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nermut MV, Fassati A. Structural analyses of purified human immunodeficiency virus type 1 intracellular reverse transcription complexes. J Virol. 2003;77:8196–8206. doi: 10.1128/JVI.77.15.8196-8206.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Warrilow D, Tachedjian G, Harrich D. Maturation of the HIV reverse transcription complex: putting the jigsaw together. Rev Med Virol. 2009;19:324–337. doi: 10.1002/rmv.627. [DOI] [PubMed] [Google Scholar]
- 7.Xu H, Franks T, Gibson G, Huber K, Rahm N, De Castillia CS, Luban J, Aiken C, Watkins S, Sluis-Cremer N, Ambrose Z. Evidence for biphasic uncoating during HIV-1 infection from a novel imaging assay. Retrovirology. 2013;10:70. doi: 10.1186/1742-4690-10-70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hübner W, Chen P, Portillo AD, Liu Y, Gordon RE, Chen BK. Sequence of Human Immunodeficiency Virus Type 1 (HIV-1) Gag Localization and Oligomerization Monitored with Live Confocal Imaging of a Replication-Competent, Fluorescently Tagged HIV-1. J Virol. 2007;81:12596–12607. doi: 10.1128/JVI.01088-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yu Z, Dobro MJ, Woodward CL, Levandovsky A, Danielson CM, Sandrin V, Shi J, Aiken C, Zandi R, Hope TJ, Jensen GJ. Unclosed HIV-1 Capsids Suggest a Curled Sheet Model of Assembly. J Mol Biol. 2013;425:112–123. doi: 10.1016/j.jmb.2012.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Padilla-Parra S, Marin M, Gahlaut N, Suter R, Kondo N, Melikyan GB. Fusion of Mature HIV-1 Particles Leads to Complete Release of a Gag-GFP-Based Content Marker and Raises the Intraviral pH. PLoS ONE. 2013;8:e71002. doi: 10.1371/journal.pone.0071002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Finkelshtein D, Werman A, Novick D, Barak S, Rubinstein M. LDL receptor and its family members serve as the cellular receptors for vesicular stomatitis virus. Proc Natl Acad Sci U S A. 2013;110:7306–7311. doi: 10.1073/pnas.1214441110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jaqaman K, Loerke D, Mettlen M, Kuwata H, Grinstein S, Schmid SL, Danuser G. Robust single-particle tracking in live-cell time-lapse sequences. Nat Methods. 2008;5:695–702. doi: 10.1038/nmeth.1237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Diaspro A, Chirico G, Usai C, Ramoino P, Dobrucki J. Photobleaching. In: Pawley JB, editor. Handb. Biol. Confocal Microsc. Springer US; 2006. pp. 690–702. [Google Scholar]
- 14.Tinevez J-Y, Dragavon J, Baba-Aissa L, Roux P, Perret E, Canivet A, Galy V, Shorte S. Chapter fifteen - A Quantitative Method for Measuring Phototoxicity of a Live Cell Imaging Microscope. In: Michael Conn P, editor. Methods Enzymol. Academic Press; 2012. pp. 291–309. [DOI] [PubMed] [Google Scholar]
- 15.Zhang Y, Hatziioannou T, Zang T, Braaten D, Luban J, Goff SP, Bieniasz PD. Envelope-Dependent, Cyclophilin-Independent Effects of Glycosaminoglycans on Human Immunodeficiency Virus Type 1 Attachment and Infection. J Virol. 2002;76:6332–6343. doi: 10.1128/JVI.76.12.6332-6343.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]