

Abstract
Aims
Lenvatinib was recently approved for the treatment of radioiodine‐refractory differentiated thyroid cancer (RR‐DTC). Here, we characterized the pharmacokinetic (PK) profile of lenvatinib and identified intrinsic and extrinsic factors that explain interindividual PK variability in humans.
Methods
This population PK analysis used pooled data from 15 clinical studies, including eight phase 1 studies in healthy subjects, four phase 1 studies in patients with solid tumours, two phase 2 studies in patients with thyroid cancer and one phase 3 study in patients with RR‐DTC.
Results
The final pooled dataset included data from 779 subjects receiving 3.2–32 mg oral lenvatinib, mainly once daily as tablets or capsules. Lenvatinib PK was best described by a three‐compartment model with linear elimination. Lenvatinib absorption was best described by simultaneous first‐ and zero‐order absorption. The population mean value for lenvatinib apparent clearance (CL/F) was 6.56 l h−1 [percent coefficient of variation (%CV) 25.5], and was independent of dose and time. The relative bioavailability of lenvatinib in capsule form was 90% vs. tablets (%CV 30.2). The final PK model included significant but marginal effects of body weight (2.8% of CL/F variation), liver‐function markers [alkaline phosphatase (−11.7%) and albumin (−6.3%)] and concomitant cytochrome P450 3A4 inducers (+30%) and inhibitors (−7.8%) on lenvatinib CL/F. Lenvatinib PK was unaffected by pH‐elevating agents, dose, age, sex, race, alanine aminotransferase, aspartate aminotransferase or bilirubin levels, or renal function.
Conclusions
The significant effects of several covariates on lenvatinib PK variability were small in magnitude, and therefore were not considered clinically relevant, or to warrant any dose adjustment.
Keywords: dose adjustment, hepatic impairment, lenvatinib, pharmacokinetics, pooled analysis, population
What is Already Known about this Subject
The pharmacokinetics of lenvatinib have previously been studied only in individual clinical trials with limited sample sizes, or in targeted pharmacokinetic studies of special populations (e.g. subjects with renal or hepatic impairment).
What This Study Add
The first population pharmacokinetic analysis for lenvatinib provides crucial insight into the intrinsic and extrinsic factors affecting lenvatinib pharmacokinetics.
The study provides useful information on the interaction of lenvatinib with pH‐elevating agents commonly used by cancer patients, based on clinical trial data and not as a prospectively designed drug–drug‐interaction study.
Introduction
Lenvatinib is an oral multikinase inhibitor of vascular endothelial growth factor receptor 1–3, fibroblast growth factor receptor 1–4, platelet‐derived growth factor receptor α, ret proto‐oncogene and v‐kit Hardy–Zuckerman 4 feline sarcoma viral oncogene homologue signalling pathways 1, 2, 3, 4. The targeting of multiple angiogenic pathways may circumvent the compensatory mechanisms that are upregulated when only a single angiogenic pathway is inhibited 5. Lenvatinib has been evaluated extensively in phase 1 studies both in healthy subjects and patients with solid tumours 6, 7, 8, 9, 10, 11, 12, 13, as well as in phase 2 studies in patients with various types of thyroid cancer (Eisai data on file) 14.
Following oral administration in humans, lenvatinib is absorbed rapidly, with the observed time for the maximum concentration to occur (t max) typically ranging between 1 h and 4 h 15. The apparent oral clearance and terminal volume of distribution ranges from 4.2 l h−1 to 7.1 l h−1 and 50.5 l to 163.0 l, respectively, with a terminal half‐life (t 1/2) of ~28 h. The contribution of renal excretion to oral clearance is minimal (~1–2%). Both the area under the plasma concentration vs. time curve (AUC) and maximum plasma concentration (C max) of lenvatinib have been shown to increase proportionally with dose 6, 7. In vitro and in vivo studies have demonstrated that lenvatinib is metabolized in both the liver and kidney, and that it is primarily excreted directly into the bile 16, 17, with ~64% of radiolabelled lenvatinib recovered in the faeces and ~25% in the urine, and only 2.5% of the administered dose recovered intact in humans 16. Lenvatinib is highly plasma protein bound (range 97.9–98.6%), primarily to albumin, and the binding is concentration independent.
Lenvatinib has been approved in the United States, Japan and Europe for the treatment of patients with radioiodine‐refractory differentiated thyroid cancer (RR‐DTC) based on the results of the phase 3 Study of (E7080) LEnvatinib in Differentiated Cancer of the Thyroid (SELECT)—a multicentre, randomized, double‐blind, placebo‐controlled study of lenvatinib (24 mg day–1) in patients with RR‐DTC 18. Although the majority of patients with differentiated thyroid cancer (DTC) can achieve a cure with standard treatment approaches—including surgery, radioactive iodine therapy and the suppression of thyroid stimulating hormone (TSH)—the prognosis is poor for patients with RR‐DTC, who have a 10‐year survival rate of only 10% 19 as they often do not respond to standard treatment 20. Data from the SELECT study demonstrated that lenvatinib treatment significantly prolonged median progression‐free survival compared with placebo [18.3 months vs. 3.6 months, respectively; hazard ratio 0.21, 99% confidence interval (CI) 0.14, 0.31; P < 0.0001] 18.
The objectives of the present study were to characterize the pharmacokinetic (PK) profile of lenvatinib and identify the intrinsic and extrinsic factors that explain interindividual variability (IIV) in the PK of lenvatinib in healthy subjects and patients with several types of cancer, based on pooled data from phase 1, 2 and 3 studies.
Methods
Patients
The current analysis was based on pooled data collected from eight phase 1 studies of lenvatinib in healthy subjects (Eisai data on file) 8, 9, 10, 11, 12, 13, four phase 1 studies of lenvatinib in patients with solid tumours (Eisai data on file) 6, 7, two phase 2 studies of lenvatinib in patients with thyroid cancers 14, 21 and the phase 3 SELECT trial of patients with RR‐DTC 18. All patients signed informed consent forms for participation in the respective trials, which were conducted in accordance with the International Conference on Harmonization Good Clinical Practice guidelines and local regulations, and approved by the independent review boards of each participating centre.
Base PK model development
The PK model was developed using the NONlinear Mixed‐Effect Modeling software (NONMEM) version 7.2.0 (ICON Development Solutions, Ellicott City, MD, USA) interfaced with PDxPop 5.0 (ICON Development Solutions). Compartment models one, two and three, with first‐, zero‐ and simultaneous zero‐ and first‐order absorption and first‐order elimination from the central compartment were tested. The process of model building started with fitting a simple PK model to the data and proceeding to more complex effects added one at a time. A two‐compartment model with simultaneous zero‐ and first‐order absorption and first‐order elimination from the central compartment was the starting point for this PK model development. Based on a graphical analysis of lenvatinib concentration–time profiles from phase 1 healthy volunteer studies, a three‐compartment model was superior to a two‐compartment model in describing the concentration–time profile following single dose of lenvatinib; this was supported by a 560‐point drop in the objective function value (OFV). IIV could not be estimated on intercompartment clearance between the central and two peripheral compartments (Q1 and Q2). Residual error was best described by a combined additive and proportional error for time after dose (TAD) ≤2 h and a separate proportional error for phase 1 clinical pharmacology studies and cancer patient studies. Estimating lag in absorption resulted in a 725‐point drop in OFV; however, the PK model was found to be sensitive to initial estimates. Hence, it was decided not to include lag time in the PK model. The effect of formulation type (capsule vs. tablet) on relative bioavailability (F1) and body weight as an allometric constant were included in the base model and resulted in a significant drop in OFV, precise parameter estimates and improvement in goodness‐of‐fit‐plots. Additionally, the effect of dose on CL/F and on F1 was also tested as part of the base model. The IIV, using an exponential error structure, which assumed log‐normal distribution for PK parameters, was also included as appropriate. The residual variability (ε) was assessed by additive, proportional and combined error structures. A residual variability model with different residual error used for healthy volunteers and patients and for early time points was found to be superior to various other models examined. All permutations of IIV and residual variability error structures were tested systematically using first‐order conditional estimation with interaction method in NONMEM.
For comparison of the full and reduced models, a P‐value of 0.01 [change in OFV of 6.68 for 1 degree of freedom (df)] was required. In addition, several criteria were considered to assess model acceptability: (i) minimization procedure converges; (ii) at least three significant digits; (iii) covariance step terminates without any error message; (iv) standard errors of the estimates less than 50%; and (v) no correlation >0.95 between parameters. Additionally, goodness‐of‐fit plots of the population and individual predicted concentrations vs. observed concentrations, as well as conditioned weighted residuals vs. population predicted concentrations and vs. time, were plotted to assess the PK model and its ability to describe the available data.
Covariate PK model development
Evaluation of associations between PK parameters and covariates were performed in a stepwise manner. First, the plots of individual Bayes post hoc estimates of eta from the base model against the covariates were generated to determine if visual trends existed in the data. Eta shrinkage was calculated and reported for each IIV parameter estimated by the model. Parameters with eta shrinkage >30% were not considered for the covariate analysis. Covariates showing a potential relationship graphically were assessed in the base model by univariate addition and ranked in descending order of change in OFV. Covariates were considered significant when P < 0.01. All significant variables were then added simultaneously to a full model. Each covariate was then re‐evaluated by deleting it from the full covariate model. For an effect to remain part of the model, a significance level of P ≤ 0.001 [ΔOFV >10.83 (1df)] was required.
The effect of each of the following intrinsic factors was investigated in relation to the PK of lenvatinib: demographics [race, sex, age, body weight, population (healthy subjects vs. patients with cancer)], Eastern Cooperative Oncology Group performance status (ECOG PS), renal function [as measured by creatinine clearance and by Common Terminology Criteria for Adverse Events v.4.0 (CTCAE) grades for renal function], liver function [as measured by levels of alanine aminotransferase (ALT), alkaline phosphatase (ALP), aspartate aminotransferase (AST), bilirubin, albumin and CTCAE grades for liver function] and thyroid function (as measured by TSH levels). In addition, the effect of several extrinsic factors were investigated in relation to the PK of lenvatinib: capsules vs. tablets, drug–drug interactions [specifically, coadministration of cytochrome P450 (CYP) 3A4 inhibitors or inducers] and coadministration of drugs that elevate gastric pH (proton pump inhibitors, H2 blockers and/or antacids).Continuous covariates were normalized by the median of the observed values and were incorporated into the PK parameter as shown in the following example for the effect of body weight on clearance:
where CL i represents the value of clearance for the i th individual, TVCL is the typical population value of the clearance, θ WGT_CL is the effect of the covariate (here, the weight) on the clearance, WGT i is the covariate value of the i th subject, WGT median is the median study population covariate value and η i_CL is the random interindividual variable, which is a normally distributed statistical error with a mean of zero and variance ω2 CL. Categorical covariates were incorporated into the PK parameters as index variables, as shown in the following example for the case of the effect of sex on clearance:
where CL i represents the value of clearance for the i th individual, SEX i is the covariate value of the i th subject, TVCL is the typical population value of clearance corresponding to one of the categories (here, for the clearance in men for SEX i = 0), θ SEXCL is the effect of the covariate (here, clearance in women compared with clearance in men) and η i_CL is the random interindividual variable that is an independent and normally distributed statistical error with a mean of zero and variance of ω2 CL.
The final PK model was evaluated using the prediction‐corrected visual predictive check 22 and bootstrap approach 23. Briefly, lenvatinib concentrations were simulated (n = 10) for 779 subjects from 15 studies using their original dosing history, covariate information and PK model. The dependent variable was then subjected to prediction correction before the statistics were calculated. For the bootstrap approach, owing to the large dataset and long runtime, 500 replicates were performed. Lenvatinib exposure for each subject was calculated as:
where individual relative bioavailability and apparent clearance (CL/F) are empirical Bayes estimates from the final population PK model for each subject.
Evaluation of the clinical implication of covariates on lenvatinib PK
To evaluate the clinical relevance of covariates identified as significant in the final PK model, the lenvatinib PK profile was simulated using parameter estimates from the final PK model, where lenvatinib was administrated as a capsule to patients with cancer, at a dose of 24 mg once daily for 15 days, to achieve steady state. Median and 90% prediction intervals (PI) of PK profiles were then compared for different values of covariates.
Results
Subject characteristics and dataset
The final pooled dataset from 15 phase 1, 2 and 3 studies for the population PK model included data from 779 subjects, of whom 56.0% were men, 70.2% were white, 59.2% had an ECOG PS ≤ 1, 42.0% had DTC, the median age was 55.0 years and the median body weight was 75.0 kg (Table 1). The majority of subjects did not receive CYP3A4 inducers (97.6%), CYP3A4 inhibitors (93.7%), H2 blockers (94.9%), proton pump inhibitors (77.5%), antacids (96.8%) or pH‐elevating agents (71.4%; Table 1). However, the number of subjects receiving pH‐elevating agents [proton pump inhibitors (n = 175), antacids (n = 25), H2 blockers (n = 40)] during the collection of PK samples was sufficient to characterize the effects of these agents on lenvatinib absorption and bioavailability. Missing covariate data were not imputed. If multiple assessments for a continuous covariate were available for an individual subject or patient, then the last available covariate value was carried forward and used. Missing categorical variables were omitted.
Table 1.
Summary of baseline demographics and categorical covariates included in the population pharmacokinetic analysis of lenvatinib
| Continuous covariates | n | Mean | SD | Median | Minimum | Maximum |
|---|---|---|---|---|---|---|
| Age (years) | 779 | 53.2 | 15.8 | 55.0 | 18.0 | 89.0 |
| Weight (kg) | 779 | 76.7 | 19.4 | 75.0 | 32.6 | 177.5 |
| Albumin ( g l −1 ) | 779 | 39.8 | 5.1 | 40.0 | 19.0 | 52.0 |
| Alkaline phosphatase ( IU l −1 ) | 779 | 104.6 | 82.1 | 77.0 | 19.0 | 752.0 |
| Alanine transaminase ( IU l −1 ) | 779 | 24.8 | 31.1 | 19.0 | 5.0 | 660.0 |
| Aspartate transaminase ( IU l −1 ) | 779 | 25.9 | 36.1 | 21.0 | 6.0 | 930.0 |
| Bilirubin (μmol l −1 ) | 779 | 9.8 | 7.7 | 8.0 | 2.0 | 101.1 |
| Creatinine clearance ( ml min −1 ) | 779 | 101.5 | 34.9 | 98.0 | 17.0 | 268.0 |
| Thyroid stimulating hormone ( mIU l −1 ) * | 327 | 0.28 | 1.04 | 0.02 | 0.002 | 14.8 |
| Categorical covariates | Category | n | % | |||
| Sex | Male | 436 | 56.0 | |||
| Female | 343 | 44.0 | ||||
| Race | White | 547 | 70.2 | |||
| Black | 73 | 9.4 | ||||
| Asian | 5 | 0.6 | ||||
| Japanese | 91 | 11.7 | ||||
| Hispanic | 6 | 0.8 | ||||
| Other | 49 | 6.3 | ||||
| American Indian or Alaska Native | 3 | 0.4 | ||||
| Native Hawaiian, Pacific Islander or other | 5 | 0.6 | ||||
| ECOG | 0 | 253 | 32.5 | |||
| 1 | 208 | 26.7 | ||||
| 2 | 19 | 2.4 | ||||
| 3 | 1 | 0.1 | ||||
| Missing | 298 | 38.3 | ||||
| Tumour type | Non‐cancer patient | 196 | 25.2 | |||
| DTC | 327 | 42.0 | ||||
| MTC | 56 | 7.2 | ||||
| ATC | 9 | 1.2 | ||||
| Other | 191 | 24.5 | ||||
| CYP3A4 inducers | Yes | 19 | 2.4 | |||
| No | 760 | 97.6 | ||||
| CYP3A4 inhibitors | Yes | 49 | 6.3 | |||
| No | 730 | 93.7 | ||||
| H2 blockers | Yes | 40 | 5.1 | |||
| No | 739 | 94.9 | ||||
| Proton pump inhibitors | Yes | 175 | 22.5 | |||
| No | 604 | 77.5 | ||||
| Antacids | Yes | 25 | 3.2 | |||
| No | 754 | 96.8 | ||||
| pH‐elevating agents | Yes | 223 | 28.6 | |||
| No | 556 | 71.4 | ||||
ATC, anaplastic thyroid cancer; CYP, cytochrome P450; DTC, differentiated thyroid cancer; ECOG, Eastern Cooperative Oncology Group; MTC, medullary thyroid cancer; SD, standard deviation.
Measured only for patients with thyroid cancer.
The PK dataset included 10 265 plasma concentrations: 5077 from full profiles from seven studies in healthy subjects, 3192 from full profiles and sparse samples from four phase 1 studies in patients with various types of cancer, 354 sparse samples from two phase 2 studies from patients with various types of thyroid cancer and 1642 sparse samples from one phase 3 study in patients with DTC.
Final PK model
The final population PK model for lenvatinib was best described by a three‐compartment model with simultaneous first‐ and zero‐order absorption and linear elimination from the central compartment parameterized for CL/F, apparent volume of central compartment (V1/F), apparent volume of peripheral compartments (V2/F and V3/F), intercompartment clearance between V2 and V3 (Q2/F and Q3/F), absorption rate constant and duration of zero‐order absorption (D1). Shrinkage for CL/F, D1 and F1 were below 30%; therefore, covariate effects were examined only on CL/F, F1 and D1 in the univariate analysis, as appropriate.
The parameter estimates for the final PK model are provided in Table 2. The population mean value for lenvatinib CL/F was estimated to be 6.56 l h−1 [percent coefficient of variation (%CV) 25.5], which was independent of dose (3.2–32.0 mg) and time. The capsule formulation had a 10.4% lower bioavailability relative to the tablet formulation. The model also included covariate functions quantifying the effects of body weight, population (healthy vs. patients with cancer), liver function markers (ALP and albumin) and concomitant administration of CYP3A4 inducers and inhibitors on lenvatinib CL/F (Table 2). Lenvatinib CL/F increased with increasing body weight (power function = 0.75). Body weight was added as an allometric constant on CL/F and volume parameters, and showed a significant effect but only explained 2.8% of the IIV on CL/F. Healthy subjects had a 15% higher lenvatinib CL/F than patients with cancer; lenvatinib CL/F decreased by 11.7% with ALP >the upper limit of normal and decreased by 16.3% with albumin levels <30 g l−1; concomitant administration of CYP3A4 inducers increased lenvatinib CL/F by 30%; and CYP3A4 inhibitors decreased lenvatinib CL/F by 7.8%. Conversely, sex, race, age, ECOG PS, renal function markers (creatinine clearance and CTCAE grades for renal function), liver function markers (ALT, AST, bilirubin and CTCAE grades for liver function) and a thyroid function marker (TSH) had no significant effect on lenvatinib CL/F (Table 2). Additionally, drugs that raise gastric pH (proton pump inhibitors, H2 blockers and antacids) had no effect on lenvatinib F1 or D1.
Table 2.
Population pharmacokinetic parameter estimates of lenvatinib from the final model
| Parameter | NONMEM population mean (95% CI) | Bootstrap median (95% CI) |
|---|---|---|
| CL/F ( l h −1 ) = Θ CL *(WGT/75) 0.75 *Θ INDU INDU *Θ INHIB INHIB *Θ ALB ALB *Θ ALP ALP *Θ TM TM | ||
| Basal CL/F in l h −1 (Θ CL ) | 6.56 (6.29, 6.83) | 6.54 (6.24, 6.89) |
| Effect of CYP3A4 inducers on CL/F (Θ INDU ) | 1.30 (1.27, 1.33) | 1.30 (1.22, 1.38) |
| Effect of CYP3A4 inhibitors on CL/F (Θ INHIB ) | 0.922 (0.902, 0.942) | 0.923 (0.898, 0.949) |
| Effect of albumin (<30 g l −1 ) on CL/F (Θ ALB ) | 0.837 (0.773, 0.901) | 0.842 (0.751, 0.946) |
| Effect of ALP on CL/F (Θ ALP ) | 0.883 (0.850, 0.916) | 0.888 (0.835, 0.956) |
| Effect of population on CL/F (Θ TM ) | 1.15 (1.09, 1.21) | 1.15 (1.09, 1.22) |
| V1/F (l) = Θ V1 *WGT/75 | ||
| Basal V1/F in l (Θ V1 ) | 49.3 (47.1, 51.5) | 49.1 (46.5, 51.8) |
| V1/F (l) = Θ V1 *WGT/75 | ||
| Basal V2/F in l (Θ V2 ) | 30.7 (28.4, 33.0) | 30.7 (28.2, 33.4) |
| V1/F (l) = Θ V1 *WGT/75 | ||
| Basal V3/F in l (Θ V3 ) | 37.1 (34.9, 39.3) | 37.2 (34.3, 40.0) |
| Q1/F ( l h −1 ) = Θ Q1 *(WGT/75) 0.75 | ||
| Basal Q1/F in l h −1 (Θ Q1 ) | 3.52 (3.29, 3.75) | 3.53 (3.28, 3.81) |
| Q2/F [ l h −1 ] = Θ Q2 *(WGT/75) 0.75 | ||
| Basal Q2/F in l h −1 (Θ Q2 ) | 0.769 (0.703, 0.835) | 0.770 (0.670, 0.863) |
| Ka ( l h −1 ) = Θ Ka | ||
| Basal Ka in l h −1 (Θ Ka ) | 1.02 (0.95, 1.09) | 1.04 (0.94, 1.14) |
| D1 (h) = Θ D1 | ||
| Basal D1 in h (Θ D1 ) | 1.22 (1.15, 1.29) | 1.23 (1.13, 1.32) |
| F1 = Θ F1 | ||
| Relative bioavailability of capsule vs. tablet formulation (Θ F1 ) | 0.896 (0.870, 0.922) | 0.894 (0.850, 0.935) |
| Interindividual variability (%CV a ) | ||
| CL/F | 25.5 | 25.3 (21.1, 29.7) |
| V1/F | 22.8 | 21.9 (1.2, 29.8) |
| V2/F | 39.0 | 38.6 (32.7, 44.5) |
| V3/F | 30.3 | 31.5 (24.6, 40.5) |
| Ka | 54.8 | 52.7 (31.5, 70.9) |
| D1 | 76.7 | 78.0 (69.6, 86.2) |
| F1 | 30.2 | 30.3 (27.7, 33.0) |
| Residual variability | ||
| Proportional (%CV ) (Clinical pharmacology studies) | 16.9 | 16.8 (14.9, 18.5) |
| Proportional (%CV) (patient studies) | 33.3 | 33.3 (31.5, 35.4) |
| Proportional (%CV) (TAD <2 h) | 48.1 | 48.0 (44.9, 51.0) |
| Additional ( ng ml −1 ) (TAD <2 h) | 7.19 | 7.60 (5.03, 9.81) |
Albumin (ALB) 0 (ALB ≥ 30 g l−1) or 1 (ALB < 30 g l−1); alkaline phosphatase (ALP) measurement (IU l−1) 0 [ALP ratio (laboratory value/upper limit of normal value) ≤1] or 1 (ALP ratio >1); CI, confidence interval; CL/F, apparent clearance; %CV, percent coefficient of variation; CYP, cytochrome P450; D1, duration of zero‐order absorption; F1, relative bioavailability of capsule‐to‐tablet formulation; INDU, CYP3A4 inducers; INHIB, CYP3A4 inhibitors; Ka, absorption rate; NONMEM, NONlinear Mixed‐Effect Modeling software; Q1, intercompartment clearance between V1 and V2; Q2, intercompartment clearance between V2 and V3; TAD, time after dose; V1/F, apparent volume of central compartment; V2/F and V3/F, apparent volume of peripheral compartment; WGT, weight (kg); TM, treatment group, 0 (cancer patients) or 1 (healthy subjects).
The % CV for both intersubject and proportional residual variability is an approximation taken as the square root of the variance*100.
Model evaluation
Simulated lenvatinib concentrations were calculated for all dosing regimens used in the pooled PK analysis and are shown with observed lenvatinib concentrations in Figure 1. Based on sampling schemes, plots were separated by phase 1 clinical pharmacology studies in healthy subjects, phase 1 studies in patients with solid tumours, and phase 2 and 3 studies in patients with thyroid cancer. The median (red line) and 5th and 95th percentiles (90% PI, grey area) of these prediction‐corrected simulated concentrations were calculated and plotted with prediction‐corrected observed lenvatinib concentration data. The majority of the observed lenvatinib concentrations fell within the 90% PI and the observations were centred around the median of the simulated data, which is indicative that the model captured the variability in the observed data adequately. There was a slight misfit at low concentrations during the absorption phase. This was expected as lag time could not be estimated using pooled data from 15 studies with varying sampling schemes during the first hour postabsorption. The results of the bootstrap method are presented in Table 2. As shown, the CIs are generally narrow and the median values of the distribution of bootstrapped parameter values were also consistent with the original parameter estimates.
Figure 1.
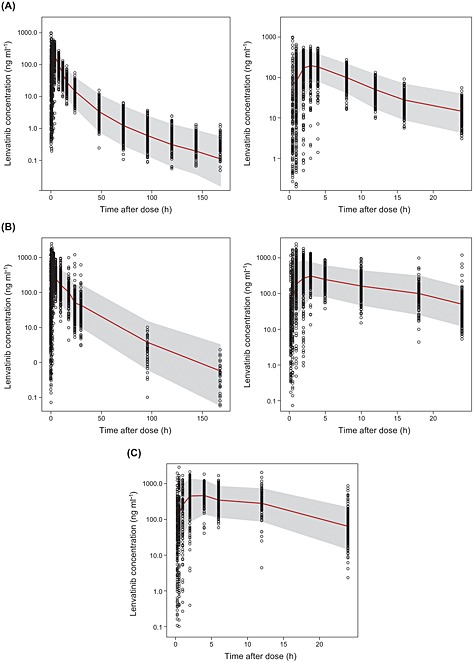
Visual predictive check of observed and model‐predicted lenvatinib concentrations: (A) following a single dose of lenvatinib administered to healthy subjects; (B) following multiple doses of lenvatinib administered to patients with solid tumours; and (C) following multiple doses of lenvatinib administered to patients with thyroid cancer. Model‐predicted median (red line), and 5th to 95th percentiles (grey shade) are plotted. For (A) and (B), the left‐hand panel shows the full profile; the right‐hand panel shows the expanded 24‐h postdose profile
Evaluation of the clinical implication of covariates on lenvatinib PK
As the final PK model included significant effects of body weight, albumin, ALP and concomitant administration of CYP3A4 inducers and inhibitors on lenvatinib CL/F, we simulated the possible clinical implications of these effects (Figure 2). The predicted median lenvatinib steady‐state concentrations and 90% PI were higher in the population with lower body weight (50 kg) compared with the heavier population (100 kg); however, there was significant overlap with the 90% PI. Similarly, the predicted median lenvatinib steady‐state concentrations and 90% PI were only slightly higher in the population receiving concomitant CYP3A4 inhibitors, which is consistent with the estimated marginal effect of a 7.8% higher CL/F. The predicted median lenvatinib steady‐state concentrations and 90% PI were lower in subjects receiving concomitant CPY3A4 inducers; however, there was also significant overlap with the 90% PI. A substantial overlap with the 90% PI was also seen for the albumin level (low albumin vs. all others) and ALP effects (high ALP level vs. all others).
Figure 2.
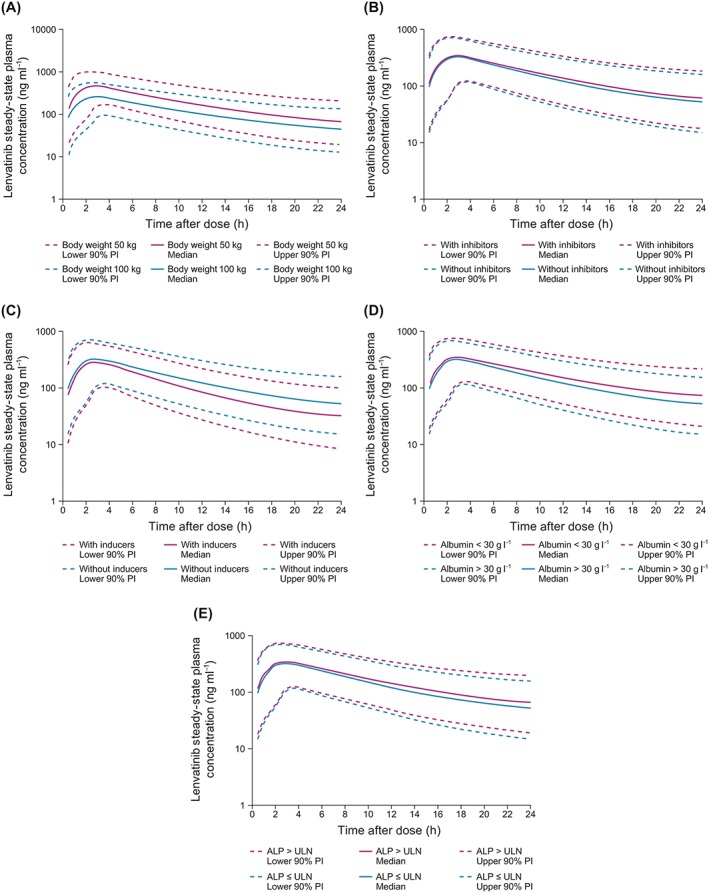
Simulated median and 90% prediction interval (PI) for lenvatinib administration to: (A) patients weighing either 50 kg or 100 kg, without concomitant cytochrome P450 (CYP) 3A4 inducers or inhibitors, and with normal alkaline phosphatase (ALP) and albumin levels; (B) patients weighing 75 kg, with and without concomitant CYP3A4 inhibitors, without CYP3A4 inducers, and with normal ALP and albumin levels; (C) patients weighing 75 kg, with and without concomitant CYP3A4 inducers, without CYP3A4 inhibitors, and with normal ALP and albumin levels; (D) patients weighing 75 kg, with albumin <30 g l−1 or >30 g l−1, without CYP3A4 inducers or inhibitors, and with normal ALP levels; (E) patients weighing 75 kg, with ALP greater than the upper limit of normal (ULN) or ≤ ULN, without CYP3A4 inducers or inhibitors, and with normal albumin levels
Lenvatinib exposure
The steady‐state exposure of lenvatinib increased proportionally with increasing dose, suggesting linear PK (Figure 3). The mean dose‐normalized AUC for patients with RR‐DTC in the SELECT study (n = 260) was 3710 ng*h ml−1 (% CV 38.3).
Figure 3.
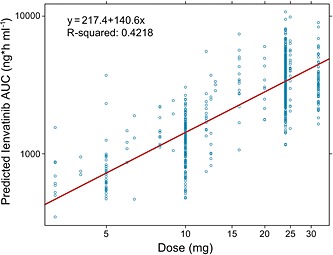
Model‐predicted lenvatinib steady‐state exposure vs. dose. AUC, area under the plasma concentration vs. time curve
Discussion
The present study characterized the population PK of lenvatinib using pooled data from 15 studies [eight phase 1 studies in healthy subjects, four phase 1 studies in patients with solid tumours, two phase 2 studies in patients with various types of thyroid cancer and one phase 3 study (SELECT) in patients with RR‐DTC]. The PK of lenvatinib was best described by a three‐compartment model with linear elimination. There was a slight misfit at low concentrations during the absorption phase. This was expected as lag time could not be estimated with combined data from 15 studies, with varying sampling schemes during the first hour postabsorption. However, overall, the lenvatinib concentration–time course was reasonably well defined by the final PK model, with good predictive performance. CL/F and exposure (AUC) of lenvatinib showed moderate‐to‐high variability. CL/F was also 15% higher and, correspondingly, the extent of exposure was slightly lower for healthy subjects than for patients with cancer. The magnitude of this small effect was within the IIV for CL/F (25.5%) and hence of no clinical relevance.
The following intrinsic factors had no significant effect on lenvatinib CL/F: dose, sex, race, age, ECOG PS, renal function markers (creatinine clearance and CTCAE grades for renal function) and a thyroid function marker (TSH). Drugs elevating gastric pH, including proton pump inhibitors, H2 blockers and antacids, also had no significant effect on lenvatinib relative bioavailability or absorption duration. Therefore, based on our retrospective population PK analysis of data pooled from 15 clinical trials, the rate and extent of lenvatinib absorption are not affected by pH‐elevating agents.
In the present study, the effect of hepatic impairment was examined in several ways: (i) using liver function markers (ALT, AST, ALP, albumin and bilirubin) as continuous variables; (ii) using CTCAE grades for liver function; and (iii) using National Cancer Institute (NCI) Organ Dysfunction Working Group criteria for hepatic dysfunction (Tables S1 and S2). CTCAE grading represents a system already in use in the clinic, and is therefore easier to implement and is sometimes more clinically meaningful, as seen in the case for albumin and liver function status. The NCI criteria have been proposed as an alternative to the Child–Pugh score for hepatic dysfunction in the field of oncology, as liver function marker data are routinely available in the clinic whereas the Child–Pugh score is not 24.
Although ALT, AST, bilirubin and CTCAE grades for liver function had no significant effects on lenvatinib CL/F, ALP and albumin reduced C/F significantly. However, the effects were minor and unlikely to be clinically relevant. Lenvatinib CL/F was reduced by 16.7% when the albumin level was <30 g l−1 and by 11.7% when ALP was greater than the upper limit of normal. These results were consistent with a previous study of patients with hepatic impairment, where the ratio of dose‐normalized AUC0–∞ geometric least square means for subjects with mild‐to‐normal and moderate‐to‐normal liver function were 1.2 and 1.1, respectively. 11. An additional variable that had a significant effect on lenvatinib CL/F was body weight but this was considered not to be clinically meaningful as it only explained 2.8% of the IIV, with substantial overlap with the 90% PI.
In conclusion, the final population PK model described here included significant effects of body weight, liver function markers (ALP and albumin) and concomitant administration of CYP3A4 inducers and inhibitors on lenvatinib CL/F. However, these effects were small and PK simulations for these effects showed a major overlap with the 90% PI for steady‐state profiles. Therefore, these effects are not considered to be clinically relevant and do not warrant dose adjustment.
Competing Interests
All authors have completed the Unified Competing Interest form at http://www.icmje.org/coi_disclosure.pdf (available on request from the corresponding author) and declare: AG and ZH have been employed by Eisai Ltd. in the previous 3 years, and RS has been employed by Eisai Inc. in the previous 3 years; BJ and JC have received honoraria and research funding from Eisai Inc. in the previous 3 years.
This study was funded by Eisai Inc. Editorial support was provided by Oxford PharmaGenesis Inc., and funded by Eisai Inc. All authors have approved the final article.
Eisai employs several of the authors of this pooled analysis. Eisai funded the conduct of this research and the editorial and writing support that was provided by Oxford PharmaGenesis, Newtown, PA, USA.
Supporting information
Table S1 National Cancer Institute Organ Dysfunction Working Group criteria for hepatic dysfunction
Table S2 The Common Terminology Criteria for Adverse Events (CTCAE) version 4.0
Supporting info item
Supporting info item
Gupta, A. , Jarzab, B. , Capdevila, J. , Shumaker, R. , and Hussein, Z. (2016) Population pharmacokinetic analysis of lenvatinib in healthy subjects and patients with cancer. Br J Clin Pharmacol, 81: 1124–1133. doi: 10.1111/bcp.12907.
References
- 1. Matsui J, Funahashi Y, Uenaka T, Watanabe T, Tsuruoka A, Asada M. Multi‐kinase inhibitor E7080 suppresses lymph node and lung metastases of human mammary breast tumor MDA‐MB‐231 via inhibition of vascular endothelial growth factor‐receptor (VEGF‐R) 2 and VEGF‐R3 kinase. Clin Cancer Res 2008; 14: 5459–65. [DOI] [PubMed] [Google Scholar]
- 2. Matsui J, Yamamoto Y, Funahashi Y, Tsuruoka A, Watanabe T, Wakabayashi T, Uenaka T, Asada M. E7080, a novel inhibitor that targets multiple kinases, has potent antitumor activities against stem cell factor producing human small cell lung cancer H146, based on angiogenesis inhibition. Int J Cancer 2008; 122: 664–71. [DOI] [PubMed] [Google Scholar]
- 3. Okamoto K, Kodama K, Takase K, Sugi NH, Yamamoto Y, Iwata M, Tsuruoka A. Antitumor activities of the targeted multi‐tyrosine kinase inhibitor lenvatinib (E7080) against RET gene fusion‐driven tumor models. Cancer Lett 2013; 340: 97–103. [DOI] [PubMed] [Google Scholar]
- 4. Tohyama O, Matsui J, Kodama K, Hata‐Sugi N, Kimura T, Okamoto K, Minoshima Y, Iwata M, Funahashi Y. Antitumor activity of lenvatinib (e7080): an angiogenesis inhibitor that targets multiple receptor tyrosine kinases in preclinical human thyroid cancer models. J Thyroid Res 2014; 2014: 638747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bergers G, Hanahan D. Modes of resistance to anti‐angiogenic therapy. Nat Rev Cancer 2008; 8: 592–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Boss DS, Glen H, Beijnen JH, Keesen M, Morrison R, Tait B, Copalu W, Mazur A, Wanders J, O'Brien JP, Schellens JH, Evans TR. A phase I study of E7080, a multitargeted tyrosine kinase inhibitor, in patients with advanced solid tumours. Br J Cancer 2012; 106: 1598–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Yamada K, Yamamoto N, Yamada Y, Nokihara H, Fujiwara Y, Hirata T, Koizumi F, Nishio K, Koyama N, Tamura T. Phase I dose‐escalation study and biomarker analysis of E7080 in patients with advanced solid tumors. Clin Cancer Res 2011; 17: 2528–37. [DOI] [PubMed] [Google Scholar]
- 8. Shumaker RC, Zhou M, Ren M, Fan J, Martinez G, Aluri J, Darpo B. Effect of lenvatinib (E7080) on the QTc interval: results from a thorough QT study in healthy volunteers. Cancer Chemother Pharmacol 2014; 73: 1109–17. [DOI] [PubMed] [Google Scholar]
- 9. Shumaker R, Aluri J, Fan J, Martinez G, Ren M, Chen K. Evaluation of the effects of formulation and food on the pharmacokinetics of lenvatinib (E7080) in healthy volunteers. Int J Clin Pharmacol Ther 2014; 52: 284–91. [DOI] [PubMed] [Google Scholar]
- 10. Shumaker R, Aluri J, Fran J, Martinez G, Thompson GA, Ren M. Effects of ketoconazole on the pharmacokinetics of lenvatinib (E7080) in healthy participants. Clin Pharmacol Drug Dev 2015; 4: 155–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Shumaker R, Aluri J, Fan J, Martinez G, Pentikis H, Ren M. Influence of hepatic impairment on lenvatinib pharmacokinetics following single‐dose oral administration. J Clin Pharmacol 2015; 55: 317–27. [DOI] [PubMed] [Google Scholar]
- 12. Shumaker R, Aluri J, Fan J, Martinez G, Thompson GA, Ren M. Effect of rifampicin in the pharmacokinetics of lenvatinib in healthy adults. Clin Drug Investig 2014; 34: 651–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lee L, D'Angelo P, Verbel D, Martinez G, Aluri J, Brimhall D. A randomized, three‐treatment, three‐period, six‐sequence‐crossover, single‐center, bioequivalence study to evaluate the impact of different 10‐mg crystalline forms on the pharmacokinetics of lenvatinib in healthy volunteers. Int J Clin Pharmacol Ther 2015; 53: 190–8. [DOI] [PubMed] [Google Scholar]
- 14. Cabanillas ME, Schlumberger M, Jarzab B, Martins RG, Pacini F, Robinson B, McCaffrey JC, Shah MH, Bodenner DL, Topliss D, Andresen C, O'Brien JP, Ren M, Funahashi Y, Allison R, Elisei R, Newbold K, Licitra LF, Sherman SI, Ball DW. A phase 2 trial of lenvatinib (E7080) in advanced, progressive, radioiodine‐refractory, differentiated thyroid cancer: a clinical outcomes and biomarker assessment. Cancer 2015; 121: 2749–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Shumaker R, Fan J, Martinez G, Chen K. Comparative bioavailability study of a 10‐mg capsule and a 10‐mg tablet of lenvatinib (E7080) in healthy subjects [abstract]. Clin Pharmacol Ther 2012; 91: S68. Abstract PII‐46. [Google Scholar]
- 16. Dubbelman AC, Rosing H, Mergui‐Roelvink M, Gupta A, Verbel D, Sellecchia R, Fan J, Thompson GA, Schumaker R, Huitema AD, Beijnen JH, Schellens JH. A mass balance study of 14C‐lenvatinib (E7080) in patients with advanced solid tumours or lymphomas [abstract]. Br J Clin Pharmacol 2013; 76: 831. [Google Scholar]
- 17. Inoue K, Asai N, Mizuo H, Fukuda K, Kusano K, Yoshimura T. Unique metabolic pathway of [14C]lenvatinib after oral administration to male cynomolgus monkey. Drug Metab Dispos 2012; 40: 662–70. [DOI] [PubMed] [Google Scholar]
- 18. Schlumberger M, Tahara M, Wirth LJ, Robinson B, Brose MS, Elisei R, Habra MA, Newbold K, Shah MH, Hoff AO, Gianoukakis AG, Kiyota N, Taylor MH, Kim SB, Krzyzanowska MK, Dutcus CE, de las Heras B, Zhu J, Sherman SI. Lenvatinib versus placebo in radioiodine‐refractory thyroid cancer. N Engl J Med 2015; 372: 621–30. [DOI] [PubMed] [Google Scholar]
- 19. Durante C, Haddy N, Baudin E, Leboulleux S, Hartl D, Travagli JP, Caillou B, Ricard M, Lumbroso JD, de Vathaire F, Schlumberger M. Long‐term outcome of 444 patients with distant metastases from papillary and follicular thyroid carcinoma: benefits and limits of radioiodine therapy. J Clin Endocrinol Metab 2006; 91: 2892–9. [DOI] [PubMed] [Google Scholar]
- 20. Busaidy NL, Cabanillas ME. Differentiated thyroid cancer: management of patients with radioiodine nonresponsive disease. J Thyroid Res 2012; 2012: 618985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Takahashi S, Tahara M, Kiyota N, Yamazaki T, Chayahara N, Nakano K, Inagaki R, Toda K, Enokida T, Minami H, Imamura Y, Sasaki T, Suzuki T, Fujino K, Dutcus C. Phase II study of lenvatinib (LEN), a multi‐targeted tyrosine kinase inhibitor, in patients (pts) with all histologic subtypes of advanced thyroid cancer (differentiated, medullary, and anaplastic) [abstract]. Ann Oncol 2014; 25: iv340–56. [Google Scholar]
- 22. Bergstrand M, Hooker AC, Wallin JE, Karlsson MO. Prediction‐corrected visual predictive checks for diagnosing nonlinear mixed‐effects models. AAPS J 2011; 13: 143–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Yafune A, Ishiguro M. Bootstrap approach for constructing confidence intervals for population pharmacokinetic parameters. I: A use of bootstrap standard error. Stat Med 1999; 18: 581–99. [DOI] [PubMed] [Google Scholar]
- 24. Ramanathan RK, Egorin MJ, Takimoto CH, Remick SC, Doroshow JH, LoRusso PA, Mulkerin DL, Grem JL, Hamilton A, Murgo AJ, Potter DM, Belani CP, Hayes MJ, Peng B, Ivy SP, National Cancer Institute Organ Dysfunction Working Group . Phase I and pharmacokinetic study of imatinib mesylate in patients with advanced malignancies and varying degrees of liver dysfunction: a study by the National Cancer Institute Organ Dysfunction Working Group. J Clin Oncol 2008; 26: 563–9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1 National Cancer Institute Organ Dysfunction Working Group criteria for hepatic dysfunction
Table S2 The Common Terminology Criteria for Adverse Events (CTCAE) version 4.0
Supporting info item
Supporting info item