

Abstract
Aim
The aim was to compare the pharmacokinetic profiles of two formulations of a combination drug product containing 0.5 mg testosterone and 50 mg sildenafil for female sexual interest/arousal disorder. The prototype (formulation 1) consists of a testosterone solution for sublingual administration and a sildenafil tablet that is administered 2.5 h later. The dual route/dual release fixed dose combination tablet (formulation 2) employs a sublingual and an oral route for systemic uptake. This tablet has an inner core of sildenafil with a polymeric time delay coating and an outer polymeric coating containing testosterone. It was designed to increase dosing practicality and decrease potential temporal non‐adherence through circumventing the relatively complex temporal dosing scheme.
Methods
Twelve healthy premenopausal subjects received both formulations randomly on separate days. Blood was sampled frequently to determine the pharmacokinetics of free testosterone, total testosterone, dihydrotestosterone, sildenafil and N‐desmethyl‐sildenafil.
Results
Formulation 2 had a higher maximum concentration (C max) for testosterone, 8.06 ng ml–1 (95% confidence interval [CI] 6.84, 9.28) and higher area under the plasma concentration–time curve (AUC), 7.69 ng ml–1 h (95% CI 6.22, 9.16) than formulation 1, 5.66 ng ml–1 (95% CI 4.63, 6.69) and 5.12 ng ml–1 h (95% CI 4.51, 5.73), respectively. Formulation 2 had a lower C max for sildenafil, 173 ng ml–1 (95% CI 126, 220) and a lower AUC, 476 ng ml–1 h (95% CI 401, 551) than formulation 1, 268 ng ml–1 (95% CI 188, 348) and 577 ng ml–1 h (95% CI 462, 692), respectively. Formulation 2 released sildenafil after 2.75 h (95% CI 2.40, 3.10).
Conclusions
The dual route/dual release fixed dose combination tablet fulfilled its design criteria and is considered suitable for further clinical testing.
What is Already Known about this Subject
Female sexual interest/arousal disorder (FSIAD) is a significant problem impacting psychological well‐being, but the pharmacotherapeutic options for this problem are lacking.
The combined, on‐demand, sublingual administration of low dose sublingual testosterone and oral administration of sildenafil is a novel pharmacotherapeutic option under development for FSIAD.
In proof‐of‐concept trials, these compounds were successfully administered via different dosage forms (sublingual and oral) at different time points (separated by 2.5 h) because of their markedly different pharmacokinetic–pharmacodynamic profiles. For future larger scale studies and the clinical practice, this raises obvious adherence issues.
What this Study Adds
A newly developed dual route/dual release fixed dose combination tablet containing testosterone and sildenafil mimics the pharmacokinetic profile of these components when they are administered as different dosage forms, 2.5 h apart.
This combination tablet is a suitable final pharmaceutical drug product that will be used in future studies.
Keywords: combination‐tablet, Lybrido, pharmacokinetics, sildenafil, testosterone
Introduction
Low sexual desire is the most common sex‐related complaint reported by women 1, 2, 3. This can cause sexual dissatisfaction, which can lower the psychological well‐being of these women 4. For low sexual desire to be regarded as a disorder, it must cause marked distress and/or interpersonal difficulties and not be better accounted for by another mental disorder, drug use (legal or illegal) or another medical condition 5. This condition, hypoactive sexual desire disorder (HSDD), has been merged with female sexual arousal disorder into a single diagnosis, female sexual interest/arousal disorder (FSIAD), in the fifth edition of the Diagnostic and Statistical Manual for Mental Disorders (DSM‐5) 6. Pharmacotherapeutic options for HSDD/FSIAD are still severely lacking, even following the recent marketing approval for flibanserin, the first approved drug for HSDD in the USA. In Europe, no prescription drugs are available for this problem.
Two pharmacotherapeutic treatments that are in development have been designed based on the hypothesis that HSDD/FSIAD may be caused and/or sustained by (at least) two different mechanisms that are independent of the prior DSM subdivision in desire and arousal disorders. The two mechanisms are based on the idea postulated in the dual control model of sexual response 7, 8 that sexual dysfunctions are caused and sustained by dysfunction in one of two separate but interacting systems, the sexual excitation and the sexual inhibition system. There is cognitive 9, 10, psychophysiological 9, 10, 11, 12, 13, subjective 10, 11, 12, neuroanatomical 14, 15 and pharmacological 9, 10, 11, 12, 15 evidence that shows that there are indeed women whose HSDD/FSIAD is caused by either a decreased propensity for sexual excitation or an increased propensity to inhibit their sexual response. The combination of testosterone and sildenafil is an on‐demand (i.e. pro re nata) therapy developed for women in whom HSDD/FSIAD is caused by a low sensitivity to sexual stimuli (i.e. decreased propensity for sexual excitation), whereas the combination of testosterone and buspirone is an on‐demand therapy developed for women in whom HSDD/FSIAD is caused by dysfunctional over‐activation of sexual inhibitory mechanisms 11, 12, 15. Both drugs have been shown to increase physiological and/or subjective indices of sexual functioning (including sexual satisfaction) as compared with placebo, in the laboratory and at home, in the respective subpopulations of HSDD 9, 10, 11, 12.
The first combination drug contains testosterone (0.5 mg), administered sublingually, and the phosphodiesterase type 5 (PDE‐5) inhibitor sildenafil (50 mg), administered orally and to be absorbed via the gastro‐intestinal tract. Sublingual testosterone is rapidly absorbed (time to maximum concentration [t max] occurs within 15 min) and circulating testosterone concentrations return to baseline values within approximately 2 h 16. The pharmacodynamic effect that sublingually administered testosterone induces, however, does not overlap with its pharmacokinetic peak profile. Instead, sublingually administered testosterone produces an increase in sexual motivation and desire (but also in other cognitive and affective functions 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29) in sexually functional women about 4 h after administration (with the window of effect between 3 to 6 h post‐dose) when the plasma testosterone concentrations have already returned to baseline 16. Thus, testosterone and sildenafil have to be released in such a timeframe that the peak plasma concentration of sildenafil largely coincides with the 4 h delay in behavioural effects of testosterone. The second combination drug contains sublingual testosterone also, and the oral 5‐HT1A receptor agonist buspirone. The release profile of the different active ingredients of this combination are also matched in such a way that the pharmacological effects of the 5‐HT1A receptor agonist coincides with the behavioural window of the pharmacodynamic effect induced by the sublingual testosterone administration 12.
In earlier clinical trials the testosterone component was administered as a sublingual solution. This was followed by oral administration of an encapsulated (to ensure blinding) PDE‐5 inhibitor or 5‐HT1A receptor agonist, 2.5 h after sublingual testosterone administration in order to let the pharmacodynamic effects of the two compounds coincide. This method was adequate for these early clinical studies, but it is unsuitable as a final pharmaceutical formulation. Self‐administering two different medications in two different dosage forms, at two different time points is complex and impractical. Such a dosing scheme could have a negative impact on medication adherence in larger scale clinical trials and clinical practice, specifically the adherence to the timing of administration of the second compound relative to testosterone administration. Therefore, the active components were formulated into single dual route/dual release fixed dose combination tablets, one for testosterone combined with sildenafil and one for testosterone combined with buspirone 30. These combination tablets deliver the testosterone component sublingually and approximately 2.5 h after the tablet has been swallowed, the sildenafil or buspirone component is released into the gastro‐intestinal tract in a pH‐independent manner. Such a single combination tablet can thus be taken on‐demand, 3 to 6 h before anticipated sexual activity.
The objective of the present comparative pharmacokinetic study was to evaluate whether the pharmacokinetic profile of the combination tablet containing testosterone and sildenafil adequately mimicked the pharmacokinetic profile of these components when administered separately. Total testosterone, free testosterone, sildenafil and sildenafil's main metabolite N‐desmethyl‐sildenafil were determined for each formulation at baseline and at regular intervals after administration. The pharmaceutical formulation principles employed in the combination tablet have been shown to give satisfactory results when comparing the two different dosage forms of the combination of testosterone and buspirone 30. It was therefore expected that the pharmacokinetic profile of the two dosage forms tested in the current study would also be comparable.
Methods
Study subjects
Eligible subjects were healthy premenopausal women of 18 to 35 years, with a body mass index (BMI) between 18 and 30 kg m– 2. Exclusion criteria included endocrine, neurological and cardiovascular conditions, hypertension, abnormal liver or renal function and a history of a hormone‐dependent malignancy. Subjects were excluded if they were on medication that could interfere with the metabolism of the study medication or otherwise confound the results of the study, i.e. medications that interfere with the metabolism of sex steroids (e.g. oral contraceptives containing anti‐androgens or (anti)androgenic progestogens) or sildenafil (e.g. nitric oxide donor compounds) or testosterone therapy within 6 months before study entry.
Women were recruited via advertisements, and via a volunteer database of the contract research organization that conducted the study (QPS, Groningen, The Netherlands). Participants were screened for eligibility approximately 4 weeks prior to study entry, after providing written informed consent. At screening, medical history was recorded, a physical examination including a 12‐lead electrocardiogram was performed, a urine pregnancy test was performed and standard biochemistry, serology and haematological laboratory parameters were assessed. Baseline levels of total testosterone, sex hormone‐binding globulin (SHBG), albumin, thyroid stimulating hormone (TSH) and follicle stimulating hormone (FSH) were also assessed at screening. Subject recruitment started on the 16 June 2011and the last visit was on the 15 July 2011.
This study was carried out in agreement with the Declaration of Helsinki (October 2008) and the International Conference on Harmonization ‐ Good Clinical Practice guidelines for clinical research. It was approved by the Medical Ethics Committee of Twente Medical School (Enschede, The Netherlands, reference number P11.05) and the Dutch Competent Authority (Centrale Commissie Mensgebonden Onderzoek, authorization number NL35616.044.11). It was registered in the European Clinical Trials Database, EudraCT number 2011–000 457‐23. The trial was retrospectively registered under International Standard Randomized Controlled Trial Number ISRCTN14616088.
Study design
This was a single centre, open label, randomized, crossover controlled study investigating the pharmacokinetic profiles of two different methods of the combined, on‐demand administration of testosterone and sildenafil. The first mode of administration, formulation 1 (F1), was the sublingual administration of a testosterone‐cyclodextrin complex containing solution (0.5 mg testosterone in 0.5 ml), followed by oral administration of a 50 mg sildenafil tablet (formulated in a gelatin capsule), 2.5 h later. The second mode of administration, formulation 2 (F2) was the dual route/dual release fixed dose combination tablet. The sublingual administration of testosterone (0.5 mg) occurred through sublingual dissolution of the tablet's solid outer coating. This was followed by oral ingestion of the remaining inner core component containing 50 mg sildenafil that was coated with a polymeric ethylcellulose‐based, pH‐independent time delay coating, designed to release the sildenafil after approximately 2.5 h in a single pulse (i.e. not sustained release).
All 12 subjects received F1 and F2 on separate admission periods and in random order based on study site entry. Each admission period had a duration of approximately 39.5 h (two overnight stays). The first subject to enter the site was allocated to treatment F1 during the first admission period and to treatment F2 during the second. The second subject to enter the site was allocated to treatment F2 during the first admission period and to treatment F1 during the second. The third subject was allocated as the first, and so on. Washout between admission periods was at least 7 days. Subjects entered the study site on the evening prior to dosing during which vital signs were checked (including ECG) and a urine drug test, a pregnancy test and alcohol breath analysis were performed. The subjects received low calorie meals and decaffeinated coffee/tea during the admission period to minimize the influence of food and drinks on pharmacokinetic parameters. Vital signs and ECG were assessed after the last blood samples were taken and prior to each discharge from site. Day of menstrual cycle was not taken into account throughout the study.
During each admission period, serial blood samples were drawn via an intravenous catheter, which was placed in the forearm, at baseline (−10 min) and at 5, 10, 15, 20, 25, 30, 60, 90, 120, 135, 145, 165, 180, 195, 210, 225, 240, 270, 300, 330, 360, 390, 450, 570, 690, 810, 930 and 1590 min after dosing. The 930 min sample was taken in the evening before subjects went to bed. The 1590 min sample was taken the next morning after breakfast. Total testosterone, free testosterone and dihydrotestosterone were assessed at the −10, 5, 10, 15, 20, 25, 30, 60, 90, 120, 145, 180, 240 and 1590 min time points; sildenafil and N‐desmethyl‐sildenafil at the −10, 10, 30, 60, 90, 120, 135, 145, 165, 180, 195, 210, 225, 240, 270, 300, 330, 360, 390, 450, 570, 690, 810, 930, 1590 min time points.
A follow‐up visit was performed 7 days after the last admission period during which subjects received a full physical check up. Vital signs, ECG and blood safety were recorded, and a pregnancy test was performed.
The study was conducted in the phase 1 unit of the contract research organization QPS (Groningen, The Netherlands), by the trained research personnel of that same unit.
Medication and dosing
Formulation 1: separate administration
Testosterone in F1 was held in a 1 mg l–1 testosterone β‐cyclodextrin complex solution. Cyclodextrin is used to increase the bioavailability of testosterone. Cyclodextrins have been used to increase the bioavailability for many different types of low soluble or low permeable pharmaceutical compounds, including androgens, for different types of barriers, including buccal membranes, in solution and in solid form (see 31 for a review). It can enhance the solubility and bioavailability of, in particular, hydrophobic compounds and enhance drug delivery through aqueous diffusion‐controlled barriers (like the membrane of the sublingual cavity) but hardly permeates the biological membranes themselves 32. The oral bioavailability β‐cyclodextrin in humans is between 0.5 and 3.3% 33. Thus, with the present method of administration for testosterone using β‐cyclodextrin it is mainly testosterone that enters the circulation.
A trained research associate administered 0.5 ml of the testosterone solution under the subjects' tongues using an Eppendorf micropipette. The subjects were instructed to keep the solution under their tongue for 60 s (which was timed by a second research associate) while moving their tongue slightly to optimize absorption. After 60 s, they swallowed the (remaining) solution. Sildenafil in F1 was encapsulated in a gelatin capsule. The encapsulated tablet was administered orally 150 min after the administration of the testosterone‐containing solution. Water (approximately 200 ml) was provided to facilitate capsule ingestion.
Formulation 2: single dual route/dual release fixed dose combination tablet.
The combination tablet is the same as described previously 30, except that the present tablet contains a different active pharmaceutical ingredient in the core, namely sildenafil instead of buspirone. The combination tablet is a menthol flavoured white tablet of 9 mm in diameter for sublingual administration followed by swallowing (oral administration). The quickly‐dissolving outer coating, obtained through film coating of the tablet from an ethanol solution, delivers β‐cyclodextrin and testosterone (0.5 mg) sublingually, and the time‐delayed release core delivers sildenafil (50 mg) about 2.5 h later. The outer coating comprises testosterone, excipients and a menthol flavour to indicate the full dissolution of the coating. The testosterone coating is designed to dissolve fully and to obtain an almost immediate and complete absorption via the mucosal membranes under the tongue. The delayed‐release core containing the sildenafil has been developed based on in vitro release studies using US Pharmacopeia (USP) dissolution method II. It is designed to release the sildenafil in a single pulse, approximately 2.5 h after oral administration. This method of delayed release is accomplished through the use of a polymer coating of ethylcellulose that allows for a slow penetration of water in a pH‐independent manner. At the predetermined time the polymer coating ruptures at the edge of the tablet. The core material is released immediately from the coating and the dissolution of the sildenafil in the surrounding fluid occurs without any delay.
For F2, the subjects were instructed to keep the tablet under the tongue for 90 s (timed by a research associate), while moving the tongue slightly to optimize absorption. After 90 s the subject was instructed to swallow the tablet as a whole, without chewing or otherwise disrupting the dosage form. If necessary the subject could take a glass of water (200 ml) to enable swallowing.
A trained research associate administered both formulations. A second research associate was responsible for quality control of the procedure and timed the sublingual testosterone exposure of both formulations.
Bioanalytical methods
Assays for pharmacokinetic comparison and their assay validations were performed by Eurofins Medinet B.V., Breda, The Netherlands. The experiments performed for method validation were based on the Bioanalytical Methods Validation in Guidance for Industry: FDA Guidance for industry: Bioanalytical Methods Validation, CDER (May 2001).
Total testosterone, free testosterone and dihydrotestosterone assays
High performance liquid chromatography with mass spectrometric detection (HPLC‐MS/MS) (API 4000, Applied Biosystems, MDS SCIEX) was used for the determination of total testosterone and dihydrotestosterone from plasma. The LLOQ for testosterone was 0.02 ng ml–1 with an intra‐assay CV of 11.0%, an inter‐assay CV of 12.8% and an overall accuracy of 97.1%. The LLOQ for dihydrotestosterone was 0.02 ng ml–1 with an intra‐assay CV of 16.0%, an inter‐assay CV < 16.0%, and an overall accuracy of 107%. Free testosterone was determined in plasma through ultra‐filtration followed by HPLC‐MS/MS. The method was validated with a lower limit of quantification (LLOQ) of 1.00 pg ml–1 for free testosterone with an intra‐assay coefficient of variation (CV) of 12.0%, an inter‐assay CV of 13.3% and an overall accuracy of 103%. The HPLC‐MS/MS assay is a reliable and sensitive method for the analysis of free testosterone and overcomes the known limitations of direct immunoassays in measurement of testosterone values in the lower range 34, 35.
Extraction method for total testosterone
The samples were vortex mixed and transferred into a clean test tube to which 20 μl internal standard was added and vortex mixed. Then methyl tertiary‐butyl ether (MTBE) was added, tubes were capped and shaken for 10 min and then centrifuged for 5 min at 2000 relative centrifugal force (rcf). The tubes were placed into a snap freezer and the bottom water layer was frozen. The supernatant was transferred into a clean tube and evaporated to dryness under a stream of nitrogen. The residue was reconstituted with mobile phase and injected for HPLC‐MS/MS analysis.
Extraction method for free testosterone
A plasma sample was transferred to a deactivated (using Triton X‐100) Amicon Ultra 4 Centrifugal Filter Unit with a cut off filter of 30 kDa. Hepes buffer was added and the mixture was homogenized and was equilibrated for 1 h at 37°C. Plasma ultrafiltrate was obtained by centrifugation of the mixture at 7500 rev min–1 at 37°C for 2 h. The yield of ultrafiltrate was approximately 90% v/v.
The ultrafiltrate samples were vortex mixed and transferred into a clean test tube to which 100 μl internal standard was added and vortex mixed. Then MTBE was added, tubes were capped and shaken for 10 min and then centrifuged for 5 min at 2000 rcf. The tubes were placed into a snap freezer and the bottom water layer was frozen. The supernatant was transferred into a clean tube and evaporated to dryness under a stream of nitrogen. The residue was reconstituted with 100 μl mobile phase and injected for HPLC‐MS/MS analysis.
Equipment
For the HPLC‐MS/MS assays an Applied Biosystem/MDS SCIEX API‐4000 triple quadrupole MS, with positive multiple reaction monitoring and ion spray (turbo spray) was used. The HPLC system and column was Shimadzu Co‐sense system and Hypersil GOLD, 50 × 2.1 mm, particle size 3 μm. Gradients of mobile phase A were methanol with 0.1% acetic acid and mobile phase B, water/methanol with 0.1% acetic acid.
Sildenafil and N‐desmethyl‐sildenafil assay
Sildenafil and its major metabolite N‐desmethyl‐sildenafil concentrations in plasma were also determined by HPLC‐MS/MS. The method was validated with a LLOQ of 1.00 ng ml–1 for sildenafil with an intra‐assay CV of 7.2%, an inter‐assay CV of 11.2% and an overall accuracy of 98.9%. The LLOQ for N‐desmethyl‐sildenafil was 0.50 ng ml–1 with an intra‐assay CV of 6.3%, an inter‐assay CV of 5.3% and an overall accuracy of 99.1%.
Extraction method for sildenafil and N‐desmethyl‐sildenafil
The samples were vortex mixed and transferred into a clean test tube to which 20 μl internal standard solution was added and vortex mixed. Then, 4 ml MTBE was added, tubes were capped and shaken for 10 min and then centrifuged for 5 min at 2000 rcf. The tubes were placed into a snap freezer and the bottom water layer was frozen. The supernatant was transferred into a clean tube and evaporated to dryness under a stream of nitrogen. The residue was reconstituted with mobile phase and injected for HPLC‐MS/MS analysis.
Equipment
For the HPLC‐MS/MS assays an Applied Biosystem/MDS SCIEX API‐4000 triple quadrupole MS, with positive Multiple Reaction Monitoring and Ion spray (Turbo spray) was used. The HPLC system and column was Shimadzu Co‐sense system and Phenomenex Kinetex, C18 100 × 2.1 mm, particle size 2.6 μm. Gradients of mobile phase A were methanol with 0.1% acetic acid and mobile phase B, water/methanol with 0.1% acetic acid.
Hormonal assays performed at screening
Standard biochemistry, serology and haematological laboratory parameters were assessed by KCL BioAnalysis, Leeuwarden, The Netherlands.
SHBG was assessed via an Elecsys® electrochemiluminescence immunoassay (ECLIA) employing a sandwich method using two monoclonal antibodies, with a Modular Analytics E170 module (Roche Diagnostics GmbH, Mannheim, Germany). The lower limit of detection was 0.350 nmol l–1. Reference ranges for SHBG were 20.0–70.0 nmol l–1.
FSH was assessed via an Elecsys® ECLIA employing a sandwich method using two monoclonal antibodies, with a Modular Analytics E170 module (Roche Diagnostics GmbH, Mannheim, Germany). Lower limit of detection was <0.100 mIU ml–1. Reference ranges for FSH were 3.0–10.0 IU l–1.
TSH was assessed via an Elecsys® ECLIA employing a sandwich method using two monoclonal antibodies, with a Modular Analytics E170 module (Roche Diagnostics GmbH, Mannheim, Germany). The lower limit of detection was 0.005 μIU ml–1. Reference ranges for TSH were 0.30–4.0 mIU l–1.
Albumin was assessed via a Tina‐quant® turbidimetric assay using anti‐albumin antibodies, with a Modular Analytics P module (Roche Diagnostics GmbH, Mannheim, Germany). The lower limit of detection was 3 g l–1. Reference ranges for albumin were 35–50 g l–1.
Statistical analysis
The present study was performed to do a pharmacokinetic comparison of a proof‐of‐concept prototype and the selected tablet for clinical development. In other words, the study was not designed to determine bioequivalence. As this study was not a bioequivalence study, the limits were used for sample size calculation only. The sample size was based on the comparison of the free testosterone of both formulations. In a previous study, a within‐subjects coefficient of variation (CV) of 21% was detected for AUC(0,230 min). Other CVs of free testosterone, total testosterone and DHT for AUC(0,230 min) and C max were lower. Therefore this CV was used for the sample size calculations. Assuming a CV of 21%, 12 evaluable subjects are required to compare two treatments with limits set to 0.75 and 1.33 (for both AUC(0,t) and C max) and at a power of 80% (β = 0.2) and α = 0.05.
Pharmacokinetic parameters including the time to maximum plasma concentration (t max), terminal half‐life (t 1/2), the maximum plasma concentration (C max) and area under the plasma concentration–time curve (AUC) were calculated based on actual and (for testosterone) baseline corrected individual concentration–time curves. AUCs were estimated using the linear trapezoidal rule. C max and t max were taken from the measured values. t 1/2 was calculated from the unweighted linear regression of the log‐transformed data determined at the elimination phase of the pharmacokinetic profile of each subject.
The AUC(0,1590 min) was determined as the area under the concentration vs. time curve from the first time point to the last time point with measurable drug concentration with a linear/log‐linear trapezoidal model. The AUC(0,∞) was calculated from the AUC(0,1590 min) by the addition of a constant (C p/λz), where C p is the last observed quantifiable concentration and λz is the elimination rate constant. C p/λz was determined by dividing the C p by λz using linear regression of C p vs. time data (standard extrapolation technique). The elimination rate constant and the corresponding elimination half‐life, was estimated by log‐linear least squares regression of the terminal part of the plasma concentration vs. time curve. The lag time (t lag) was determined as the first time point with a measurable concentration in plasma indicating the onset of absorption. The pharmacokinetic parameters were analyzed using the Watson 7.2 Bioanalytical LIMS software (Thermo Electron Corporation‐Philadelphia‐USA).
The demographic baseline levels of total and free testosterone, dihydrotestosterone, SHBG and albumin were calculated by taking the mean of F1 and F2. For the baseline corrected pharmacokinetic parameters the raw data of each subject was taken as baseline. Paired‐samples t‐tests were used to test the difference between the F1 and F2 pharmacokinetic parameters if the data were normally distributed. For non‐normally distributed data, Wilcoxon signed rank tests were used. For all analyses, a (two‐sided) P value <0.05 was considered statistically significant. Statistical analyses were performed in SPSS 21.0 (IBM SPSS Statistics for Windows, Version 21.0. Armonk, NY,USA: IBM Corp). The ratios of geometric least square means and the corresponding 90% confidence intervals (CI) of the C max and AUC(0,1590 min) were computed, with F2 as reference. This was performed with R‐software version 3.2.2 (R Foundation for Statistical Computing, Vienna, Austria).
Results
The baseline characteristics and hormone concentrations of the 12 study participants are outlined in Table 1. Of the 12 study participants, 11 subjects were of Western European descent, one subject was of mixed Western European‐Asian descent, and mean age was 23.3 years. Nine women were on hormonal contraception, none of which contained anti‐androgens or (anti)androgenic progestogens. Baseline levels of testosterone, SHBG and albumin were all in the expected range for premenopausal women (see Table 1). TSH and FSH were also in the expected range for premenopausal women.
Table 1.
Baseline and clinical characteristics of the participants
| Characteristic | Value (n = 12) |
|---|---|
| Age (years) | 23 (19–34) |
| Race (n) | |
| Western European descent | 11 |
| Mixed Western European – Asian descent | 1 |
| BMI (kg m–2) | 22.4 ± 2.5 |
| Contraception (n) | |
| hormonal | 9 |
| combined oral contraceptive pill | 8 |
| IUD (progestagen) | 1 |
| non‐hormonal | 3 |
| Total testosterone (ng ml–1) | 0.21 ± 0.12 |
| Free testosterone (pg ml–1) | 1.86 ± 0.76 |
| Dihydrotestosterone (ng ml–1) | 0.096 ± 0.078 |
| SHBG (nmol l–1) | 87.9 ± 39.0 |
| Albumin (g l–1) | 39.4 ± 2.60 |
Age is given in median (range). BMI and hormonal values are mean ± SD. To convert total testosterone to nmol l–1, multiply by 3.467. BMI, body mass index; IUD, intrauterine device; SHBG, sex hormone‐binding globulin.
For one subject, the pre‐dose plasma concentrations of testosterone, free testosterone and dihydrotestosterone in F2 were extremely high while the 10 min sample was significantly lower, suggesting sample mix‐up. For this reason, the results of this subject were excluded from PK calculations for the F2 dosing group with the analysis of testosterone, dihydrotestosterone and free testosterone. For another subject, the free testosterone result of the last time point during F1 dosing was questionable. For this reason this subject was not included in the free testosterone PK calculations for the F1 dosing group (see Figure 1 for CONSORT flow diagram).
Figure 1.
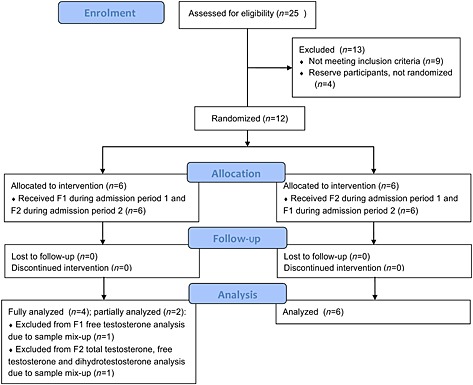
CONSORT flow diagram
Pharmacokinetic results
Testosterone, free testosterone and dihydrotestosterone
Pharmacokinetic results of the two administrations show that for both products, testosterone was rapidly absorbed with a total testosterone t max of 14 min for F1 and 15 min for F2 (0.229 and 0.250 h, respectively). Half‐life was 37 min for F1 and 38 min for F2 (0.615 and 0.629 h, respectively). Free testosterone reached the maximum concentration after 15 min for F1 and F2 (0.250 and 0.242 h, respectively) with a half‐life of 39 min for F1 and 36 min for F2 (0.652 and 0.593 h, respectively). Dihydrotestosterone reached its maximum concentration in 26 min for F1 and in 29 min for F2 (0.438 and 0.485 h, respectively) with a half‐life of 108 min for F1 and 84 min for F2 (1.80 and 1.40 h, respectively).
C max was significantly higher for total testosterone (P = 0.001), free testosterone (P = 0.008) and dihydrotestosterone (P = 0.001) after F2 administration compared with F1 dosing. This difference between the two dosing methods was also apparent in the 90% CI of the C max ratios (1.44 [1.29, 1.62], 1.58 [1.31, 1.90] and 1.30 [1.18, 1.43], respectively) that were outside the set limits (0.75, 1.33). Furthermore, the average AUC with F2 dosing was significantly higher for total testosterone (P = 0.002) and for free testosterone (P = 0.022) compared with the F1 dosing. The 90% CI of the AUC ratios were 1.44 (1.30, 1.60) for total testosterone, 1.68 (1.23, 2.28) for free testosterone, and 1.13 (0.90, 1.43), also outside the set limits. Finally, the t max of dihydrotestosterone following F1 dosing was significantly shorter than for F2 (P = 0.041). The pharmacokinetic parameters of total and free testosterone, and dihydrotestosterone after the different modes of administration, and the ratios and corresponding 90% CI of the C max and AUC(0,1590 min) are summarized in Table 2.
Table 2.
Pharmacokinetic parameters for total testosterone, free testosterone and dihydrotestosterone after F1 and F2 administration
| Analyte | Total testosterone | Free testosterone | Dihydrotestosterone | ||||
|---|---|---|---|---|---|---|---|
| Formulation | F1 | F2 | F1 | F2 | F1 | F2 | |
| n | 12 | 11 | 11 | 11 | 12 | 11 | |
| C max (ng ml –1 ) | Mean | 5.66 | 8.06* | 0.0300 | 0.0476* | 0.492 | 0.645* |
| 95% CI | 4.63, 6.69 | 6.84, 9.28 | 0.0230, 0.0370 | 0.0361, 0.0591 | 0.397, 0.587 | 0.508, 0.782 | |
| Ratio† | 1.44 | 1.58 | 1.30 | ||||
| 90% CI | 1.29, 1.62 | 1.31, 1.90 | 1.18, 1.43 | ||||
| AUC(0,1590 min) (ng ml –1 h) | Mean | 5.12 | 7.69* | 0.0276 | 0.0449* | 1.07 | 1.22 |
| 95% CI | 4.51, 5.73 | 6.22, 9.16 | 0.0177, 0.0375 | 0.0321, 0.0577 | 0.79, 1.35 | 0.88, 1.56 | |
| Ratio† | 1.44 | 1.68 | 1.13 | ||||
| 90% CI | 1.30, 1.60 | 1.23, 2.28 | 0.90, 1.43 | ||||
| t max (h) | Mean | 0.229 | 0.250 | 0.250 | 0.242 | 0.438 | 0.485* |
| 95% CI | 0.193, 0.265 | 0.212, 0.288 | 0.212, 0.288 | 0.201, 0.283 | 0.397, 0.479 | 0.465, 0.505 | |
| t 1/2 (h) | Mean | 0.615 | 0.629 | 0.652 | 0.593 | 1.80 | 1.40 |
| 95% CI | 0.554, 0.676 | 0.577, 0.681 | 0.536, 0.768 | 0.529, 0.657 | 1.23, 2.37 | 0.90, 1.90 | |
P < 0.05, value at F2 is significantly different from F1.For all calculations the pre‐dose concentration is subtracted from the determined concentration after dosing.
Ratio of geometric least square means. AUC(0,1590 min), area under the curve from time 0 to 1590 min; CI, confidence interval; C max, maximum concentration; F1, formulation 1; F2, formulation 2; t max, time to maximum concentration; t ½, half‐life.
The mean concentrations of testosterone, free testosterone and dihydrotestosterone measured after sublingual administration of a single dose of 0.5 mg testosterone (F1) and after the fixed dose combination tablet (F2) administration are shown in Figures 2, 3 and 4.
Figure 2.
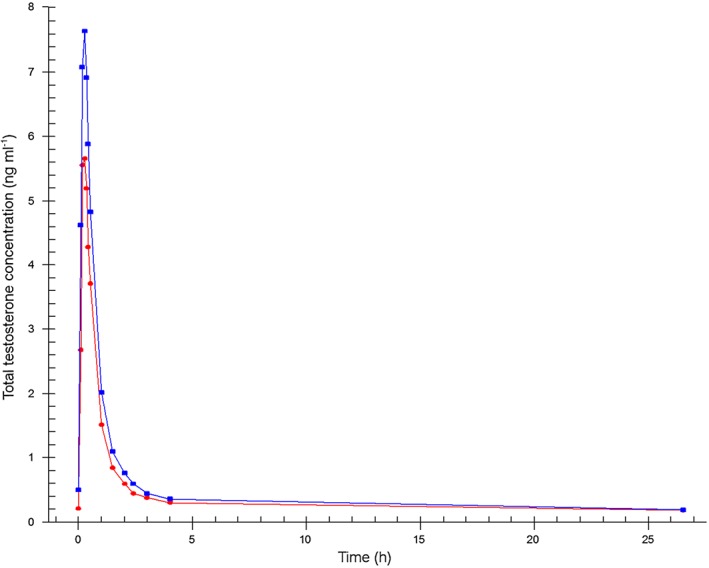
Mean total testosterone plasma concentration–time profiles.  Formulation 1: separate dosage forms prototype,
Formulation 1: separate dosage forms prototype,  Formulation 2: dual‐route/dual‐release fixed‐dose combination tablet
Formulation 2: dual‐route/dual‐release fixed‐dose combination tablet
Figure 3.
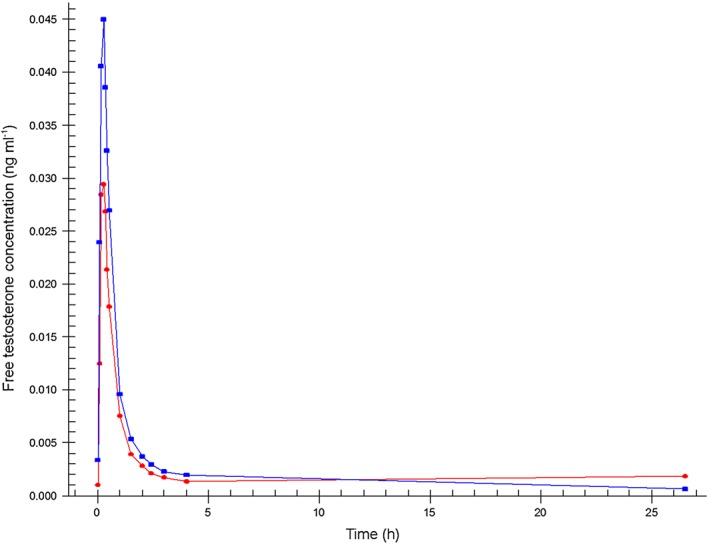
Mean free testosterone plasma concentration–time profiles.  Formulation 1: separate dosage forms prototype,
Formulation 1: separate dosage forms prototype,  Formulation 2: dual‐route/dual‐release fixed‐dose combination tablet
Formulation 2: dual‐route/dual‐release fixed‐dose combination tablet
Figure 4.
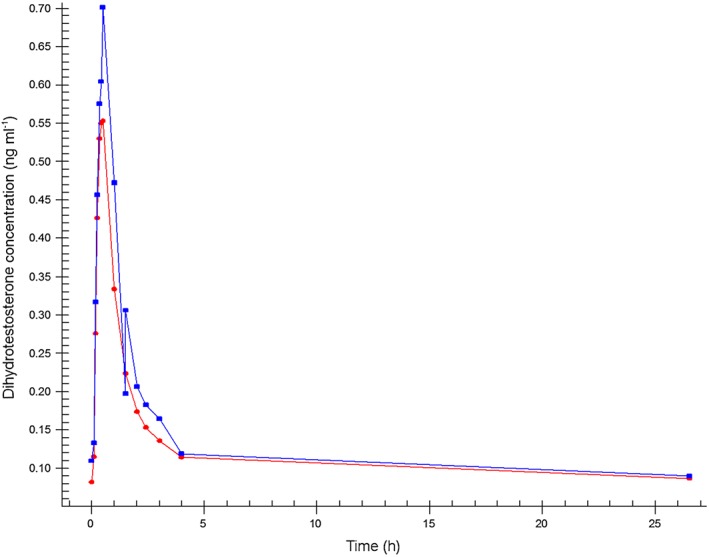
Mean dihydrotestosterone plasma concentration–time profiles.  Formulation 1: separate dosage forms prototype,
Formulation 1: separate dosage forms prototype,  Formulation 2: dual‐route/dual‐release fixed‐dose combination tablet
Formulation 2: dual‐route/dual‐release fixed‐dose combination tablet
Sildenafil and N‐desmethyl‐sildenafil
Pharmacokinetic results of the two modes of administration show that sildenafil was absorbed with a t max of 3 h and 53 min for F1 and 3 h and 6 min for F2 (3.88 and 3.10 h, respectively), and with a half‐life of 3 h and 52 min for F1 and 4 h and 58 min for F2 (3.87 and 4.96 h, respectively). Sildenafil t lag (median) was 3 h after F1 and 2 h and 45 min after F2 administration (3.00 and 2.75 h, respectively). Since for F1 the encapsulated tablet was taken after 150 min (2.5 h) the in vivo dissolution and absorption of sildenafil took 30 min. The in vivo lag time for F2 was 165–30 = 135 min, which was well in line with in vitro results of the tablet. N‐desmethyl‐sildenafil reached the maximum concentration after 4 h for F1 and after 3 h and 20 min for F2 (4.00 and 4.34 h, respectively) with a half‐life of 5 h and 13 min for F1 and 7 h and 4 min for F2 (5.21 and 7.07 h, respectively).
C max and AUC were not significantly different between the F1 and F2 administration either for sildenafil or for N‐desmethyl‐sildenafil. The 90% CI of the C max ratios (0.67 [0.43, 1.05] and 0.74 [0.53, 1.04], respectively) deviated from the 0.75, 1.33 limits, as did the 90% CI of the AUC ratio of sildenafil (0.84 [0.72, 0.99]). The 90% CI of the AUC ratio of N‐desmethyl‐sildenafil was within the set limits (0.85 [0.75, 0.96]). There was a significant difference between the N‐desmethyl‐sildenafil half‐lives of the two administration methods (P = 0.041). The pharmacokinetic parameters of sildenafil and its primary metabolite N‐desmethyl‐sildenafil after the F1 and F2 modes of administration, and the ratios and corresponding 90% CI of the C max and AUC(0,1590 min) are summarized in Table 3.
Table 3.
Pharmacokinetic parameters for sildenafil and N‐desmethyl sildenafil after either F1 or F2 administration
| Analyte | Sildenafil | N‐desmethyl sildenafil | |||
|---|---|---|---|---|---|
| formulation | F1 | F2 | F1 | F2 | |
| n | 12 | 12 | 12 | 12 | |
| C max (ng ml –1 ) | Mean | 268 | 173 | 55.5 | 42.7 |
| 95% CI | 188, 348 | 126, 220 | 44.1, 66.9 | 32.3, 53.1 | |
| Ratio† | 0.67 | 0.74 | |||
| 90% CI | 0.43, 1.05 | 0.53, 1.04 | |||
| AUC(0,1590 min) (ng ml –1 h) | Mean | 577 | 476 | 194 | 155 |
| 95% CI | 462, 692 | 401, 551 | 143, 245 | 127, 183 | |
| Ratio† | 0.84 | 0.85 | |||
| 90% CI | 0.71, 0.99 | 0.75, 0.96 | |||
| AUC(0,∞) (ng ml –1 h) | Mean | 596 | 500 | 203 | 171 |
| 95% CI | 481, 711 | 423, 577 | 151, 255 | 139, 203 | |
| t max (h) | Mean | 3.88 | 3.10 | 4.00 | 3.34 |
| 95% CI | 3.27, 4.49 | 2.74, 3.46 | 3.28, 4.72 | 2.89, 3.79 | |
| t lag (h) | Mean | 3.00 | 2.75 | 3.29 | 2.78 |
| 95% CI | 2.72, 3.28 | 2.40, 3.10 | 2.94, 3.64 | 2.37, 3.19 | |
| t 1/2 (h) | Mean | 3.87 | 4.96 | 5.21 | 7.07* |
| 95% CI | 2.72, 5.02 | 3.82, 6.10 | 4.55, 5.87 | 5.79, 8.35 | |
P < 0.05, value at F2 is significantly different from F1.
Ratio of geometric least square means. AUC(0,1590 min), area under the curve from time 0 to 1590 min; AUC(0,∞), area under the curve from time 0 to infinity; C max, maximum concentration; t lag, time to first measurable quantity; t max, time to maximum concentration; t ½, half‐life.
The mean concentration–time profiles of sildenafil and N‐desmethyl‐sildenafil measured after oral administration of a single dose of sildenafil (50 mg) using the F1 and F2 modes of administration are shown in Figures 5, 6.
Figure 5.
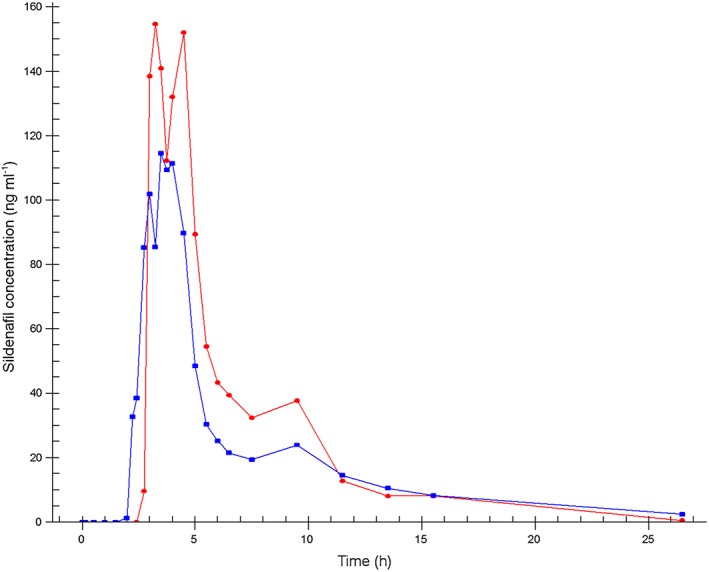
Mean sildenafil plasma concentration–time profiles.  Formulation 1: separate dosage forms prototype,
Formulation 1: separate dosage forms prototype,  Formulation 2: dual‐route/dua‐release fixed‐dose combination tablet
Formulation 2: dual‐route/dua‐release fixed‐dose combination tablet
Figure 6.
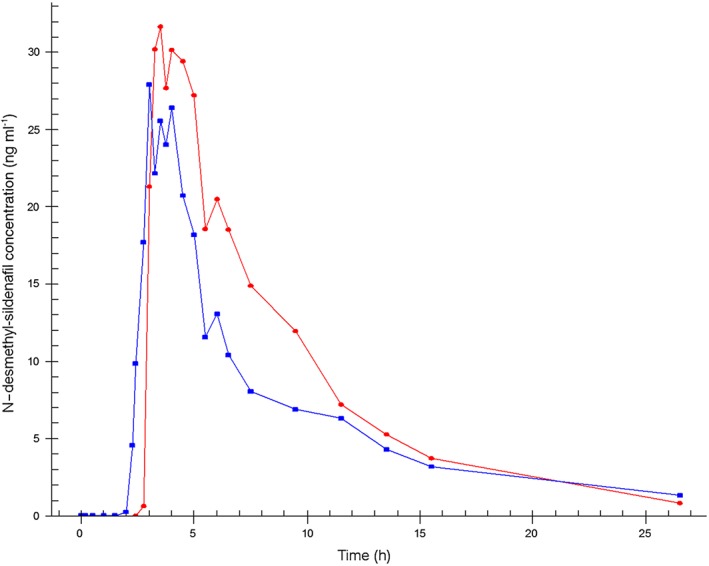
Mean N‐desmethyl‐sildenafil plasma concentration–time profiles.  Formulation 1: separate dosage forms prototype,
Formulation 1: separate dosage forms prototype,  Formulation 2: dual‐route/dual‐release fixed‐dose combination tablet
Formulation 2: dual‐route/dual‐release fixed‐dose combination tablet
The two formulations were well tolerated by all volunteers.
Discussion
This study demonstrates that the pharmacokinetic profile of two different drugs (testosterone, 0.5 mg, and sildenafil, 50 mg) administered in a single fixed dose combination via two different routes and with two different time vs. release profiles, is comparable but not bioequivalent to the separate administration of the two drugs administered 2.5 h apart and in separate dosage forms. This observation of non‐bioequivalence of the proof‐of‐concept formulation and the clinical formula is not meaningful since the clinical development started with a dose‐finding study of the new final fixed dose combination tablet. This is in line with a previous study in which the pharmacokinetic profiles of a single fixed dose combination tablet containing testosterone and buspirone and its separate administration equivalent were compared 30. For that combination tablet, the same rapid outer coating dissolution technology and inner core delayed‐release technology were employed.
In both formulations, sublingual administration of testosterone resulted in an almost immediate and steep increase in circulating total and free testosterone concentrations. Time to maximum concentration was 10 to 20 min. Total and free testosterone reached baseline concentrations again after approximately 2.5 h. These observations are in line with previous studies 16, 30, 36.
There was an apparent difference in the C max of testosterone between the two formulations. C max of total testosterone following liquid dosing of testosterone (F1) was consistent with the reported C max of this dosage form found in previous studies 16, 30, 36. However, the C max and AUC of total and free testosterone were higher for the fixed dose combination tablet (F2). This indicates a very fast and more complete absorption from the solid polymeric matrix. Since there is no delay or difference in absorption profile for the two formulations, the in vivo dissolution of testosterone from the tablet coating is not likely to be the rate‐limiting step in the absorption process. The bioavailability of testosterone from the tablet is higher than from the liquid, which was also observed between the two formulations described by van Rooij et al. 30. This is likely due to a higher testosterone concentration gradient for testosterone in saliva from the drug originating from the tablet coating as compared with the administered testosterone liquid leading to a more complete absorption. Secondly, it should also be taken into account that a certain volume of the liquid testosterone (0.5 ml) may have leaked away to the oesophagus and been swallowed which would certainly lower the bioavailability from the liquid dosage form.
Dihydrotestosterone reached maximum concentration within 30 min and returned to baseline levels within 4 h, consistent with previous pharmacokinetic studies 30, 36. There was a small difference between the two formulations in t max of dihydrotestosterone in the present study (26 min for F1 vs. 29 min for F2), but this is unlikely to be clinically relevant. No such differences were observed for total testosterone or free testosterone and differences in (speed of) metabolism from testosterone to dihydrotestosterone between the two different dosing methods were not likely.
In the previous study of van Rooij et al., it was estimated that a 0.50 mg testosterone cyclodextrin solution had an absolute bioavailability of approximately 70%. These observations and the current results that show that the new combination tablet with the solid form coating of testosterone has both a higher C max and AUC, indicate that the absolute bioavailability of this tablet is above 70%, and close to 80%, as was described earlier for the combination tablet containing testosterone and buspirone 30.
The current study also investigated a new delayed release matrix coated core that was designed to release the full dose of sildenafil at once after approximately 150 min (specified range of 120–240 min), thereby mimicking the time lag that was applied between the separate administration of the two active compounds. The results showed that the in vivo rupture time of the tablet was within the set specifications. t lag and t max for both sildenafil and its main metabolite N‐desmethyl‐sildenafil were comparable for both modes of administration. There was, as expected, a high variability between subjects for the sildenafil concentrations due to the high first pass effect.
The average C max was slightly (non‐significantly) lower for the combination tablet as compared with the encapsulated tablet of F1 that was administered separately 150 min after the administration of the testosterone dose. This is probably caused by a difference in rate of absorption within different parts of the gastro‐intestinal tract. The separately administered gelatin capsule dissolves almost immediately in the stomach and thus absorption starts in the stomach and first part of the small intestine. The fixed dose combination tablet releases its drug load after an approximately 150 min longer passage time through the gastrointestinal tract and absorption therefore starts further down in the small intestines, where the available surface for absorption is smaller than in the upper small intestines. However, the AUC of N‐desmethyl sildenafil, the main first pass metabolite of sildenafil, does not suggest a significant incomplete absorption of sildenafil, but rather a more extensive first‐pass effect of the sildenafil released from the fixed dose combination tablet that releases the drug further down in the gastrointestinal tract. This slight difference was also observed in the previous study that investigated the combination tablet containing testosterone and buspirone 30.
The present method of testosterone administration increases the brain's sensitivity to sexual stimuli from approximately 3 h up until approximately 6 h after administration 15. Previous research suggests that a dose of 0.75 mg testosterone, administered sublingually using the same liquid solution as used in F1, will not further increase the brain's sensitivity to sexual stimuli than a dose of 0.5 mg 36. Therefore it is not expected that the increased bioavailability of testosterone of F2 will further increase the brain's sensitivity to sexual stimuli as compared with F1.
The lower bioavailability of sildenafil in F2 (approximately 80% of F1) is not expected to be clinically relevant. The effectivity of the combination of testosterone and sildenafil for the treatment of FSIAD is based on testosterone's ability to increase the brain's sensitivity to sexual stimuli, and sildenafil's ability to increase genital vasocongestion (see 15 for a full description of the mechanism of effect). Sildenafil at a dose of 25 mg has a lower C max and AUC than observed in the present study 37, but it is clinically effective in men with erectile dysfunction 38. Sildenafil has the same physiological effect in women as it does in men 39 but when administered without testosterone it is not an effective treatment for FSIAD 9, 10. So, if 1] a 25 mg dose induces a physiological effect in men, 2] sildenafil has a comparable physiological effect in women as in men, and 3] the C max and AUC of the present 50 mg dose in F2 is substantially higher than observed for the 25 mg dose, it is likely that the present dose of 50 mg in F2 (i.e. combined with sublingual testosterone) will be adequate to ensure the combination drug's efficacy in treating FSIAD. This remains to be confirmed in future research.
The study did not take into account cyclical and diurnal variation of testosterone. Also, no post‐menopausal women were included and of the 12 premenopausal women, nine were on hormonal contraceptives. This raises the question as to how generalizable the total testosterone, free testosterone and DHT results of this study are to these other situations and populations. Total testosterone increases approximately 25% in the ovulatory phase as compared with the mid‐follicular phase 40, and a decrease in total testosterone concentrations which is smaller in magnitude has been observed in post‐menopausal women 41. The testosterone administration tested in the present study increases total testosterone concentrations 25 to 40‐fold compared with baseline levels. Thus, it is unlikely that cyclical, diurnal and menopausal status related variations in circulating testosterone levels will impact the present results and conclusions as such fluctuations would account for less than 1% of the observed C max in total testosterone. Hormonal contraceptives are known to induce an increase in circulating SHBG levels and, consequentially, a decrease in circulating free testosterone 42. This may have impacted the observed free testosterone C max and AUC of the current study, but not those of testosterone and DHT as these assessments are compiled of SHBG‐bound and unbound concentrations. If free testosterone C max and AUC are influenced by use of hormonal contraceptives in this study, it is unlikely that this influences the ratio of these parameters between the two formulations (i.e. if hormonal contraception decreases free testosterone levels following F1, it will do so in the same manner and magnitude in F2). The possible impact on free testosterone is thus unlikely to impact the conclusions of the study because the goal was to compare the PK of two different formulations with each other and each subject served as their own control.
In conclusion, the absorption of testosterone and sildenafil and the time delay for the release of sildenafil after administration of the dual route/dual release fixed dose combination tablet was adequate. The newly developed tablet may therefore be a convenient and suitable formulation that increases dosing practicality and decreases potential temporal non‐adherence through circumventing the relatively complex temporal dosing scheme in future clinical trials and in a daily practice setting for treating FSIAD.
Competing Interests
All authors have completed the Unified Competing Interest form at www.icmje.org/coi_disclosure.pdf (available on request from the corresponding author) and declare JB, KvR, LdL, HF, HK and AT had support from Emotional Brain for the submitted work, JB, KvR, LdL, HK, BO and AT have owned shares and/or stock options in Emotional Brain in the previous 3 years and JB, LdL and HF have a patent WO/2012/158 030 pending. JB and AT have a patent WO/2007/055 563 issued. AT has a patent WO/2005/107 810 issued.
This research was funded by Emotional Brain BV. The authors acknowledge Anko Eissens for his assistance in the development of the combination tablet. The authors acknowledge the principle investigator, Tjeert Mensinga, and the personnel of QPS Netherlands for their excellent care of the subjects. The authors also acknowledge Eurofins employees Lorraine Jacobs for her help in the calculation of the pharmacokinetic parameters and Rudi Segers, Robert van der Wegen and George Wouters for their excellent bioanalytical work.
Author Contributions
JB, KvR, LdL, HF, HK, BO and AT wrote the manuscript. JB, KvR, LdL and AT designed and executed the study. JB, KvR and LdL analyzed the data. LdL, HF and AT developed the tablet and contributed analytical tools.
Bloemers, J. , van Rooij, K. , de Leede, L. , Frijlink, H. W. , Koppeschaar, H. P. F. , Olivier, B. , and Tuiten, A. (2016) Single dose sublingual testosterone and oral sildenafil vs. a dual route/dual release fixed dose combination tablet: a pharmacokinetic comparison. Br J Clin Pharmacol, 81: 1091–1102. doi: 10.1111/bcp.12887.
References
- 1. Laumann EO, Paik A, Rosen RC. Sexual dysfunction in the United States: prevalence and predictors. JAMA 1999; 281: 537–44. [DOI] [PubMed] [Google Scholar]
- 2. Fugl‐Meyer KS. Sexual disabilities and sexual problems In: Sex in Sweden, ed Lewin B. Stockholm: The National Institute of Public Health, 2000; 199–215. [Google Scholar]
- 3. Shifren JL, Monz BU, Russo PA, Segreti A, Johannes CB. Sexual problems and distress in United States women: prevalence and correlates. Obstet Gynecol 2008; 112: 970–8. [DOI] [PubMed] [Google Scholar]
- 4. Davison SL, Bell RJ, LaChina M, Holden SL, Davis SR. The relationship between self‐reported sexual satisfaction and general well‐being in women. J Sex Med 2009; 6: 2690–7. [DOI] [PubMed] [Google Scholar]
- 5. American Psychiatric Association . Diagnostic and Statistical Manual of Mental Disorders (DSM‐IV), 4th Edition, Washington DC: American Psychiatric Association, 2000. [Google Scholar]
- 6. American Psychiatric Association . Diagnostic and Statistical Manual of Mental Disorders, 5th Edition, Arlington, VA: American Psychiatric Publishing, 2013. [Google Scholar]
- 7. Bancroft J. Central inhibition of sexual response in the male: a theoretical perspective. Neurosci Biobehav Rev 1999; 23: 763–84. [DOI] [PubMed] [Google Scholar]
- 8. Janssen E, Bancroft J. The dual control model: the role of sexual inhibition and excitation in sexual arousal and behaviour In: The psychophysiology of sex, ed Janssen E. Bloomington: Indiana University Press, 2007; 197. [Google Scholar]
- 9. van der Made F, Bloemers J, Yassem WE, Kleiverda G, Everaerd W, van Ham D, Olivier B, Koppeschaar H, Tuiten A. The influence of testosterone combined with a PDE5‐inhibitor on cognitive, affective, and physiological sexual functioning in women suffering from sexual dysfunction. J Sex Med 2009; 6: 777–90. [DOI] [PubMed] [Google Scholar]
- 10. Van der Made F, Bloemers J, Van Ham D, El Yassem W, Kleiverda G, Everaerd W, Olivier B, Tuiten A. Childhood sexual abuse, selective attention for sexual cues and the effects of testosterone with or without vardenafil on physiological sexual arousal in women with sexual dysfunction: A pilot study. J Sex Med 2009; 6: 429–39. [DOI] [PubMed] [Google Scholar]
- 11. Poels S, Bloemers J, van Rooij K, Goldstein I, Gerritsen J, van Ham D, van Mameren F, Chivers M, Everaerd W, Koppeschaar H, Olivier B, Tuiten A. Toward personalized sexual medicine (part 2): testosterone combined with a PDE5 inhibitor increases sexual satisfaction in women with HSDD and FSAD, and a low sensitive system for sexual cues. J Sex Med 2013; 10: 810–23. [DOI] [PubMed] [Google Scholar]
- 12. van Rooij K, Poels S, Bloemers J, Goldstein I, Gerritsen J, van Ham D, van Mameren F, Chivers M, Everaerd W, Koppeschaar H, Olivier B, Tuiten A. Toward personalized sexual medicine (part 3): testosterone combined with a Serotonin1A receptor agonist increases sexual satisfaction in women with HSDD and FSAD, and dysfunctional activation of sexual inhibitory mechanisms. J Sex Med 2013; 10: 824–37. [DOI] [PubMed] [Google Scholar]
- 13. Gerritsen J, van der Made F, Bloemers J, van Ham D, Kleiverda G, Everaerd W, Olivier B, Levin R, Tuiten A. The clitoral photoplethysmograph: a new way of assessing genital arousal in women. J Sex Med 2009; 6: 1678–87. [DOI] [PubMed] [Google Scholar]
- 14. Bloemers J, Scholte HS, van Rooij K, Goldstein I, Gerritsen J, Olivier B, Tuiten A. Reduced gray matter volume and increased white matter fractional anisotropy in women with hypoactive sexual desire disorder. J Sex Med 2014; 11: 753–67. [DOI] [PubMed] [Google Scholar]
- 15. Bloemers J, van Rooij K, Poels S, Goldstein I, Everaerd W, Koppeschaar H, Chivers M, Gerritsen J, van Ham D, Olivier B, Tuiten A. Toward personalized sexual medicine (part 1): integrating the ‘dual control model’ into differential drug treatments for hypoactive sexual desire disorder and female sexual arousal disorder. J Sex Med 2013; 10: 791–809. [DOI] [PubMed] [Google Scholar]
- 16. Tuiten A, Van Honk J, Koppeschaar H, Bernaards C, Thijssen J, Verbaten R. Time course of effects of testosterone administration on sexual arousal in women. Arch Gen Psychiatry 2000; 57: 149–53 discussion 155–6. [DOI] [PubMed] [Google Scholar]
- 17. Postma A, Meyer G, Tuiten A, van Honk J, Kessels RP, Thijssen J. Effects of testosterone administration on selective aspects of object‐location memory in healthy young women. Psychoneuroendocrinology 2000; 25: 563–75. [DOI] [PubMed] [Google Scholar]
- 18. Aleman A, Bronk E, Kessels RP, Koppeschaar HP, van Honk J. A single administration of testosterone improves visuospatial ability in young women. Psychoneuroendocrinology 2004; 29: 612–7. [DOI] [PubMed] [Google Scholar]
- 19. Schutter DJ, van Honk J. Decoupling of midfrontal delta‐beta oscillations after testosterone administration. Int J Psychophysiol 2004; 53: 71–3. [DOI] [PubMed] [Google Scholar]
- 20. van Honk J, Schutter DJ, Hermans EJ, Putman P, Tuiten A, Koppeschaar H. Testosterone shifts the balance between sensitivity for punishment and reward in healthy young women. Psychoneuroendocrinology 2004; 29: 937–43. [DOI] [PubMed] [Google Scholar]
- 21. van Honk J, Peper JS, Schutter DJ. Testosterone reduces unconscious fear but not consciously experienced anxiety: implications for the disorders of fear and anxiety. Biol Psychiatry 2005; 58: 218–25. [DOI] [PubMed] [Google Scholar]
- 22. van Honk J, Schutter DJ. Testosterone reduces conscious detection of signals serving social correction: implications for antisocial behavior. Psychol Sci 2007; 18: 663–7. [DOI] [PubMed] [Google Scholar]
- 23. van Honk J, Tuiten A, Hermans E, Putman P, Koppeschaar H, Thijssen J, Verbaten R, van Doornen L. A single administration of testosterone induces cardiac accelerative responses to angry faces in healthy young women. Behav Neurosci 2001; 115: 238–42. [DOI] [PubMed] [Google Scholar]
- 24. Hermans EJ, Putman P, van Honk J. Testosterone administration reduces empathetic behavior: a facial mimicry study. Psychoneuroendocrinology 2006; 31: 859–66. [DOI] [PubMed] [Google Scholar]
- 25. Hermans EJ, Putman P, Baas JM, Gecks NM, Kenemans JL, van Honk J. Exogenous testosterone attenuates the integrated central stress response in healthy young women. Psychoneuroendocrinology 2007; 32: 1052–61. [DOI] [PubMed] [Google Scholar]
- 26. Hermans EJ, Ramsey NF, van Honk J. Exogenous testosterone enhances responsiveness to social threat in the neural circuitry of social aggression in humans. Biol Psychiatry 2008; 63: 263–70. [DOI] [PubMed] [Google Scholar]
- 27. Bos PA, Terburg D, van Honk J. Testosterone decreases trust in socially naive humans. Proc Natl Acad Sci U S A 2010; 107: 9991–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Eisenegger C, Naef M, Snozzi R, Heinrichs M, Fehr E. Prejudice and truth about the effect of testosterone on human bargaining behaviour. Nature 2010; 463: 356–9. [DOI] [PubMed] [Google Scholar]
- 29. Bos PA, van Honk J, Ramsey NF, Stein DJ, Hermans EJ. Testosterone administration in women increases amygdala responses to fearful and happy faces. Psychoneuroendocrinology 2013; 38: 808–17. [DOI] [PubMed] [Google Scholar]
- 30. van Rooij K, de Leede L, Frijlink HW, Bloemers J, Poels S, Koppeschaar H, Olivier B, Tuiten A. Pharmacokinetics of a prototype formulation of sublingual testosterone and a buspirone tablet, versus an advanced combination tablet of testosterone and buspirone in healthy premenopausal women. Drugs R&D 2014; 14: 125–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Loftsson T, Brewster ME. Pharmaceutical applications of cyclodextrins: effects on drug permeation through biological membranes. J Pharm Pharmacol 2011; 63: 1119–35. [DOI] [PubMed] [Google Scholar]
- 32. Frijlink HW, Eissens AC, Schoonen AJM, Lerk CF. The effects of cyclodextrins on drug absorption II. In vivo observations. Int J Pharm 1990; 64: 195–205. [Google Scholar]
- 33. Loftsson T, Brewster ME. Pharmaceutical applications of cyclodextrins: basic science and product development. J Pharm Pharmacol 2010; 62: 1607–21. [DOI] [PubMed] [Google Scholar]
- 34. Labrie F, Belanger A, Belanger P, Berube R, Martel C, Cusan L, Gomez J, Candas B, Castiel I, Chaussade V, Deloche C, Leclaire J. Androgen glucuronides, instead of testosterone, as the new markers of androgenic activity in women. J Steroid Biochem Mol Biol 2006; 99: 182–8. [DOI] [PubMed] [Google Scholar]
- 35. Miller KK, Rosner W, Lee H, Hier J, Sesmilo G, Schoenfeld D, Neubauer G, Klibanski A. Measurement of free testosterone in normal women and women with androgen deficiency: comparison of methods. J Clin Endocrinol Metab 2004; 89: 525–33. [DOI] [PubMed] [Google Scholar]
- 36. van Rooij K, Bloemers J, de Leede L, Goldstein I, Lentjes E, Koppeschaar H, Olivier B, Tuiten A. Pharmacokinetics of three doses of sublingual testosterone in healthy premenopausal women. Psychoneuroendocrinology 2012; 37: 773–81. [DOI] [PubMed] [Google Scholar]
- 37. Nichols DJ, Muirhead GJ, Harness JA. Pharmacokinetics of sildenafil after single oral doses in healthy male subjects: absolute bioavailability, food effects and dose proportionality. Br J Clin Pharmacol 2002; 53 (Suppl 1): 5S–12S. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Goldstein I, Lue TF, Padma‐Nathan H, Rosen RC, Steers WD, Wicker PA. Oral sildenafil in the treatment of erectile dysfunction. Sildenafil Study Group. N Engl J Med 1998; 338: 1397–404. [DOI] [PubMed] [Google Scholar]
- 39. Laan E, van Lunsen RH, Everaerd W, Riley A, Scott E, Boolell M. The enhancement of vaginal vasocongestion by sildenafil in healthy premenopausal women. J Womens Health Gend Based Med 2002; 11: 357–65. [DOI] [PubMed] [Google Scholar]
- 40. Salonia A, Pontillo M, Nappi RE, Zanni G, Fabbri F, Scavini M, Daverio R, Gallina A, Rigatti P, Bosi E, Bonini PA, Montorsi F. Menstrual cycle‐related changes in circulating androgens in healthy women with self‐reported normal sexual function. J Sex Med 2008; 5: 854–63. [DOI] [PubMed] [Google Scholar]
- 41. Spencer JB, Klein M, Kumar A, Azziz R. The age‐associated decline of androgens in reproductive age and menopausal Black and White women. J Clin Endocrinol Metab 2007; 92: 4730–3. [DOI] [PubMed] [Google Scholar]
- 42. Wiegratz I, Kutschera E, Lee JH, Moore C, Mellinger U, Winkler UH, Kuhl H. Effect of four different oral contraceptives on various sex hormones and serum‐binding globulins. Contraception 2003; 67: 25–32. [DOI] [PubMed] [Google Scholar]