

INTRODUCTION
Magnetic resonance imaging (MRI) is a powerful non-invasive tool in the hands of clinicians to diagnose specific medical conditions affecting the structure of the brain that is usually displayed in an image format. MRI is also a valuable tool in the hands of clinical researchers trying to understand morphologic changes associated with disease. MRI takes advantage of the most abundant chemical in the brain, water, and its two hydrogen nuclei per water molecule. Because of its high concentration in the molar range, modern structural MR images are clear and crisp, showing exquisite structural detail down into the submillimeter range. Manufacturers of MR equipment have focused on optimizing image quality since the first images of a human were obtained (in this initial attempt, a finger) in 1977 by Sir Peter Mansfield and Andrew A. Maudsley (Mansfield, 1977; Mansfield and Maudsley, 1977). Many different MR methodologies were derived from the principles and lessons learned while developing structural MRI into the invaluable diagnostic tool it is today: perfusion imaging allows cerebral blood flow to be measured without the use of an external contrast agent; diffusion imaging allows measurement and visualization of water diffusion along the fibers connecting cortical brain regions; and functional MRI allows mapping cortical brain activity based on blood flow needs at a location of neural activity in response to a cognitive task.
Recent years have shown an explosion of research activity around these MR methodologies that all take advantage of different physical properties of the water molecules in the brain. As powerful as these methods are (and all of them as applied to brain changes associated with alcohol use disorders (AUD) are described in other chapters), they do not tell us about anything else than water in different structural and molecular environments and how these environments are potentially affected by the specific substance use behaviors of an individual with AUD, including, but not limited to, the chronic use of alcohol. What happens in and to the brain associated with these chronic behaviors should be evaluated with methods that go beyond their sequelae or influence on brain water.
Many brain chemicals are of critical importance in alcohol-related neuropathology and in fact are targeted by different measurement methodologies: molecular imaging methods such as positron emission tomography (PET) and single-photon emission computed tomography (SPECT) study various receptors present in the brain at very low concentrations (in the nanomolar range). In order to create a measurable signal, however, it is necessary to inject specifically synthesized short-lived radioactive tracers into the blood stream of the individual undergoing such studies.
Magnetic resonance spectroscopy (MRS) on the other hand does not require the use of an exogenous agent to yield a signal from chemicals in the brain that are present in millimolar concentrations and that have been shown to correlate to brain function and behavior. This methodology can be applied to yield images of metabolite distributions from throughout the brain or to yield quantitative information on different chemicals within a preselected brain region. MRS has the additional advantage that it can be performed on the very same equipment used for routine MRI, even in the same session as the MRI exam. Depending on what specific type of MRS equipment and experiment is used, up to 18 different chemicals can be measured from selectable regions of almost the entire brain by analyzing their respective signals within a known frequency range.
In this chapter, the first section describes neurochemicals, that is, hydrogen-bearing metabolites that are most commonly measured by in vivo MRS (therefore called proton MRS or 1H MRS), continues to summarize our knowledge on their general functions in the brain, and ends with a brief description of basic human 1H MRS methodology. The second section discusses brain 1H MRS findings, both cross-sectional and longitudinal, published in the biomedical research and clinical literature of individuals in treatment and of treatment-naïve individuals with AUD; the neuropathology of uncomplicated alcohol dependence is briefly summarized as a basis for interpreting the in vivo findings. The third section critically evaluates the influence of the most common substance use comorbidities in AUD – chronic cigarette smoking and illicit substance use – on regional brain metabolite levels. The fourth section describes two recent research directions: the search for neurobiologic correlates of relapse, and neuroimaging genetics, which investigates genetic influences on brain metabolite levels; the section finishes with ideas about future directions for 1H MRS in AUD, based on current research, knowledge gaps, and perceived insufficiencies in the reviewed literature.
NEUROCHEMICALS MEASURED BY 1H MRS AND BASIC MRS METHODS
MRS yields spectra with several “peaks” or “resonances” that represent signals from different neurochemicals spread out across a frequency range that neighbors the water resonance imaged in MRI. The frequency range is commonly expressed as parts per million (ppm), a frequency scale that is independent of the magnetic field strength of the MR scanner used for the spectral acquisition. The intensities of these peaks (i.e., their integrals or fitted curve areas) are directly proportional to the concentration of the signal-generating metabolites, and are modulated by some physical parameters of the measured metabolites (relaxation times) and experimental parameters. As such, “curve fitting” and proper calibration of the signal intensities allow quantitation of metabolite levels in absolute concentrations (mmol/kg or mmol/L), in absolute “institutional units,” or as levels relative to a reference compound in the same brain region presumed to be stable and unaffected by disease status (metabolite ratio). There are advantages and disadvantages associated with either quantitation approach. Only the first quantitation approach properly accounts for potential differences in relaxation times between individuals or groups of individuals, but these metabolite- and region-specific parameters are very time consuming to measure. Absolute quantitation also requires careful accounting for measured amounts of tissue, cerebrospinal fluid, and water signal in the spectroscopy volumes, as well as for differences of potential receiver and transmitter gains between individual measurements. Quantitation in “institutional units” usually does not correct for potential relaxation time differences, but calculates absolute regional metabolite concentrations relative to the strong water signal obtained from the same region. Calculating metabolite ratios is procedurally and computationally least demanding as it does not require the above-mentioned corrections to individual metabolite signals; however, interpretation of findings can be problematic when both nominator and denominator change as a function of the disease under investigation. (For further technical details, see Jansen et al., 2006.)
In vivo 1H MRS can measure the intracellular levels of as many as 18 metabolites in healthy brain parenchyma at ultra-high magnetic field strengths of 7 or 9.4 T (Tkác et al., 2001; Mekle et al., 2009; Marjanska et al., 2012). However, some of these are not observable at the lower field strengths typically used for clinical research MRS (between 1.5 and 4 T). Thus, 1H MRS studies in the alcohol literature have focused on a subset of metabolites including the neuronal metabolite N-acetylaspartate (NAA), the glial metabolite myo-inositol (mIno), choline-containing compounds (Cho) such as glycerophosphorylcholine and phosphorylcholine, creatine metabolites (Cr) (the sum of creatine and phosphocreatine), and more recently, glutamate (Glu) and γ-aminobutyric acid (GABA), which are critical for neurotransmission (Fig. 19.1). Alcohol itself can be detected in the brain after alcohol consumption (Fein and Meyerhoff, 2000) and there is a body of work that describes the “visibility” of the alcohol signal in the brain as a function of membrane rigidity and chronic tolerance to alcohol; however, these studies are not reviewed here and the interested reader is referred to Kroenke et al. (2013). Here we give a brief description of the commonly detected proton metabolites, their role in alcohol dependence, and how they have furthered our understanding of the neurobiology of alcohol dependence and addiction in general. For an extensive description of MRS methodology, the biochemical roles of these metabolites, and a consensus paper on the use of 1H MRS in the clinical setting, see Govindaraju et al. (2000), Vion-Dury et al. (1994), and Öz et al. (2014).
Fig. 19.1.
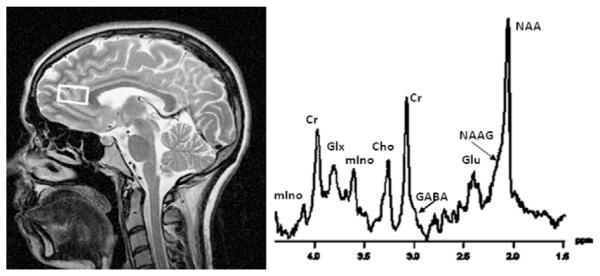
Single-volume 1H magnetic resonance (MR) spectrum (right) obtained from the anterior cingulate cortex (see volume of interest on corresponding MR image, left) at 4 T with a 25-ms echo time. The main spectral resonances are labeled and their peak areas can be quantitated via curve fitting; they are proportional to the absolute concentration of the metabolites from which they originate. NAA, N-acetylaspartate; NAAG, N-acetylaspartyl glutamate; Glu, glutamate; Glx, glutamate + glutamine; GABA, gamma-aminobutyric acid (which is usually quantitated via J-editing from different spectra); Cr, creatine + phosphocreatine; Cho, choline-containing metabolites; mIno, myo-inositol.
NAA is an amino acid found in high concentrations in all structural elements of neurons and it is virtually absent in mature glial cells (Moffett et al., 1991; Simmons et al., 1991; Benarroch, 2008). The “NAA” peak, the strongest peak in an in vivo spectrum of a healthy brain, is composed of overlapping signals from NAA and the much less concentrated N-acetylaspartylglutamate (NAAG, which acts to regulate glutamate and dopamine release). NAA is synthesized in neuronal mitochondria and exported to the cytosol; it flows out into the extracellular space and diffuses to oligodendrocytes, where it is rapidly hydrolyzed (Baslow, 2003a). NAA acts as an organic osmolyte controlling cellular water distribution (Baslow, 2003b), provides acetate for myelin lipid synthesis in oligodendrocytes, and facilitates energy metabolism in neuronal mitochondria. Abnormalities in NAA synthesis, transport, or breakdown (i.e., mitochondrial and neuronal dysfunction) as well as overall loss of neuronal soma size may contribute to a compromised steady-state concentration that is measurable by 1H MRS. Because of these functions in the brain, the NAA signal is generally considered a biomarker of neuronal viability or integrity (Miller, 1991; Vion-Dury et al., 1994; De Stefano et al., 1995; Hugg et al., 1996; Sullivan, 2000; Schuff et al., 2001; Baslow and Guilfoyle, 2007; Rigotti et al., 2007). Studies have shown that NAA reduction is reversible in various pathologies, which suggests that NAA does not necessarily reflect the density or concentration of neurons per se but more the plasticity of neuronal components and the pathologic processes affecting the metabolic functioning of neurons (Bertolino et al., 2001), particularly neuronal bioenergetics (Pan and Takahashi, 2005; Baslow and Guilfoyle, 2007). NAA is distributed fairly homogeneously throughout the brain, at a concentration of approximately 10 mmol/L, very similar in gray matter and white matter; NAAG (around 2 mmo/L) exhibits higher concentrations in white matter than in gray matter (Pouwels and Frahm, 1998; Haga et al., 2009). NAA tends to decrease throughout the brain with normal aging (Ende et al., 2000; Haga et al., 2009; Maudsley et al., 2012), even after correcting for age-related gray-matter loss (Maudsley et al., 2012).
The “Cho” peak in a 1H MRS spectrum represents the trimethyl ammonium resonance from phosphocholine and glycerophosphocholine, with free choline and the neurotransmitter acetylcholine contributing less than 5% (Khiat et al., 2000). Phosphatidylcholine, the main constituent of myelin and cell membranes, is invisible to in vivo MRS due to its restricted molecular mobility. The detectable Cho are intermediates of membrane synthesis and breakdown (Pettegrew et al., 1987, 1990) with a marked regional variability in the brain (highest concentrations in the cerebellum and in rostral cerebrum) (Pouwels and Frahm, 1998). The Cho concentration is between 1.5 and 2.5 mmol/L and increases with age (Haga et al., 2009; Maudsley et al., 2012; Zahr et al., 2013). The Cho signal reflects cell membrane turnover and density (Miller et al., 1996), myelin content (Ross and Bluml, 2001) and/or cell types associated with tissue inflammation (Brenner et al., 1993; Venkatesh et al., 2001).
The “Cr” peak in a spectrum represents creatine and phosphocreatine. The Cr signal is a marker of brain energy metabolism, reflecting overall bioenergetics of neuronal and glial tissues (Balestrino et al., 2002; Ferguson et al., 2002). The metabolite concentration calculated from the Cr MRS signal is about 9 mmol/L, with the highest levels seen in cerebellum, paralleling the distribution of creatine kinase and energy-requiring processes in the brain (Pouwels and Frahm, 1998). As Cr has an important buffer capacity in cellular energy metabolism, its brain concentration is not necessarily stable or invariant under pathologic conditions. Cr levels are dependent on brain activity (Ke et al., 2002) and age (Haga et al., 2009; Maudsley et al., 2012; Zahr et al., 2013) and vary across diseases or pathologic states (Ross and Bluml, 2001; Rosen and Lenkinski, 2007) including alcohol dependence (Durazzo et al., 2010a; Mon et al., 2012; Abé et al., 2013). Therefore, Cr cannot be considered a useful internal concentration reference (Ross and Michaelis, 1994; Sartorius et al., 2008) and the common practice of using the Cr peak signal as an internal concentration reference for other metabolite levels from the same region of interest (e.g., NAA/Cr, Cho/Cr) is problematic (Lee et al., 2007), although experimentally straightforward and methodologically appealing when comparing metabolite measures across individuals. The Cr, NAA, and Cho resonances are single peaks and can therefore be detected rather readily at all field strengths and any practical echo time, an experimental parameter that is critical for optimal detection of other metabolites.
mIno, a carbohydrate that structurally resembles glucose, is a constituent of phosphatidylinositol, an important component of the phospholipid bilayer of cell membranes. mIno is synthesized by and resides predominantly in brain glia, and it is incapable of crossing the blood–brain barrier (Brand et al., 1993). This simple cyclic carbohydrate has been suggested to be a glial marker, to regulate the cellular osmotic environment and maintain cell volume (Brand et al., 1993; Schweinsburg et al., 2000; Ross and Bluml, 2001), and to be involved in intracellular second-messenger system functioning (Fisher et al., 2002). A larger than normal “mIno” peak in an in vivo spectrum therefore can reflect glial activation, gliosis, neuroinflammation, an osmotic response to cell shrinkage, or a combination of such processes (Rosen and Lenkinski, 2007). The mIno signal has a complex appearance (i.e., it is not a single resonance, but a coupled multiplet) and its MRS detection is therefore optimal at the shortest echo times practically possible.
Glu, the major excitatory neurotransmitter in the human brain and an important metabolite in all cells, is a mediator of synaptic plasticity and as such implicated in the initiation and maintenance of all addictive disorders (Kalivas and O’Brien, 2008). In a neurotransmitter cycle between neurons and glia, Glu is released from synaptic vesicles of neurons (where it is highly concentrated) into the cytoplasm, actively transported into surrounding glial cells, where it is converted to glutamine (Gln), and then released into the extracellular space to be taken up again by neurons, where Glu is resynthesized. The MR-detectable Glu signal derives mainly from intracellular (including vesicular) and cytosolic pools (Kam and Nicoll, 2007) and is thought to be linked to neurotransmitter activity. Although Glu is more highly concentrated in the brain than NAA (about 11 mmol/L in gray matter, less in white matter) (Pouwels and Frahm, 1998), its MRS detection sensitivity is relatively low: the Glu signal is quite complex, being dispersed over a wide frequency range and overlapping with equally complicated Gln and other resonances. However, with the advent of static magnetic field strengths of 3 T and higher, with optimal echo times (Schubert et al., 2004), specialized acquisition schemes that simplify the Glu signal (Hurd et al., 2004), and with spectral fitting routines that model the complex signal, the measurement of regionally specific levels of Glu (and in some cases also Gln) has become less exceptional.
GABA is the main inhibitory neurotransmitter in the mammalian brain. The entire metabolically active pool of GABA has a concentration of approximately 1.2 mmol/L in human cortex (Rothman et al., 1993; Hetherington et al., 1998), very near the detection limit of in vivo MRS. The neurotransmitter pool is a relatively small fraction of the entire pool and is in dynamic equilibrium with it. Abnormalities of GABAergic neurotransmission are implicated in many disorders, including AUD, and in neurologic and psychiatric diseases such as epilepsy, mood and anxiety disorders, and schizophrenia. Due to its relatively low concentration and small MRS signal in close proximity to strong singlet resonances, in vivo GABA quantification is technically challenging. Special MRS methods are used that require the subtraction of two spectra from the same brain region acquired with slightly different experimental parameters (Rothman et al., 1993; Kaiser et al., 2008). Although large cerebral molecules (lipids and peptides) may contribute to the commonly detected GABA signal obtained by difference spectroscopy (Choi et al., 2007) and although chronic heavy drinking is known to be associated with brain lipid alterations, changes of the intensity of the detected MRS signal are largely ascribed to changes in GABA (Xin et al., 2010).
The metabolite signal intensities are proportional to metabolite concentrations and their quantification is either in absolute terms or relative to a cerebral concentration standard. Relative or absolute metabolite concentrations can be derived from spectra that are obtained either from one preselected brain region at a time in the case of single-voxel spectroscopy (SVS) or from multiple brain regions simultaneously in the case of MR spectroscopic imaging (MRSI). During a typical MRS examination, SVS spectra are obtained from rectilinear (block-shaped) volumes of interest (VOI) placed in one to four different brain regions (e.g., anterior cingulate cortex (ACC), parietal white matter, cerebellum, thalamus) that each have to be selected before the patient examination begins. The typical VOI size is 4–16 cc and each spectrum requires 2–12 minutes of acquisition time, excluding experimental optimization. The 10 000 times stronger water signal (used to reconstruct high-resolution structural MRIs) is suppressed in MRS by frequency-selective saturation, so that the weak signals from metabolites can be detected from volumes that are much larger than the small voxels that make up an MRI. SVS spectra typically have relatively high signal-to-noise ratios, which facilitates the integration (curve fitting) of metabolite peaks needed for metabolite quantitation. MRSI, conceptually a combination of MRI and MRS, allows imaging of metabolite distributions from either several slices or a three-dimensional volume covering most of the brain. MRSI obtains many hundreds of spectra simultaneously from VOIs that are approximately 1 cc in size within about 15–45 minute. MRSI spectra are typically of lower signal-to-noise ratio than SVS spectra, but they allow evaluation of the distribution of metabolites throughout the imaged regions. In addition, MRSI spectra can be reconstructed from brain regions after the patient has left the MR scanner, allowing greater flexibility for regional data analyses and postacquisition explorations. A major challenge of MRSI is the methodologically and computationally more demanding processing of the MRSI data set compared to that for SVS data. Most scanner manufacturers provide good SVS-processing capabilities, but MRSI-processing methods are still rudimentary and often require offline processing with non-commercial software.
1H MRS OF ALCOHOL USE DISORDERS
Most 1H MRS studies of AUD were performed in alcohol-dependent individuals recruited after acute withdrawal from substance abuse treatment centers (often referred to as “treatment seekers”). The study participants were usually receiving standard care within the rigid structures of inpatient or outpatient treatment that did not include the use of pharmacotherapies (except perhaps disulfiram, which is not centrally active). These studies were therefore purely observational (except for that of Umhau et al., 2010), which describes neuropharmacologic research), providing a snapshot of the brain after withdrawal at various durations of abstinence from alcohol. This research has shown that duration of abstinence has a significant effect on brain metabolite concentrations, as the brain adapts to the sudden absence of alcohol from the system. This realization spurred longitudinal studies aimed at understanding brain plasticity associated with abstinence. Across published studies, the age of the alcohol-dependent study participants varies – though most subjects were in their late 40s – and so does the duration of chronic alcohol consumption over lifetime before study. Duration of abstinence and age, which is usually highly correlated with duration of drinking, are important factors that influence brain metabolite concentrations, and they need to be carefully considered when comparing 1H MRS results across different studies. Table 19.1 lists 1H MRS studies published in peer-reviewed journals on the effects of chronic alcohol consumption in patients with AUD. The study participants are referred to as “alcoholics” in case of treatment seekers and “treatment-naïve heavy drinkers” when referring to individuals with AUD who were recruited from the community. To the best of our knowledge, Table 19.1 contains a complete list of all 34 original 1H MRS studies of AUD published in the English research literature as of chapter completion (February 2013); reviews or symposia proceedings are not included.
Table 19.1.
Original reports of human in vivo 1H magnetic resonance spectroscopy in alcohol use disorder (up to February 2013)
| Authors, year of publication |
Study groups | Duration of abstinence |
Age (mean ± sd) |
Major findings (all findings statistically significant unless otherwise stated) |
|---|---|---|---|---|
| Fein et al., 1994 | 11 alcoholics, 9 controls | 3–24 months | 65 ± 9 | Greater frontal than parietal cortical NAA loss in alcoholics vs controls |
| Martin et al., 1995 | 10 alcoholics, 9 controls | 0–28 days | 44 ± 9 | Increase of Cho/NAA in cerebellar vermis with abstinence |
| Jagannathan et al., 1996 | 10 alcoholics, 27 controls | 1 month | 45 ± 4 | Low NAA/Cr and NAA/Cho in cerebellum, frontal lobe, and thalamus |
| Seitz et al., 1999 | 11 alcoholics, 12 controls | 4 days | 41 ± 11 | Low NAA/Cr at TE = 135 ms and low Cho/Cr at TE = 5 ms |
| Behar et al., 1999 | 5 alcoholics, 10 controls | 5 weeks | 35 ± 7 | Low GABA + homocarnosine in occipital lobe |
| Schweinsburg et al., 2000 | 9 alcoholics, 5 controls | 5 weeks (n = 4) and ≥ 1.5 years (n = 5) |
46 ± 7 | High mI in anterior cingulate cortex and thalamus at 5 weeks. NAA, Cho, and Cr not altered. mI normal at 6 years |
| Schweinsburg et al., 2001 | 37 alcoholics, 15 controls | 1 month | 39 ± 9 | Low NAA in frontal WM, high mI in WM |
| Bendszus et al., 2001 | 17 alcoholics, 12 controls | 2 days and 5 weeks | 40 ± 8 | Low NAA/Cr in cerebellum and frontal lobe, low Cho/Cr in cerebellum (days 3–6) and normalized values after 5 weeks |
| O’Neill et al., 2001 | 8 treatment-naïve heavy drinkers, 12 alcoholics |
2 years (alcoholics) | 43 ± 7 | NAA, Cr, and Cho throughout brain similar between groups |
| Parks et al., 2002 | 31 alcoholics, 12 controls | 4 days and 3 months | 41 ± 9 | Low cerebellar NAA and Cho. NAA increase over 3 months (n = 11) |
| Schweinsburg et al., 2003 | 25 alcoholics (11 F, 17 M),25 controls (12 F, 13 M) |
40 days (F), 26 days (M) |
43 ± 8 (F) 36 ± 6 (M) |
Low frontal white-matter NAA in female and male alcoholics. Low frontal gray-matter NAA only in female alcoholics |
| Meyerhoff et al., 2004 | 46 treatment-naïve heavy drinkers, 52 controls |
12 hours | 41 ± 9 | Low NAA in frontal WM of heavy drinkers vs controls (driven by those without a positive family history of alcoholism, associated with lower executive and working memory functions and lower frontal P3b amplitudes). High Cr, ml, and Cho in parietal GM (but not WM) of heavy drinkers vs controls |
| Durazzo et al., 2004 | 24 alcoholics, 26 controls | 1 week | 50 ± 6 | Low NAA and Cho in frontal lobe, low Cho in thalamus and parietal lobe. Low NAA throughout brain of smoking vs non-smoking alcoholics. Normal Cr and mI |
| Ende et al., 2005 | 33 alcoholics (12 female), 30 controls (10 female) |
3 weeks, 3 months and 6 months |
45 ± 9 | Low Cho in cerebellum and frontal lobe, trend to low NAA in frontal WM; Cho increases over 3 months (n = 14); no longitudinal NAA changes over 3 and 6 months (n = 11) |
| Ende et al., 2006 | 42 social drinkers | ~40 ± 10 | Greater alcohol consumption correlates with higher frontal Cho | |
| Mason et al., 2006 | 12 alcoholics, 8 controls | 1 week and 1 month | 39 ±8 | No GABA group differences in occipital gray matter at 1 week. Higher GABA in non-smoking vs smoking alcoholics. GABA decrease in non- smoking alcoholics over 1 month |
| Durazzo et al., 2006a | 25 alcoholics, 29 controls | 1 week and 4 weeks | 49 ± 7 | NAA and Cho increase in frontal and parietal lobes over 4 weeks. mI and Cr in frontal white matter increase over 4 weeks. Changes more pronounced in non-smoking alcoholics |
| Lee et al., 2007 | 13 alcoholics (smokers), 18 controls (non-smokers) |
16 days | 33 ± 3 | Low Cho and Cr, high Glu/Cr, normal NAA in the anterior cingulate cortex (high Glu/Cr due to low Cr?). High Glu measures correlate with better short-term memory and attention, but greater past alcohol consumption. No metabolic differences in insula |
| Bartsch et al., 2007 | 15 alcoholics, 10 controls | 5 days and 6 weeks | 44 ± 8 | Low frontomesial NAA and low cerebellar Cho. Both increase over 6–7 weeks |
| Durazzo et al., 2008 | 70 alcoholics | 1 month | 51 ± 9 | Low -3NAAintemporal gray matter and frontal white matter and low Cho in frontal gray matter predict relapse within 6–12 months of treatment |
| Gazdzinski et al., 2008a | 35 alcoholics, 32 heavy drinkers |
1 week (alcoholics) | 44 ± 8 | Low NAA, Cho, and mI throughout the brain of treatment-seeking alcoholics vs non-treated heavy drinkers |
| Gazdzinski et al., 2008b | 24 alcoholics, 14 controls | 1 week and 4 weeks | 49 ± 9 | Low NAA and Choin medial temporal lobe. NAA and Cho increase in non- smoking alcoholics over 4 weeks |
| Wang et al., 2009 | 48 alcoholics (26 smoking, 22 non-smoking), 26 controls |
1 month | 49 ± 8 | Only in smoking alcoholics, NAA lower in frontal WM region of low fractional anisotropy (FA) than in controls. No NAA differences in frontal WM with normal FA |
| Durazzo et al., 2010b | 51 alcoholics 26 controls | 1 week | 48 ± 9 | Low NAA and Cr in brain reward system of those who relapse within 6–12 months of treatment vs abstainers and controls |
| Gazdzinski et al., 2010 | 54 alcoholics | Approx. 1 month | 50 ± 8 | Higher BMI associated with lower NAA, Cho, Cr, and m-Ino in frontal lobe, subcortical nuclei, and vermis. Increased alcohol intake related to elevated m-Ino in multiple brain regions and to smaller volumes of frontal and temporal GM |
| Umhau et al., 2010 | 33 alcoholics (15 acamprosate, 18 placebo) |
Days 4 and 25 of study medication |
33 ± 1 | Longitudinal decrease of Glu/Cr in anterior cingulate cortex of those treated with acamprosate. No significant change in any metabolite concentrations in those treated with placebo. |
| Modi et al., 2011 | 9 alcoholics, 13 controls | 1 week | 44 ± 7 | High Cho/Cr in occipital lobe (high Cho? low Cr?) |
| Yeo et al., 2011 | 146 treatment-naïve heavy drinkers |
≤3 weeks | 32 ± 9 | A specific type of genetic mutation (copy number variation) correlated with NAA, Glx, and Cr in anterior cingulate cortex |
| Thoma et al., 2011 | 7 treatment-naïve heavy drinkers, 6 alcoholics, 17 controls |
>1 year (alcoholics) | 34 ± 7 | Significant substance use and psychiatric comorbidity. In anterior cingulate cortex low Glu and high Gln in both drinking groups. Related to drinking severity. No differences between drinking groups |
| Durazzo et al., 2013 | 76 alcoholics (43 smoking, 33 non-smoking), 42 controls (non- smoking) |
1 week | 51 ± 9 (alcoholics), 45 ± 9 (controls) |
Lower NAA in smoking vs non-smoking alcoholics and controls in the DLPFC, insula, superior corona radiata, and the total brain reward system. Differences between alcoholic groups not mediated by alcohol consumption, hypertension, psychiatric comorbidities |
| Abé et al., 2013 | 40 alcoholics, 28 polysubstance users, 16 controls |
1 month | 49 ± 9 | No significant metabolic differences between abstinent alcoholics and controls. In the DLPFC of alcohol-dependent polysubstance users, significantly lower concentrations of NAA, Cho, and mI than both alcoholics and controls. Cr was significantly lower in polysubstance users compared to alcoholics |
| Hermann et al., 2012 | 47 alcoholics, 57 controls | 1 day and 14 days | 46 ± 1 | High Glu at 1 day in prefrontal cortex of alcoholics vs controls. Normal Glu at 14 days. Confirmed in parallel rat studies |
| Mon et al., 2012 | 44 alcoholics, 16 controls | 9 days and 5 weeks | 51 ± 9 | Low NAA, Cho, Cr, and Glu in anterior cingulate cortex at 1 week recover to normal levels at 5 weeks. GABA and mI normal at both times. No abnormalities or changes in DLPFC and POC. Higher cortical mI at 1 week correlates with worse cognition and greater alcohol consumption |
| Xia et al., 2012 | 49 treatment-naïve heavy drinkers, 45 controls (all non-smoking) |
0 | 46 ±6 | Low NAA/Cr in prefrontal GM and WM and worse executive function. NAA levels and executive skills influenced by different SNPs of the metabotropic glutamate receptor 3 (GMR3) gene. Some of these genotypic differences also found in controls |
NAA, N-acetylaspartate; Cho, choline-containing metabolites; Cr, creatine; TE, echo time; GABA, γ-aminobutyric acid; mI, WM, white matter; BMI, body mass index; m-Ino, myo-inositol; GM, gray matter; Glx, glutamate; Gln, glutamine; DLPFC, dorsolateral prefrontal cortex; POC, parieto-occipital cortex; SNP, single nucleotide polymorphism.
Cross-sectional 1H MRS
1H MRS studies have generally described lower NAA and Cho levels in several brain regions of detoxified alcohol-dependent individuals compared to nonalcoholic controls, suggesting relatively widespread neuronal, glial, and membrane abnormalities. The first MRS study to measure NAA levels in abstinent alcohol-dependent individuals employed 1H MRSI (Fein et al., 1994) and suggested neuronal injury in the frontal cortex. Most subsequent studies employed SVS in the frontal lobes and cerebellum of alcoholics after 3–40 days of sobriety. Several groups reported low NAA levels in mostly frontal cortices and white matter (Jagannathan et al., 1996; Seitz et al., 1999; Bendszus et al., 2001; Parks et al., 2002; Durazzo et al., 2004, 2013; Ende et al., 2005; Wang et al., 2009; Mon et al., 2012), suggesting neuronal injury, atrophied dendrites and/or axons, or derangement of neuronal metabolism in brain regions most vulnerable to chronic alcohol abuse as indicated by neuropathology (see below). However, not all studies report lower mesial frontal cortical NAA in recently detoxified alcohol-dependent individuals (Bartsch et al., 2007; Yeo et al., 2013), and this has been related to lower severity of dependence and “uncomplicated, not heavily nicotine abusing alcoholics” (Bartsch et al., 2007), younger age and shorter duration of heavy drinking, and sex (Schweinsburg et al., 2003). In the latter study, alcohol-dependent men and women of similar age, with similar length of “alcoholism” and with comparable alcohol consumption quantities had equally low NAA levels in the frontal white matter. However, only women had significantly lower NAA in an ACC gray-matter region. This careful study suggests a greater vulnerability of frontal gray-matter neurons to chronic alcohol consumption in women than men.
Studies also revealed lower Cho in the cerebellum (Bendszus et al., 2001; Parks et al., 2002; Ende et al., 2005; Bartsch et al., 2007), frontal gray matter (Lee et al., 2007), thalamus, and parietal lobe (Durazzo et al., 2004) relative to light-drinking controls. Lower than normal Cho may reflect less membrane turnover, lower glial cell activity including demyelination, and/or undetected or subclinical pathologies such as thiamine deficiency and liver cirrhosis (Zahr et al., 2010). Only one of several groups who measured mIno in treatment seekers reported elevated mIno in the frontal lobe and in thalamic gray matter of 1-month-abstinent alcohol-dependent individuals using single-volume MRS (Schweinsburg et al., 2000, 2001); all others cited above showed normal or low mIno levels. High mIno levels in early abstinence suggest astrogliosis and/or osmotic changes. Cortical mIno concentrations are correlated positively with measures of learning and memory in treatment seekers (Mon et al., 2012), whereas NAA deficits in frontal white matter and levels of NAA, Cho, and mIno in the cerebellum have been related to lower neuro-cognitive and motor functioning (Bendszus et al., 2001; Parks et al., 2002). One recent study reported higher Cho/Cr in the occipital lobe of alcohol-dependent patients (Modi et al., 2011), but it is unclear if this finding related to high Cho and/or low Cr concentrations. Two groups using absolute quantitation approaches recently reported significantly lower Cr levels in frontal gray matter of 1-week-abstinent treatment seekers (Mon et al., 2012), who relapsed within the year after treatment (Durazzo et al., 2010a), and in smoking alcoholics compared to non-smoking controls (Lee et al., 2007). These studies demonstrate that Cr, an indicator of general cellular high-energy metabolism and buffer of high-energy metabolism, may be decreased in alcohol dependence, a finding not necessarily surprising considering parallel findings of reduced membrane and glial turnover, neuronal injury, and reduced glucose metabolism/consumption in alcohol dependence. The overall pattern of low NAA, low Cho, and low Cr observed in abstinent alcohol-dependent individuals signals a general compromise of bioenergetics and metabolism of brain tissue with potential loss of cells (both neurons and glia), lower neuronal viability, and reduced membrane turnover. This spectral pattern is distinctly different from common neuropathologies studied by 1H MRS (e.g., brain tumors, dementia, epilepsy, human immunodeficiency virus (HIV) infection, multiple sclerosis), which all show low NAA together with high Cho, high mIno, or both. Such patterns are generally interpreted to reflect compromised neuronal viability in the presence of gliosis or pronounced neuroinflammation (Öz et al., 2014). Because the observed brain metabolites have more than one function in the brain (depending on the pathology under investigation, drinking status – active or sober – as well as length of alcohol abstinence), their functional selectivity is not yet fully understood, making data interpretation indefinite.
For the interpretation of MRS findings in AUD, researchers lean heavily on neuropathologic reports from individuals with uncomplicated AUD. Some (Kril et al., 1996; Harding et al., 1997; Korbo, 1999; Kril and Halliday, 1999), but not all (Jensen and Pakkenberg, 1993; Fabricius et al., 2007), postmortem studies reveal neuronal loss mainly in the dorsolateral frontal cortex and the cerebellum as well as glial loss in the hippocampi. Others demonstrate reduced density and size of glial cells in the dorsolateral prefrontal cortex (Miguel-Hidalgo et al., 2002) and lower neuronal and glial cell density in the orbitofrontal cortex (Miguel-Hidalgo et al., 2006). Loss of dendritic arbor and shrinkage of neuronal cell body volume or neuronal death and Wallerian degeneration of myelinated axons (Harper and Kril, 1989; Schwab and Bartholdi, 1996) occur primarily in the frontal lobes, especially the superior frontal cortex (Harper, 2009). Anterior white-matter loss may involve disturbances in both myelin and axonal integrity (Harper, 2009). The mechanisms of chronic alcohol-associated brain injury and neurocognitive dysfunction are hypothesized to involve glutamate and homocysteine-induced excitotoxicity, reduced levels of brain-derived neurotrophic factors, increased oxidative stress and free radical levels, neuroinflammation, thiamine and other nutritional deficiencies, increased aldehyde levels, hepatic dysfunction, and genetic vulnerability (for review, see Durazzo and Meyerhoff, 2007). These mechanisms may work independently or together in AUD to affect cellular oxidative phosphorylation and alter cerebral cellular structures or organelles, membrane phospholipids, myelin, DNA, gene expression, protein synthesis, and cellular metabolism. Associations between MRS-determined metabolite concentrations and neurocognitive deficits are frequently observed (Meyerhoff et al., 2004; Durazzo and Meyerhoff, 2007; Meyerhoff and Durazzo, 2008), demonstrating the functional relevance and clinical significance of their measurements. Thus, in vivo 1H MRS, through its unique sensitivity to functionally significant and specific cellular changes and causative as well as mediating agents of brain injury, contributes considerably to our understanding of mechanistic brain alterations in individuals with AUD and of metabolic neuroplasticity during sustained abstinence from alcohol.
Longitudinal 1H MRS during alcohol abstinence
The neuropathologic correlates of extended abstinence in neocortical and subcortical gray matter are increases of dendritic arbor and neuronal cell body volume as well as myelin structure changes and tissue density increases (Dlugos and Pentney, 1997). These changes are associated with partial reversal of brain atrophy on MRI (Pfefferbaum et al., 1995; Durazzo and Meyerhoff, 2007). Underlying these structural changes are basic metabolic and cellular processes during abstinence from alcohol that may be elucidated by longitudinal 1H MRS. Neuroplasticity in the form of metabolic recovery occurs in the frontal lobes and cerebellum within just a few weeks of abstinence. Importantly, these metabolic improvements have been associated with functional improvements. Three early studies (Martin et al., 1995; Bendszus et al., 2001; Parks et al., 2002) reported increases in frontal and cerebellar lobar NAA/Cr, cerebellar lobar Cho/Cr, and Cho/NAA in the cerebellar vermis after varying durations of abstinence (3–9 weeks). After a few weeks, a higher frontal NAA/Cr ratio was related to better auditory-verbal learning and memory and increased cerebellar vermis NAA/Cr correlated positively with attention/concentration. Cho/NAA in the cerebellar vermis did not recover completely after 3 months, suggesting that this might indicate continued compromise of cerebellar vermis tissue (Parks et al., 2002), consistent with neuropathologic findings (Harper, 1998). Several studies using absolute molar quantitation showed significant increases of NAA and Cho (Ende et al., 2005; Durazzo et al., 2006a; Bartsch et al., 2007; Gazdzinski et al., 2008b; Mon et al., 2012) in cortical, subcortical, and white-matter regions over 4–9 weeks of abstinence, which correlated significantly with cognitive improvements (Durazzo et al., 2006a; Bartsch et al., 2007). The longitudinal metabolic improvements and their correlations with cognitive recovery were most prominent in non-smoking alcoholics or in alcoholics consuming fewer than 10 cigarettes per day and suggest increasing neuronal viability (recovery of neuronal size, dendritic sprouting, regeneration, or synaptogenesis) during sustained abstinence from alcohol and membrane changes, including potentially remyelination.
One study reported persistently low NAA in frontal white matter between 3 weeks and 6 months of abstinence, together with significant Cho increases (Ende et al., 2005). Although in this study NAA may have recovered somewhat before the first assessment, this suggests low-level but persistent neuronal injury in abstinent alcohol-dependent individuals. Significant increases over 4 weeks of abstinence were also reported for mIno and Cr in the frontal white matter, primarily in non-smoking alcoholics (Durazzo et al., 2006a), and for Cho and Cr in the ACC (Mon et al., 2012). Finally, and although not a true longitudinal study, the only group reporting high regional mIno concentrations in 1-month-abstinent alcoholics reported normal mIno levels in another group of alcoholics abstinent for an average of 6 years (Schweinsburg et al., 2000, 2001).
Taken together, these longitudinal 1H MRS studies reflect brain plasticity after cessation of chronic alcohol consumption. Converging evidence suggests that the brain of an alcohol-dependent individual in abstinence recovers from neuronal injury, membrane and osmotic abnormalities toward the normal control range over a period of 1–3 months of sustained abstinence, even after 20–30 years of chronic alcohol consumption. These metabolic improvements are paralleled by recovery of cognitive abilities. Correlations between changes in metabolite levels and changes of cognitive performance during abstinence have been observed, but corresponding analyses are uncommon as they require complete data sets from longitudinal assessments. (For a review of research on cognitive recovery and MRS measures during alcohol abstinence, see Durazzo and Meyerhoff, 2007.)
GABA and Glu concentrations in alcohol use disorders
Although quantitative GABA and Glu measures give limited insight into molecular mechanisms, measuring their basal concentrations in psychiatric disorders promises to help with psychiatric diagnosis, may inform pharmacotherapy, and can monitor patient response to drug therapy (Agarwal and Renshaw, 2012; Maddock and Buonocore, 2012). GABA and Glu levels demonstrate functional significance relevant to AUD: extracellular Glu levels measured by microdialysis relate to drug seeking and drug taking in animals, whereas MRS-determined cortical Glu (Zahr et al., 2008) and GABA relate to cognition (Abé et al., 2013), depressive states (Sanacora et al., 2004), and extroversion (Goto et al., 2010) in humans. In non-alcoholic controls, medial pre- frontal cortex Glu concentrations correlated negatively with sensation seeking and positively with impulsivity scores (Gallinat et al., 2007a; Hoerst et al., 2010), and ACC Glu/Cr levels related to cognitive control (Falkenberg et al., 2012). In alcohol dependence, a small early study demonstrated low GABA in the occipital lobe at 5 weeks of abstinence (Behar et al., 1999). A larger follow-up study by the same research group (Mason et al., 2006) showed no significant occipital cortex GABA deficits at 1 week of abstinence compared with non-alcoholic controls, but further decreases over 1 month of abstinence in non-smoking alcoholics only. In addition, GABA levels were higher in non-smoking vs smoking alcoholics at 1 week of abstinence. These studies demonstrate changing GABA levels with duration of abstinence and modulation by smoking status. A recent larger high-field MRS study found largely normal GABA levels in frontal and parieto-occipital cortices (POCs) of 50-year-old alcoholics after both 9 days and 5 weeks of abstinence relative to non-smoking healthy controls, with little, if any, modulating effects of cigarette smoking on GABA levels (Mon et al., 2012).
The concentration of Glu in the ACC was significantly higher relative to light or non-drinking controls at 1 day after last alcoholic drink during withdrawal, when alcohol was still detectable in the brain of many study participants (Hermann et al., 2012) and significantly lower than in controls at 9 days of abstinence (Mon et al., 2012). ACC Glu levels on average decreased significantly between day 1 and 14 of abstinence (Hermann et al., 2012) but behaved quite differently across individuals, decreasing in 15/27 cases, increasing in 4/27 and staying unchanged in 8/27 cases. Between 9 and 32 days of abstinence (Mon et al., 2012), initially low Glu levels in ACC and POC increased significantly on average, but also showed variability across individuals in the ACC, increasing in 6/11 cases, decreasing in 1, and staying unchanged in 4/11 cases. The ACC Glu increases in this study were accompanied by significant NAA and Cr recovery, signaling reversal of compromised prefrontal gray-matter bioenergetics or metabolism. Similarly, ACC Glu/Cr tended to increase in 18 alcoholics between 4 and 25 days of abstinence, with no significant NAA ratio changes (Umhau et al., 2010). In another report of Glu levels in 16-day abstinent alcoholics, ACC Glu concentration was normal (Glu/Cr was elevated, but this was due to significantly decreased Cr concentration) (Lee et al., 2007). Taken together, these reports confirm the view that Glu homeostasis is restored during early sobriety, with Glu levels fluctuating dynamically around levels found in controls and normalizing within a few weeks of abstinence. Similar cortical Glu dynamics have been observed in rat alcohol administration protocols (Hermann et al., 2012).
While prefrontal Glu levels are transiently increased during withdrawal, excitotoxic injury may occur (Prendergast et al., 2000; Harris et al., 2003; De Witte, 2004). As studies show that Glu levels decrease rapidly and homeostasis is restored within a few weeks after withdrawal, a single short period of elevated Glu may not promote significant neuronal injury. However, typical alcoholics cycle through relapse/remit phases for much of their drinking lives, so that prefrontal neuronal injury from withdrawal-induced Glu elevations may accumulate over the lifespan. Such an assertion is consistent with the observation that repeated detoxifications are associated with greater loss of control over future drinking (Duka et al., 2011) and with delayed, if not reduced, recovery from executive dysfunction (Durazzo and Meyerhoff, 2007; Loeber et al., 2010).
High Glu levels during withdrawal were hypothesized to be related to future relapse (Mann and Hermann, 2010). If indeed so, acamprosate, one of the three pharmaceuticals approved by the Food and Drug Administration to treat alcoholism and thought to facilitate Glu homeostasis, could be used to reduce relapse most effectively in those with particularly high Glu levels during withdrawal. This hypothesis, however, was not confirmed (Hermann, unpublished observations), in part probably because prefrontal Glu levels early in abstinence are transient and state-dependent and do not reflect the long-term challenges to sobriety faced during recovery from alcoholic drinking. Nevertheless, this idea is an example of how 1H MRS can be used to identify potential subgroups of patients in whom certain medications may be more effective than others or to monitor the efficacy of treatment that mitigates some metabolite imbalance. Case in point is a study that found decreasing Glu/Cr in the ACC of alcohol-dependent individuals treated with acamprosate between 4 and 25 days of abstinence (Umhau et al., 2010). Placebo treatment on the other hand was associated with a trend to a Glu/Cr increase over the same interval. Prefrontal NAA/Cr and NAA/Cho did not change significantly during therapy in either medication group of this study. The MRS measure of Glu was not correlated with cerebrospinal fluid levels of glutamate, suggesting that these glutamate measures reflect different pools. As MRS measures of Glu (and GABA) have been shown to correlate with behavior and cognition, they may in fact better reflect the neurotransmitter pool). Constituting neuropharmacologic research, this report is the only 1H MRS study of AUD to date that was not mainly observational. In vivo Glu and GABA measurements will improve our understanding of substance use behavior and relapse, expanding the role of 1H MRS in neuropsychiatric disease (Maddock and Buonocore, 2012).
Treatment-seeking vs treatment-naïve alcohol-dependent individuals
Less than 10% of individuals with AUD seek treatment at some time in their lives (Fein and Landman, 2005; Hasin et al., 2007; Moss et al., 2007). Nevertheless, most of what we know about the effects of AUD on the human brain has been gleaned from studies of these treatment seekers recruited from substance use treatment programs (Fein et al., 2002); moreover, only a diminishingly small fraction of these individuals volunteer to participate in MRS research. Thus, it can be easily understood that the research data obtained from these convenience samples are heavily biased and may not realistically represent the state of the brain in the much larger population of individuals with AUD who never seek treatment in their lives. Studying treatment seekers thus can only generate conclusions with questionable relevance to the majority of individuals with AUD. We therefore review in this section the limited research that has been conducted in treatment-naïve individuals recruited from the community. As opposed to abstinent treatment seekers, they actively consume alcohol at the time of study (but are usually studied with basically 0% blood alcohol concentration several hours after their last alcoholic drink), and have not had a chance to recover from any potential brain abnormalities during the abstinence period before MRS study. Furthermore, treatment-seeking vs treatment-naïve heavy drinkers generally have more severe medical, psychiatric, and substance use comorbidities that may confound brain MRS measures in AUD widely thought to reflect effects from chronic drinking; they also show greater alcohol consumption over lifetime (>50%) and more periods of abstinence than their treatment-naive counterparts (Fein and Landman, 2005). Therefore, it may be reasonable to expect that treatment seekers in early abstinence have generally greater brain abnormalities than active heavy drinkers and that both do not simply represent a continuum of AUD on a common trajectory or drinking scale.
A case in point is an early 1H MRSI study that demonstrated that NAA, Cho, and Cr concentrations throughout the brain of treatment-naïve heavy drinkers were statistically equivalent to those in alcohol-dependent individuals, who had been abstinent – and thus had had time to recover from their (presumed) metabolite deficits – for an average of 2 years (O’Neill et al., 2001). Relative to light-drinking controls, metabolite abnormalities in community-dwelling, treatment-naive heavy drinkers (most of them alcohol-dependent) were less pronounced and demonstrated a pattern different from that reported in recently detoxified treatment seekers: we reported lower NAA in frontal white and parietal gray matter, as well as higher parietal gray matter Cr, Cho, and mIno relative to healthy controls (Meyerhoff et al., 2004; Gazdzinski et al., 2008a), while another recent study found lower-than-normal NAA/Cr in prefrontal gray and white matter (Xia et al., 2012). As weak as the metabolite level abnormalities were, they correlated with poorer performances on executive skills, working memory and processing speed (Fig. 19.2) as well as with lower frontal P300b amplitudes (Meyerhoff et al., 2004; Xia et al., 2012). Furthermore, family history of alcohol problems (FHA) and drinking pattern (binge vs non-binge) as well as age and sex modulated the brain MRS abnormalities in treatment-naïve heavy drinkers (Meyerhoff et al., 2004). Relative to treatment seekers in their mid-40s, NAA, Cho, and mIno concentrations in multiple brain regions of treatment-naïve individuals of similar age were higher and parietal and thalamic atrophy less pronounced after correcting for differences in drinking severity (Gazdzinski et al., 2008a). The only study that assessed Glu levels in treatment-naïve heavy drinkers showed low Glu and high Gln in the ACC compared to light-drinking controls (Thoma et al., 2011). However, the study included only 7 heavy drinkers, their Glu levels were similar to those in 6 1-year-abstinent alcoholics, and significant substance use and psychiatric comorbidity were present in this relatively young cohort (mean age 35 years) that either or both conditions could have affected metabolite measures. In a larger follow-up study of these heavy drinkers (most of them dependent, with a mean age of 31 years, and studied 3 days after their last alcohol intake), metabolite levels in the ACC overall (i.e., a multivariate general linear model including NAA, Cr, Cho, mIno, and Glx) were not different between treatment-naïve and treatment-seeking heavy drinkers after correction for greater and longer alcohol consumption in the treatment seekers (Yeo et al., 2013). More surprisingly though, in the model comparing all metabolite levels between the combined groups of heavy drinkers and age-matched controls, all metabolite levels were weakly elevated in the heavy drinkers and related to more years of heavy drinking.
Fig. 19.2.

Relationships of regional gray matter (GM) and white matter (WM) metabolite concentrations (arbitrary units) with measures of cognitive functioning in treatment-naïve heavy drinkers recruited from the community (here, mI ¼ myo-inositol). NAA, N-acetylaspartate. (Adapted from Meyerhoff et al., 2004.)
In a study of social drinkers, greater alcohol consumption, i.e., up to 2 drinks/day, was correlated with higher Cho measures (Ende et al., 2006). Although no interpretation was provided, the finding suggests that Cho levels, at least in the light-to-moderate drinking range typical for control groups in the alcohol MR research literature, are influenced by drinking severity and altered physical properties of the metabolites contributing to the Cho signal (i.e., relaxation times). This observation may explain some of the discrepant Cho findings in the AUD literature.
Altogether, most studies suggest that treatment-naïve (i.e., currently drinking) heavy drinkers have measurable neuronal injury (low NAA) in some brain regions with functional (cognitive) ramifications, and some suggest higher-than-normal energy metabolism (high Cr) and osmotic changes (mIno). Importantly, the regional pattern and magnitude of metabolite levels are different from those of treatment seekers found especially after several decades of drinking, suggesting that we cannot generalize our knowledge of brain abnormalities in abstinent alcoholics to the bulk of individuals with AUD who are not seeking treatment (Fein and Landman, 2005; Durazzo and Meyerhoff, 2007). Alcohol use patterns describing frequency and quantity of use over lifetime are different between these AUD populations and treatment seekers present with greater neuropsychiatric (Di Sclafani et al., 2008) and substance use comorbidities and potentially with greater subclinical liver disease and neuropathology. These factors, individually and in concert, have measurable effects on MRS-observable brain metabolite levels and should influence the interpretation of any MRS data obtained from individuals with AUD.
EFFECTS OF COMMON SUBSTANCE USE COMORBIDITIES ON 1H MRS MEASURES IN ALCOHOL USE DISORDERS
Most 1H MRS studies of AUD described in Table 19.1 ostensibly include participants with dependence on or abuse of alcohol only (unless otherwise specified in the patient characterization section). In most of these studies, screening procedures excluded individuals with other substance dependence and medical and psychiatric comorbidities that can also affect the outcome measures (see Chapter 33 on psychiatric comorbidities in AUD). However, AUD researchers in general have allowed abuse of other substances as defined by standard diagnostic criteria (Diagnostic and Statistical Manual of Mental Disorders or International Classification of Diseases), pathology that is common in alcoholics such as hypertension and depressive symptomatology, as well as other more subclinical pathology. The reader of these research articles is advised to inquisitively study the participant characterization section to appreciate the comorbid conditions that may have affected the studies’ outcome measures. Such examination reveals that some research participants described in the reports listed in Table 19.1 may have been better characterized as polysubstance abusers rather than simply alcohol-dependent individuals.
More recent attempts have been undertaken to more formally understand the brain changes in alcohol-dependent individuals as a function of the most common substance use comorbidities, the chronic misuse of tobacco and illicit substances. This research has been spurred by treatment providers, who have pointed out that the “pure” alcoholic has become less common over the years and that comorbid substance abuse is the clinical reality today. In fact, the US-based National Epidemiologic Survey on Alcohol and Related Conditions (NESARC) finds a number of premorbid and comorbid disorders associated with AUD (Mertens et al., 2003; Stinson et al., 2005; Mansell et al., 2006; Hasin et al., 2007). These contribute considerable variability to the pattern and magnitude of the observed neurobiologic and neurocognitive abnormalities. One of the most common substance use comorbidities in AUD is chronic cigarette smoking (Daeppen et al., 2000; John et al., 2003; Room, 2004). It is recognized that today, more than half of individuals in substance abuse treatment are not only alcohol-dependent, but are also abusing or dependent on illicit drugs, such as cannabis, psychostimulants, heroin, and prescription drugs (Medina et al., 2004; Kedia et al., 2007). For a summary of 1H MRS data on “pure” psychostimulant and polydrug users we refer the reader to an excellent review (Licata and Renshaw, 2010). Below, we review the published literature on the differential effects of AUD with and without tobacco or substance use comorbidities on brain 1H MRS measures.
Smoking comorbidity
An estimated 60–80% of individuals with AUD smoke cigarettes chronically (Romberger and Grant, 2004; Durazzo et al., 2007). Emerging research suggests that chronic smoking, independent of AUD, is associated with abnormalities in brain morphology, cerebral blood flow, neurochemistry, and neurocognition that are similar to those reported in AUD (for review, see Durazzo and Meyerhoff, 2007). We investigated the effects of concurrent chronic cigarette smoking on lobar and subcortical metabolite concentrations in alcohol-dependent individuals (Durazzo et al., 2004) and on longitudinal brain metabolite changes during short-term abstinence from alcohol (Durazzo et al., 2006a). Smoking alcoholics compared with their non-smoking counterparts at 1 week of abstinence had lower NAA in frontal white matter, parietal gray matter, and lenticular nuclei and lower NAA and Cho in the midbrain. Alcohol dependence, independent of smoking, was associated with lower Cho in the thalami and parietal lobes and lower frontal lobe NAA and Cho, consistent with other reports (see above). The regional Cr or mIno levels were not influenced by drinking or smoking status. Greater nicotine dependence and more cigarettes smoked per day were correlated with lower NAA in thalamic and lenticular nuclei. Cerebellar vermis NAA was positively correlated with visuomotor scanning speed (smokers) and visuospatial learning and memory (non-smokers). Largely consistent with metabolic findings in non-alcoholic chronic smokers (Gallinat et al., 2007b), our 1H MRSI findings suggest that chronic smoking compounds alcohol-induced neuronal injury and cell membrane injury in the frontal lobes of persons with AUD and that it has independent adverse effects on neuronal viability and cell membrane turnover/synthesis in the cerebellar vermis and midbrain.
In a related study of a similar patient population (Durazzo et al., 2013), we evaluated if the widespread smoking-related metabolite abnormalities described above (Durazzo et al., 2004) were specific to the cortical and subcortical components of the extended brain reward/executive oversight system (BREOS), a network of overlapping circuitry that is critically involved in the development and maintenance of all forms of addictive disorders. Smoking alcoholics had significantly lower NAA concentrations than either non-smoking alcoholics or controls in the dorsolateral prefrontal cortex (DLPFC), insula, white matter of the superior corona radiata, and the BREOS overall (Fig. 19.3); NAA deficits were non-significant when averaged over spectroscopy volumes from the entire frontal cortex or entire frontal white-matter lobe. These focal metabolic differences, similar to observed cortical thinning (Durazzo et al., 2013), were not mediated by alcohol consumption and common medical or psychiatric comorbidities. Also of note are the normal NAA levels in non-smoking alcoholics that were independent of the lower drinking severity in the non-smokers; unaltered NAA levels were also noted in the ACC of alcoholics who smoked fewer than 10 cigarettes per day (Bartsch et al., 2007).
Fig. 19.3.
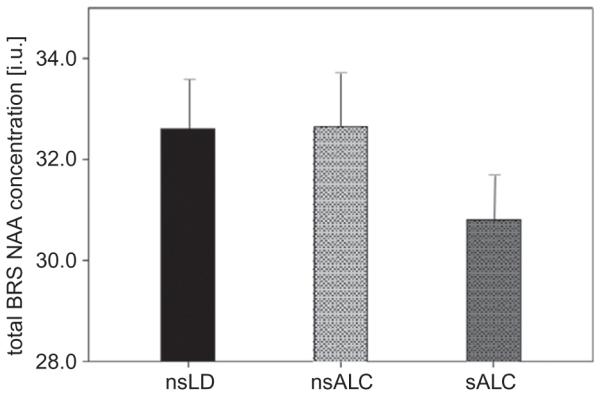
Absolute N-acetylaspartate (NAA) concentrations (arbitrary units) averaged over cortical brain regions of the brain reward system (BRS) in non-smoking light/non-drinking controls (nsLD), non-smoking (nsALC) and smoking recovering alcohol-dependent individuals (sALC) at 5 weeks of abstinence from alcohol. (Adapted from Durazzo et al., 2013.)
Thus, chronic smoking in alcohol dependence specifically is associated with significant NAA abnormalities in critical anterior brain regions that are implicated in the development and maintenance of addictive disorders. The smoking-associated metabolic brain abnormalities may be clinically significant as they are accompanied by worse performance on cognitive tests that require fast and flexible processing, including set shifting, processing speed, and cognitive efficiency (see Friend et al., 2005; Durazzo et al., 2006b; Glass et al., 2006 and references cited therein). The metabolite–cognition relationships are often different in pattern or magnitude between smoking and non-smoking alcoholics (Durazzo et al., 2004, 2006a; Gazdzinski et al., 2008b), thereby giving clues to the detrimental impact of chronic smoking on functional neurocircuitry.
Over approximately 1 month of abstinence from alcohol, frontal and parietal NAA and Cho increased significantly (Durazzo et al., 2006a) (see section on longitudinal 1H MRS during alcohol abstinence). Although not strongly decreased at baseline, mIno and Cr levels in frontal white matter also increased during abstinence. Specifically, non-smokers showed wide-spread increases of all metabolite levels, whereas increases in smokers were much less pronounced and seen in fewer brain regions. At 1 month of abstinence, white-matter NAA in the area of the anterior corona radiata was lower than normal in smoking but not in non-smoking alcoholics (Wang et al., 2009). In non-smokers, metabolite gains were related to improvements in cognition, while smoking alcoholics showed significantly fewer relationships of this kind (Durazzo et al., 2006a). Furthermore, the longer the abstainers had smoked, the smaller the longitudinal increases in regional NAA and Cho concentrations during recovery. Similarly, in a study of medial temporal lobe (including hippocampus), NAA and Cho levels increased and mIno tended to increase over 1 month of abstinence in non-smokers but not in smokers (Gazdzinski et al., 2008b).
1H MRS showed that brain GABA concentrations are also influenced by cigarette smoking in healthy controls (Zhu and Chiappinelli, 1999; Epperson et al., 2005) and during abstinence from alcohol in alcohol-dependent individuals (Mason et al., 2006). At 1 week of abstinence, GABA levels in the occipital cortex were higher in alcohol-dependent non-smokers than smokers. Two weeks later, GABA levels had decreased to similar levels in both groups. Higher GABA in early alcohol abstinence may reflect compensation for reduced cortical benzodiazepine-GABAA receptor function thought to contribute to alcohol tolerance and withdrawal, while the subsequent GABA decrease may reflect “normalization” of GABAA receptor function with sobriety (see review in Cosgrove et al., 2011). A recent larger high-field MRS study found normal GABA levels in dorsolateral, medial prefrontal, and POCs at both 9 days and 5 weeks of abstinence, with little, if any, effects of cigarette smoking on regional cortical GABA concentrations (Mon et al., 2012).
Together, the reviewed material suggests that chronic cigarette smoking affects both neurobiology and neurocognition. If confirmed in additional human studies and animal models, we need to consider that smoking and non-smoking individuals with AUD may differ in the nature or extent of their response to pharmacologic and behavioral interventions designed to promote abstinence from alcohol. Thus, effective treatment may have to consider the smoking status of the treatment-seeking individual.
Other substance use comorbidity
In short-term abstinent individuals with comorbid AUD and cocaine dependence, frontal lobe NAA/Cr and Cho/Cr were normal, but GABA/Cr was lower than in controls and similar to levels found in “pure” cocaine dependence (Ke et al., 2004). The study did not include “pure” alcohol-dependent individuals. Using high magnetic field strength (4 T) 1H MRS in three cortical brain regions (ACC, DLPFC, POC), we compared 40 alcohol-dependent individuals and 28 age-matched alcohol-dependent polysubstance users at 1 month of abstinence from alcohol, psychostimulants, and cannabis to 16 drug-free controls. As reported earlier (Mon et al., 2012), after this period of abstinence metabolite levels in “pure” alcoholics and controls were statistically equivalent. However, the polysubstance users at 1 month of abstinence had lower concentrations of NAA, Cho, and mIno in the DLPFC compared to “pure” alcoholics and controls, while Cr concentration was lower compared to alcoholics only; these metabolite levels were normal in the ACC and POC. GABA, largely normal in alcohol dependence throughout abstinence (Mon et al., 2012), tended to be selectively lower in the ACC of polysubstance users compared with both alcoholics and controls (mean difference of 17–20%), and GABA levels in the DLPFC related negatively to greater cocaine consumption. The metabolic group differences in our study were unrelated to age, alcohol consumption, body mass index, smoking status, depression, and anxiety symptomatologies. It appears that, of the cortical regions examined in these polysubstance users, the DLPFC is most strongly compromised. This cortical region is in charge of self-control, -monitoring, and -censoring (i.e., critical for response inhibition, working memory, reasoning, problem solving, planning, organization, and set shifting) and several cortical metabolite concentrations were related to corresponding measures of cognitive performance. By contrast, “pure” alcohol-dependent individuals at 1 week of abstinence show NAA, Cho, and Cr reductions only in the ACC (Mon et al., 2012), a region important for decision making, organizing and integrating information. Together, these findings demonstrate regionally and functionally different as well as longer-lasting metabolic deficits in alcohol-dependent individuals with than without comorbid illicit substance dependence. This metabolic signature may therefore serve as MRS biomarker in substance abusers and suggests targets for pharmacologic and behavioral treatment specific to polysubstance abuse. For example, the GABA data support the view that efforts to increase GABA levels in substance users through topiramate treatment should be a promising treatment possibility (Johnson, 2005).
All but one (Glu) of the measured metabolite levels were lower in the DLPFC of polysubstance users relative to individuals with alcohol dependence only (Abé et al., 2013). Thus, in addition to dose-related cellular injury to neurons, we have to consider alternative interpretations of the reductions, such as presumed associated neuronal or glial edema related to substance use; the associated higher tissue water content might lead to overall lower metabolite concentrations. In addition, osmotic imbalance and/or cell swelling may contribute to the observed MRS differences in the DLPFC of polysubstance users. Finally, DLPFC mIno correlated positively with days of abstinence, ranging from 10 to 45 days. This suggests that at least some brain metabolic abnormalities in alcohol-dependent polysubstance users recover with extended abstinence, even if slower than in “pure” alcohol dependence.
These studies demonstrate that chronic comorbid tobacco and illicit drug use each have significant and unique effects on metabolite concentrations and related cognitive performance in alcohol dependence. While studies in “pure” alcohol-dependent individuals have contributed to our understanding of the detrimental brain changes associated with an alcohol dependence diagnosis, the knowledge gained from these studies cannot necessarily be generalized to brain changes in the majority of alcohol-dependent individuals who have comorbid substance dependence. For MR methods, including 1H MRS, to become generally useful clinical tools, we have to study the clinical reality of polysubstance use and adopt our analytical and statistical tools to understand the complexities of brain changes associated with concurrent use of several licit and illicit substances.
CURRENT AND FUTURE 1H MRS RESEARCH IN ALCOHOL USE DISORDERS
In this section, we touch on a few newly emerging lines of 1H MRS research in AUD. As we have much neuro-imaging data from individuals in early treatment and evidence that these relate to cognitive function, researchers have asked to what degree the observed neurobiologic and cognitive brain abnormalities are associated with relapse propensity after treatment or to what degree they may predict relapse. If significant correlates of treatment outcomes were identified, the critical brain phenotypes could become prime candidates for selective therapeutic targeting or cognitive and behavioral manipulations. In addition, as neuroimaging research has related brain structure and task-induced brain activity to specific genotypes, and as those who can maintain long-term abstinence appear to have some resilience to MRS-detectable brain abnormalities not seen in future relapsers (see below), efforts are also underway to evaluate if MRS measures in AUD are influenced by specific genes and common functional polymorphisms. Furthermore, the relative dearth of significant correlations between alcohol consumption and regional metabolite concentrations (assuming the consumption measures are good estimates of the true drinking behavior) is used to argue for the influence of common psychiatric and/or medical comorbidities on brain metabolite levels: these comorbidities are part and parcel of AUD so that excluding for them would eliminate the majority of our target population from study (e.g., depression, anxiety, stress, hypertension), rendering the research clinically irrelevant, or they are subclinical (e.g., liver dysfunction, metabolic syndrome) and therefore not detected at screening. In addition, certain personality traits commonly associated with substance abuse (e.g., impulsivity and anxiety) have been shown to affect brain metabolite levels directly (e.g., Glu in ACC of healthy controls (Hoerst et al., 2010; Grimm et al., 2012) and substance-dependent individuals (Schmaal et al., 2012)) and may also contribute to the large variance of metabolite concentrations in AUD samples. Thus, AUD is not a phenomenon only explained by chronic heavy drinking; therefore, for a better understanding of the metabolic brain abnormalities and associated brain functions in AUD, meaningful future study designs have to take salient confounds of MRS measures into account. The interested reader is referred to Chapter 33 on comorbidities in AUD, which details the literature on influences of psychiatric and medical comorbidities as well as personality traits on neuroimaging measures in AUD.
Here, we briefly summarize the applicability of proton MRS to the prediction of relapse and the relatively new field of neuroimaging genetics in AUD, and we outline future directions in MRS research.
1H MRS correlates of relapse in AUD
Most individuals treated for AUD relapse within 6 months following treatment (e.g., Dawson et al., 2007; reviewed in Durazzo et al., 2010a). While the neuropsychologic, psychiatric, sociodemographic, and behavioral correlates of relapse in AUD have been studied extensively, potential neurobiologic factors as objective predictors of successful abstinence in AUD are not as well understood. Identifying 1H MRS-detectable factors that distinguish abstainers from relapsers will help understand aspects of neurobiology that drive the often lifelong relapse/remit cycle so characteristic of alcohol dependence (Maisto and Connors, 2006; Zywiak et al., 2006; Dawson et al., 2007) and that are critical for maintaining long-term abstinence. Treatment-seeking alcohol-dependent individuals who relapsed within 3 weeks of study demonstrated lower than normal cerebellar NAA and Cho at 3–5 days of abstinence, whereas cerebellar and frontal metabolite levels were not reduced in individuals who relapsed after 3 weeks of abstinence (Parks et al., 2002). While these specific early findings were not confirmed by others, they demonstrate an attempt to link 1H MRS measures to treatment outcome. We recently combined 1H MR spectroscopic imaging with assessment of comorbid psychiatric disorders and comprehensive cognitive testing in individuals at 1 month of abstinence from alcohol (Durazzo et al., 2008). The study participants were classified into abstainers (no alcohol consumption) and relapsers (any alcohol consumption) based on their drinking behavior between 6 and 12 months after treatment and then contrasted on the regional metabolite concentrations and other outcome measures obtained at 1 month of abstinence. Independent classifiers were NAA in frontal white and temporal gray matter, Cho in frontal gray matter, processing speed, and comorbid unipolar mood disorder. Combined, these measures accurately classified 83% of abstainers and 90% of relapsers into their respective groups, accounting for 72% of the variance in drinking status at follow-up. In a largely similar population studied at 1 week of abstinence, 1H MRSI metabolite levels in regions of the extended BREOS were related to relapse (Durazzo et al., 2010a): Future relapsers had lower NAA than future abstainers in DLPFC, ACC, insula, superior corona radiata, and cerebellar vermis and lower Cr in similar BREOS regions. Of note, baseline metabolite concentrations of future abstainers did not differ significantly from those in controls, although these individuals had consumed similar amounts of alcohol over their lifetimes as the metabolically abnormal relapsers. Moreover, regional baseline NAA levels and several measures of posttreatment alcohol consumption were strongly correlated in relapsers. Together these data suggest that metabolic abnormalities in anterior brain regions and specifically those in the BREOS early in treatment – in brain structures that subserve reward-related processes and behaviors – are associated with treatment outcome, even relapse severity. These MRS findings are consistent with other neurobiologic abnormalities in the ACC and DLPFC found to be associated with increased risk of relapse to substance use (Baler and Volkow, 2006; Goldstein et al., 2009; Durazzo et al., 2010b; Heatherton and Wagner, 2011; Volkow et al., 2011). Whether these MRS classifiers are useful in predicting relapse needs to be tested in independent samples. Clearly, more research is needed to understand how these brain abnormalities in treatment seekers contribute to treatment outcomes (Loeber et al., 2010), so that specific treatments can be developed that help mitigate these select abnormalities. Finally, the fact that regional MRS measures in abstainers did not differ significantly from those in non-alcoholic controls, whereas those in relapsers with similar long-term drinking histories did, suggests that other yet unexplored factors drive drinking behavior after treatment. A more comprehensive and integrated assessment is necessary to understand salient mechanisms contributing to relapse in AUD. One of those factors relating to vulnerability and resilience to relapse is neurogenetics.
Neuroimaging genetics
Neuroimaging genetics (or genomics) is an evolving field that aims to explain some of the variance observed in neuroimaging measures through genetic influences. Twin studies have shown that NAA, mIno, Cr, and Cho levels show substantial heritability, as high as 72% for NAA in the posterior cingulate cortex (Batouli et al., 2012), demonstrating the high degree of genetic determination of metabolite levels. Other recent reports have linked specific functional single nucleotide polymorphisms (SNPs) of certain genes to regional metabolite levels in healthy controls and other pathologies (e.g., brain-derived neurotrophic factor (BDNF) and regional NAA concentrations (Stern et al., 2008; Gallinat et al., 2010; Gruber et al., 2012)). Recent observations also support the possibility that genetic background contributes to the variance of 1H MRS measures within alcohol-dependent groups: (1) the common finding that measures of alcohol drinking severity do not correlate well with 1H MRS outcome measures, after correcting for the influences of age and common comorbidities, suggests that other factors must influence these measures; (2) the intriguing finding that brain metabolite levels in individuals in early treatment who can maintain long-term abstinence do not differ significantly from non-alcoholic control populations, while those in future relapsers with similar drinking severity do (Durazzo et al., 2010a) (see above); and (3) results of abnormal brain structure and cognitive processing in alcohol-naïve adolescents with a positive FHA, which are similar to abnormalities detected in adult alcoholics (Hanson et al., 2010; Herting et al., 2010; for review, see Tessner and Hill, 2010).
An early study suggested genetic influences on MRS metabolite measures by demonstrating that the low frontal white-matter NAA levels in treatment-naïve heavy drinkers vs controls were driven by those with a negative FHA (Meyerhoff et al., 2004). This suggested a protective effect of a positive FHA on frontal lobe neuronal integrity, consistent with the interpretation of higher frontal glucose metabolism in alcohol-naïve individuals with a positive vs a negative FHA (Volkow et al., 2006). Although this is in apparent contrast to the protective effect of a positive FHA on brain structure and cognition described above (Hanson et al., 2010; Herting et al., 2010), the studies together demonstrate a complex modulation of different phenotypes by genetic factors. Also among heavy drinkers, rare copy number deletions, a measure of overall genetic mutation load, correlated with NAA, Glx, and Cr levels in ACC, suggesting genetic influences on prefrontal cellular oxidative phosphorylation (Yeo et al., 2011). In another study of treatmentnaïve heavy drinkers (Xia et al., 2012), low NAA/Cr in a prefrontal volume including ACC and adjacent white matter was driven by drinkers with the A/A genotype for metabotropic glutamate receptor 3 (GMR3) SNP rs6465084. Similar genotypic differences were observed in controls. Furthermore, alcoholic T-carriers for a different GMR3 SNP had lower NAA/Cr in prefrontal GM and poorer executive function than the A/A genotype. As the prefrontal NAA levels correlated with executive function, a common finding in AUD, cognitive performance in AUD may also be influenced by this genotype. Cognitive function in recently abstinent alcoholics has been shown to be affected by catechol-O-methyltransferase genotype (Durazzo et al., 2012). Furthermore, preliminary analyses indicate that the different alleles of the BDNF SNP rs6265 are not only associated with variable tissue volume in the frontal lobe (Mon et al., 2013) and hippocampi during abstinence from alcohol, but also with differential long-term metabolic recovery of frontal white-matter NAA and Cho (Meyerhoff et al., 2011).
While there is strong evidence for genetic factors influencing a person’s risk for developing AUD (Bierut et al., 1998; Schuckit et al., 2001; Kendler et al., 2003; Mayfield et al., 2008), few studies have related genetics to relapse vulnerability (Bauer et al., 2007; Pinto et al., 2008; Wojnar et al., 2009; Hashimoto et al., 2011). The genes shown to be involved in risk to relapse also critically influence impulsivity, reward sensitivity, and inhibition (Boettiger et al., 2007; Mier et al., 2010). These complex relationships may be mediated through brain characteristics that can be assessed by MR methods, especially 1H MRS with its ability to measure metabolites reflecting specific cell types and tissue metabolism (Wojnar et al., 2009). Establishing links among functional genetic SNPs, brain neuroimaging phenotypes, and behavior will help build a mechanistic foundation for individual differences in behavior and related psychiatric disease, as has been described (Hariri, 2009). In this context, 1H MRS should be evaluated as a potential intermediate brain phenotype (endophenotype) of AUD, linking genotype to substance use behavior and psychopathology (Gottesman and Gould, 2003; Meyerhoff and Durazzo, 2008; Hutchison, 2010). With critical genetic effects on MRS-detectable metabolite measures forthcoming, research in MRS genetics will offer clues to the functional significance of genetic differences in AUD, and 1H MRS can potentially influence the future of clinical management of AUD via monitoring the efficacy of pharmacologic and behavioral interventions as a function of genotype.
Knowledge gaps and outlook
Brain 1H MRS has contributed significantly to our understanding of the pathophysiology and biologic basis of AUD at the tissue level and it has helped demonstrate and expand our understanding of the neuroplastic processes active during recovery from chronic drinking. 1H MRS has contributed significantly to the realization that brain changes in AUD are not associated solely with chronic heavy drinking, but with numerous conditions that are highly comorbid with chronic alcohol consumption. Possibly because of these complexities, retrospective cross-sectional MRS studies have not been able to clarify drinking patterns that are most damaging to the brain in the long term. We also still do not know how to interpret some of the observed metabolic brain alterations in chronic heavy drinkers – this is an area where carefully designed mechanistic animal studies can help inform human research in a mutually crossfertilizing way – and to what degree the observed metabolite abnormalities may be consistent with neuroinflammation (Sullivan and Zahr, 2008) or associated with chronic stress (Sinha, 2012).
Longitudinal studies during abstinence from alcohol have demonstrated some metabolic changes consistent with considerable brain plasticity even after several decades of chronic drinking. However, because of the difficulties associated with studying large numbers of abstinent alcohol-dependent individuals over extended periods of documented alcohol abstinence, it is unclear if brain metabolite levels recover completely into the “normal” control range or even if “recovered” alcohol-dependent individuals ought to be expected to display metabolite levels similar to those observed in age-matched non-alcoholic controls. Some of the metabolite abnormalities detected in alcohol dependence may be present before the development of AUD, may have little to do with heavy drinking per se, and should therefore not be expected to recover into the “normal” range anyway. Supporting this assertion are recent studies in alcohol-naïve adolescents with a positive FHA that show structure and function different from those of their FHA-negative counterparts, suggesting the possibility also of FHA-associated MRS abnormalities before significant alcohol exposure.
In addition, more longitudinal studies are needed to further understanding of risk factors versus consequences of excessive alcohol use and abuse. Such research may include studies during recovery from heavy drinking and during development of alcohol problems in adolescence and early adulthood as well as prospective studies of disease progression in non-treatment seekers. Any such studies need to be carefully controlled for comorbidities and personality traits that can affect common 1H MRS outcome measures in a way both similar to that of chronic heavy drinking and distinctly different (Abé et al., 2013). Although alcohol dependence is more common in men (5.4%) than women (2.3%) (Grant et al., 2004), women have been found to be specifically sensitive to the neurobiologic and neurocognitive effects of chronic alcohol consumption at equivalent drinking severity levels (Mann et al., 1992; Parsons and Nixon, 1998; Sullivan et al., 2004). More data are needed to understand gender effects on 1H MRS metabolite levels and their changes with heavy drinking and abstinence.
Understanding metabolic recovery processes, linking specific neurobiologic abnormalities and their cognitive correlates as well as their changes during abstinence to successful treatment outcome will help identify novel pharmacologic targets or pinpoint specific networks (e.g., frontal cortex exerting control over automatic behavior) that can be strengthened via cognitive behavioral therapy or cognitive remediation; such approaches are reminiscent of harnessing the brain’s intrinsic regenerative powers seen with rehabilitation therapy after a stroke or traumatic brain injury. 1H MRS can further help monitor treatment effects and clarify neuroplastic processes, monitor changes in levels of the neurotransmitters GABA and Glu that are common pharmacologic targets in AUD treatment research, and it can provide biomarkers of neurobiologic improvements as correlates of cognitive recovery. The clinical value of 1H MRS will increase substantially if it can be demonstrated that a few select in vivo metabolite measures can increase our abilities to predict successful treatment outcome, as this would be one way of focusing treatment resources on those at greatest risk of relapse and repeated treatments. While this sounds expensive, we should recognize that early detection of Alzheimer’s disease these days considers PET imaging a viable tool for screening those at greatest risk for the disease.
Most 1H MRS studies have been unable to demonstrate strong correlations between high quantity/frequency of alcohol consumption and brain metabolic abnormalities. This perceived lack of specificity may have hindered 1H MRS to become a more useful tool in understanding brain effects of AUD. However, the absence of such correlations should not be a reason for lack of concern about chronic heavy drinking nor for lack of a role of 1H MRS in yielding useful biomarkers of AUD to help inform better treatment approaches. Substantial evidence suggests that the brain of an alcohol-dependent individual is metabolically abnormal, and that abstinence from chronic consumption can help reverse some of the brain injury. Nevertheless, stopping alcohol intake is not the panacea for optimal brain health. It is rather outside circumstances that “form” the alcohol-dependent individual, personality characteristics, common comorbidities, genetic background, lifestyle (exercise, diet, and nutrition), and living environment that all add up to an individual’s neurometabolic profile. This has been woefully underestimated in the past, when brain alterations in alcohol dependence have often been understood as (or expected to be) straightforward consequences of chronic alcohol consumption. If we are to explicate these complex relationships in individuals struggling with AUD, we as researchers and program officials need to be better prepared to study complex relationships with appropriate scientific rigor that deal with the unique challenges of studying complex populations, including recruitment, diagnostic assessment, reproducibility, generalizability, and design of relevant clinical trials. Isolating chronic drinking experimentally has no doubt taught us a lot about brain abnormalities in alcohol dependence; however, studying our target population out of their social context does little to provide clinically useful biomarkers for informing and designing efficient and safe treatments that succeed in the real world. 1H MRS has a unique role to play here as part of the non-invasive MR armamentarium that can be applied repeatedly without known harm and at comparatively low overall cost (when applied judiciously) to studying salient brain changes associated with injury and recovery from addiction for the purpose of informing better and more efficient treatments.
ACKNOWLEDGMENTS
This review was made possible through support from NIH R01 AA10788 (DJM).
REFERENCES
- Abé C, Mon A, Durazzo TC, et al. Polysubstance and alcohol dependence: unique differences in magnetic resonance-derived brain metabolite concentrations. Drug Alcohol Depend. 2013;130:30–37. doi: 10.1016/j.drugalcdep.2012.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agarwal N, Renshaw PF. Proton MR spectroscopy-detectable major neurotransmitters of the brain: biology and possible clinical applications. AJNR Am J Neuroradiol. 2012;33:595–602. doi: 10.3174/ajnr.A2587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baler RD, Volkow ND. Drug addiction: the neurobiology of disrupted self-control. Trends Mol Med. 2006;12:559–566. doi: 10.1016/j.molmed.2006.10.005. [DOI] [PubMed] [Google Scholar]
- Balestrino M, Lensman M, Parodi M, et al. Role of creatine and phosphocreatine in neuronal protection from anoxic and ischemic damage. Amino Acids. 2002;23:221–229. doi: 10.1007/s00726-001-0133-3. [DOI] [PubMed] [Google Scholar]
- Bartsch AJ, Homola G, Biller A, et al. Manifestations of early brain recovery associated with abstinence from alcoholism. Brain. 2007;130:36–47. doi: 10.1093/brain/awl303. [DOI] [PubMed] [Google Scholar]
- Baslow MH. Brain N-acetylaspartate as a molecular water pump and its role in the etiology of Canavan disease: a mechanistic explanation. J Mol Neurosci. 2003a;21:185–190. doi: 10.1385/jmn:21:3:185. [DOI] [PubMed] [Google Scholar]
- Baslow MH. N-acetylaspartate in the vertebrate brain: metabolism and function. Neurochem Res. 2003b;28:941–953. doi: 10.1023/a:1023250721185. [DOI] [PubMed] [Google Scholar]
- Baslow MH, Guilfoyle DN. Using proton magnetic resonance imaging and spectroscopy to understand brain “activation”. Brain Lang. 2007;102:153–164. doi: 10.1016/j.bandl.2006.06.119. [DOI] [PubMed] [Google Scholar]
- Batouli SA, Sachdev PS, Wen W, et al. The heritability of brain metabolites on proton magnetic resonance spectroscopy in older individuals. Neuroimage. 2012;62:281–289. doi: 10.1016/j.neuroimage.2012.04.043. [DOI] [PubMed] [Google Scholar]
- Bauer LO, Covault J, Harel O, et al. Variation in GABRA2 predicts drinking behavior in project MATCH subjects. Alcohol Clin Exp Res. 2007;31:1780–1787. doi: 10.1111/j.1530-0277.2007.00517.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behar KL, Rothman DL, Petersen KF, et al. Preliminary evidence of low cortical GABA levels in localized 1H-MR spectra of alcohol-dependent and hepatic encephalopathy patients. Am J Psychiatry. 1999;156:952–954. doi: 10.1176/ajp.156.6.952. [DOI] [PubMed] [Google Scholar]
- Benarroch EE. N-acetylaspartate and N-acetylaspartyl-glutamate: neurobiology and clinical significance. Neurology. 2008;70:1353–1357. doi: 10.1212/01.wnl.0000311267.63292.6c. [DOI] [PubMed] [Google Scholar]
- Bendszus M, Weijers HG, Wiesbeck G, et al. Sequential MR imaging and proton MR spectroscopy in patients who underwent recent detoxification for chronic alcoholism: correlation with clinical and neuropsychological data. AJNR Am J Neuroradiol. 2001;22:1926–1932. [PMC free article] [PubMed] [Google Scholar]
- Bertolino A, Callicott JH, Mattay VS, et al. The effect of treatment with antipsychotic drugs on brain N-acetylaspartate measures in patients with schizophrenia. Biol Psychiatry. 2001;49:39–46. doi: 10.1016/s0006-3223(00)00997-5. [DOI] [PubMed] [Google Scholar]
- Bierut LJ, Dinwiddie SH, Begleiter H, et al. Familial transmission of substance dependence: alcohol, marijuana, cocaine, and habitual smoking: a report from the Collaborative Study on the Genetics of Alcoholism. Arch Gen Psychiatry. 1998;55:982–988. doi: 10.1001/archpsyc.55.11.982. [DOI] [PubMed] [Google Scholar]
- Boettiger CA, Mitchell JM, Tavares VC, et al. Immediate reward bias in humans: fronto-parietal networks and a role for the catechol-O-methyltransferase 158(Val/ Val) genotype. J Neurosci. 2007;27:14383–14391. doi: 10.1523/JNEUROSCI.2551-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brand A, Richter-Landsberg C, Leibfritz D. Multinuclear NMR studies on the energy metabolism of glial and neuronal cells. Dev Neurosci. 1993;15:289–298. doi: 10.1159/000111347. [DOI] [PubMed] [Google Scholar]
- Brenner RE, Munro PM, Williams SC, et al. The proton NMR spectrum in acute EAE: the significance of the change in the Cho:Cr ratio. Magn Reson Med. 1993;29:737–745. doi: 10.1002/mrm.1910290605. [DOI] [PubMed] [Google Scholar]
- Choi C, Bhardwaj PP, Kalra S, et al. Measurement of GABA and contaminants in gray and white matter in human brain in vivo. Magn Reson Med. 2007;58:27–33. doi: 10.1002/mrm.21275. [DOI] [PubMed] [Google Scholar]
- Cosgrove KP, Esterlis I, Mason GF, et al. Neuroimaging insights into the role of cortical GABA systems and the influence of nicotine on the recovery from alcohol dependence. Neuropharmacology. 2011;60:1318–1325. doi: 10.1016/j.neuropharm.2011.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daeppen JB, Smith TL, Danko GP, et al. Clinical correlates of cigarette smoking and nicotine dependence in alcohol-dependent men and women. The Collaborative Study Group on the Genetics of Alcoholism. Alcohol Alcohol. 2000;35:171–175. doi: 10.1093/alcalc/35.2.171. [DOI] [PubMed] [Google Scholar]
- Dawson DA, Goldstein RB, Grant BF. Rates and correlates of relapse among individuals in remission from DSM-IV alcohol dependence: a 3-year follow-up. Alcohol Clin Exp Res. 2007;31:2036–2045. doi: 10.1111/j.1530-0277.2007.00536.x. [DOI] [PubMed] [Google Scholar]
- De Stefano N, Matthews PM, Arnold DL. Reversible decreases in N-acetylaspartate after acute brain injury. Magn Reson Med. 1995;34:721–727. doi: 10.1002/mrm.1910340511. [DOI] [PubMed] [Google Scholar]
- De Witte P. Imbalance between neuroexcitatory and neuroinhibitory amino acids causes craving for ethanol. Addict Behav. 2004;29:1325–1339. doi: 10.1016/j.addbeh.2004.06.020. [DOI] [PubMed] [Google Scholar]
- Di Sclafani V, Finn P, Fein G. Treatment-naive active alcoholics have greater psychiatric comorbidity than normal controls but less than treated abstinent alcoholics. Drug Alcohol Depend. 2008;98:115–122. doi: 10.1016/j.drugalcdep.2008.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dlugos CA, Pentney RJ. Morphometric evidence that the total number of synapses on Purkinje neurons of old F344 rats is reduced after long-term ethanol treatment and restored to control levels after recovery. Alcohol Alcohol. 1997;32:161–172. doi: 10.1093/oxfordjournals.alcalc.a008250. [DOI] [PubMed] [Google Scholar]
- Duka T, Trick L, Nikolaou K, et al. Unique brain areas associated with abstinence control are damaged in multiply detoxified alcoholics. Biol Psychiatry. 2011;70:545–552. doi: 10.1016/j.biopsych.2011.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durazzo TC, Meyerhoff DJ. Neurobiological and neurocognitive effects of chronic cigarette smoking and alcoholism. Front Biosci. 2007;12:4079–4100. doi: 10.2741/2373. [DOI] [PubMed] [Google Scholar]
- Durazzo TC, Gazdzinski S, Banys P, et al. Cigarette smoking exacerbates chronic alcohol-induced brain damage: a preliminary metabolite imaging study. Alcohol Clin Exp Res. 2004;28:1849–1860. doi: 10.1097/01.alc.0000148112.92525.ac. [DOI] [PubMed] [Google Scholar]
- Durazzo TC, Gazdzinski S, Banys P, et al. Brain metabolite concentrations and neurocognition during short-term recovery from alcohol dependence: preliminary evidence of the effects of concurrent chronic cigarette smoking. Alcohol Clin Exp Res. 2006a;30:539–551. doi: 10.1111/j.1530-0277.2006.00060.x. [DOI] [PubMed] [Google Scholar]
- Durazzo TC, Rothlind JC, Gazdzinski S, et al. A comparison of neurocognitive function in nonsmoking and chronically smoking short-term abstinent alcoholics. Alcohol. 2006b;39:1–11. doi: 10.1016/j.alcohol.2006.06.006. [DOI] [PubMed] [Google Scholar]
- Durazzo TC, Gazdzinski S, Meyerhoff DJ. The neurobiological and neurocognitive consequences of chronic cigarette smoking in alcohol use disorders. Alcohol Alcohol. 2007;42:174–185. doi: 10.1093/alcalc/agm020. [DOI] [PubMed] [Google Scholar]
- Durazzo TC, Gazdzinski S, Yeh PH, et al. Combined neuroimaging, neurocognitive and psychiatric factors to predict alcohol consumption following treatment for alcohol dependence. Alcohol Alcohol. 2008;43:683–691. doi: 10.1093/alcalc/agn078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durazzo TC, Pathak V, Gazdzinski S, et al. Metabolite levels in the brain reward pathway discriminate those who remain abstinent from those who resume hazardous alcohol consumption after treatment for alcohol dependence. J Stud Alcohol Drugs. 2010a;71:278–289. doi: 10.15288/jsad.2010.71.278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durazzo T, Gazdzinski S, Mon A, et al. Cortical perfusion in alcohol-dependent individuals during short-term abstinence: relationships to resumption of hazardous drinking after treatment. Alcohol. 2010b;44:201–210. doi: 10.1016/j.alcohol.2010.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durazzo T, Hutchison K, Fryer S, et al. Associations of cigarette smoking and polymorphisms in brain-derived neurotrophic factor and catechol-O-methyltransferase with neurocognition in alcohol dependent individuals during early abstinence. Front Pharmacol. 2012;3(178) doi: 10.3389/fphar.2012.00178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durazzo TC, Mon A, Gazdzinski S, et al. Chronic cigarette smoking in alcohol dependence: associations with cortical thickness and N-acetylaspartate levels in the extended brain reward system. Addict Biol. 2013;18:379–391. doi: 10.1111/j.1369-1600.2011.00407.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ende G, Braus DF, Walter S, et al. Effects of age, medication, and illness duration on the N-acetyl aspartate signal of the anterior cingulate region in schizophrenia. Schizophr Res. 2000;41:389–395. doi: 10.1016/s0920-9964(99)00089-4. [DOI] [PubMed] [Google Scholar]
- Ende G, Welzel H, Walter S, et al. Monitoring the effects of chronic alcohol consumption and abstinence on brain metabolism: a longitudinal proton magnetic resonance spectroscopy study. Biol Psychiatry. 2005;58:974–980. doi: 10.1016/j.biopsych.2005.05.038. [DOI] [PubMed] [Google Scholar]
- Ende G, Walter S, Welzel H, et al. Alcohol consumption significantly influences the MR signal of frontal choline-containing compounds. Neuroimage. 2006;32:740–746. doi: 10.1016/j.neuroimage.2006.03.049. [DOI] [PubMed] [Google Scholar]
- Epperson CN, O’Malley S, Czarkowski KA, et al. Sex, GABA, and nicotine: the impact of smoking on cortical GABA levels across the menstrual cycle as measured with proton magnetic resonance spectroscopy. Biol Psychiatry. 2005;57:44–48. doi: 10.1016/j.biopsych.2004.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabricius K, Pakkenberg H, Pakkenberg B. No changes in neocortical cell volumes or glial cell numbers in chronic alcoholic subjects compared to control subjects. Alcohol Alcohol. 2007;42:400–406. doi: 10.1093/alcalc/agm007. [DOI] [PubMed] [Google Scholar]
- Falkenberg LE, Westerhausen R, Specht K, et al. Resting-state glutamate level in the anterior cingulate predicts blood-oxygen level-dependent response to cognitive control. Proc Natl Acad Sci U S A. 2012;109:5069–5073. doi: 10.1073/pnas.1115628109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fein G, Landman B. Treated and treatment-naive alcoholics come from different populations. Alcohol. 2005;35:19–26. doi: 10.1016/j.alcohol.2004.10.007. [DOI] [PubMed] [Google Scholar]
- Fein G, Meyerhoff DJ. Ethanol in human brain by magnetic resonance spectroscopy: correlation with blood and breath levels, relaxation, and magnetization transfer. Alcohol Clin Exp Res. 2000;24:1227–1235. [PMC free article] [PubMed] [Google Scholar]
- Fein G, Meyerhoff DJ, Di Sclafani V, et al. 1H magnetic resonance spectroscopic imaging separates neuronal from glial changes in alcohol-related brain atrophy. Chapter in NIAAA Research Monograph No. 27/Alcohol and Glial Cells. 1994:227–241. [Google Scholar]
- Fein G, Di Sclafani V, Cardenas VA, et al. Cortical gray matter loss in treatment-naive alcohol dependent individuals. Alcohol Clin Exp Res. 2002;26:558–564. [PMC free article] [PubMed] [Google Scholar]
- Ferguson KJ, MacLullich AM, Marshall I, et al. Magnetic resonance spectroscopy and cognitive function in healthy elderly men. Brain. 2002;125:2743–2749. doi: 10.1093/brain/awf278. [DOI] [PubMed] [Google Scholar]
- Fisher SK, Novak JE, Agranoff BW. Inositol and higher inositol phosphates in neural tissues: homeostasis, metabolism and functional significance. J Neurochem. 2002;82:736–754. doi: 10.1046/j.1471-4159.2002.01041.x. [DOI] [PubMed] [Google Scholar]
- Friend KB, Malloy PF, Sindelar HA. The effects of chronic nicotine and alcohol use on neurocognitive function. Addict Behav. 2005;30:193–202. doi: 10.1016/j.addbeh.2004.04.020. [DOI] [PubMed] [Google Scholar]
- Gallinat J, Kunz D, Lang UE, et al. Association between cerebral glutamate and human behaviour: the sensation seeking personality trait. Neuroimage. 2007a;34:671–678. doi: 10.1016/j.neuroimage.2006.10.004. [DOI] [PubMed] [Google Scholar]
- Gallinat J, Lang UE, Jacobsen LK, et al. Abnormal hippocampal neurochemistry in smokers: evidence from proton magnetic resonance spectroscopy at 3 T. J Clin Psychopharmacol. 2007b;27:80–84. doi: 10.1097/JCP.0b013e31802dffde. [DOI] [PubMed] [Google Scholar]
- Gallinat J, Schubert F, Bruhl R, et al. Met carriers of BDNF Val66Met genotype show increased N-acetylaspartate concentration in the anterior cingulate cortex. Neuroimage. 2010;49:767–771. doi: 10.1016/j.neuroimage.2009.08.018. [DOI] [PubMed] [Google Scholar]
- Gazdzinski S, Durazzo TC, Weiner MW, et al. Are treated alcoholics representative of the entire population with alcohol use disorders? A magnetic resonance study of brain injury. Alcohol. 2008a;42:67–76. doi: 10.1016/j.alcohol.2008.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gazdzinski S, Durazzo TC, Yeh PH, et al. Chronic cigarette smoking modulates injury and short-term recovery of the medial temporal lobe in alcoholics. Psychiatry Res. 2008b;162:133–145. doi: 10.1016/j.pscychresns.2007.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gazdzinski S, Durazzo TC, Mon A, et al. Body mass index is associated with brain injury in alcohol dependence – a multimodal magnetic resonance study. Alcohol Clin Exp Res. 2010;34:2089–2096. doi: 10.1111/j.1530-0277.2010.01305.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glass JM, Adams KM, Nigg JT, et al. Smoking is associated with neurocognitive deficits in alcoholism. Drug Alcohol Depend. 2006;82:119–126. doi: 10.1016/j.drugalcdep.2005.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein RZ, Craig AD, Bechara A, et al. The neurocircuitry of impaired insight in drug addiction. Trends Cogn Sci. 2009;13:372–380. doi: 10.1016/j.tics.2009.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goto N, Yoshimura R, Moriya J, et al. Critical examination of a correlation between brain gamma-aminobutyric acid (GABA) concentrations and a personality trait of extroversion in healthy volunteers as measured by a 3 Tesla proton magnetic resonance spectroscopy study. Psychiatry Res. 2010;182:53–57. doi: 10.1016/j.pscychresns.2009.11.002. [DOI] [PubMed] [Google Scholar]
- Gottesman II, Gould TD. The endophenotype concept in psychiatry: etymology and strategic intentions. Am J Psychiatry. 2003;160:636–645. doi: 10.1176/appi.ajp.160.4.636. [DOI] [PubMed] [Google Scholar]
- Govindaraju V, Young K, Maudsley AA. Proton NMR chemical shifts and coupling constants for brain metabolites. NMR Biomed. 2000;13:129–153. doi: 10.1002/1099-1492(200005)13:3<129::aid-nbm619>3.0.co;2-v. [DOI] [PubMed] [Google Scholar]
- Grant BF, Dawson DA, Stinson FS, et al. The 12-month prevalence and trends in DSM-IV alcohol abuse and dependence: United States, 1991–1992 and 2001–2002. Drug Alcohol Depend. 2004;74:223–234. doi: 10.1016/j.drugalcdep.2004.02.004. [DOI] [PubMed] [Google Scholar]
- Grimm S, Schubert F, Jaedke M, et al. Prefrontal cortex glutamate and extraversion. Soc Cogn Affect Neurosci. 2012;7:811–818. doi: 10.1093/scan/nsr056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gruber O, Hasan A, Scherk H, et al. Association of the brain-derived neurotrophic factor val66met polymorphism with magnetic resonance spectroscopic markers in the human hippocampus: in vivo evidence for effects on the glutamate system. Eur Arch Psychiatry Clin Neurosci. 2012;262:23–31. doi: 10.1007/s00406-011-0214-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haga KK, Khor YP, Farrall A, et al. A systematic review of brain metabolite changes, measured with 1H magnetic resonance spectroscopy, in healthy aging. Neurobiol Aging. 2009;30:353–363. doi: 10.1016/j.neurobiolaging.2007.07.005. [DOI] [PubMed] [Google Scholar]
- Hanson KL, Medina KL, Nagel BJ, et al. Hippocampal volumes in adolescents with and without a family history of alcoholism. Am J Drug Alcohol Abuse. 2010;36:161–167. doi: 10.3109/00952991003736397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding AJ, Wong A, Svoboda M, et al. Chronic alcohol consumption does not cause hippocampal neuron loss in humans. Hippocampus. 1997;7:78–87. doi: 10.1002/(SICI)1098-1063(1997)7:1<78::AID-HIPO8>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- Hariri AR. The neurobiology of individual differences in complex behavioral traits. Annu Rev Neurosci. 2009;32:225–247. doi: 10.1146/annurev.neuro.051508.135335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harper C. The neuropathology of alcohol-specific brain damage, or does alcohol damage the brain? J Neuropathol Exp Neurol. 1998;57:101–110. doi: 10.1097/00005072-199802000-00001. [DOI] [PubMed] [Google Scholar]
- Harper C. The neuropathology of alcohol-related brain damage. Alcohol Alcohol. 2009;44:136–140. doi: 10.1093/alcalc/agn102. [DOI] [PubMed] [Google Scholar]
- Harper C, Kril J. Patterns of neuronal loss in the cerebral cortex in chronic alcoholic patients. J Neurol Sci. 1989;92:81–89. doi: 10.1016/0022-510x(89)90177-9. [DOI] [PubMed] [Google Scholar]
- Harris BR, Gibson DA, Prendergast MA, et al. The neurotoxicity induced by ethanol withdrawal in mature organotypic hippocampal slices might involve cross-talk between metabotropic glutamate type 5 receptors and N-methyl-D-aspartate receptors. Alcohol Clin Exp Res. 2003;27:1724–1735. doi: 10.1097/01.ALC.0000093601.33119.E3. [DOI] [PubMed] [Google Scholar]
- Hashimoto JG, Forquer MR, Tanchuck MA, et al. Importance of genetic background for risk of relapse shown in altered prefrontal cortex gene expression during abstinence following chronic alcohol intoxication. Neuroscience. 2011;173:57–75. doi: 10.1016/j.neuroscience.2010.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasin DS, Stinson FS, Ogburn E, et al. Prevalence, correlates, disability, and comorbidity of DSM-IV alcohol abuse and dependence in the United States: results from the National Epidemiologic Survey on Alcohol and Related Conditions. Arch Gen Psychiatry. 2007;64:830–842. doi: 10.1001/archpsyc.64.7.830. [DOI] [PubMed] [Google Scholar]
- Heatherton TF, Wagner DD. Cognitive neuroscience of self-regulation failure. Trends Cogn Sci. 2011;15:132–139. doi: 10.1016/j.tics.2010.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hermann D, Weber-Fahr W, Sartorius A, et al. Translational magnetic resonance spectroscopy reveals excessive central glutamate levels during alcohol withdrawal in humans and rats. Biol Psychiatry. 2012;71:1015–1021. doi: 10.1016/j.biopsych.2011.07.034. [DOI] [PubMed] [Google Scholar]
- Herting MM, Schwartz D, Mitchell SH, et al. Delay discounting behavior and white matter microstructure abnormalities in youth with a family history of alcoholism. Alcohol Clin Exp Res. 2010;34:1590–1602. doi: 10.1111/j.1530-0277.2010.01244.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hetherington HP, Newcomer BR, Pan JW. Measurements of human cerebral GABA at 4.1 T using numerically optimized editing pulses. Magn Reson Med. 1998;39:6–10. doi: 10.1002/mrm.1910390103. [DOI] [PubMed] [Google Scholar]
- Hoerst M, Weber-Fahr W, Tunc-Skarka N, et al. Glutamate levels in the anterior cingulate cortex correlate with self-reported impulsivity in patients with borderline personality disorder and healthy controls. Arch Gen Psychiatry. 2010;67:946–954. doi: 10.1001/archgenpsychiatry.2010.93. [DOI] [PubMed] [Google Scholar]
- Hugg JW, Kuzniecky RI, Gilliam FG, et al. Normalization of contralateral metabolic function following temporal lobectomy demonstrated by h-1 magnetic resonance spectroscopic imaging. Ann Neurol. 1996;40:236–239. doi: 10.1002/ana.410400215. [DOI] [PubMed] [Google Scholar]
- Hurd R, Sailasuta N, Srinivasan R, et al. Measurement of brain glutamate using TE-averaged PRESS at 3T. Magn Reson Med. 2004;51:435–440. doi: 10.1002/mrm.20007. [DOI] [PubMed] [Google Scholar]
- Hutchison KE. Substance use disorders: realizing the promise of pharmacogenomics and personalized medicine. Annu Rev Clin Psychol. 2010;6:577–589. doi: 10.1146/annurev.clinpsy.121208.131441. [DOI] [PubMed] [Google Scholar]
- Jagannathan NR, Desai NG, Raghunathan P. Brain metabolite changes in alcoholism: an in vivo proton magnetic resonance spectroscopy (MRS) study. Magn Reson Imaging. 1996;14:553–557. doi: 10.1016/0730-725x(96)00048-3. [DOI] [PubMed] [Google Scholar]
- Jansen JF, Backes WH, Nicolay K, et al. 1H MR spectroscopy of the brain: absolute quantification of metabolites. Radiology. 2006;240:318–332. doi: 10.1148/radiol.2402050314. [DOI] [PubMed] [Google Scholar]
- Jensen GB, Pakkenberg B. Do alcoholics drink their neurons away? Lancet. 1993;342:1201–1204. doi: 10.1016/0140-6736(93)92185-v. [DOI] [PubMed] [Google Scholar]
- John U, Meyer C, Rumpf HJ, et al. Strength of the relationship between tobacco smoking, nicotine dependence and the severity of alcohol dependence syndrome criteria in a population-based sample. Alcohol Alcohol. 2003;38:606–612. doi: 10.1093/alcalc/agg122. [DOI] [PubMed] [Google Scholar]
- Johnson BA. Recent advances in the development of treatments for alcohol and cocaine dependence: focus on topiramate and other modulators of GABA or glutamate function. CNS Drugs. 2005;19:873–896. doi: 10.2165/00023210-200519100-00005. [DOI] [PubMed] [Google Scholar]
- Kaiser LG, Young K, Meyerhoff DJ, et al. A detailed analysis of localized J-difference GABA editing: theoretical and experimental study at 4 T. NMR Biomed. 2008;21:22–32. doi: 10.1002/nbm.1150. [DOI] [PubMed] [Google Scholar]
- Kalivas PW, O’Brien C. Drug addiction as a pathology of staged neuroplasticity. Neuropsychopharmacology. 2008;33:166–180. doi: 10.1038/sj.npp.1301564. [DOI] [PubMed] [Google Scholar]
- Kam K, Nicoll R. Excitatory synaptic transmission persists independently of the glutamate-glutamine cycle. J Neurosci. 2007;27:9192–9200. doi: 10.1523/JNEUROSCI.1198-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ke Y, Cohen BM, Lowen S, et al. Biexponential transverse relaxation (T(2)) of the proton MRS creatine resonance in human brain. Magn Reson Med. 2002;47:232–238. doi: 10.1002/mrm.10063. [DOI] [PubMed] [Google Scholar]
- Ke Y, Streeter CC, Nassar LE, et al. Frontal lobe GABA levels in cocaine dependence: a two-dimensional, J-resolved magnetic resonance spectroscopy study. Psychiatry Res. 2004;130:283–293. doi: 10.1016/j.pscychresns.2003.12.001. [DOI] [PubMed] [Google Scholar]
- Kedia S, Sell MA, Relyea G. Monoversus polydrug abuse patterns among publicly funded clients. Subst Abuse Treat Prev Policy. 2007;2:33. doi: 10.1186/1747-597X-2-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kendler KS, Prescott CA, Myers J, et al. The structure of genetic and environmental risk factors for common psychiatric and substance use disorders in men and women. Arch Gen Psychiatry. 2003;60:929–937. doi: 10.1001/archpsyc.60.9.929. [DOI] [PubMed] [Google Scholar]
- Khiat A, Bard C, Lacroix A, et al. Recovery of the brain choline level in treated Cushing’s patients as monitored by proton magnetic resonance spectroscopy. Brain Res. 2000;862:301–307. doi: 10.1016/s0006-8993(00)02147-8. [DOI] [PubMed] [Google Scholar]
- Korbo L. Glial cell loss in the hippocampus of alcoholics. Alcohol Clin Exp Res. 1999;23:164–168. [PubMed] [Google Scholar]
- Kril JJ, Halliday GM. Brain shrinkage in alcoholics: a decade on and what have we learned? Prog Neurobiol. 1999;58:381–387. doi: 10.1016/s0301-0082(98)00091-4. [DOI] [PubMed] [Google Scholar]
- Kril JJ, Harding AJ, Wong A, et al. Is the hippocampus damaged in alcoholics with memory loss? Alcohol Clin Exp Res. 1996;20:76A. [Google Scholar]
- Kroenke CD, Flory GS, Park B, et al. Chronic ethanol (EtOH) consumption differentially alters gray and white matter EtOH methyl (1) H magnetic resonance intensity in the primate brain. Alcohol Clin Exp Res. 2013;37:1325–1332. doi: 10.1111/acer.12097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee E, Jang DP, Kim JJ, et al. Alteration of brain metabolites in young alcoholics without structural changes. Neuroreport. 2007;18:1511–1514. doi: 10.1097/WNR.0b013e3282ef7625. [DOI] [PubMed] [Google Scholar]
- Licata SC, Renshaw PF. Neurochemistry of drug action: insights from proton magnetic resonance spectroscopic imaging and their relevance to addiction. Ann N Y Acad Sci. 2010;1187:148–171. doi: 10.1111/j.1749-6632.2009.05143.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loeber S, Duka T, Welzel Marquez H, et al. Effects of repeated withdrawal from alcohol on recovery of cognitive impairment under abstinence and rate of relapse. Alcohol Alcohol. 2010;45:541–547. doi: 10.1093/alcalc/agq065. [DOI] [PubMed] [Google Scholar]
- Maddock RJ, Buonocore MH. MR spectroscopic studies of the brain in psychiatric disorders. Curr Top Behav Neurosci. 2012 doi: 10.1007/7854_2011_197. (epub ahead of print) PubMed PMID: 22294088. [DOI] [PubMed] [Google Scholar]
- Maisto SA, Connors GJ. Relapse in the addictive behaviors: integration and future directions. Clin Psychol Rev. 2006;26:229–231. doi: 10.1016/j.cpr.2005.11.009. [DOI] [PubMed] [Google Scholar]
- Mann K, Hermann D. Individualised treatment in alcohol-dependent patients. Eur Arch Psychiatry Clin Neurosci. 2010;260(Suppl 2):S116–S120. doi: 10.1007/s00406-010-0153-7. [DOI] [PubMed] [Google Scholar]
- Mann K, Batra A, Gunthner A, et al. Do women develop alcoholic brain damage more readily than men? Alcohol Clin Exp Res. 1992:1052–1056. doi: 10.1111/j.1530-0277.1992.tb00698.x. [DOI] [PubMed] [Google Scholar]
- Mansell D, Penk W, Hankin CS, et al. The illness burden of alcohol-related disorders among VA patients: the veterans health study. J Ambul Care Manage. 2006;29:61–70. doi: 10.1097/00004479-200601000-00007. [DOI] [PubMed] [Google Scholar]
- Mansfield P. Multi-planar image formation using NMR spin echoes. J Phys C: Solid State Phys. 1977;10:L55–L58. [Google Scholar]
- Mansfield P, Maudsley AA. Medical imaging by NMR. Br J Radiol. 1977;50:188–194. doi: 10.1259/0007-1285-50-591-188. [DOI] [PubMed] [Google Scholar]
- Marjanska M, Auerbach EJ, Valabregue R, et al. Localized 1H NMR spectroscopy in different regions of human brain in vivo at 7 T: T2 relaxation times and concentrations of cerebral metabolites. NMR Biomed. 2012;25:332–339. doi: 10.1002/nbm.1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin PR, Gibbs SJ, Nimmerrichter AA, et al. Brain proton magnetic resonance spectroscopy studies in recently abstinent alcoholics. Alcohol Clin Exp Res. 1995;19:1078–1082. doi: 10.1111/j.1530-0277.1995.tb00992.x. [DOI] [PubMed] [Google Scholar]
- Mason GF, Petrakis IL, de Graaf RA, et al. Cortical gamma-aminobutyric acid levels and the recovery from ethanol dependence: preliminary evidence of modification by cigarette smoking. Biol Psychiatry. 2006;59:85–93. doi: 10.1016/j.biopsych.2005.06.009. [DOI] [PubMed] [Google Scholar]
- Maudsley AA, Govind V, Arheart KL. Associations of age, gender and body mass with 1H MR-observed brain metabolites and tissue distributions. NMR Biomed. 2012;25:580–593. doi: 10.1002/nbm.1775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayfield RD, Harris RA, Schuckit MA. Genetic factors influencing alcohol dependence. Br J Pharmacol. 2008;154:275–287. doi: 10.1038/bjp.2008.88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medina KL, Shear PK, Schafer J, et al. Cognitive functioning and length of abstinence in polysubstance dependent men. Arch Clin Neuropsychol. 2004;19:245–258. doi: 10.1016/S0887-6177(03)00043-X. [DOI] [PubMed] [Google Scholar]
- Mekle R, Mlynarik V, Gambarota G, et al. MR spectroscopy of the human brain with enhanced signal intensity at ultrashort echo times on a clinical platform at 3T and 7T. Magn Reson Med. 2009;61:1279–1285. doi: 10.1002/mrm.21961. [DOI] [PubMed] [Google Scholar]
- Mertens JR, Lu YW, Parthasarathy S, et al. Medical and psychiatric conditions of alcohol and drug treatment patients in an HMO: comparison with matched controls. Arch Intern Med. 2003;163:2511–2517. doi: 10.1001/archinte.163.20.2511. [DOI] [PubMed] [Google Scholar]
- Meyerhoff DJ, Durazzo TC. Proton magnetic resonance spectroscopy in alcohol use disorders: a potential new endophenotype? Alcohol Clin Exp Res. 2008;32:1146–1158. doi: 10.1111/j.1530-0277.2008.00695.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyerhoff D, Blumenfeld R, Truran D, et al. Effects of heavy drinking, binge drinking, and family history of alcoholism on regional brain metabolites. Alcohol Clin Exp Res. 2004;28:650–661. doi: 10.1097/01.ALC.0000121805.12350.CA. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyerhoff DJ, Durazzo TC, Hutchison K, et al. Associations between BDNF Val66Met genotype and brain metabolite concentrations in alcohol dependence. Alcohol Clin Exp Res. 2011;35(134A) [Google Scholar]
- Mier D, Kirsch P, Meyer-Lindenberg A. Neural substrates of pleiotropic action of genetic variation in COMT: a meta-analysis. Mol Psychiatry. 2010;15:918–927. doi: 10.1038/mp.2009.36. [DOI] [PubMed] [Google Scholar]
- Miguel-Hidalgo JJ, Wei J, Andrew M, et al. Glia pathology in the prefrontal cortex in alcohol dependence with and without depressive symptoms. Biol Psychiatry. 2002;52:1121–1133. doi: 10.1016/s0006-3223(02)01439-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miguel-Hidalgo JJ, Overholser JC, Meltzer HY, et al. Reduced glial and neuronal packing density in the orbitofrontal cortex in alcohol dependence and its relationship with suicide and duration of alcohol dependence. Alcohol Clin Exp Res. 2006;30:1845–1855. doi: 10.1111/j.1530-0277.2006.00221.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller BL. A review of chemical issues in 1H NMR spectroscopy: N-Acetyl-L-aspartate, creatine and choline. NMR Biomed. 1991;4:47–52. doi: 10.1002/nbm.1940040203. [DOI] [PubMed] [Google Scholar]
- Miller BL, Chang L, Booth R, et al. In vivo 1H MRS choline: correlation with in vitro chemistry/histology. Life Sci. 1996;58:1929–1935. doi: 10.1016/0024-3205(96)00182-8. [DOI] [PubMed] [Google Scholar]
- Modi S, Bhattacharya M, Kumar P, et al. Brain metabolite changes in alcoholism: localized proton magnetic resonance spectroscopy study of the occipital lobe. Eur J Radiol. 2011;79:96–100. doi: 10.1016/j.ejrad.2009.11.003. [DOI] [PubMed] [Google Scholar]
- Moffett JR, Namboodiri MA, Cangro CB, et al. Immunohistochemical localization of N-acetylaspartate in rat brain. Neuroreport. 1991;2:131–134. doi: 10.1097/00001756-199103000-00005. [DOI] [PubMed] [Google Scholar]
- Mon A, Durazzo T, Meyerhoff DJ. Glutamate, GABA, and other cortical metabolite concentrations during early abstinence from alcohol and their associations with neurocognitive changes. Drug Alcohol Depend. 2012;125:27–36. doi: 10.1016/j.drugalcdep.2012.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mon A, Durazzo TC, Gazdzinski S, et al. Brain-derived neurotrophic factor (BDNF) genotype is associated with lobar gray and white matter volume recovery in abstinent alcohol dependent individuals. Genes Brain Behav. 2013;12:98–107. doi: 10.1111/j.1601-183X.2012.00854.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moss HB, Chen CM, Yi HY. Subtypes of alcohol dependence in a nationally representative sample. Drug Alcohol Depend. 2007;91:149–158. doi: 10.1016/j.drugalcdep.2007.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Neill J, Cardenas VA, Meyerhoff DJ. Effects of abstinence on the brain: quantitative magnetic resonance imaging and magnetic resonance spectroscopic imaging in chronic alcohol abuse. Alcohol Clin Exp Res. 2001;25:1673–1682. [PubMed] [Google Scholar]
- Öz G, Alger JR, Barker PB, et al. Clinical proton MR spectroscopy in central nervous system disorders: The MRS Consensus Group. Radiology. 2014;279:658–679. doi: 10.1148/radiol.13130531. 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan JW, Takahashi K. Interdependence of N-acetyl aspartate and high-energy phosphates in healthy human brain. Ann Neurol. 2005;57:92–97. doi: 10.1002/ana.20317. [DOI] [PubMed] [Google Scholar]
- Parks MH, Dawant BM, Riddle WR, et al. Longitudinal brain metabolic characterization of chronic alcoholics with proton magnetic resonance spectroscopy. Alcohol Clin Exp Res. 2002;26:1368–1380. doi: 10.1097/01.ALC.0000029598.07833.2D. [DOI] [PubMed] [Google Scholar]
- Parsons OA, Nixon SJ. Cognitive functioning in sober social drinkers: a review of the research since 1986. J Stud Alcohol. 1998;59:180–190. doi: 10.15288/jsa.1998.59.180. [DOI] [PubMed] [Google Scholar]
- Pettegrew JW, Kopp SJ, Minshew NJ, et al. 31P nuclear magnetic resonance studies of phosphoglyceride metabolism in developing and degenerating brain: preliminary observations. J Neuropathol Exp Neurol. 1987;46:419–430. doi: 10.1097/00005072-198707000-00002. [DOI] [PubMed] [Google Scholar]
- Pettegrew JW, Panchalingam K, Withers G, et al. Changes in brain energy and phospholipid metabolism during development and aging in the Fischer 344 rat. J Neuropathol Exp Neurol. 1990;49:237–249. doi: 10.1097/00005072-199005000-00005. [DOI] [PubMed] [Google Scholar]
- Pfefferbaum A, Sullivan EV, Mathalon DH, et al. Longitudinal changes in magnetic resonance imaging brain volumes in abstinent and relapsed alcoholics. Alcohol Clin Exp Res. 1995;19:1177–1191. doi: 10.1111/j.1530-0277.1995.tb01598.x. [DOI] [PubMed] [Google Scholar]
- Pinto E, Reggers J, Gorwood P, et al. The short allele of the serotonin transporter promoter polymorphism influences relapse in alcohol dependence. Alcohol Alcohol. 2008;43:398–400. doi: 10.1093/alcalc/agn015. [DOI] [PubMed] [Google Scholar]
- Pouwels PJ, Frahm J. Regional metabolite concentrations in human brain as determined by quantitative localized proton MRS. Magn Reson Med. 1998;39:53–60. doi: 10.1002/mrm.1910390110. [DOI] [PubMed] [Google Scholar]
- Prendergast MA, Harris BR, Mayer S, et al. Chronic, but not acute, nicotine exposure attenuates ethanol withdrawal-induced hippocampal damage in vitro. Alcohol Clin Exp Res. 2000;24:1583–1592. [PubMed] [Google Scholar]
- Rigotti DJ, Inglese M, Gonen O. Whole-brain N-acetylaspartate as a surrogate marker of neuronal damage in diffuse neurologic disorders. AJNR Am J Neuroradiol. 2007;28:1843–1849. doi: 10.3174/ajnr.A0774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romberger DJ, Grant K. Alcohol consumption and smoking status: the role of smoking cessation. Biomed Pharmacother. 2004;58:77–83. doi: 10.1016/j.biopha.2003.12.002. [DOI] [PubMed] [Google Scholar]
- Room R. Smoking and drinking as complementary behaviours. Biomed Pharmacother. 2004;58:111–115. doi: 10.1016/j.biopha.2003.12.003. [DOI] [PubMed] [Google Scholar]
- Rosen Y, Lenkinski RE. Recent advances in magnetic resonance neurospectroscopy. Neurotherapeutics. 2007;4:330–345. doi: 10.1016/j.nurt.2007.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross B, Bluml S. Magnetic resonance spectroscopy of the human brain. Anat Rec. 2001;265:54–84. doi: 10.1002/ar.1058. [DOI] [PubMed] [Google Scholar]
- Ross B, Michaelis T. Clinical applications of magnetic resonance spectroscopy. Magn Reson Q. 1994;10:191–247. [PubMed] [Google Scholar]
- Rothman DL, Petroff OA, Behar KL, et al. Localized 1H NMR measurements of gamma-aminobutyric acid in human brain in vivo. Proc Natl Acad Sci U S A. 1993;90:5662–5666. doi: 10.1073/pnas.90.12.5662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanacora G, Gueorguieva R, Epperson CN, et al. Subtype-specific alterations of gamma-aminobutyric acid and glutamate in patients with major depression. Arch Gen Psychiatry. 2004;61:705–713. doi: 10.1001/archpsyc.61.7.705. [DOI] [PubMed] [Google Scholar]
- Sartorius A, Lugenbiel P, Mahlstedt MM, et al. Proton magnetic resonance spectroscopic creatine correlates with creatine transporter protein density in rat brain. J Neurosci Methods. 2008;172:215–219. doi: 10.1016/j.jneumeth.2008.04.028. [DOI] [PubMed] [Google Scholar]
- Schmaal L, Veltman DJ, Nederveen A, et al. N-acetylcysteine normalizes glutamate levels in cocaine-dependent patients: a randomized crossover magnetic resonance spectroscopy study. Neuropsychopharmacology. 2012;37:2143–2152. doi: 10.1038/npp.2012.66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schubert F, Gallinat J, Seifert F, et al. Glutamate concentrations in human brain using single voxel proton magnetic resonance spectroscopy at 3 Tesla. Neuroimage. 2004;21:1762–1771. doi: 10.1016/j.neuroimage.2003.11.014. [DOI] [PubMed] [Google Scholar]
- Schuckit MA, Edenberg HJ, Kalmijn J, et al. A genome-wide search for genes that relate to a low level of response to alcohol. Alcohol Clin Exp Res. 2001;25:323–329. [PubMed] [Google Scholar]
- Schuff N, Ezekiel F, Gamst AC, et al. Region and tissue differences of metabolites in normally aged brain using multislice 1H magnetic resonance spectroscopic imaging. Magn Reson Med. 2001;45:899–907. doi: 10.1002/mrm.1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwab ME, Bartholdi D. Degeneration and regeneration of axons in the lesioned spinal cord. Physiol Rev. 1996;76:319–370. doi: 10.1152/physrev.1996.76.2.319. [DOI] [PubMed] [Google Scholar]
- Schweinsburg BC, Taylor MJ, Videen JS, et al. Elevated myo-inositol in gray matter of recently detoxified but not long-term alcoholics: a preliminary MR spectroscopy study. Alcohol Clin Exp Res. 2000;24:699–770. [PubMed] [Google Scholar]
- Schweinsburg BC, Taylor MJ, Alhassoon OM, et al. Chemical pathology in brain white matter of recently detoxified alcoholics: a 1H magnetic resonance spectroscopy investigation of alcohol-associated frontal lobe injury. Alcohol Clin Exp Res. 2001;25:924–934. [PubMed] [Google Scholar]
- Schweinsburg BC, Alhassoon OM, Taylor MJ, et al. Effects of alcoholism and gender on brain metabolism. Am J Psychiatry. 2003;160:1180–1183. doi: 10.1176/appi.ajp.160.6.1180. [DOI] [PubMed] [Google Scholar]
- Seitz D, Widmann U, Seeger U, et al. Localized proton magnetic resonance spectroscopy of the cerebellum in detoxifing alcoholics. Alcohol Clin Exp Res. 1999;23:158–163. [PubMed] [Google Scholar]
- Simmons ML, Frondoza CG, Coyle JT. Immunocytochemical localization of N-acetyl-aspartate with monoclonal antibodies. Neuroscience. 1991;45:37–45. doi: 10.1016/0306-4522(91)90101-s. [DOI] [PubMed] [Google Scholar]
- Sinha R. How does stress lead to risk of alcohol relapse? Alcohol Res Curr Rev. 2012;34:432–440. [PMC free article] [PubMed] [Google Scholar]
- Stern AJ, Savostyanova AA, Goldman A, et al. Impact of the brain-derived neurotrophic factor Val66Met polymorphism on levels of hippocampal N-acetyl-aspartate assessed by magnetic resonance spectroscopic imaging at 3 Tesla. Biol Psychiatry. 2008;64:856–862. doi: 10.1016/j.biopsych.2008.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stinson FS, Grant BF, Dawson DA, et al. Comorbidity between DSM-IV alcohol and specific drug use disorders in the United States: results from the National Epidemiologic Survey on Alcohol and Related Conditions. Drug Alcohol Depend. 2005;80:105–116. doi: 10.1016/j.drugalcdep.2005.03.009. [DOI] [PubMed] [Google Scholar]
- Sullivan EV. NIAAA Research Monograph No. 34: Human brain vulnerability to alcoholism: Evidence from neuroimaging studies. In: Noronha A, Eckardt M, Warren K, editors. Review of NIAAA’s neuroscience and behavioral research portfolio. National Institute on Alcohol Abuse and Alcoholism; Bethesda, MD: 2000. pp. 473–508. [Google Scholar]
- Sullivan EV, Zahr NM. Neuroinflammation as a neurotoxic mechanism in alcoholism: commentary on “Increased MCP-1 and microglia in various regions of human alcoholic brain”. Exp Neurol. 2008;213:10–17. doi: 10.1016/j.expneurol.2008.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan EV, Rosenbloom M, Serventi KL, et al. Effects of age and sex on volumes of the thalamus, pons, and cortex. Neurobiol Aging. 2004;25:185–192. doi: 10.1016/s0197-4580(03)00044-7. [DOI] [PubMed] [Google Scholar]
- Tessner KD, Hill SY. Neural circuitry associated with risk for alcohol use disorders. Neuropsychol Rev. 2010;20:1–20. doi: 10.1007/s11065-009-9111-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thoma R, Mullins P, Ruhl D, et al. Perturbation of the glutamate-glutamine system in alcohol dependence and remission. Neuropsychopharmacology. 2011;36:1359–1365. doi: 10.1038/npp.2011.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tkác I, Andersen P, Adriany G, et al. In vivo 1H NMR spectroscopy of the human brain at 7 T. Magn Reson Med. 2001;46:451–456. doi: 10.1002/mrm.1213. [DOI] [PubMed] [Google Scholar]
- Umhau JC, Momenan R, Schwandt ML, et al. Effect of acamprosate on magnetic resonance spectroscopy measures of central glutamate in detoxified alcohol-dependent individuals: a randomized controlled experimental medicine study. Arch Gen Psychiatry. 2010;67:1069–1077. doi: 10.1001/archgenpsychiatry.2010.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkatesh SK, Gupta RK, Pal L, et al. Spectroscopic increase in choline signal is a nonspecific marker for differentiation of infective/inflammatory from neoplastic lesions of the brain. J Magn Reson Imaging. 2001;14:8–15. doi: 10.1002/jmri.1144. [DOI] [PubMed] [Google Scholar]
- Vion-Dury J, Meyerhoff DJ, Cozzone PJ, et al. What might be the impact on neurology of the analysis of brain metabolism by in vivo magnetic resonance spectroscopy? J Neurol. 1994;241:354–371. doi: 10.1007/BF02033352. [DOI] [PubMed] [Google Scholar]
- Volkow ND, Wang GJ, Begleiter H, et al. High levels of dopamine D2 receptors in unaffected members of alcoholic families: possible protective factors. Arch Gen Psychiatry. 2006;63:999–1008. doi: 10.1001/archpsyc.63.9.999. [DOI] [PubMed] [Google Scholar]
- Volkow ND, Wang G-J, Fowler JS, et al. Addiction: Beyond dopamine reward circuitry. Proc Natl Acad Sci. 2011;108:15037–15042. doi: 10.1073/pnas.1010654108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang JJ, Durazzo TC, Gazdzinski S, et al. MRSI and DTI: a multimodal approach for improved detection of white matter abnormalities in alcohol and nicotine dependence. NMR Biomed. 2009;22:516–522. doi: 10.1002/nbm.1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wojnar M, Brower KJ, Strobbe S, et al. Association between Val66Met brain-derived neurotrophic factor (BDNF) gene polymorphism and post-treatment relapse in alcohol dependence. Alcohol Clin Exp Res. 2009;33:693–702. doi: 10.1111/j.1530-0277.2008.00886.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia Y, Ma D, Hu J, et al. Effect of metabotropic glutamate receptor 3 genotype on N-acetylaspartate levels and neurocognition in non-smoking, active alcoholics. Behav Brain Funct. 2012;8:42. doi: 10.1186/1744-9081-8-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xin L, Mlynarik V, Lei H, et al. Influence of regional macromolecule baseline on the quantification of neuro-chemical profile in rat brain; Paper read at ISMRM; Stockholm, Sweden. 2010. [Google Scholar]
- Yeo RA, Gangestad SW, Gasparovic C, et al. Rare copy number deletions predict individual variation in human brain metabolite concentrations in individuals with alcohol use disorders. Biol Psychiatry. 2011;70:537–544. doi: 10.1016/j.biopsych.2011.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeo RA, Thoma RJ, Gasparovic C, et al. Neurometabolite concentration and clinical features of chronic alcohol use: a proton magnetic resonance spectroscopy study. Psychiatry Res. 2013;211:141–147. doi: 10.1016/j.pscychresns.2012.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zahr NM, Mayer D, Pfefferbaum A, et al. Low striatal glutamate levels underlie cognitive decline in the elderly: evidence from in vivo molecular spectroscopy. Cereb Cortex. 2008;18:2241–2250. doi: 10.1093/cercor/bhm250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zahr NM, Mayer D, Rohlfing T, et al. Brain injury and recovery following binge ethanol: evidence from in vivo magnetic resonance spectroscopy. Biol Psychiatry. 2010;67:846–854. doi: 10.1016/j.biopsych.2009.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zahr NM, Mayer D, Rohlfing T, et al. In vivo glutamate measured with magnetic resonance spectroscopy: behavioral correlates in aging. Neurobiol Aging. 2013;34:1265–1276. doi: 10.1016/j.neurobiolaging.2012.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu PJ, Chiappinelli VA. Nicotine modulates evoked GABAergic transmission in the brain. J Neurophysiol. 1999;82:3041–3045. doi: 10.1152/jn.1999.82.6.3041. [DOI] [PubMed] [Google Scholar]
- Zywiak WH, Stout RL, Trefry WB, et al. Alcohol relapse repetition, gender, and predictive validity. J Subst Abuse Treat. 2006;30:349–353. doi: 10.1016/j.jsat.2006.03.004. [DOI] [PubMed] [Google Scholar]