

Abstract
Aims
One barrier contributing to the lack of pharmacokinetic (PK) data in paediatric populations is the need for serial sampling. Analysis of clinically obtained specimens and data may overcome this barrier. To add evidence for the feasibility of this approach, we sought to determine PK parameters for fentanyl in children after cardiac surgery using specimens and data generated in the course of clinical care, without collecting additional blood samples.
Methods
We measured fentanyl concentrations in plasma from leftover clinically‐obtained specimens in 130 paediatric cardiac surgery patients and successfully generated a PK dataset using drug dosing data extracted from electronic medical records. Using a population PK approach, we estimated PK parameters for this population, assessed model goodness‐of‐fit and internal model validation, and performed subset data analyses. Through simulation studies, we compared predicted fentanyl concentrations using model‐driven weight‐adjusted per kg vs. fixed per kg fentanyl dosing.
Results
Fentanyl clearance for a 6.4 kg child, the median weight in our cohort, is 5.7 l h–1 (2.2–9.2 l h–1), similar to values found in prior formal PK studies. Model assessment and subset analyses indicated the model adequately fit the data. Of the covariates studied, only weight significantly impacted fentanyl kinetics, but substantial inter‐individual variability remained. In simulation studies, model‐driven weight‐adjusted per kg fentanyl dosing led to more consistent therapeutic fentanyl concentrations than fixed per kg dosing.
Conclusions
We show here that population PK modelling using sparse remnant samples and electronic medical records data provides a powerful tool for assessment of drug kinetics and generation of individualized dosing regimens.
Keywords: analgesia, opportunistic research, paediatrics, population pharmacokinetics, post‐operative pain, pragmatic research
What is Already Known about this Subject
Pharmacokinetic (PK) parameters are largely unknown for paediatric populations.
A major barrier to paediatric PK research is the need for serial blood sampling.
What this Study Adds
We estimated fentanyl PK parameters for children using only remnant plasma specimens and electronic medical records data.
Model‐driven weight‐adjusted per kg fentanyl dosing led to more consistent therapeutic fentanyl concentrations than fixed per kg dosing in simulations.
PK modelling using clinically‐generated specimens and data is a feasible and powerful tool for personalized dosing.
Introduction
Fentanyl is a synthetic opioid pain reliever FDA approved for children over 2 years of age for perioperative analgesia. It is commonly used off‐label in younger children and for pain management over the course of recovery after surgery and is typically given intravenously through continuous infusion and/or intermittent bolus dosing. Due to benefits such as rapid analgesia, haemodynamic stability and low frequency of side effects, fentanyl is one of the analgesic agents of choice after cardiac surgery in children 1.
Fentanyl pharmacokinetic (PK) data for paediatric patients are limited. Studies to date indicate that fentanyl clearance is age dependent 2, 3, 4, 5, affected by cardiac surgery 6, 7 and highly variable 3, 4, 5, 8, 9, 10. One barrier to further study of the factors influencing this variability is the need for serial invasive blood sampling for PK analysis. Some parents may not provide consent for their child to participate due to the pain of additional blood draws, and the smallest or most critically ill children may be excluded from PK studies. To overcome this obstacle, it has been proposed that population PK modelling can be applied to residual (‘remnant’) clinical specimens, i.e. plasma left over after clinical testing is complete 11, 12. In contrast to traditional PK, population PK methods allow analysis of sparse sampling data obtained at variable time points after drug dosing in diverse patient populations (Figure 1) 13, 14. This methodology also assesses the impact of covariates such as demographic, clinical, or genetic factors on PK parameters, enabling development of individualized dosing regimens based on those factors.
Figure 1.
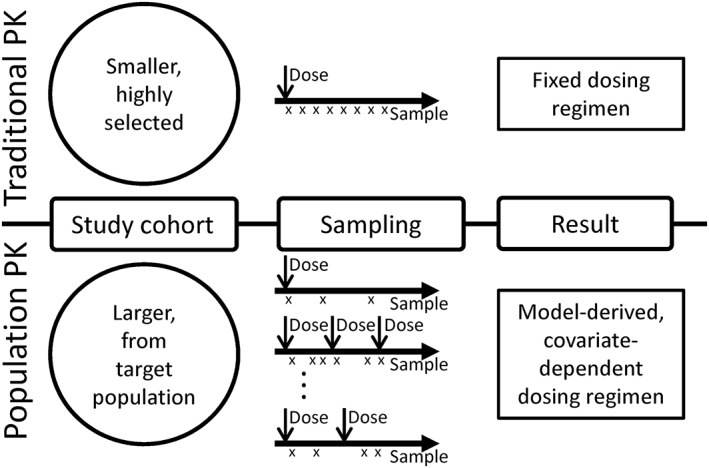
Traditional vs. population pharmacokinetic (PK) analyses. In traditional PK studies (top), small cohorts of patients, often healthy volunteers, are administered a drug. Serial samples are obtained to determine drug concentration at precise times after the dose, from which PK parameters are calculated. These parameters determine fixed dosing regimens for the study drug. In population PK studies (bottom), larger cohorts of patients are required and often include patients who are being treated with the study drug as part of clinical care. Fewer samples are obtained from each patient and are obtained at variable times after drug doses are given. These data are used to determine model based dosing, incorporating covariates that are determined to influence drug kinetics
In this study, we collected plasma from remnant clinical specimens for a cohort of paediatric participants hospitalized for cardiac surgery. Using measured fentanyl concentrations from the remnant specimens and dosing data from electronic medical records (EMRs), we were able to generate a complex dataset for population PK modelling. We performed population PK analyses for fentanyl, determined covariates affecting fentanyl PK and simulated fentanyl concentrations to compare model‐driven weight‐adjusted per kg vs. fixed per kg dosing regimens. Our findings indicate this approach is feasible and may be useful for dose individualization.
Methods
Study design
This study was approved by the Vanderbilt University Medical Center (VUMC) Institutional Review Board. Eligible participants were any patient with congenital heart disease scheduled to undergo a corrective or palliative operative procedure at our institution. Parents provided written consent for their child's participation, and informed assent was also obtained when appropriate. Enrolment with remnant specimen collection began in July of 2012 and is ongoing. Data analyzed for this study were collected prior to May 28, 2014. All study participants were admitted to the paediatric cardiac intensive care unit (ICU) after surgery. Participants were excluded from the analysis if their surgery was cancelled, if there was missing fentanyl dosing or covariate data or if they did not survive to hospital discharge, as large fentanyl doses may have been given as part of end‐of‐life care. When participants had multiple surgeries, only specimens obtained following the first procedure were used. Specimens were excluded if inadequate internal standard concentrations were detected and insufficient volume remained to repeat analysis or if they were obtained before any documented fentanyl dosing. Analgesic and sedative selection and dosing were determined by the primary clinical team and were not impacted by study enrollment. Clinical teams were not blinded to study enrollment, but were blinded to fentanyl concentration data obtained through this study.
Data collection
Demographic data and medical history were documented at the time of study enrollment. Surgical and clinical data were extracted from the EMR. Fentanyl dosing, including scheduled boluses, as‐needed intermittent boluses, and continuous infusions after post‐operative admission to the intensive care unit were determined from the EMR and the VUMC Enterprise Data Warehouse. The Enterprise Data Warehouse contains an electronic copy of both nurse administration and pharmacy operational data, enabling the computation of administered drug amounts over specific time periods. Study data were collected and managed using REDCap electronic data capture tools, a secure, web‐based application hosted at VUMC 15.
Sample collection and analysis
For the purposes of drug concentration analysis, all remnant plasma specimens ≥100 μL from blood obtained for clinical testing of electrolyte or basic metabolic panels in study participants were obtained from the VUMC Clinical Chemistry Laboratory. After retrieval, remnant specimens were stored at –20°C until sample processing for drug concentration analysis. Sample preparation and mass spectrometry analysis are described in the Supporting Information.
Data process
Serum creatinine concentration was a time‐varying covariate, which was matched with each fentanyl concentration data point. When serum creatinine measurement was not available at the same time when fentanyl concentration was measured, we selected the serum creatinine concentration measured at the closest time to that concentration data within 7 days (168 h). Within individuals, weight varied very little within the time frame of available concentration data and hence most weight data were the same as the baseline demographic measurements. However, whenever additional weight measurements were available, weight measurements obtained at the same time of concentration data point were used. Measures of albumin concentration were available for only 37% of participants, precluding use of albumin concentration as covariate.
Population PK analysis
We performed population PK analysis of fentanyl for children using a non‐linear mixed effects model implemented by NONMEM® version 7.3 15. First, we chose the base model by comparing one‐ and two‐compartment PK models without covariates, assuming a combined additive and proportional residual error model and lognormal distribution for the random effects PK parameters. The first‐order conditional estimation (FOCE) method with interaction was used for the estimation. Parameter estimates as well as the 95% bootstrapped confidence intervals (CIs) for the main population PK parameters such as total clearance (CL, l h–1), volume of distribution for the central compartment (V 1, l), inter‐compartmental clearance (Q, l h–1) and volume of distribution for the peripheral compartment (V 2, l) were generated. A two compartment model was chosen as the base model, which described the fentanyl PK substantially better than a one compartment model. A model with random‐effects for all main PK parameters (i.e. CL, V 1, Q and V 2) did not converge well due to a limited number of participants and sparse sampling and hence random‐effects were assumed only for CL and V 1 in the final model.
Covariate model building was performed using individual specific PK parameters estimated from the base model. Both graphical and statistical methods were considered with the following candidate covariates, which we chose a priori based on previous research and biological plausibility: weight, age, sex, Society of Thoracic Surgery–European Association for Cardio‐Thoracic Surgery (STAT) Congenital Heart Surgery Mortality score 16, cardiac bypass time, length of ICU stay and serum creatinine. Inclusion of a maturation factor as a function of age was tested to assess improvement in model fit 17. We also fited an allometric theory‐based model by fixing the allometric scaling parameters in model 1 at the corresponding theoretical values, that is the scaling parameters for CL and Q were fixed at 0.75 (i.e. θ2 = θ6 = 0.75) and those for V 1 and V 2 were fixed at 1 (i.e. θ4 = θ8 = 1).
Model selection was performed based on the objective function (–2 log likelihood) along with the number of parameters, which would approximately follow χ2 distribution. The χ2 statistics of 3.84, 6.63, and 10.83 with 1 degree of freedom correspond to P values of 0.05, 0.01 and 0.001, respectively. We considered the objective function value decrease of 10.83 to be significant model improvement 18.
Model assessment
To check the PK model, we examined goodness‐of‐fit plots using population and individual predicted fentanyl plasma concentrations and residual plots using conditional weighted residuals (CWRES). In addition, we performed internal model validation using bootstrap methods 19, 20. As a measure of model validation, the predicted fentanyl plasma concentrations were used. Specifically, the participants' data were bootstrapped with replacement and the bootstrapped participants' data were used as a training set to develop a model and the remaining (i.e. unselected) participants' data were held out as a validation set. The estimated parameters from the developed model were used to predict fentanyl plasma concentrations in the held out validation set. This whole process was repeated 1000 times. The 95% predicted region and the median of predicted fentanyl plasma concentrations were calculated along the time points within a 3 h window and plotted over time. For predictive checking, the 95% region where 95% of the observed fentanyl plasma concentrations lie and the data points were overlaid on the 95% predicted region as well as the median predicted line as a function of time. We used the programming language R version 3.1.3 for covariate building, model checking and model validation 21. Allometric scaling analysis methods are described in the Supporting Information.
Subset data analyses
Subset data analyses were performed in order to examine the impact of PK parameters estimated based on data from three subsets of the cohort: (1) data from participants with weight <40 kg [n = 121, excluding nine participants (7%) whose weight was relatively high compared with the remainder of the cohort], (2) data from participants who received intravenous bolus doses only [n = 93, excluding 37 participants (28%) who received both bolus and infusion fentanyl doses] and (3) data where fentanyl concentrations were above the lower limit of quantification (LLOQ) [total specimens = 504, excluding 575 specimens (53%) below LLOQ]. We fit the PK models using the subset data and compared the estimates of PK parameters with those obtained from the full data.
Simulated fentanyl concentration profiles
We performed a simulation study to explore potential benefit of model based dosing compared with fixed per kg infusions. Fentanyl concentrations were predicted using two different fixed per kg infusion rates and one weight‐adjusted per kg dosing rate derived from our final model in hypothetical children weighing 2.5, 6.4, and 20 kg. The weight‐adjusted per kg dosing rate for a hypothetical child with specific weight, wgti, was derived from our final model using the equation, infusion dose = C target x CLi, where C target is the target plasma concentration and CLi was calculated from our model by CLi = θ1 × (wgti /70)θ2 based on the parameter estimates (i.e. θ1 = 49.0 l h–1 and θ2 = 0.9, see Table 2). For example, to achieve the target plasma concentration of 2 μg l–1 in a hypothetical child weighing 6.4 kg, CLi = 49.0 l h–1 × (6.4 kg/70 kg)0.9 = 5.7 l h–1, and hence the infusion dose = 2 μg l–1 × 5.7 l h–1 = 11.4 μg h–1, yielding the weight‐adjusted per kg dosing rate of 11.4 μg h–1 6.4 kg–1 = 1.78 μg kg–1 h–1. Applying the same calculation to different weights, the weight‐adjusted per kg dosing rates for hypothetical children weighing 2.5 and 20 kg were 1.95 μg kg–1 h–1 and 1.59 μg kg–1 h–1, respectively.
Table 2.
PK model parameter estimates
| Base model* | Covariate model | ||||
|---|---|---|---|---|---|
| Model 1 (free allometric scaling parameters) | Model 2 (fixed allometric scaling parameters) | ||||
| Parameters | Estimates (SE) | Parameters | Estimates (SE) | Parameters | Estimates (SE) |
| (Obj = ‐2197) | (Obj = ‐2321) | (Obj = ‐2285) | |||
| CL | CL = θ1 (wgt/70)θ2 | CL = θ1 (wgt/70)θ2 | |||
| 8.4 (0.9) [4.7, 9.9]† | θ1 | 49.0 (8.2) [19.3, 79.2] | θ1 | 39.6 (2.2) [33.2, 46.1] | |
| θ2 | 0.9 (0.1) [0.4, 1.1] | θ2 = 0.75 | |||
| V 1 | V 1 = θ3 (wgt/70)θ4 | V 1 = θ3 (wgt/70)θ4 | |||
| 86.5 (13.9) [14.8, 145.2] | θ3 | 198.8 (107.8) [28.1, 659.4] | θ3 | 529.1 (72.1) [255.8, 893.3] | |
| θ4 | 0.7 (0.2) [0.001, 1.1] | θ4 = 1 | |||
| Q | 0.5 (0.2) [0.1, 3.5] | Q = θ5 (wgt/70)θ6 | Q = θ5 (wgt/70)θ6 | ||
| θ5 | 35.3 (17.0) [3.9, 332.5] | θ5 | 7.8 (2.9) [2.0, 26.5] | ||
| θ6 | 1.3 (0.2) [0.5, 2.5] | θ6 = 0.75 | |||
| V 2 | 9.0 (2.9) [4.8, 67.8] | V 2 = θ7 (wgt/70)θ8 | V 2 = θ7 (wgt/70)θ8 | ||
| θ7 | 520.9 (181.3) [87.4, 2323.1] | θ7 | 227.3 (56.8) [140.0, 554.1] | ||
| θ8 | 1.2 (0.2) [0.1, 1.8] | θ8 = 1 | |||
| Ω2 CL (%CV) | 103 (13) [56, 133] | Ω2 CL (%CV) | 70 (13) [43, 130] | Ω2 CL (%CV) | 64 (13) [38, 97] |
| Ω2 V1 (%CV) | 118 (12) [47, 176] | Ω2 V1 (%CV) | 64 (19) [40, 178] | Ω2 V1 (%CV) | 73 (16) [35, 165] |
| σ2 proportional (%CV ) | 48 (5) [40, 90] | σ2 proportional (%CV) | 52 (5) [40, 104] | σ2 proportional (%CV) | 49 (5) [40, 80] |
| σ2 additive (ng ml–1) | 0.01 (0.01) [0.00, 0.02] | σ2 additive (ng ml–1) | 0.01 (0.01) [0.00, 0.02] | σ2 additive (ng ml–1) | 0.01 (0.01) [0.00, 0.02] |
The estimates from the base model using the entire cohort are presented.
The 95% bootstrap confidence intervals (CIs) are presented in square brackets.
SE, the standard error; Obj, the objective function value; CL, total clearance (l h–1); Q, intercompartmental clearance (l h–1); V 1, volume of distribution for the central compartment (l); V 2, volume of distribution for the peripheral compartment (l); CV, coefficient of variation; wgt, body weight in kg; Ω2 CL and Ω2 V1, the variance for ηi CL and ηi V1, respectively; σ2 proportional and σ2 additive, the proportional and additive residual error variance, respectively.
To compare the two fixed per kg dosing rates to the weight‐adjusted per kg dosing regimens, we simulated 200 different sets of values for CL, V 1, Q, and V 2 using the estimates of population means for CL, V 1, Q, and V 2 for each hypothetical child weight (2.5, 6.4 and 20 kg) and the variances for CL and V 1 (i.e. Ω2 CL and Ω2 V1) from an allometric theory‐based model (i.e. model 2, see Table 2). This allometric theory‐based model was chosen as the underlying true model in simulations, which is different from our final model, to provide a more fair comparison. Under each assumed model, the plasma concentrations were predicted as if each of dosing regimen were given: fixed per kg infusions at 1 μg kg–1 h–1 (standard fentanyl starting dose) and 2.67 μg kg–1 h–1 (based on the median total fentanyl dose given in the first day in our cohort), and the weight‐adjusted per kg dosing rate for hypothetical children of each weight. The predicted fentanyl concentrations were compared with a proposed target range of 1–3 ng ml–1, concentrations associated with analgesic effect and minimal respiratory depression 3, 5, 22, 23, 24, 25. The proportion of simulated patients within this target range was calculated over time. Additional post hoc simulations are described in the Supporting Information.
Results
Study population and specimens
We collected 1321 residual plasma specimens from 140 participants. Three of the 140 participants were excluded due to in‐hospital mortality, one for cancellation of surgery and five due to missing dosing or covariate data. A total of 61 specimens, including all specimens from one participant, were excluded due to low internal standard on initial analysis and insufficient volume to repeat analysis (n = 1) or because the specimen was drawn before fentanyl was given (n = 60). The final study population (n = 130, with 1079 specimens) is described in Table 1. Clinical and demographic data for the subsets of participants used for subset data analysis, namely those with weight <40 kg (n = 121) and those receiving only bolus doses of fentanyl (n = 93) are indicated in Table S1 and the primary surgical procedures performed in the cohort and both subsets are shown in Table S2. All fentanyl concentrations were determined using a multiplexed 16 drug assay developed for this purpose. Assay performance and fentanyl stability are described in the Supporting Information.
Table 1.
Study cohort
| Entire cohort | |
|---|---|
| n | 130 |
| Age (months)* | 5.9 (1.2–28.4) |
| Weight (kg)* | 6.4 (3.7–11.6) |
| Male sex† | 72 (55%) |
| Race† | |
| White | 104 (80%) |
| Black | 11 (8%) |
| American Indian or Alaska Native | 1 (1%) |
| Asian | 1 (1%) |
| Other | 6 (5%) |
| Unknown | 7 (5%) |
| Median serum creatinine (mg dl–1)* | 0.4 (0.4 – 0.5) |
| STAT score† | |
| 1 | 26 (20%) |
| 2 | 50 (38%) |
| 3 | 25 (19%) |
| 4 | 18 (14%) |
| 5 | 11 (8%) |
| Cardiac bypass time (min)* | 111 (67–160) |
| Length of ICU hospitalization (days)* | 4 (2–8) |
| Fentanyl dose (μg kg–1 day–1)* | 64 (35–98) |
| Number of specimens per participant* | 6 (4–10) |
Median (interquartile range);
Number (%)
Population PK model
The results of the primary analysis, including the base model and the final covariate model based on data from the entire cohort, are presented in Table 2. Parameter estimates and the 95% bootstrapped CIs for CL, V 1, Q, and V 2 were first generated for the base model, without covariates. The PK parameters varied substantially among individual participants. The coefficients of variation (CV) for CL and V 1 were 103% and 118%, respectively.
Among the covariates we considered, participant weight substantially explained inter‐individual variability for both CL and V 1 and improved model fit; including weight reduced the objective function by 124 (from –2197 to –2321). Other a priori defined covariates (age, gender, STAT score 16, cardiac bypass time, length of ICU stay and serum creatinine) and inclusion of a maturation factor minimally improved model fit (i.e. decreased the objective function by <5). Thus the final covariate model included only the participant's weight. The main part of the model is presented as follows:
where CLi and V 1i are the individual‐specific PK parameters corresponding to CL and V 1, wgti is participant weight in kilograms (kg), and ηi CL and ηi V1 are random variables explaining inter‐individual variability for CL and V 1 which follow a normal distribution with mean zero and variance of Ω2 CL and Ω2 V1. Q and V 2 are inter‐compartmental clearance and volume of distribution for the peripheral compartment, respectively. The θs in the equations denote model parameters as typically used in statistical models.
For the final covariate model, model 1, we estimated CL, V 1, Q and V 2 as 5.7 l h–1, 37.3 l, 1.6 l h–1 and 29.5 l for a child with weight of 6.4 kg, the median weight of the entire cohort. The estimates of CL, V 1, Q and V 2 in terms of a standard population with weight of 70 kg are shown in Table 2 [49.0 l h–1 (CL = θ1), 198.8 l (V 1 = θ3), 35.3 l h–1 (Q = θ5) and 520.9 l (V 2 = θ7), respectively]. The CVs for CL and V 1 were reduced to 70% and 64%, respectively, for the final model.
The allometric theory‐based model with fixed allometric scaling parameters is presented in Table 2 under model 2. Model 2 substantially increased the objective function by 36 compared to Model 1 (–2285 vs. –2321), which corresponds to p value of 2.9 x 10–7 with 4 degrees of freedom, supporting model 1 as the final model.
Goodness‐of‐fit plots are shown in Figure S1 A and B, including plots of the all measured fentanyl concentrations (inset) and those <5 ng/mL, which represent 95% of the data collected (main graph). The large variability in population predicted values shown in Figure S1A is significantly reduced in the individual predicted values as shown in Figure S1B. Observed vs. predicted values are not systematically above or below the diagonal line of equality, indicating the model does not systematically over‐ or under‐predict concentrations, although there were few outlying predictions.
The CWRES were also examined as model diagnostics, where symmetric distribution around the zero line indicated good model fit. For the most significant covariate, weight (kg), obvious model misspecification was not identified (Figure S1C) although less variability was shown in a range of large weight where only a small percent of data are available. Likewise, the plot for CWRES vs. serum creatinine appeared to be symmetrically distributed (Figure S1D). When the CWRES were plotted against other covariates, no obvious trends were observed. Thus, the goodness‐of‐fit and CWRES plots support the adequacy of our final model to fit these data.
We also performed predictive model checking over time and Figure S2 shows a range of data up to 2 weeks, where most of observed concentrations lie, restricting to observed concentrations >0 ng ml–1 in a log scale. The 95% predicted region (grey shaded) in the plots covers the 95% region (pink shaded) of observed fentanyl concentration well, again indicating the model was adequate to describe these data.
Subset data analyses of population PK model
We performed subset data analyses using two subsets of the participants, those with weight <40 kg and those who received only bolus fentanyl dosing. Table S3 summarizes the parameter estimates and their standard errors from the allometric theory‐based model fitted with the two subsets of data. Parameter estimates from the entire cohort and the subset with weight <40 kg were highly similar to each other, but appeared to differ from the bolus only subset. However, when fentanyl concentrations were predicted for a hypothetical patient (weight 6.4 kg, continuous infusion of fentanyl at 10 μg h–1 for 5 days, with and without an additional three bolus doses of 50 μg) using each of the three sets of parameter estimates, the predicted profiles were nearly identical (Figure S3). We also assessed a subset of samples above the LLOQ, since the concentration of fentanyl for some samples (53%) was below the LLOQ of the mass spectrometry assay. All resulting parameter estimates were very similar (Figure S3).
Simulated fentanyl concentration profiles
Figure 2 shows 20 randomly selected predicted plasma concentration profiles during and after fixed per kg infusions at 1 μg kg–1 h–1 (standard starting dose) and 2.67 μg kg–1 h–1 (based on the median total fentanyl dose given in the first day in our cohort), as well as model‐driven weight‐adjusted per kg dosing for hypothetical children weighing 2.5 kg (Figure 2A), 6.4 kg (Figure 2B) and 20 kg (Figure 2C) in simulations. For easier comparison among dosing regimens, Figure 2D–F present the percent of predicted plasma concentrations falling within the target range of 1–3 ng ml–1 at each time point from 6 to 48 h (until infusion stopped) for hypothetical children of each weight. The lower fixed dose infusion was the worst for most scenarios, often failing to reach therapeutic concentrations of fentanyl, although it performed better than the higher fixed dose after longer infusion for larger children. On the other hand, the higher fixed dose infusion performed better than the weight‐adjusted per kg dosing at the beginning of infusion, and was similar for the smallest children during steady‐state, but after 20 h concentrations often were above the target range. The results of additional simulations are shown in Figure S4. Overall, using a dose calculated based on the population PK model estimates performed better than the fixed dose infusions, resulting in fentanyl concentrations within the target range more often across the range of patient weights after reaching steady state.
Figure 2.
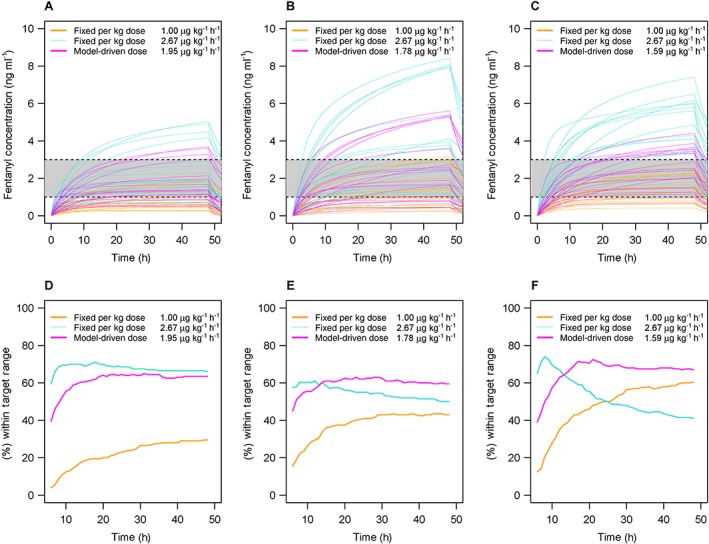
Simulated fentanyl concentration profiles with fixed and model based dosing. The shaded region designates the therapeutic range of 1–3 ng ml–1. Twenty randomly selected predicted fentanyl concentrations profiles for hypothetical 2.5 kg (A), 6.4 kg (B) and 20 kg (C) children are shown over time. For each hypothetical child, simulations are shown for fixed infusion rates of 1 μg kg–1 h–1 (orange lines) and 2.67 μg kg–1 h–1 (light blue lines) given from 0 to 48 h, then stopped. Simulated fentanyl concentrations resulting from model‐driven weight‐adjusted per kg dosing given from 0 to 48 h are shown for each hypothetical child (pink lines). The lower panels present the percent of predicted plasma concentrations falling within the target range of 1–3 ng ml–1 at each time point from 6 to 48 h (until infusion stopped) for hypothetical children weighing 2.5 kg (D), 6.4 kg (E) and 20 kg (F) based on 1 μg kg –1h–1 (orange lines) and 2.67 μg kg h–1 (light blue lines) and model‐driven weight‐adjusted per kg dosing (pink lines)
Discussion
We successfully generated a fentanyl PK dataset using drug concentration data from remnant clinical specimens and dose information directly obtained from EMRs, and we used this dataset to perform population PK analysis. Although we did not identify new clinical covariates impacting fentanyl PK for children after cardiac surgery, we confirmed previously estimated fentanyl PK parameters and the non‐linear association of fentanyl PK with patient weight. This is one of few studies successfully reproducing a paediatric PK profile. Our work supports the feasibility of using remnant clinical specimens and EMR data.
The approach demonstrated here, using data generated during routine clinical care for population PK modeling, is especially relevant for studies in paediatric therapeutics. Traditional PK studies done to date, especially in neonates, have demonstrated unique drug distribution profiles among young patients. However, serial sampling for research is difficult and, for a subset of patients, potentially harmful. This is not the first study to utilize remnant clinical samples for research. Previous studies have used combinations of remnant (scavenged), opportunistic (drawn at the same time as clinical blood draws) and/or timed (drawn for research) samples to determine PK parameters for anti‐infective drugs in infants and children 12, 26, 27, 28, 29, 30, 31, 32. One prior study of ciprofloxacin models derived from remnant vs. timed samples found similar results 12. Our study is the largest cohort collected to date and is comprised solely of remnant specimens. In addition, we utilized clinical data and dosing information from the EMR, i.e. ‘re‐using’ this clinically‐generated data. This pragmatic PK design represents an efficient approach to further study of the clinical and genetic factors contributing to individual variability in drug exposure and response in children. These data may also be valuable for validation of semi‐physiologic models 33, another potential approach to improve paediatric dosing.
Evidence for the validity of our approach is provided by our subset data analyses, allometric scaling analysis and comparison of our data to prior reports. Our study design enabled us to enroll participants with few exclusion criteria and no pre‐specified fentanyl dosing regimen. While this more closely represents the patient population of children who undergo cardiac surgery than most clinical trials, it resulted in a much larger and more heterogeneous cohort that may be influenced by extreme values. Our analyses of subsets of data based on participant, dosing or drug concentration criteria resulted in PK parameter estimates similar to those found in the entire cohort, validating our findings and indicating that this strategy may be applied to either heterogeneous populations or smaller population subsets. The 95% CIs of allometric scaling parameters in the final model included the corresponding theoretical values, supporting that the relationships between body size and PK parameters are similar to those expected based on allometric theory (additional information in Supporting Information). Comparison of our PK parameter estimates with those from prior reports with respect to the most commonly reported parameter, CL, requires conversion of the reported values to l h–1 and using the median participant weight from our cohort (6.4 kg). Using this approach, CLs reported in Ginsberg et al. 34 and Koren et al. 7 (7.3 l h–1 and 5.0 l h–1, respectively) are similar to the 5.7 l h–1 and 6.6 l h–1 from our final model with allometric scaling parameters being free (model 1) and the one with allometric scaling parameters being fixed at the theoretical values (model 2), respectively.
Simulation studies allowed us to compare fixed per kg infusions with the use of a model‐driven weight‐adjusted per kg dosing algorithm. Predicted fentanyl concentrations during fixed per kg infusions were not consistently within the therapeutic concentration range for patients of different weights due to the nonlinear relationship of fentanyl PK with weight. Model‐driven weight‐adjusted per kg dosing resulted in a higher proportion of simulated 6.4 kg and 20 kg patients within target range. Furthermore, model‐driven dosing led to more consistent fentanyl concentrations in the therapeutic range across the size spectrum. With the wide use of EMRs and computerized order entry, the implementation of model based dosing is now feasible and allows personalization of drug dosing based on covariates influencing drug kinetics, once models are appropriately externally validated in large data sets. More precise determination of target concentrations of fentanyl to achieve analgesia or sedation and the individual factors influencing drug response (pharmacodynamics) could be incorporated into these dosing calculators.
The major study limitations are the relatively small sample size (for a population PK study) and a heterogeneous patient population. Hence the PK parameters could not be precisely estimated and have wide CIs. The small sample size and wide age distribution may also be the reasons that the inclusion of a maturation factor as a function of age did not significantly improve the model fit, and the 95% CI of parameter estimate was very wide, including 0. Accrual of additional patients is underway to allow refinement of the PK model and potential inclusion of a maturation factor. In addition, EMR data are not as extensive or accurate as those collected from well controlled clinical studies with prospective data collection. Our drug dosing data collection is facilitated by our institution using a bar‐code based electronic medication administration record, where healthcare providers are directed to scan the drug and the patient's wrist band at the time of drug administration. However, delays in drug administration and/or documentation may occur in clinical practice and may add to the uncertainty in our dataset. Further, important covariates that could be a source of variation on PK parameters may not have been measured. For example, albumin concentrations are known to impact on clearance of many drugs, but albumin concentrations are infrequently obtained in clinical settings. Only 48 participants in our dataset had albumin data available. Depending on the specific covariate, remnant samples may be used to collect these data, or specific sampling may be necessary from the participant. Importantly, this model includes only fentanyl PK and did not assess pharmacodynamic variability, which may lead to differences in individual analgesia or sedation in response to fentanyl exposures in the target concentration range.
These data provide further proof‐of‐principle that population PK analysis using remnant clinical specimens and EMR data is a feasible approach. This strategy of coupling EMR data to biologic samples that would otherwise be discarded provides a method to overcome barriers to studying drug kinetics and response in populations where traditional PK studies are not or cannot be performed, including neonatal and pediatric patients.
Competing Interests
All authors have completed the Unified Competing Interest form at http://www.icmje.org/coi_disclosure.pdf (available on request from the corresponding author) and declare SLV had support from Pharmaceutical Research and Manufacturers of America Foundation and CTSA award KL2 TR 000446 from the NIH National Center for Advancing Translational Sciences, LC had support from NIH National Institute on Aging grant AG034412, AHS had support from the American Heart Association award 12CRP10560001, RC reports grants from NIH National Institute of General Medical Sciences 5R01 GM058008‐16 and 2P41 GM103391‐05, all authors were supported by NIH National Center for Advancing Translational Sciences UL1 TR000445 and there are no financial relationships with any organizations that might have an interest in the submitted work in the previous 3 years and no other relationships or activities that could appear to have influenced the submitted work.
The authors thank the Vanderbilt Clinical Laboratory staff for their assistance in obtaining the remnant samples for this research, the patients and families who participated in this study and Dan Roden, MD, for his support of this project and critical review of the manuscript.
SLV is supported by an Early Career Award from the Pharmaceutical Research and Manufacturers of America Foundation and CTSA award KL2 TR 000446 from the NIH National Center for Advancing Translational Sciences. This work was supported in part by NIH National Institute on Aging grant AG034412 (LC), American Heart Association 12CRP10560001 (AHS) and NIH National Center for Advancing Translational Sciences UL1 TR000445 (Vanderbilt CTSA).
Contributors
All authors participated in critical review and revision of the final manuscript and approved the final manuscript draft. SLV: wrote manuscript, designed research, performed research and analyzed data. MDM: performed research and contributed analytical tools. BH: designed research, performed research and contributed analytical tools. CB: analyzed data. KC: performed research. JO: performed research. AHS: performed research. PJK: performed research. AW: performed research. RMC: designed research and contributed analytical tools. LC: wrote manuscript, designed research and analyzed data.
Supporting information
Table S1 Study Cohort Subsets
Table S2 Operative Procedures Performed on Study Participants
Table S3 Subset data analyses
Figure S1 Population PK Model Diagnostic Plots
Figure S2 Predictive Model Check
Figure S3 Comparison of Predicted Concentrations over Time from Subset Analyses
Figure S4 Additional Simulated Fentanyl Concentration Profiles With Fixed and Model Based Dosing
Supporting info item
Van Driest, S. L. , Marshall, M. D. , Hachey, B. , Beck, C. , Crum, K. , Owen, J. , Smith, A. H. , Kannankeril, P. J. , Woodworth, A. , Caprioli, R. M. , and Choi, L. (2016) Pragmatic pharmacology: population pharmacokinetic analysis of fentanyl using remnant samples from children after cardiac surgery. Br J Clin Pharmacol, 81: 1165–1174. doi: 10.1111/bcp.12903.
References
- 1. Saarenmaa E, Huttunen P, Leppäluoto J, Meretoja O, Fellman V. Advantages of fentanyl over morphine in analgesia for ventilated newborn infants after birth: A randomized trial. J Pediatr 1999; 134: 144–50. [DOI] [PubMed] [Google Scholar]
- 2. Singleton MA, Rosen JI, Fisher DM. Plasma concentrations of fentanyl in infants, children and adults. Can J Anaesth J Can Anesth 1987; 34: 152–5. [DOI] [PubMed] [Google Scholar]
- 3. Katz R, Kelly HW. Pharmacokinetics of continuous infusions of fentanyl in critically ill children. Crit Care Med 1993; 21: 995–1000. [DOI] [PubMed] [Google Scholar]
- 4. Gauntlett IS, Fisher DM, Hertzka RE, Kuhls E, Spellman MJ, Rudolph C. Pharmacokinetics of fentanyl in neonatal humans and lambs: effects of age. Anesthesiology 1988; 69: 683–7. [DOI] [PubMed] [Google Scholar]
- 5. Saarenmaa E, Neuvonen PJ, Fellman V. Gestational age and birth weight effects on plasma clearance of fentanyl in newborn infants. J Pediatr 2000; 136: 767–70. [PubMed] [Google Scholar]
- 6. Lynn A, Nespeca MK, Bratton SL, Strauss SG, Shen DD. Clearance of morphine in postoperative infants during intravenous infusion: the influence of age and surgery. Anesth Analg 1998; 86: 958–63. [DOI] [PubMed] [Google Scholar]
- 7. Koren G, Goresky G, Crean P, Klein J, MacLeod SM. Pediatric fentanyl dosing based on pharmacokinetics during cardiac surgery. Anesth Analg 1984; 63: 577–82. [PubMed] [Google Scholar]
- 8. Wheeler M, Birmingham PK, Lugo RA, Heffner CL, Coté CJ. The pharmacokinetics of the intravenous formulation of fentanyl citrate administered orally in children undergoing general anesthesia. Anesth Analg 2004; 99: 1347–51. [DOI] [PubMed] [Google Scholar]
- 9. Koehntop DE, Rodman JH, Brundage DM, Hegland MG, Buckley JJ. Pharmacokinetics of fentanyl in neonates. Anesth Analg 1986; 65: 227–32. [PubMed] [Google Scholar]
- 10. Collins JJ, Dunkel IJ, Gupta SK, Inturrisi CE, Lapin J, Palmer LN, Weinstein SM, Portenoy RK. Transdermal fentanyl in children with cancer pain: feasibility, tolerability, and pharmacokinetic correlates. J Pediatr 1999; 134: 319–23. [DOI] [PubMed] [Google Scholar]
- 11. Laughon MM, Benjamin DK, Capparelli EV, Kearns GL, Berezny K, Paul IM, Wade K, Barrett J, Smith PB, Cohen‐Wolkowiez M. Innovative clinical trial design for pediatric therapeutics. Expert Rev Clin Pharmacol 2011; 4: 643–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Leroux S, Turner MA, Guellec CB‐L, Hill H, van den Anker JN, Kearns GL, Jacqz‐Aigrain E, Zhao W, TINN (Treat Infections in NeoNates) and GRiP (Global Research in Paediatrics) Consortiums . Pharmacokinetic studies in neonates: The utility of an opportunistic sampling design. Clin Pharmacokinet 2015; 54: 1273–85. [DOI] [PubMed] [Google Scholar]
- 13. Kiang TKL, Sherwin CMT, Spigarelli MG, Ensom MHH. Fundamentals of population pharmacokinetic modelling: modelling and software. Clin Pharmacokinet 2012; 51: 515–25. [DOI] [PubMed] [Google Scholar]
- 14. Sherwin CMT, Kiang TKL, Spigarelli MG, Ensom MHH. Fundamentals of population pharmacokinetic modelling: validation methods. Clin Pharmacokinet 2012; 51: 573–90. [DOI] [PubMed] [Google Scholar]
- 15. Beal S, Sheiner L, Boeckmann A, Bauer R. NONMEM User's Guides. Ellicott City, MD: Icon Development Solutions; 2009. [Google Scholar]
- 16. Jacobs ML, O'Brien SM, Jacobs JP, Mavroudis C, Lacour‐Gayet F, Pasquali SK, Welke K, Pizarro C, Tsai F, Clarke DR. An empirically based tool for analyzing morbidity associated with operations for congenital heart disease. J Thorac Cardiovasc Surg 2013; 145: 1046–1057.e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Anderson BJ, Holford NHG. Tips and traps analyzing pediatric PK data. Paediatr Anaesth 2011; 21: 222–37. [DOI] [PubMed] [Google Scholar]
- 18. Beal SL. Commentary on significance levels for covariate effects in NONMEM. J Pharmacokinet Pharmacodyn 2002; 29: 403–10 discussion 411–412. [DOI] [PubMed] [Google Scholar]
- 19. Efron B. Estimating the error rate of a prediction rule: improvement on cross‐validation. J Am Stat Assoc 1983; 78: 316–31. [Google Scholar]
- 20. Efron B, Tibshirani RJ. An introduction to the bootstrap. CRC press, New York, NY; 1994. [Google Scholar]
- 21. R Core Team . R: A Language and Environment for Statistical Computing. Vienna, Austria: R Foundation for Statistical Computing; 2014. [Google Scholar]
- 22. Foster D, Upton R, Christrup L, Popper L. Pharmacokinetics and pharmacodynamics of intranasal versus intravenous fentanyl in patients with pain after oral surgery. Ann Pharmacother 2008; 42: 1380–7. [DOI] [PubMed] [Google Scholar]
- 23. Hertzka RE, Gauntlett IS, Fisher DM, Spellman MJ. Fentanyl‐induced ventilatory depression: effects of age. Anesthesiology 1989; 70: 213–8. [DOI] [PubMed] [Google Scholar]
- 24. Gourlay GK, Kowalski SR, Plummer JL, Cousins MJ, Armstrong PJ. Fentanyl blood concentration‐analgesic response relationship in the treatment of postoperative pain. Anesth Analg 1988; 67: 329–37. [PubMed] [Google Scholar]
- 25. Woodhouse A, Mather LE. The minimum effective concentration of opioids: a revisitation with patient controlled analgesia fentanyl. Reg Anesth Pain Med 2000; 25: 259–67. [DOI] [PubMed] [Google Scholar]
- 26. Zhao W, Zhang D, Storme T, Baruchel A, Declèves X, Jacqz‐Aigrain E. Population pharmacokinetics and dosing optimization of teicoplanin in children with malignant haematological disease. Br J Clin Pharmacol 2015; 80: 1197–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Gonzalez D, Melloni C, Yogev R, Poindexter BB, Mendley SR, Delmore P, Sullivan JE, Autmizguine J, Lewandowski A, Harper B, Watt KM, Lewis KC, Capparelli EV, Benjamin DK, Cohen‐Wolkowiez M, Best Pharmaceuticals for Children Act ‐ Pediatric Trials Network Administrative Core Committee . Use of opportunistic clinical data and a population pharmacokinetic model to support dosing of clindamycin for premature infants to adolescents. Clin Pharmacol Ther 2014; 96: 429–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Tremoulet A, Le J, Poindexter B, Sullivan JE, Laughon M, Delmore P, Salgado A, Ian‐U Chong S, Melloni C, Gao J, Benjamin DK, Capparelli EV, Cohen‐Wolkowiez M, Administrative Core Committee of the Best Pharmaceuticals for Children Act ‐ Pediatric Trials Network . Characterization of the population pharmacokinetics of ampicillin in neonates using an opportunistic study design. Antimicrob Agents Chemother 2014; 58: 3013–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Zhao W, Hill H, le Guellec C, Neal T, Mahoney S, Paulus S, Castellan C, Kassai B, van den Anker JN, Kearns GL, Turner MA, Jacqz‐Aigrain E, TINN Consortium . Population pharmacokinetics of ciprofloxacin in neonates and young infants less than three months of age. Antimicrob Agents Chemother 2014; 58: 6572–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Wade KC, Wu D, Kaufman DA, Ward RM, Benjamin DK, Sullivan JE, Ramey N, Jayaraman B, Hoppu K, Adamson PC, Gastonguay MR, Barrett JS, National Institute of Child Health and Development Pediatric Pharmacology Research Unit Network . Population pharmacokinetics of fluconazole in young infants. Antimicrob Agents Chemother 2008; 52: 4043–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Cohen‐Wolkowiez M, Ouellet D, Smith PB, James LP, Ross A, Sullivan JE, Walsh MC, Zadell A, Newman N, White NR, Kashuba ADM, Benjamin DK. Population pharmacokinetics of metronidazole evaluated using scavenged samples from preterm infants. Antimicrob Agents Chemother 2012; 56: 1828–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Cohen‐Wolkowiez M, Benjamin DK, Ross A, James LP, Sullivan JE, Walsh MC, Zadell A, Newman N, White NR, Kashuba ADM, Ouellet D. Population pharmacokinetics of piperacillin using scavenged samples from preterm infants. Ther Drug Monit 2012; 34: 312–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Encinas E, Calvo R, Lukas JC, Vozmediano V, Rodriguez M, Suarez E. A predictive pharmacokinetic/pharmacodynamic model of fentanyl for analgesia/sedation in neonates based on a semi‐physiologic approach. Paediatr Drugs 2013; 15: 247–57. [DOI] [PubMed] [Google Scholar]
- 34. Ginsberg B, Howell S, Glass PS, Margolis JO, Ross AK, Dear GL, Shafer SL. Pharmacokinetic model‐driven infusion of fentanyl in children. Anesthesiology 1996; 85: 1268–75. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1 Study Cohort Subsets
Table S2 Operative Procedures Performed on Study Participants
Table S3 Subset data analyses
Figure S1 Population PK Model Diagnostic Plots
Figure S2 Predictive Model Check
Figure S3 Comparison of Predicted Concentrations over Time from Subset Analyses
Figure S4 Additional Simulated Fentanyl Concentration Profiles With Fixed and Model Based Dosing
Supporting info item