

Abstract
The effects of diclofenac (Dic), an acetic acid derivative‐type nonsteroidal anti‐inflammatory drug, were examined on the function of transient receptor potential (TRP) melastatin (TRPM) 3 (TRPM3) in human embryonic kidney 293 cell‐line (HEK293) cells with recombinant human TRPM3 isoforms (TRPM31325, TRPM3‐3, TRPM3‐9, and TRPM3‐S) and in a neuroblastoma cell line human neuroblastoma IMR‐32 cells (IMR‐32 cells) derived from human peripheral neurons. TRPM3 responses evoked by pregnenolone sulfate (PregS) were effectively inhibited by Dic in a concentration‐dependent manner in Ca2+ measurement and electrophysiological assays. The apparent IC 50 for PregS‐induced Ca2+ response of TRPM31325, TRPM3‐3, and TRPM3‐9 was calculated to be 18.8, 42.5, and 7.1 μmol/L, respectively. The TRPM3‐dependent Ca2+ responses evoked by nifedipine, another TRPM3 agonist, were also significantly inhibited by Dic. In contrast, aceclofenac, an acetoxymethyl analog of Dic, had no effects on PregS‐induced TRPM3 responses. Constitutive channel activity of TRPM3‐S without TRPM3 agonists was substantially inhibited by Dic, ruling out the possibility of interaction of Dic against TRPM3 agonists to the channel binding sites. Moreover, Dic reversibly inhibited TRPM3 single‐channel activity recorded in excised outside‐out patches without affecting the channel conductance. In differentiated neuronal IMR‐32 cells with endogenous TRPM3, Dic inhibited PregS‐evoked Ca2+ responses with an apparent IC 50 of 17.1 μmol/L. Taken together, our findings demonstrate that Dic inhibits human TRPM3 without interacting with the channel pore.
Keywords: Diclofenac, neuroblastoma, pain, TRPM3
Abbreviations
- Afc
aceclofenac
- Asp
aspirin
- BrdU
5‐bromo‐2′‐deoxyuridine
- COX
cyclo‐oxygenase
- Dic
diclofenac
- Etd
etodolac
- GSK
GSK1016790A
- HEK293
human embryonic kidney 293 cell‐line
- HEPES
4‐(2‐hydroxyethyl)‐1‐piperazine ethane sulfonic acid
- Ibu
ibuprofen
- IMR‐32 cells
human neuroblastoma IMR‐32 cells
- Ind
indomethacin
- Lox
loxoprofen
- MEM
minimum essential medium
- Men
menthol
- Nfm
niflumic acid
- Nif
nifedipine
- nIMR‐32 cells
neuronal IMR‐32 cells
- NSAID
nonsteroidal anti‐inflammatory drug
- pIC50
negative logarithm of the apparent IC50
- Pil
piloxicam
- PregS
pregnenolone sulfate
- PVDF
polyvinylidene difluoride
- TBS
tris‐buffered saline
- TRP
transient receptor potential
- TRPA
transient receptor potential ankyrin
- TRPC
transient receptor potential canonical
- TRPM
transient receptor potential melastatin
- TRPV
transient receptor potential vanilloid
Introduction
An acetic acid‐type, nonsteroidal anti‐inflammatory drug (NSAID), diclofenac (Dic), is commonly used as a potent painkiller, which exhibits inhibitory action on cyclo‐oxygenase (COX). Dic relieves nociception for a relatively long time and hence is widely prescribed as an antiacute and chronic pain medication, in particular, when these are accompanied by inflammation. Although the wide safety margin of Dic has been established, the possible adverse effects of this drug have been reported on gastrointestinal, hepatic, renal, and cardiovascular organs (Bort et al. 1999; Hickey et al. 2001; McGettigan and Henry 2006). Among them, the gastrointestinal complications are the most common when NSAIDs including Dic are clinically used. Therefore, selective COX‐2 inhibitors had been introduced although increased risks of heart attacks by rofecoxib have been identified later (Kearney et al. 2006; Trelle et al. 2011). Dic has a moderate preference to inhibit human COX‐1 (1.6 times potent for COX‐1; Jett et al. 1999) than COX‐2, and hence is better than indomethacin (Ind, 25 times potent for COX‐1) in the point of view of incidence of gastrointestinal complaints. Despite the short lifetime of Dic in blood, the action of one single dose is relatively long and it has been shown that Dic is one of the most potent NSAIDs. One rational reason to explain this discrepancy is that Dic has multiple molecular targets possibly contributing to its antinociceptive actions besides the COX inhibition. Indeed, some ion channels, which are possibly involved in nociception, are modulated by Dic, for example, inhibition of voltage‐dependent Na+ channel (Lee et al. 2003a), activation of ATP‐dependent K+ channel (Alves et al. 2004), and activation of Ca2+‐dependent K+ channel (Ortiz et al. 2002, 2003). However, since effective dose against these ion channels are relatively high, it is not clear if these modulations can contribute to its antinociceptive action in vivo.
Transient receptor potential melastatin 3 (TRPM3) is a Ca2+‐permeable nonselective cation channel expressed in neuronal and non‐neuronal cells. Although TRPM3 is activated by noxious heat and by sphingolipids and neurosteroids at relatively high concentrations (Grimm et al. 2005; Wagner et al. 2008; Majeed et al. 2010; Drews et al. 2013; Vriens et al. 2014), its functional roles are still unclear. In in vitro animal studies, change in TRPM3 activity by a neurosteroid, pregnenolone sulfate (PregS), modified the functions of pancreatic β cells and vascular arteries (Wagner et al. 2008; Naylor et al. 2010). However, transgenic mice lacking TRPM3 had normal function in pancreas, but showed suppression of sensitivity to noxious heat (Vriens et al. 2011). On the other hand, it has been recently shown that some clinical drugs and natural compounds substantially inhibit TRPM3 (Klose et al. 2011; Majeed et al. 2011; Straub et al. 2013a,b). These pharmacological approaches are helpful for better understanding of function of TRPM3 in vivo and can provide new clinical and nonclinical tools.
In this study, we therefore aimed to identify a clinical drug, which affects TRPM3 channels that are heterologously expressed in human embryonic kidney 293 cell‐line (HEK293) cells and endogenously expressed in human neuroblastoma IMR‐32 (IMR‐32) cells, a neuroblastoma cell line derived from human peripheral neurons. According to the difference in N‐ and C‐termini of the amino acid sequence, human TRPM3 has several isoforms (Oberwinkler et al. 2005; see also a review Ohya et al. 2016). Among them, we used four human TRPM3s in this study (Fig. 1A): the longest TRPM3‐9, the short C‐terminal TRPM31325, the short N‐terminal TRPM3‐3, and the short N‐ and C‐terminal TRPM3‐S. Since TRPM31325 is the most commonly used human variant (Naylor et al. 2010; Majeed et al. 2011, 2012), we here compared pharmacological characteristics of TRPM31325 with other three human variants. We found that a NSAID, Dic, effectively inhibited human TRPM3 isoforms with Ca2+ measurement as well as whole‐cell and single‐channel assays, reaching the important conclusion that Dic is an antagonist of TRPM3 that does not interact with the channel pore.
Figure 1.
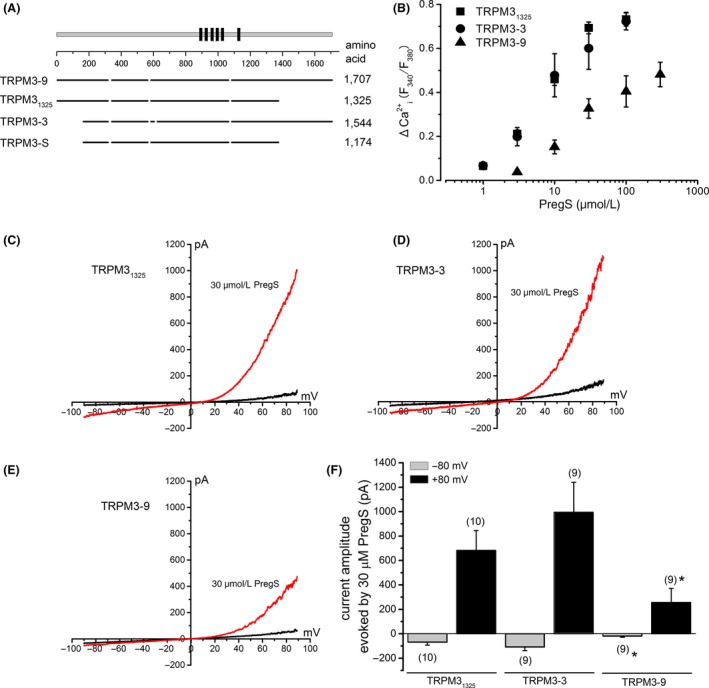
PregS‐evoked TRPM3‐mediated responses. (A) Scheme of human TRPM3 isoforms used in this study. (B) The ΔCa2+ i induced by the application of PregS at a concentration range between 1 and 100 μmol/L (3–300 μmol/L for TRPM3‐9) is summarized as concentration–response relationships (square symbols, six independent experiments for TRPM31325; circle symbols, six independent experiments for TRPM3‐3; triangle symbols, 10 independent experiments for TRPM3‐9). (C–E) Current and voltage (I‐V) relationships of TRPM31325 (C), TRPM3‐3 (D), and TRPM3‐9 (E) evoked by ramp voltage command pulses were shown in the absence and presence of 30 μmol/L PregS. Ramp voltage command pulses for 400 msec were applied every 10 sec at a holding potential of −10 mV. (F) Comparison of PregS‐evoked current amplitude in HEK‐TRPM3s cells. Bars represent the mean ± SEM from 9 to 10 cells. * versus TRPM31325 and TRPM3‐3. PregS, pregnenolone sulfate; TRPM, transient receptor potential melastatin; HEK, human embryonic kidney.
Materials and Methods
Cell culture
HEK293 and IMR‐32 cells obtained from the Health Science Research Resources Bank and ATCC, respectively, were maintained in Dulbecco's modified minimum essential medium (MEM) for HEK293 cells (Sigma‐Aldrich, St. Louis, MO) and in MEM with nonessential amino acid solution (Sigma‐Aldrich), both supplemented with 10% heat‐inactivated fetal calf serum (Gibco™, ThermoFisher Scientific, Yokohama, Japan), streptomycin (100 μg/mL, Meiji Seika Pharma Co., Ltd.), and penicillin G (100 U/mL, Meiji Seika Pharma Co., Ltd., Tokyo, Japan). When differentiated to neuronal IMR‐32 cells (nIMR‐32), IMR‐32 cells were treated with 5‐bromo‐2′‐deoxyuridine (BrdU, 5 μmol/L, Wako, Osaka, Japan) and then cultured for 12 days in BrdU‐containing medium that was exchanged every 48 h (Hatano et al. 2013).
Recombinant expression of TRP channels in HEK293 cells
HEK293 cells with 40–60% confluency were transfected with the pcDNA3.1/neo(+) plasmid with human TRPM3s (TRPM31325, TRPM3‐3, TRPM3‐9, and TRPM3‐S), TRPM2, TRPM8, or TRP vanilloid 4 (TRPV4) using lipofectamine 2000 (Invitrogen, Carlsbad, CA). All experiments were performed within 72 h of transfection. To detect the expression of TRPM3s in HEK293 cells for electrophysiological experiments, cells were cotransfected with pAcGFP1 (Takara Bio Inc., Shiga, Japan) and all GFP‐positive cells responded to PregS in HEK‐TRPM31325, HEK‐TRPM3‐3, and HEK‐TRPM3‐S cells. In contrast, 30% HEK293 cells transfected with TRPM3‐9 and GFP were sensitive to PregS.
Quantitative PCR
Real‐time quantitative PCR was performed with the use of SYBR Green Chemistry on a Thermal Cycler Dice Real Time System (Takara Bio, Inc.) as described previously (Suzuki et al. 2014). Transcriptional quantification of gene products was normalized to β‐actin and each cDNA sample was tested in triplicate. Briefly, after an initial 30‐sec activation of Ex Taq™ DNA polymerase (Takara Bio, Inc.) at 95°C, the program used for quantitative PCR amplification included a 15‐sec denaturation step at 95°C, a 60‐sec annealing, and an extension step at 60°C (for 45 cycles). This was followed by a final dissociation step (15 sec at 95°C, 30 sec at 60°C, and 15 sec at 95°C). The oligonucleotide sequences of primers specific for human TRPM3 and β‐actin (sense and antisense: 5′ to 3′) were GGACCAGCTGTTGGTGACTATACA and CCATCCGAAATACCGTAATCAATT, and ACCGAGCGCGGCTACA and CAGCCGTGGCCATCTCTT, respectively.
Western blotting
After HEK293 cells were lysed in 50 μL lysis buffer ([in mmol/L] NaCl 150, ethylenediamine tetraacetic acid (EDTA) 5, Tris‐HCl 50 [pH 8.0]) including 0.5% sodium deoxycholate, 1% NP‐40, 0.1% sodium dodecyl sulfate (SDS), and protease inhibitors (Hatano et al. 2012), the cell lysates were incubated on ice for 30 min with vortexing every 5 min and then centrifuged at 20,000g for 30 min at 4°C. Each lysate (10 μg) was separated on an 8% polyacrylamide gel and proteins were then transferred to a polyvinylidene difluoride (PVDF) membrane. Nonspecific binding of antibodies was blocked by incubation for 2 h in Tris‐buffered saline (TBS) containing 5% skim milk and 0.1% Tween‐20. The PVDF membrane was subsequently incubated with the first antibody with 1:1000 dilution (rabbit anti‐TRPM3; Abcam, Cambridge Science Park, Cambridge, U.K.) overnight at 4°C. The blot was then washed three times with washing TBS buffer containing 0.1% Tween‐20 and secondary antibody with 1:10,000 dilution (IgG‐HRP, Jackson Immuno Research Laboratories Inc., West Grove, PA) was added to the PVDF membrane. Blots were washed again and detection reagents (Millipore, Billerica, MA) were added to generate a chemiluminescence signal. To determine the relative quantity of TRPM3s to β‐actin in each sample, the PVDF membrane was also exposed to mouse anti‐β‐actin monoclonal antibody (1:2000 dilution, Sigma‐Aldrich). Finally, gels were scanned with an LAS‐3000 mini apparatus (Fujifilm, Tokyo, Japan).
Patch clamp experiments
Whole‐cell and single‐channel patch clamp experiments were performed as described previously (Suzuki et al. 2014). The resistance of electrodes was 2.5–5 MΩ in whole‐cell and outside‐out patch configurations when filled with the pipette solution ([in mmol/L] Cs‐aspartate 110, CsCl 30, MgCl2 1, 4‐(2‐hydroxyethyl)‐1‐piperazine ethane sulfonic acid (HEPES) 10, ethylene glycol tetraacetic acid (EGTA) 1, Na2ATP 2 [adjusted to pH 7.2 with CsOH]). Membrane currents and voltage signals were digitized onto a computer using an analog‐digital converter (PCI6229, National Instruments Japan, Tokyo, Japan). Data acquisition and analysis of whole‐cell currents and excised outside‐out patch currents were performed using WinWCP4.5 and WinEDR3.38, respectively, developed by Dr. John Dempster (University of Strathclyde, U.K.). The liquid junction potential between the pipette and bath solutions (−10 mV) was corrected in the present experiments. A ramp voltage protocol from −110 mV to +90 mV for 400 msec was applied every 10 sec from a holding potential of −10 mV. The leak current component was not subtracted from the recorded currents. A standard HEPES‐buffered bathing solution ([in mmol/L] NaCl 137, KCl 5.9, CaCl2 2.2, MgCl2 1.2, glucose 14, HEPES 10 [adjusted to pH 7.4 with NaOH]) was used. All experiments were performed at 25 ± 1°C.
Measurement of the Ca2+ fluorescence ratio
HEK293 cells and IMR‐32 cells were loaded with 10 μmol/L Fura2‐AM (Dojindo, Kumamoto, Japan) in the standard HEPES solution for 30 min at 25 ± 1°C. After cells were superfused with the standard HEPES solution for 10 min, Fura‐2 fluorescence signals were measured using Ca2+‐imaging system driven by simple PCI software 6.0 (Hamamatsu Photonics, Hamamatsu, Japan). The frequency of image acquisition was set every 10 sec. The efficacy of gene transfection was similar in HEK293 cells, but not identical from cell to cell. To reduce the variation, we collected 15 single cells in one coverslip for analysis and repeated the same experiment with other independent coverslips. In each analysis, a whole‐cell area was chosen as a region of interest to average the fluorescence ratio of the area.
Chemicals and reagents
The following drugs were used: PregS (Sigma‐Aldrich), nifedipine (Nif, Sigma‐Aldrich), Dic (Wako), aspirin (Asp, Wako), ibuprofen (Ibu, Wako), loxoprofen (Lox, Wako), Ind (Wako), etodolac (Etd, Wako), piloxicam (Pil, Wako), niflumic acid (Nfm, Sigma‐Aldrich), aceclofenac (Afc, Tokyo Chemical Industry Co., Ltd., Tokyo, Japan), GSK1016790A (GSK; Sigma‐Aldrich), hydrogen peroxide (H2O2, Sigma‐Aldrich), and menthol (Men, Wako). Each drug was dissolved in the vehicle recommended by the manufacturer.
Data analysis
The data are expressed as mean ± SEM. The statistical significance between two or multiple groups was analyzed using nonpaired Student's t tests or Tukey's multiple comparison tests, respectively. The statistical significance at P < 0.05 and <0.01 is indicated in figures as *, # and **, ##, respectively. To determine the apparent IC50 of Dic against TRPM3 agonist‐induced Ca2+ responses (Figs. 2D, 4E, 8F), a set of data under each experimental condition was fitted to a sigmoid curve with Origin 9.1J (OriginLab Corp, Northampton, MA). In Figure 4E, because the elevation of [Ca2+]i by the second application of Nif in the absence of Dic was reduced to 0.93 ± 0.07 (six independent experiments) and 0.87 ± 0.05 (four independent experiments) in HEK‐TRPM31325‐ and HEK‐TRPM3‐9 cells, we set the fractional maximum of a sigmoid curve fitting equation to 0.93 and 0.87, respectively.
Figure 2.
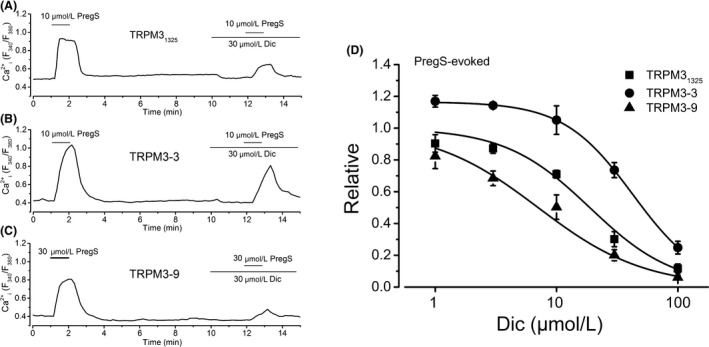
The effect of Dic on PregS‐induced Ca2+ responses of HEK‐TRPM3s cells. (A–C) PregS at 10 μmol/L (for HEK‐TRPM31325 [A] and HEK‐TRPM3‐3 [B]) or 30 μmol/L (for HEK‐TRPM3‐9 [C]) was applied twice and 30 μmol/L Dic was added before the second application of PregS. (D) Change in PregS‐induced Ca2+ response by Dic at a concentration range between 1 and 100 μmol/L is summarized as concentration–response relationships. The amplitude of change in the second PregS‐induced Ca2+ response with Dic was normalized to that in the first without Dic (square symbols, six independent experiments for TRPM31325; circle symbols, six independent experiments for TRPM3‐3; triangle symbols, six independent experiments for TRPM3‐9). Data were fitted to a sigmoid curve to determine the apparent IC 50 of Dic against PregS‐induced Ca2+ response of each TRPM3 isoform. Dic, diclofenac; PregS, pregnenolone sulfate; HEK, human embryonic kidney; TRPM, transient receptor potential melastatin.
Figure 4.
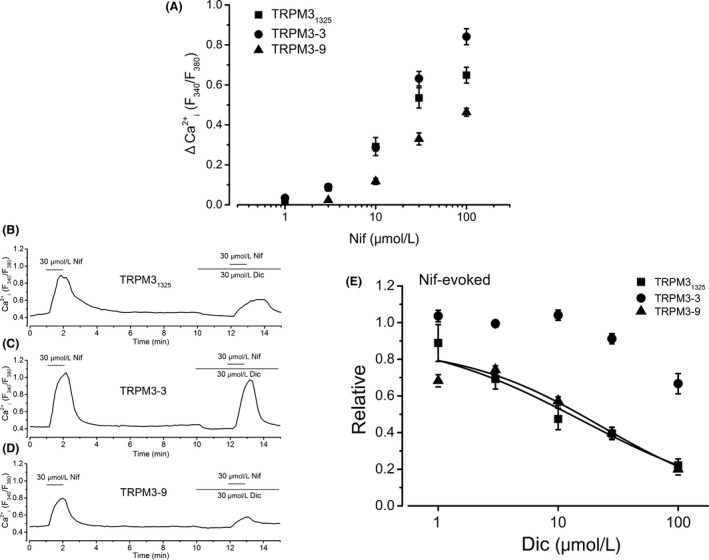
Nif‐induced TRPM3‐mediated Ca2+ response and the effect of Dic. (A) The ΔCa2+ i evoked by the application of Nif at a concentration range between 1 and 100 μmol/L is summarized as concentration–response relationships (square symbols, six independent experiments for TRPM31325; circle symbols, six independent experiments for TRPM3‐3; triangle symbols, six independent experiments for TRPM3‐9). (B–D) Nif at 30 μmol/L was applied twice and 30 μmol/L Dic was added before the second application of Nif. (E) Change in Nif‐induced Ca2+ response by Dic at a concentration range between 1 and 100 μmol/L is summarized as concentration–response relationships. The amplitude of change in the second Nif‐induced Ca2+ response with Dic was normalized to that in the first without Dic (square symbols, six to eight independent experiments for TRPM31325; circle symbols, four to seven independent experiments for TRPM3‐3; triangle symbols, five to seven independent experiments for TRPM3‐9). Data were fitted to a sigmoid curve to determine the apparent IC 50 of Dic against Nif‐induced Ca2+ response of TRPM31325 and TRPM3‐9 isoforms. Nif, nifedipine; TRPM, transient receptor potential melastatin; Dic, diclofenac.
Figure 8.
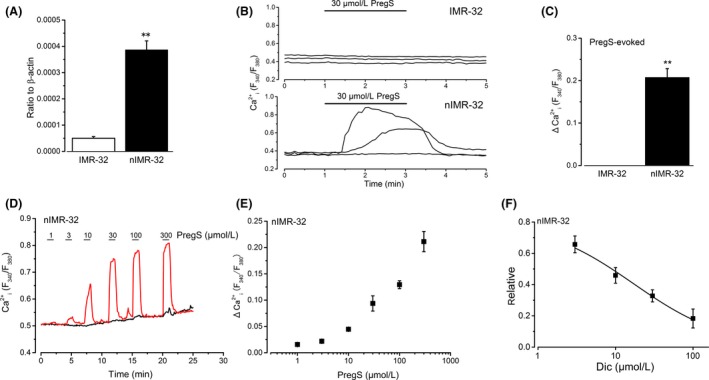
Induction of TRPM3 in differentiated human neuroblastoma IMR‐32 cells and effects of Dic. Non‐neuronal IMR‐32 cells were differentiated to nIMR‐32 cells with BrdU treatment (see Materials and Methods). (A) To confirm quantitative induction of TRPM3, TRPM3 mRNA transcripts were compared between IMR‐32 and nIMR‐32 cells (six independent experiments). ** versus IMR‐32. (B and C) The Ca2+ response (ΔCa2+ i) to 30 μmol/L PregS was shown in representative three IMR‐32 and nIMR‐32 cells, and the pooled data were summarized as a bar graph (C, four independent experiments for IMR‐32 and nIMR‐32 cells, respectively). ** versus IMR‐32. (D and E) Cumulative application of PregS between 1 and 300 μmol/L to nIMR‐32 cells. Typical traces (D, one responsive and another nonresponsive cell to PregS) and summarized data of responsive cells (E, four independent experiments) were shown. (F) Application of 3–100 μmol/L Dic to nIMR‐32 cells significantly inhibited PregS‐induced Ca2+ response. Bars represent the mean ± SEM (five to eight independent experiments). Data were fitted to a sigmoid curve to determine the apparent IC 50 of Dic against PregS‐induced Ca2+ responses. TRPM, transient receptor potential melastatin; IMR‐32, human neuroblastoma IMR‐32; Dic, diclofenac; nIMR‐32, neuronal IMR‐32; BrdU, 5‐bromo‐2′‐deoxyuridine; PregS, pregnenolone sulfate.
Results
Expression in HEK293 cells transfected with TRPM31325, TRPM3‐3, TRPM3‐9, and TRPM3‐S plasmid (HEK‐TRPM31325, HEK‐TRPM3‐3, HEK‐TRPM3‐9, and HEK‐TRPM3‐S) was confirmed by three assays; TRPM3s mRNA transcripts expression (not shown), TRPM3s protein expression (Fig. S1A), and TRPM3s channel functions (Fig. 1, Fig. S1B, and Fig. 6). Although the protein expression level of TRPM3‐9 was relatively low, application of PregS, a putative steroidal TRPM3 agonist, induced elevation of intracellular Ca2+ concentration (Ca2+ i) in these HEK‐TRPM3s cells in a concentration‐dependent manner (Fig. 1B, see also Fig. S1B). Moreover, PregS evoked typical outward‐rectifying TRPM3 membrane currents in each HEK‐TRPM3s cell (Fig. 1C–E, see also Fig. 6C for HEK‐TRPM3‐S). Consistent to the protein expression data, both Ca2+ and current responses of HEK‐TRPM3‐9 cells to PregS were significantly smaller than those of HEK‐TRPM31325 and HEK‐TRPM3‐3 cells (Fig. 1B and F).
Figure 6.
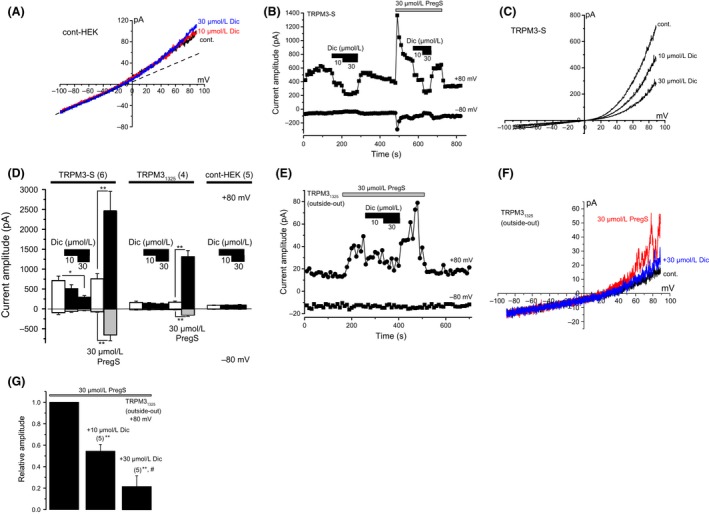
Inhibitory mechanisms of Dic on TRPM3. (A) Effects of Dic on membrane currents in a wild‐type control HEK293 cell. (B and C) Effects of Dic on constitutive spontaneous TRPM3‐S‐mediated currents, which were recorded in the absence of TRPM3 agonists. Change in current amplitude of TRPM3‐S at −80 mV and +80 mV was plotted against time (B). I‐Vs evoked by ramp voltage command pulses were shown in the absence (cont) and presence of 10 and 30 μmol/L Dic in a wild‐type control (A) and a TRPM3‐S expressing HEK293 cell (C). Ramp voltage command pulses for 400 msec were applied every 10 sec at a holding potential of −10 mV. A dash‐line in (A) exhibits leak‐like currents. (D) Comparison of change in current amplitude in the absence of TRPM3 agonists by 10 and 30 μmol/L Dic among HEK‐TRPM3‐S, HEK‐TRPM31325, and wild‐type control HEK293 cells. To confirm the expression of TRPM3 channels in HEK‐TRPM3‐S and HEK‐TRPM31325 cells, 30 μmol/L PregS was applied to these cells. Bars represent the mean ± SEM from four to six cells. (E–G) Effects of Dic on PregS‐evoked single TRPM31325 channel currents, which were recorded in excised outside‐out patches. Change in peak channel current amplitude of TRPM31325 at −80 mV and +80 mV was plotted against time (E). I‐Vs evoked by ramp voltage command pulses were also shown (F) in the absence of both PregS and Dic (cont), and presence of 30 μmol/L PregS with (+30 μmol/L Dic) or without Dic (30 μmol/L PregS). Ramp voltage command pulses for 400 msec were applied every 10 sec at a holding potential of −10 mV. (G) The amplitude of peak PregS‐evoked single‐channel currents at +80 mV was summarized before and during application of 10 and 30 μmol/L Dic. Bars represent the mean ± SEM from five cells. ** versus without Dic. # versus with 10 μmol/L Dic. Dic, diclofenac; TRPM, transient receptor potential melastatin; HEK293, human embryonic kidney 293 cell‐line; PregS, pregnenolone sulfate.
Using HEK293 cells with human TRPM31325, we simply screened eight NSAIDs (propionic acid derivatives: Ibu and Lox; acetic acid derivatives: Etd and Dic; an indole derivative: Ind; a salicylate: Asp; an enolic acid derivative: Pil, and a fenamate: Nfm, Fig. S2). In particular, we focused on nonfenamate (nonanthranilic acid derivatives) NSAIDs because it has been already shown that some fenamates effectively inhibit TRPM3 (Klose et al. 2011). Seven NSAIDs except Dic failed to affect a basal Ca2+ level of HEK293‐TRPM31325 cells in the absence of TRPM3 agonists, but application of 10 μmol/L Dic substantially inhibited the Ca2+ level (Fig. S2A and B). Therefore, we systematically examined the inhibitory effects of Dic on PregS‐induced Ca2+ response of TRPM3s as shown in Figure 2. After constructing a concentration–response curve of PregS‐induced Ca2+ response (Fig. 2D), the negative logarithm of the apparent IC50 (pIC50) of Dic needed to inhibit the PregS‐induced Ca2+ response of each TRPM3 isoform was 4.73 ± 0.04 for TRPM31325, 4.37 ± 0.01 for TRPM3‐3, and 5.15 ± 0.05 for TRPM3‐9. In the whole‐cell recording mode, application of 10 and 30 μmol/L Dic to HEK‐TRPM3s cells also inhibited 30 μmol/L PregS‐evolved inward and outward currents (Fig. 3A–I). However, these inhibitions were immediately reversed after withdrawal of Dic (Fig. 3A, D, and G). Moreover, comparison of the inhibition between −80 mV and +80 mV revealed that Dic affects TRPM3s channels in a voltage‐independent manner (Fig. 3C, F, and I).
Figure 3.
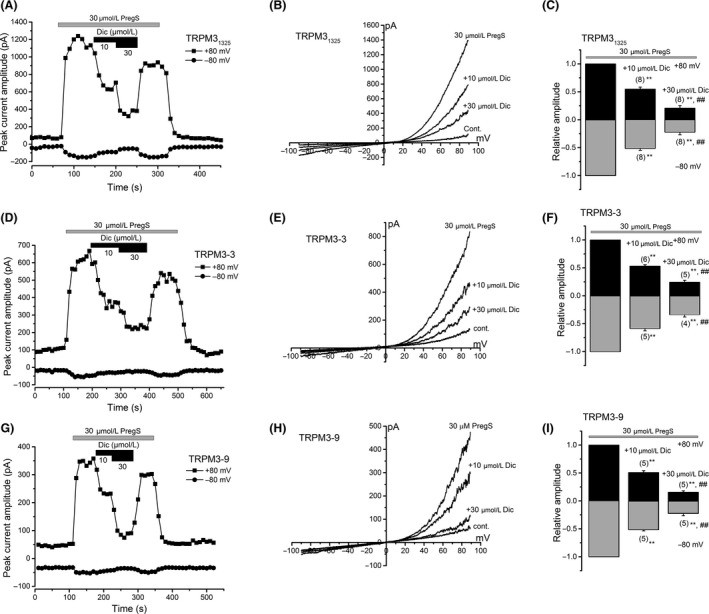
(A–C) Change in current amplitude of TRPM31325 at −80 mV and +80 mV was plotted against time (A). I‐Vs evoked by ramp voltage command pulses were also shown in the control and with or without 10 and 30 μmol/L Dic in the presence of 30 μmol/L PregS (B). Ramp voltage command pulses for 400 msec were applied every 10 sec at a holding potential of −10 mV. Comparison of relative change in PregS‐evoked current amplitude by 10 and 30 μmol/L Dic in HEK‐TRPM31325 cells (C). Peak current amplitude at −80 mV and +80 mV was pooled and summarized in a bar graph. Bars represent the mean ± SEM from eight cells. (D–F) As for (A–C) except TRPM31325 to TRPM3‐3. Bars represent the mean ± SEM from four to six cells. (G–I) As for (A–C) except TRPM31325 to TRPM3‐9. Bars represent the mean ± SEM from five cells. ** versus without Dic. ## versus with 10 μmol/L Dic. TRPM, transient receptor potential melastatin; Dic, diclofenac; PregS, pregnenolone sulfate; HEK, human embryonic kidney.
Since an organic Ca2+ antagonist, Nif, can activate TRPM3 (Wagner et al. 2008), we next tested the inhibitory effects of Dic on Nif‐induced Ca2+ response in HEK‐TRPM3s cells (Fig. 4). Nif at a concentration range between 1 and 100 μmol/L induced Ca2+ response in HEK‐TRPM3s cells (Fig. 4A). Application of Dic inhibited 30 μmol/L Nif‐induced Ca2+ responses in a concentration‐dependent manner in each HEK‐TRPM3s cell (Fig. 4B). However, compared with PregS, Nif response of TRPM3‐3 was rather resistant to Dic (Fig. 4E vs. Fig. 2D); pIC50 of Dic needed to inhibit Nif‐induced Ca2+ response of TRPM3s was 4.80 ± 0.06 and 4.64 ± 0.1 for TRPM31325 and TRPM3‐9, respectively, while <4 for TRPM3‐3 (Fig. 4E). To confirm the resistance of Nif response against Dic in HEK‐TRPM3‐3 cells, we used a different experimental protocol for the assay. We applied 100 μmol/L Dic to HEK‐TRPM3‐3 cells in which PregS (10 μmol/L) or Nif (30 μmol/L) induced sustainable Ca2+ responses (Fig. S3). Even after using this protocol, Nif‐evoked Ca2+ response of TRPM3‐3 was clearly resistant to Dic in comparison with PregS response. Although having different potency to inhibit TRPM3 among the isoforms and between agonists, Dic could be an antagonist of human TRPM3 channel.
Afc is a glycolic acid ester of Dic that is metabolized to Dic in the human body. Since Afc has a chemical structure similar to that of Dic (Fig. 5A), its effects on PregS‐induced Ca2+ response were examined in HEK‐TRPM31325 cells. As shown in Figure 5B and C, Afc had no effects on the response even at 100 μmol/L. In addition, we tested whether Dic affects TRPM2, TRPM8, and TRPV4 because pharmacological profile of fenamates against TRPM3 is distinguishable according to the inhibitory effect on these TRPs (Klose et al. 2011). We used H2O2 (1 mmol/L) and Men (100 μmol/L) for the activation of TRPM2 and TRPM8, and GSK (3 nmol/L) as a TRPV4 agonist, to evoke TRPM2, TRPM8, and TRPV4 response, respectively, in Ca2+ measurement assays (Fig. 5D). However, in HEK293 cells transfected successfully with TRPM2, TRPM8, and TRPV4, 100 μmol/L Dic failed to inhibit all the agonist‐induced Ca2+ responses. These results suggest that among these TRPs, TRPM3 is the putative target for Dic.
Figure 5.
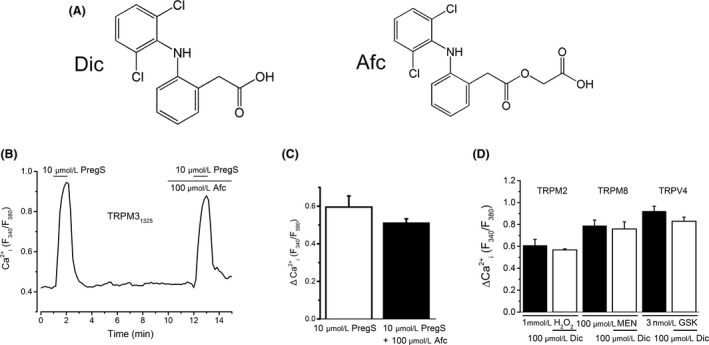
No effects of a glycolic acid ester of Dic (Afc) on TRPM3 and Dic on TRPM2, TRPM8, and TRPV4. (A) Chemical structures of Dic and Afc. (B and C) Effects of 100 μmol/L Afc on PregS‐induced Ca2+ response in HEK‐TRPM31325 cells: a representative trace (B) and summary of change in Ca2+ response in the absence and presence of Afc (C, three independent experiments). (D) Summary of effects of 100 μmol/L Dic on the Ca2+ response to TRPM2 (four independent experiments), TRPM8 (five independent experiments), and TRPV4 (four independent experiments). Hydrogen peroxide (H2O2; 1 mmol/L), menthol (Men; 100 μmol/L), and GSK1016790 (GSK; 3 nmol/L) were applied to evoke TRPM2‐, TRPM8‐, and TRPV4‐mediated response, respectively. Dic, diclofenac; Afc, aceclofenac; TRPM, transient receptor potential melastatin; TRPV, transient receptor potential vanilloid; PregS, pregnenolone sulfate; HEK, human embryonic kidney.
One possible Dic‐induced inhibitory mechanism against TRPM3 is to antagonize the binding of PregS and Nif to the channel. Since we found that the short isoform TRPM3‐S has a relatively large constitutive spontaneous TRPM3 channel activity even without PregS, we examined the effects of Dic on the spontaneous channel activity of TRPM3‐S. To rule out that Dic has nonspecific inhibitory effects on membrane currents in wild‐type control HEK293 cells (cont‐HEK), we applied Dic to these cells without expression of TRPM3s. As shown in Figure 6A (see also Fig. 6D), 10 and 30 μmol/L Dic did not affect the small basal currents evoked by ramp voltage commands. Dic also had little effects on the basal currents from HEK‐TRPM31325 without PregS (Fig. 6D). In contrast, 10 and 30 μmol/L Dic significantly inhibited the outwardly rectifying spontaneously active TRPM3 currents in HEK‐TRPM3‐S cells in the absence of PregS (Fig. 6B–D), suggesting that Dic inhibits TRPM3 channel with the independence of agonist binding.
Ion channel functions may be modulated by factors, which are intracellularly produced in intact cells. To exclude the possibility that such factors might influence the channel inhibition of TRPM3 by Dic, we tested whether Dic directly inhibits TRPM3 channel in membrane patches excised from HEK‐TRPM31325 cells in an outside‐out configuration. As shown in Figure 6E–G, application of 10 μmol/L and 30 μmol/L Dic inhibited the PregS‐induced channel activity at +80 mV in excised outside‐out patches. On the other hand, the single‐channel conductance of TRPM31325 in the presence of 10 μmol/L (77.7 ± 5.7 pS, four independent experiments, Fig. 7A–C) and 30 μmol/L Dic (86.6 ± 3.3 pS, four independent experiments, Fig. 7A–C), which was calculated between +70 and +100 mV, was not significantly different from that in the absence of Dic (87.9 ± 3.1 pS, five independent experiments, Fig. 7A–C). In contrast, the PregS‐induced open probability of TRPM31325 was inhibited by Dic in a concentration‐dependent manner (Fig. 7D and E). Taken together, Dic inhibits TRPM3 channel in a membrane‐delimited manner without interacting with the channel pore.
Figure 7.
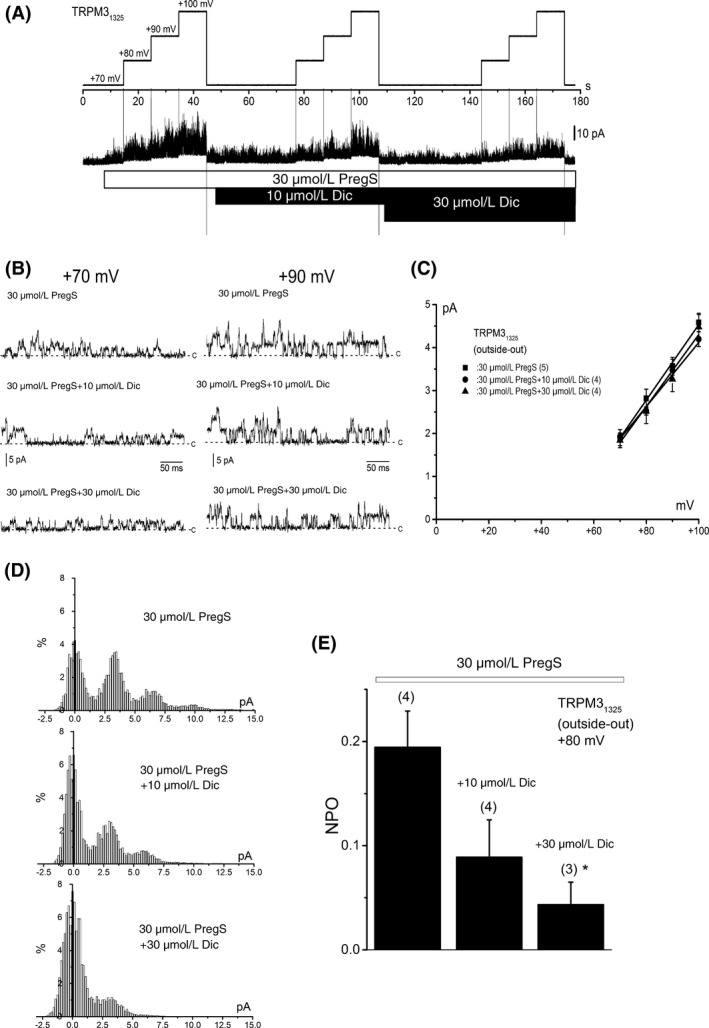
Channel conductance and open probability of TRPM31325 with or without Dic. (A) A representative single‐channel current trace in an outside‐out patch excised from a HEK‐TRPM31325 cell. The channel activity was increased by 30 μmol/L PregS and thereafter 10 and 30 μmol/L Dic were applied. During application of PregS and PregS plus Dic, the holding potential was changed from +70 to +80, +90, and +100 mV to determine the channel conductance. In lower panels, traces at +70 and +90 mV were shown in a higher time resolution (B). The “c” indicates the channel close level. I‐Vs of PregS‐induced single‐channel currents with or without Dic (C, PregS; five cells, +10 μmol/L Dic; four cells +30 μmol/L Dic; four cells). The channel conductance was calculated to fit a set of data in each cell to a linear regression line. (D) Amplitude histograms at +80 mV for 5 sec in the presence of 30 μmol/L PregS, 30 μmol/L PregS plus 10 μmol/L Dic, and 30 μmol/L PregS plus 30 μmol/L Dic from the same cell as shown in (A). The bin width is 0.163 pA. (E) The open probability of PregS‐evoked single‐channel currents was summarized before and during application of 10 and 30 μmol/L Dic. Bars represent the mean ± SEM from three to four cells. * versus without Dic. TRPM, transient receptor potential melastatin; Dic, diclofenac; HEK, human embryonic kidney; PregS, pregnenolone sulfate.
It is important to test whether Dic can inhibit TRPM3 endogenously expressed in human tissues, as it is widely used for treatments with pain. Successfully, we found that ~20% IMR‐32 cells express TRPM3 when they are differentiated to nIMR‐32 (Hatano et al. 2013). Based on real‐time PCR, we confirmed the mRNA expression of TRPM3 in IMR‐32 cells and found that the expression in nIMR‐32 cells was significantly higher than that in non‐neuronal IMR‐32 cells (Fig. 8A). Consistently, application of 30 μmol/L PregS evoked a Ca2+ response in some nIMR‐32 cells, but not in non‐neuronal IMR‐32 cells (Fig. 8B and C). Application of PregS between 1 and 300 μmol/L evoked Ca2+ responses in nIMR‐32 cells in a concentration‐dependent manner (Fig. 8D and E). Dic at a concentration range between 3 and 100 μmol/L, effectively inhibited PregS (30 μmol/L)‐induced Ca2+ response in nIMR‐32 cells with pIC50 of 4.77 ± 0.04 (Fig. 8F), while did not affect the Ca2+ response of cells insensitive to PregS (relative amplitude of second Pregs‐evoked against first PregS‐evoked: 0.72 ± 0.23 and 0.91 ± 0.11 with and without 100 μmol/L Dic, six and eight independent experiments, respectively).
Discussion
In this study, we demonstrated that the acetic acid derivative NSAID, Dic effectively inhibits human TRPM3 expressed in differentiated nIMR‐32 cells and in HEK293 cells with recombinant TRPM3s in a concentration‐dependent manner. In contrast, a glycolic acid ester of Dic, Afc, even at 100 μmol/L had no effects on TRPM3. Dic also inhibited the spontaneous constitutive activity of TRPM3 channel currents without treatment with TRPM3 agonists. Moreover, Dic reduced TRPM3 single‐channel activity recorded in excised outside‐out patches without affecting channel conductance. Taken together, our findings demonstrate that Dic inhibits human TRPM3 channel without interacting with the channel pore.
All four human TRPM3 isoforms used in this study were effectively activated by PregS higher than 1–3 μmol/L (Fig. 1 and Fig. S1). However, the physiological relevance of PregS as an endogenous TRPM3 ligand is not clear, because the concentration is relatively high. Therefore, unidentified natural ligands, which are produced in mammals, could be a potent TRPM3 activator. On the other hand, heat stimulation activates mouse TRPM3 (Vriens et al. 2011), suggesting that heat is a potential endogenous activator of TRPM3. Inconsistently, Uchida et al. (2016) showed that mouse TRPM3 reconstituted in lipid bilayers lacked the strong intrinsic temperature sensitivity. The reason of this discrepancy is not clear and further extensive studies are required for finding endogenous TRPM3 ligands.
Many members of NSAID group exert influence on ion channels including TRPs. Fenamates inhibited TRP canonical (TRPC3, TRPC4, and TRPC5), TRPM (TRPM2, TRPM3, and TRPM5), and TRPV4 (Lee et al. 2003b; Ullrich et al. 2005; Kraft et al. 2006; Klose et al. 2011; Chen et al. 2012; Jiang et al. 2012), while activated TRPC6 (Inoue et al. 2001) and TRP ankyrin (TRPA1, Hu et al. 2010). Among nonfenamate NSAIDs examined in this study, only Dic inhibited TRPM3 responses and the apparent IC50 values of Dic against agonist‐induced Ca2+ responses of TRPM3s (7.1–42.5 μmol/L) were comparable to those of fenamates (6.6–123.5 μmol/L, Klose et al. 2011). Owing to the similarity of IC50 against human TRPM3‐9 (7.1 μmol/L by Dic in this study) and mouse TRPM3α2 (mTRPM3α2, mouse ortholog of human TRPM3‐9, 6.6 μmol/L by mefenamic acid; Klose et al. 2011) and the resistance to TRPM2 and TRPV4 responses (>100 μmol/L), Dic has pharmacological profiles quite similar to mefenamic acid. Consistently, TRPM3 current responses recovered instantaneously after washout of Dic and mefenamic acid. On the other hand, although Klose et al. (2011) had proposed that fenamates might represent channel pore inhibitors, Dic did not change the conductance of TRPM3 channel in our study (Fig. 7). Moreover, inhibition of each TRPM3 isoform by Dic was voltage‐independent (Fig. 3), hence suggesting that Dic may not bind to deep pore of the channel.
It has been shown that a variety of ion channels including TRPs are affected by Dic (see review in Gwanyanya et al. 2012). TRPA1 channels heterologously expressed in xenopus oocytes were activated by Dic, where the EC50 was relatively high (~210 μmol/L, Hu et al. 2010). In podocytes of freshly isolated glomeruli, Dic at 500 μmol/L decreased TRPC6‐like channel activity without affecting the channel conductance (Ilatovskaya et al. 2011). In addition, Dic inhibited TRPC4 and TRPC5 in a concentration‐dependent manner; the IC50 for TRPC4 and TRPC5 was 138 and 170 μmol/L, respectively (Jiang et al. 2012). In contrast, Dic had no effects on capsaicin‐induced ileum relaxation in guinea pig (Fujimoto et al. 2006) and Men‐evoked mouse hyperthermia (Tajino et al. 2007), strongly suggesting that Dic has little effects on TRPV1 and TRPM8. It is consistent that TRPM8 heterologously expressed in HEK293 cells was resistant to Dic (100 μmol/L) in our study. Therefore, among these TRPs, Dic selectively inhibits human TRPM3s.
Straub et al. (2013b) have found that some citrus fruit flavanones significantly inhibit mTRPM3α2. In particular, isosakuranetin was selective and displayed an IC50 of 50 nmol/L against PregS‐activated mTRPM3α2 responses (Straub et al. 2013a). On the other hand, clinically used thiazolidinediones inhibited human TRPM31325, IC50 of 12 μmol/L for troglitazone and <0.1 and 5–10 μmol/L for rosiglitazone (Majeed et al. 2011). In our study, Dic inhibited human TRPM31325, TRPM3‐3, TRPM3‐9, and TRPM3‐S. Moreover, N‐terminus deletion isoforms TRPM3‐3 and TRPM3‐S were rather more resistant to Dic. In our knowledge, this is the first report to show that human neuronal cells functionally express PregS‐sensitive TRPM3. However, we have not determined the isoform of TRPM3 expressed in nIMR‐32 cells because the cells with TRPM3 (~20%) were not sufficient for western blotting. Although human synovial fibroblasts express TRPM3, the type of isoform is not clear (Ciurtin et al. 2010). Nevertheless, because the physiological functions of TRPM3 are not well defined yet, substances that specifically affect TRPM3 or are clinically used are expected to contribute to identifying the role of TRPM3 in vivo, in particular in humans.
The potential significance of the fact that Dic inhibits channels putatively involved in nociception is not clear. The peak plasma concentrations of Dic can be 5–10 μmol/L under clinical short intake conditions (Scheidel et al. 1993; Winek et al. 2001). On the other hand, Dic easily binds to plasma albumin, and dietary constituents and coadministrated medications can affect the plasma level. The effects of 10–30 μmol/L Dic on TRPM3s reversed immediately on wash in this study, so unless these concentrations are sustained, it is highly unlikely that the channel will be inhibited by much for long. Therefore, a part of TRPM3 in several organs including neurons is possibly inhibited by administrated Dic for short time, but further study will be required for the understanding of the contribution of Dic to TRPM3‐dependent antinociception.
In summary, we have shown that clinically and widely used Dic was identified as an antagonist of TRPM3. This inhibition was shown in human TRPM3 isoforms heterologously expressed in HEK293 cells as well as in differentiated human neuronal cells endogenously expressing TRPM3. Molecular and clinical approaches of Dic will provide further insights into the pathophysiological role of TRPM3.
Author Contributions
K. Muraki participated in research design. K. Muraki, H. Suzuki, E. Sasaki, A. Nakagawa, and N. Hatano conducted experiments. Y. Muraki conducted molecular cloning. K. Muraki, H. Suzuki, E. Sasaki, A. Nakagawa, Y. Muraki, and N. Hatano performed data analysis. K. Muraki wrote or contributed to the writing of the manuscript.
Disclosures
None declared.
Supporting information
Figure S1. (A) Protein expression of four human TRPM3 isoforms in HEK293 cells transfected with TRPM31325, TRPM3‐3, TRPM3‐9, and TRPM3‐S. TRPM3 proteins were assayed by western blotting. As a control, β‐actin protein was detected. A representative image from two independent experiments is shown. (B) PregS‐induced Ca2+ response in HEK293 cells transfected with TRPM3‐S. The ΔCa2+ i induced by the application of PregS at a concentration range between 1 and 100 μmol/L is summarized as a concentration–response relationship (three independent experiments).
Figure S2. Simple screening of eight NSAIDs used against TRPM3‐dependent response in the absence of TRPM3 agonists. (A) A representative Ca2+ response of a HEK‐TRPM31325 cell when Dic, Ind, and PregS were applied without TRPM3 agonists. (B) HEK‐TRPM31325 cells were exposed to Ibu, Lox, Dic, Ind, Asp, Pil, Etd, and Nfm and the corresponding Ca2+ response was summarized. To confirm the expression of TRPM31325 in these cells, 10 μmol/L PregS was applied at the end of each experiment. The changes in Ca2+ response to each NSAID (three independent experiments) and PregS (27 independent experiments) were summarized in a bar graph.
Figure S3. Effects of Dic on TRPM3‐3. HEK‐TRPM3‐3 cells had a sustained Ca2+ response to PregS (10 μmol/L), Nif (30 μmol/L) or plus 100 μmol/L Dic. The change in the response was summarized as a bar graph (six independent experiments for PregS‐evoked and four independent experiments for Nif‐evoked, respectively). ** versus PregS‐ or Nif‐evoked. ## versus PregS+100 μmol/L Dic.
Acknowledgements
This work was supported by Grants‐in‐Aid for Scientific Research to K. M. from the Japanese Society of Promotion and Science (JSPS). We thank Dr. J. Dempster (University of Strathclyde, UK) for developing the electrophysiology software (WinWCP and WinEDR).
Suzuki H., Sasaki E., Nakagawa A., Muraki Y., Hatano N., Muraki K., Diclofenac, a nonsteroidal anti‐inflammatory drug, is an antagonist of human TRPM3 isoforms, Pharma Res Per, 4(3), 2016, e00232, doi: 10.1002/prp2.232
References
- Alves DP, Tatsuo MA, Leite R, Duarte ID (2004). Diclofenac‐induced peripheral antinociception is associated with ATP‐sensitive K+ channels activation. Life Sci 74: 2577–2591. [DOI] [PubMed] [Google Scholar]
- Bort R, Ponsoda X, Jover R, Gomez‐Lechon MJ, Castell JV (1999). Diclofenac toxicity to hepatocytes: a role for drug metabolism in cell toxicity. J Pharmacol Exp Ther 288: 65–72. [PubMed] [Google Scholar]
- Chen GL, Zeng B, Eastmond S, Elsenussi SE, Boa AN, Xu SZ (2012). Pharmacological comparison of novel synthetic fenamate analogues with econazole and 2‐APB on the inhibition of TRPM2 channels. Br J Pharmacol 167: 1232–1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciurtin C, Majeed Y, Naylor J, Sukumar P, English AA, Emery P, et al. (2010). TRPM3 channel stimulated by pregnenolone sulphate in synovial fibroblasts and negatively coupled to hyaluronan. BMC Musculoskelet Disord 11: 111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drews A, Mohr F, Rizun O, Wagner TF, Dembla S, Rudolph S, et al. (2013). Structural requirements of steroidal agonists of transient receptor potential melastatin 3 (TRPM3) cation channels. Br J Pharmacol 171: 1019–1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujimoto S, Mori M, Tsushima H, Kunimatsu M (2006). Capsaicin‐induced, capsazepine‐insensitive relaxation of the guinea‐pig ileum. Eur J Pharmacol 530: 144–151. [DOI] [PubMed] [Google Scholar]
- Grimm C, Kraft R, Schultz G, Harteneck C (2005). Activation of the melastatin‐related cation channel TRPM3 by D‐erythro‐sphingosine [corrected]. Mol Pharmacol 67: 798–805. [DOI] [PubMed] [Google Scholar]
- Gwanyanya A, Macianskiene R, Mubagwa K (2012). Insights into the effects of diclofenac and other non‐steroidal anti‐inflammatory agents on ion channels. J Pharm Pharmacol 64: 1359–1375. [DOI] [PubMed] [Google Scholar]
- Hatano N, Itoh Y, Suzuki H, Muraki Y, Hayashi H, Onozaki K, et al. (2012). Hypoxia‐inducible factor‐1alpha (HIF1alpha) switches on transient receptor potential ankyrin repeat 1 (TRPA1) gene expression via a hypoxia response element‐like motif to modulate cytokine release. J Biol Chem 287: 31962–31972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatano N, Suzuki H, Muraki Y, Muraki K (2013). Stimulation of human TRPA1 channels by clinical concentrations of the antirheumatic drug auranofin. Am J Physiol Cell Physiol 304: C354–C361. [DOI] [PubMed] [Google Scholar]
- Hickey EJ, Raje RR, Reid VE, Gross SM, Ray SD (2001). Diclofenac induced in vivo nephrotoxicity may involve oxidative stress‐mediated massive genomic DNA fragmentation and apoptotic cell death. Free Radic Biol Med 31: 139–152. [DOI] [PubMed] [Google Scholar]
- Hu H, Tian J, Zhu Y, Wang C, Xiao R, Herz JM, et al. (2010). Activation of TRPA1 channels by fenamate nonsteroidal anti‐inflammatory drugs. Pflugers Arch 459: 579–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ilatovskaya DV, Levchenko V, Ryan RP, Cowley AW Jr, Staruschenko A (2011). NSAIDs acutely inhibit TRPC channels in freshly isolated rat glomeruli. Biochem Biophys Res Commun 408: 242–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue R, Okada T, Onoue H, Hara Y, Shimizu S, Naitoh S, et al. (2001). The transient receptor potential protein homologue TRP6 is the essential component of vascular alpha(1)‐adrenoceptor‐activated Ca(2 + )‐permeable cation channel. Circ Res 88: 325–332. [DOI] [PubMed] [Google Scholar]
- Jett MF, Ramesha CS, Brown CD, Chiu S, Emmett C, Voronin T, et al. (1999). Characterization of the analgesic and anti‐inflammatory activities of ketorolac and its enantiomers in the rat. J Pharmacol Exp Ther 288: 1288–1297. [PubMed] [Google Scholar]
- Jiang H, Zeng B, Chen GL, Bot D, Eastmond S, Elsenussi SE, et al. (2012). Effect of non‐steroidal anti‐inflammatory drugs and new fenamate analogues on TRPC4 and TRPC5 channels. Biochem Pharmacol 83: 923–931. [DOI] [PubMed] [Google Scholar]
- Kearney PM, Baigent C, Godwin J, Halls H, Emberson JR, Patrono C (2006). Do selective cyclo‐oxygenase‐2 inhibitors and traditional non‐steroidal anti‐inflammatory drugs increase the risk of atherothrombosis? Meta‐analysis of randomised trials. BMJ 332: 1302–1308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klose C, Straub I, Riehle M, Ranta F, Krautwurst D, Ullrich S, et al. (2011). Fenamates as TRP channel blockers: mefenamic acid selectively blocks TRPM3. Br J Pharmacol 162: 1757–1769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraft R, Grimm C, Frenzel H, Harteneck C (2006). Inhibition of TRPM2 cation channels by N‐(p‐amylcinnamoyl)anthranilic acid. Br J Pharmacol 148: 264–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HM, Kim HI, Shin YK, Lee CS, Park M, Song JH (2003a). Diclofenac inhibition of sodium currents in rat dorsal root ganglion neurons. Brain Res 992: 120–127. [DOI] [PubMed] [Google Scholar]
- Lee YM, Kim BJ, Kim HJ, Yang DK, Zhu MH, Lee KP, et al. (2003b). TRPC5 as a candidate for the nonselective cation channel activated by muscarinic stimulation in murine stomach. Am J Physiol Gastrointest Liver Physiol 284: G604–G616. [DOI] [PubMed] [Google Scholar]
- Majeed Y, Agarwal AK, Naylor J, Seymour VA, Jiang S, Muraki K, et al. (2010). Cis‐isomerism and other chemical requirements of steroidal agonists and partial agonists acting at TRPM3 channels. Br J Pharmacol 161: 430–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majeed Y, Bahnasi Y, Seymour VA, Wilson LA, Milligan CJ, Agarwal AK, et al. (2011). Rapid and contrasting effects of rosiglitazone on transient receptor potential TRPM3 and TRPC5 channels. Mol Pharmacol 79: 1023–1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majeed Y, Tumova S, Green BL, Seymour VA, Woods DM, Agarwal AK, et al. (2012). Pregnenolone sulphate‐independent inhibition of TRPM3 channels by progesterone. Cell Calcium 51: 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGettigan P, Henry D (2006). Cardiovascular risk and inhibition of cyclooxygenase: a systematic review of the observational studies of selective and nonselective inhibitors of cyclooxygenase 2. JAMA 296: 1633–1644. [DOI] [PubMed] [Google Scholar]
- Naylor J, Li J, Milligan CJ, Zeng F, Sukumar P, Hou B, et al. (2010). Pregnenolone sulphate‐ and cholesterol‐regulated TRPM3 channels coupled to vascular smooth muscle secretion and contraction. Circ Res 106: 1507–1515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oberwinkler J, Lis A, Giehl KM, Flockerzi V, Philipp SE (2005). Alternative splicing switches the divalent cation selectivity of TRPM3 channels. J Biol Chem 280: 22540–22548. [DOI] [PubMed] [Google Scholar]
- Ohya S, Kito H, Hatano N, Muraki K (2016). Recent advances in therapeutic strategies that focus on the regulation of ion channel expression. Pharmacol Ther pii: S0163‐7258(16)00030‐9. doi: 10.1016/j.pharmthera.2016.02.001 [Epub ahead of print]. [DOI] [PubMed] [Google Scholar]
- Ortiz MI, Torres‐Lopez JE, Castaneda‐Hernandez G, Rosas R, Vidal‐Cantu GC, Granados‐Soto V (2002). Pharmacological evidence for the activation of K(+) channels by diclofenac. Eur J Pharmacol 438: 85–91. [DOI] [PubMed] [Google Scholar]
- Ortiz MI, Granados‐Soto V, Castaneda‐Hernandez G (2003). The NO‐cGMP‐K+ channel pathway participates in the antinociceptive effect of diclofenac, but not of indomethacin. Pharmacol Biochem Behav 76: 187–195. [DOI] [PubMed] [Google Scholar]
- Scheidel B, Blume H, Walter K, von Nieciecki A, Babej‐Dolle RM (1993). Biological availability of gastric juice‐resistant coated diclofenac preparations. 1. Bioavailability study following a single administration of a multiple‐unit formulation in comparison with a single‐unit formulation. Arzneimittelforschung 43: 1211–1215. [PubMed] [Google Scholar]
- Straub I, Krugel U, Mohr F, Teichert J, Rizun O, Konrad M, et al. (2013a). Flavanones that selectively inhibit TRPM3 attenuate thermal nociception in vivo. Mol Pharmacol 84: 736–750. [DOI] [PubMed] [Google Scholar]
- Straub I, Mohr F, Stab J, Konrad M, Philipp SE, Oberwinkler J, et al. (2013b). Citrus fruit and fabacea secondary metabolites potently and selectively block TRPM3. Br J Pharmacol 168: 1835–1850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki H, Hatano N, Muraki Y, Itoh Y, Kimura S, Hayashi H, et al. (2014). The NADPH oxidase inhibitor diphenyleneiodonium activates the human TRPA1 nociceptor. Am J Physiol Cell Physiol 307: C384–C394. [DOI] [PubMed] [Google Scholar]
- Tajino K, Matsumura K, Kosada K, Shibakusa T, Inoue K, Fushiki T, et al. (2007). Application of menthol to the skin of whole trunk in mice induces autonomic and behavioral heat‐gain responses. Am J Physiol Regul Integr Comp Physiol 293: R2128–R2135. [DOI] [PubMed] [Google Scholar]
- Trelle S, Reichenbach S, Wandel S, Hildebrand P, Tschannen B, Villiger PM, et al. (2011). Cardiovascular safety of non‐steroidal anti‐inflammatory drugs: network meta‐analysis. BMJ 342: c7086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uchida K, Demirkhanyan L, Asuthkar S, Cohen A, Tominaga M, Zakharian E (2016) Stimulation‐dependent gating of TRPM3 channel in planar lipid bilayers. FASEB J 30: 1306–1316. pii: fj.15‐281576 [Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ullrich ND, Voets T, Prenen J, Vennekens R, Talavera K, Droogmans G, et al. (2005). Comparison of functional properties of the Ca2+‐activated cation channels TRPM4 and TRPM5 from mice. Cell Calcium 37: 267–278. [DOI] [PubMed] [Google Scholar]
- Vriens J, Owsianik G, Hofmann T, Philipp SE, Stab J, Chen X, et al. (2011). TRPM3 is a nociceptor channel involved in the detection of noxious heat. Neuron 70: 482–494. [DOI] [PubMed] [Google Scholar]
- Vriens J, Held K, Janssens A, Toth BI, Kerselaers S, Nilius B, et al. (2014). Opening of an alternative ion permeation pathway in a nociceptor TRP channel. Nat Chem Biol 10: 188–195. [DOI] [PubMed] [Google Scholar]
- Wagner TF, Loch S, Lambert S, Straub I, Mannebach S, Mathar I, et al. (2008). Transient receptor potential M3 channels are ionotropic steroid receptors in pancreatic beta cells. Nat Cell Biol 10: 1421–1430. [DOI] [PubMed] [Google Scholar]
- Winek CL, Wahba WW, Winek CL Jr, Balzer TW (2001). Drug and chemical blood‐level data 2001. Forensic Sci Int 122: 107–123. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. (A) Protein expression of four human TRPM3 isoforms in HEK293 cells transfected with TRPM31325, TRPM3‐3, TRPM3‐9, and TRPM3‐S. TRPM3 proteins were assayed by western blotting. As a control, β‐actin protein was detected. A representative image from two independent experiments is shown. (B) PregS‐induced Ca2+ response in HEK293 cells transfected with TRPM3‐S. The ΔCa2+ i induced by the application of PregS at a concentration range between 1 and 100 μmol/L is summarized as a concentration–response relationship (three independent experiments).
Figure S2. Simple screening of eight NSAIDs used against TRPM3‐dependent response in the absence of TRPM3 agonists. (A) A representative Ca2+ response of a HEK‐TRPM31325 cell when Dic, Ind, and PregS were applied without TRPM3 agonists. (B) HEK‐TRPM31325 cells were exposed to Ibu, Lox, Dic, Ind, Asp, Pil, Etd, and Nfm and the corresponding Ca2+ response was summarized. To confirm the expression of TRPM31325 in these cells, 10 μmol/L PregS was applied at the end of each experiment. The changes in Ca2+ response to each NSAID (three independent experiments) and PregS (27 independent experiments) were summarized in a bar graph.
Figure S3. Effects of Dic on TRPM3‐3. HEK‐TRPM3‐3 cells had a sustained Ca2+ response to PregS (10 μmol/L), Nif (30 μmol/L) or plus 100 μmol/L Dic. The change in the response was summarized as a bar graph (six independent experiments for PregS‐evoked and four independent experiments for Nif‐evoked, respectively). ** versus PregS‐ or Nif‐evoked. ## versus PregS+100 μmol/L Dic.