

Abstract
Translational research is defined as the process of applying ideas, insights and discoveries generated through basic scientific inquiry to treatment or prevention of human diseases. Although precise information is lacking, several lines of evidence attest that up to 95% early-phase studies may not translate into tangible outcomes for improving clinical management. Major theoretical hurdles exist in the translational process, but is it also undeniable that many studies may have failed for practical reasons, such as the use of inappropriate diagnostic testing for evaluating efficacy, effectiveness or safety of a given medical intervention, or poor quality in laboratory testing. This can generate biased test results and result in misconceptions during data interpretation, eventually leading to no clinical benefit, possible harm, and a waste of valuable resources. From a genuine economic perspective, it can be estimated that over 10 million euros of funding may be lost each year in clinical trials in the European Union due to preanalytical and analytical problems. These are mostly attributions to the heterogeneity of current guidelines and recommendations for the testing process, to the poor evidence base for basic pre-analytical, analytical and post-analytical requirements in clinical trials, and to the failure to thoughtfully integrate the perspectives of clinicians, patients, nurses and diagnostic companies in laboratory best practices. The most rational means for filling the gap between what we know and what we practice in clinical trials cannot discount the development of multidisciplinary teams including research scientists, clinicians, nurses, patients associations and representative of in vitro diagnostic (IVD) companies, who should actively interplay and collaborate with laboratory professionals to adapt and disseminate evidence-based recommendations about biospecimen collection and management into the research settings, from preclinical to phase III studies.
Keywords: Clinical trials, quality, preanalytical variability, safety
Overview on clinical trials
Clinical trials are conventionally defined as studies carried out in clinical research. These biomedical or behavioral research studies involve human participants. They are designed to answer specific questions about biomedical or behavioral interventions, thus entailing new treatments (e.g., drugs, vaccines, dietary choices and supplements, changes in life styles, medical devices and biomarkers). They may also address medical interventions requiring further study and comparison for validation. Clinical trials and preclinical studies are usually classified in various stages, according to primary goals and number of participants (Table 1) (1).
Table 1. Classification of clinical trials.
| Stage | Primary goal | No. of subjects |
|---|---|---|
| Preclinical | Analysis in non-human subjects, to preliminary evaluate efficacy, toxicity and pharmacokinetics | – |
| Phase 0 | Preliminary evaluation of efficacy, effectiveness and safety in healthy volunteers | 10–20 |
| Phase I | Preliminary evaluation of efficacy, effectiveness and safety in healthy volunteers | 20–100 |
| Phase II | Evaluation of efficacy, effectiveness and safety in patients | 100–300 |
| Phase III | Evaluation of efficacy, effectiveness and safety in patients | >300 |
| Phase IV | Postmarketing surveillance | All subjects |
Translational research is defined as the process of applying ideas, insights and discoveries generated through basic scientific inquiry to treatment or prevention of human diseases (2). Although there are no definitive figures about the number of studies that are lost during the translational process from the bench to the bedside, an interesting analysis of 101 studies published in high-profile journals revealed that only 27 of them were capable to generate randomized clinical trials, and only five of these finally yielded a licensed clinical application (3). This evidence inherently underscores that as many as 95% early-phase studies may not translate into tangible improvements in clinical management.
Major obstacles challenge effective translational medicine; basic science discoveries do not always lead to clinical studies, and many of such studies do not always translate into changes in medical practice and health care policy. A major emphasis has been placed on the fact that studies in model systems often fail to reproduce the complexity of disease, as well as on the challenges of dealing with often highly heterogeneous groups of patients. So far, relatively little attention has been focused on another explanation: the fact that many studies may have failed to meet the expected outcomes for other reasons, such as the use of inappropriate diagnostic testing for evaluating efficacy, effectiveness and safety of a given medical intervention, or even for poor quality in laboratory testing, which may eventually generate biased test results and misconceptions during data interpretation.
It has also been clearly established that the evaluation of the potential clinical impact of biomarkers in early-phase studies requires the accurate definition of levels of biomarker performance for validation, before they can be used in medical practice and health care policy (4). Biomarkers meeting these preliminary performance criteria should be subsequently be evaluated in studies that can estimate their real impact on patient outcomes (5,6). Even more importantly than for clinical trials involving innovative drugs or treatments, biomarkers studies require that evidence-based criteria are implemented and strictly followed throughout the total testing process, which spans from sample collection (i.e., the pre-analytical phase), through sample analysis (i.e., the analytical phase), to data interpretation (i.e., the post-analytical phase) (7,8). As for routine laboratory diagnostics, something that goes wrong throughout the testing process may ultimately impair the generation of reliable data and, consequently, may even jeopardize one or more outcomes in the study.
Laboratory medicine and patient safety in clinical trials
Laboratory diagnostics plays a foremost role in clinical studies, wherein biomarkers are used for the screening, diagnosis, prognostication and therapeutic monitoring of many (virtually each) human disorders. However, laboratory tests are also used for safety reasons, to timely and accurately identify whether an innovative drug or a new medical or surgical treatment produce side-effects and complications.
Drug-induced liver injury has an estimated incidence comprised between 10–15 per 10,000 persons exposed to different types of drugs, accounting for accounts for approximately 10% of all cases of acute hepatitis (9). Hepatotoxicity, defined as mild to moderate impairment of liver function, has been the most frequent single cause of safety-related drug marketing withdrawals over the past 50 years. Paradigmatic cases of US withdrawals include iproniazid, ticrynafen, benoxaprofen, bromfenac, troglitazone and nefazodone, whereas ibufenac, perhexiline alpidem were subjected to withdrawal by the European Union (EU) for the same reason (10).
The US Department of Health and Human Services, the US Food and Drug Administration (FDA), the US Center for Drug Evaluation and Research (CDER) and the US Center for Biologics Evaluation and Research (CBER) have released a joint document providing guidance to assist the pharmaceutical industry and other investigators who are conducting new drug development in assessing the potential for a drug to cause severe liver injury (10). According to this guidance document, hepatocellular injury is suspected in the presence of an increase of serum or plasma values of some liver function tests, namely bilirubin, alanine aminotransferase (ALT), aspartate aminotransferase (AST) and/or lactate dehydrogenase (LDH). A significant elevation of liver enzymes [i.e., approximately 10× to 15× the upper limit of the reference range (ULR)] is regarded as a specific index of potential hepatotoxicity, whereas the presence of an increase over 1,000 U/L of aminotransferases and >2× the ULR of bilirubin raises major concern, immediate withdrawal of the drug and even interruption of the clinical trial. Overall, drugs suspected of causing hepatocellular injury are conventionally identified as those showing an incidence of 3-fold or greater elevations above the URL of ALT or AST; among trial subjects showing these abnormalities, an increase of total serum bilirubin >2× URL deserve special focus for potential of developing serious liver injury. According to this evidence, appropriate testing and analysis of liver function tests in clinical trials is mandatory to timely identify drugs that may cause hepatocellular injury and safeguard the safety of the study cohort.
A similar approach has also been endorsed by the European Medicines Agency (EMA), wherein a panel of biomarkers should be measured in preclinical studies for identification of hepatocellular or hepatobiliary injury. The panel should include at least two of the following: ALT, AST, LDH, sorbitol dehydrogenase (SDH) and glutamate dehydrogenase (GDH). Similarly, at least two of the following serum parameters should be measured for identification of hepatobiliary injury: alkaline phosphatase (ALP), γ-glutamyltransferase (GGT), and total bilirubin (11). Accordingly, increases in serum ALT levels of 2–4× the URL may raise concern as reflecting potential hepatic injury, whereas an increase >3–5× the URL is considered and adverse event even in the lack of clear histologic changes.
Inappropriate sample collection in clinical trials: clinical issues
Sample management in clinical trials entails a series of activities that are quite similar to those used in routine diagnostic testing (Figure 1), but some peculiarities remain. In multicenter clinical trials, sample aliquoting, (long-term) storage and retrieval are frequently required before the specimens can be shipped to the many testing facilities (i.e., clinical and research laboratories) that are in charge of analyzing conventional and innovative biomarkers (12). Sample shipment is also an increasing need of many clinical laboratories participating in large networks, wherein an economically-driven reorganization of laboratories services is leading the way to a hierarchical consolidation of smaller laboratories into larger facilities (i.e., according to the so-called “hub-and-spoke model”) (13). Therefore, fulfillment of validated criteria for aliquoting, transport and storage of biological materials is vital for routine laboratory diagnostics, but may also be even more essential in clinical trials.
Figure 1.
The total testing process in clinical studies.
If one considers the potential impact of inappropriate sample collection on analytes that are usually tested in clinical trials, the ensuing picture is somehow concerning. Spurious hemolysis, the leading cause of unsuitable specimens in clinical laboratories, dramatically impacts test results of many analytes, especially those displaying a higher concentration in the intra-cellular than in the extra-cellular space. Figure 2 clearly shows the behavior of AST, ALT, LDH and bilirubin in human serum samples with increasing concentration of cell-free hemoglobin due to spurious erythrocyte injury (i.e., for inappropriate collection, mixing, storage and/or transportation), as measured with some specific laboratory instrumentation (14). At a cell-free hemoglobin concentration of 5.1 g/L, that is not so rare in the hemolyzed specimens received in clinical laboratories (16), the value of LDH exceeds the 3× URL. At a cell-free hemoglobin concentration of 10.3 g/L, that may be also due to partial freezing of whole blood during sample shipment (e.g., for direct contact with ice) (17), the value of AST exceeds the 3× URL. Importantly, at this threshold of cell-free hemoglobin, the serum concentration of ALT is also 1.65 the URL, a value that may alert clinicians about potential hepatocellular toxicity of the drug under investigation (Figure 2). In an additional study (15), the values of bilirubin and LDH and were found to increase above the 3× URL at a cell-free hemoglobin concentration of 3.0 and 6.0 g/L, respectively (Figure 2). These are two hemolysis thresholds that can also be occasionally observed in hemolyzed samples referred for laboratory testing. Rather understandably, when spurious elevation of liver function tests due to spurious hemolysis rather than to real hepatocellular injury is not promptly recognized by the investigators, this may then potentially lead to misinterpretation of tests results and, in the worst scenario, safety-related drug withdrawal. Importantly, this issue is not only limited to human studies, but also strongly impact preclinical studies, wherein the chance of collecting hemolyzed serum or plasma samples from animals is enormously amplified and even more neglected (18).
Figure 2.
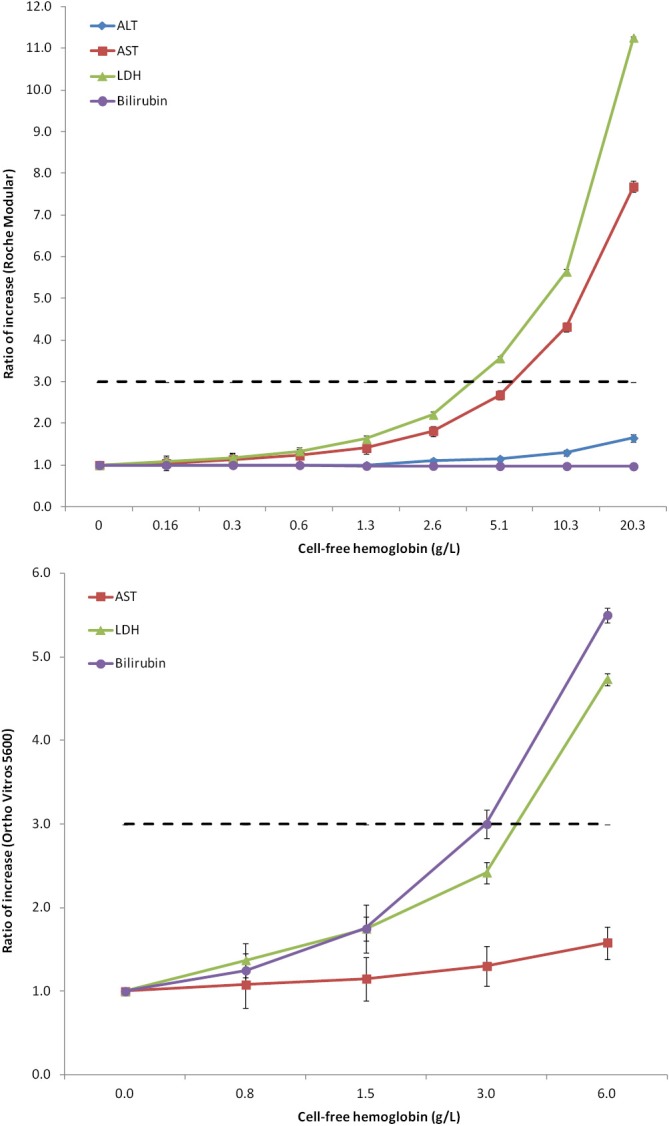
Variation of alanine aminotransferase (ALT), aspartate aminotransferase (AST), lactate dehydrogenase (LDH) and bilirubin as a function of spurious hemolysis (i.e., concentration of cell-free hemoglobin) in two separate studies using Roche Modular (14) and Ortho Vitros 5600 (15).
Additional sources of variability have been clearly described, and which may introduce inappropriate variability or bias in test results. These substantially include the fasting status (ALT, AST and total bilirubin exhibit considerable and clinically significant biochemical changes in the postprandial state) (19), venous stasis (i.e., maintaining the tourniquet over 3 min increase the measured values of certain analytes by over 10%) (20), low sample volume (e.g., collection of samples containing less than 90% of the blood tube nominal volumes impairs data generated with routine and specialized coagulation testing) (21), patient posture (e.g., there is a huge difference of test results—up to 15%—when samples are collected from patients in standing position or without waiting a sufficient time for enabling equilibration of body liquids between blood vessels and the extra-vascular space compared to blood samples collected from patients bedridden) (22).
The heterogeneity of the material used for collecting blood samples is another peculiar issue in multicenter clinical studies. When multiple collecting centers are enrolled, it is predictable that different blood collection devices and blood tubes may be used. There is now clear evidence that these materials—especially blood tubes which may differ in size, composition, additives—may introduce a substantial bias in test results, thus jeopardizing the comparability of data produced in samples collected using different materials (23,24).
Inappropriate sample collection in clinical trials: economic issues
In the European Union, as many as 5,549 clinical trials have been registered in Eudract in 2014, totaling over 1,000,000 patients, with an overall expenditure exceeding 10 billion euros and a cost of laboratory analyses of approximately 1 billion euros (25). Given current regulations, one can expect that the vast majority of these patients have been submitted to some kind of laboratory analyses, and even to repeated testing to establish the safety profile or the effectiveness of a given drug or medical treatment. The numbers of unsuitable specimens that are usually received in clinical laboratory, and which cannot be processed to ensure that the quality of data is preserved, are approximately 0.3–1.0% of the total blood samples. The vast majority of these, over 90%, are unsuitable due to incorrect procedures for collection and/or transportation. Indeed, these figures are probably magnified in the clinical trial setting, wherein blood collection is typically carried out at different sites and sample transportation over long distances is more commonplace than for routine laboratory testing. By simple translation of actual figures for routine diagnostic testing into the clinical trial setting, it can be (optimistically) assumed that over 10,000 blood samples may be unsuitable for testing, thus generating the need for recollection (when timely recognized as unsuitable) or retesting (which generates incremental costs). From a genuine economic perspective, this implies that up to 10 million euros of funding may be lost each year in clinical trials in the EU due to receipt of unsuitable blood samples in testing facilities.
Filling the gap between what we know and what we practice
Though there is no doubt that many recommendations and guidelines exist for the appropriate management of the testing process for routine laboratory diagnostics (i.e., “what we know”), the available documents often differ in many aspects regarding the various phases of sample collection, handling, preparation for testing, storage and shipment (26). Notably, there is also little evidence that these basic requirements are clearly defined as mandatory quality prerequisites in clinical trials (i.e., “what we practice”). This aspect has been emphasized in previous articles (27-29), which suggest that a gap still remains between the best (evidence-based) laboratory practice and the way samples are collected and managed in many clinical trials. In fact, some investigators are not so familiar with extra-analytical requirements, and preanalytical issues are often overlooked in study designs mainly due to insufficient awareness or exclusion of laboratory professionals in the early phases of project design. Another important issue is represented by the fact that the existing guidance documents for quality testing are mainly focused on routine rather than on research setting (26). As stated above, biomarker testing in clinical trials often develops quite differently from routine assessment, differing substantially for type of resources employed, testing sites, patient cohorts and, last but not least, outcomes and/or endpoints. It is also undeniable that patients’, clinicians’, nurses’ and diagnostic companies’ perspectives have not been thoughtfully integrated in laboratory best practices.
These unquestionable drawbacks (Table 2) make it difficult to harmonize/standardize practices across clinical trials and even within a single multicenter study. Therefore, the most rational approach for filling the gap between what we know and what we practice in clinical studies cannot discount the development of multidisciplinary teams including research scientists, clinicians, nurses, patients associations and representative of in vitro diagnostic (IVD) companies, who should actively interplay and collaborate with laboratory professionals to adapt and widely disseminate evidence-based recommendations for biospecimens collection and management into the research settings, from preclinical to phase III studies. Accordingly, the forthcoming steps may entail (I) survey of the many existing evidence-based recommendations for preanalytical management of biospecimens; (II) development of quality indicators for monitoring sample quality throughout the total testing process of clinical trials, from sample collection to sample analysis; (III) generation of an evidence-based document aimed for promoting standardization of best practices across clinical trials; (IV) generation of an evidence-based guidance document for researchers and practitioners, with real-life examples, on how to set outcome-based pre-analytical and analytical performance criteria for specific applications of clinical studies; (V) spreading guidelines by targeted dissemination, and training of pertinent stakeholders. These include patients and patient associations, researchers and university students, physicians, laboratory professionals, phlebotomists and nurses, trainers in charge of delivering courses to physicians, IVD companies, companies manufacturing drugs or medical devices, contract research organizations, regulatory boards, national or supranational institutions, standardization organizations as well as national governments.
Table 2. Major hurdles for establishing high quality laboratory testing in clinical trials.
| Heterogeneity of current guidelines and recommendations for the testing process |
| Little evidence exists that basic pre-analytical, analytical and post-analytical requirements are defined as mandatory quality prerequisites in clinical trials |
| Current guidelines and recommendations are specifically designed for routine laboratory testing |
| Clinicians’, patients’, nurses’ and diagnostic companies’ perspectives are not thoughtfully integrated in laboratory best practices |
Conclusions
There is now widespread recognition of the fact that many promising treatments or biomarkers are “lost in translation” on their route from basic research to routine practice due to unwarranted bias emerging from analytical and especially extra-analytical activities (30). Rather understandably, when something goes wrong in any phase of the total testing process (Figure 1), the results obtained on these samples will also fail. We hence believe that the development of best practice recommendations for laboratory diagnostics in clinical trials and their endorsement by a multidisciplinary team involving all the stakeholders of laboratory services will be crucial for improving the reliability and quality of trial findings. Such recommendations can help standardizing data sharing among trials and may decrease the waste of resources necessary for sample recollection/retesting. Additional benefits include standardization of handling and transport of biological samples, which would contribute to reduce the time to start projects and facilitate the approval in different countries. Contextually, sample transportation following a unified recommendation may help improve the activity of sample transport companies, thus ultimately decreasing the overall expenditure of the investigation. In summary, we firmly support the creation of a supranational initiative or consortium aimed to develop and disseminate evidence-based recommendations about biospecimen collection and management into the research settings.
Acknowledgements
None.
Footnotes
Conflicts of Interest: The authors have no conflicts of interest to declare.
References
- 1.U.S. National Library of Medicine. ClinicalTrials.gov - Clinical Trial Phases. Available online: https://www.nlm.nih.gov/services/ctphases.html, access 5 April 2016.
- 2.Mankoff SP, Brander C, Ferrone S, et al. Lost in Translation: Obstacles to Translational Medicine. J Transl Med 2004;2:14. 10.1186/1479-5876-2-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lost in clinical translation. Nat Med 2004;10:879. 10.1038/nm0904-879 [DOI] [PubMed] [Google Scholar]
- 4.Lippi G, Jansen-Duerr P, Viña J, et al. Laboratory biomarkers and frailty: presentation of the FRAILOMIC initiative. Clin Chem Lab Med 2015;53:e253-5. 10.1515/cclm-2015-0147 [DOI] [PubMed] [Google Scholar]
- 5.Lippi G, Mattiuzzi C. The biomarker paradigm: between diagnostic efficiency and clinical efficacy. Pol Arch Med Wewn 2015;125:282-8. [DOI] [PubMed] [Google Scholar]
- 6.Pepe MS, Janes H, Li CI, et al. Clin Chem 2016. [Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Banfi G, Lippi G. The impact of preanalytical variability in clinical trials: are we underestimating the issue? Ann Transl Med 2016;4:59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bossuyt PM, Cohen JF, Gatsonis CA, et al. STARD 2015: updated reporting guidelines for all diagnostic accuracy studies. Ann Transl Med 2016;4:85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shapiro MA, Lewis JH. Causality assessment of drug-induced hepatotoxicity: promises and pitfalls. Clin Liver Dis 2007;11:477-505, v. 10.1016/j.cld.2007.06.003 [DOI] [PubMed] [Google Scholar]
- 10.US Food and Drug Administration. Guidance for Industry. Drug-Induced Liver Injury: Premarketing Clinical Evaluation. Available online: http://www.fda.gov/downloads/Drugs/.../Guidances/UCM174090.pdf, access 5 April 2016.
- 11.European Medicines Agency. Reflection paper on non-clinical evaluation of drug-induced liver injury (DILI). Available online: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2010/07/WC500094591.pdf, access 5 April 2016.
- 12.Erusalimsky JD, Grillari J, Grune T, et al. In Search of 'Omics'-Based Biomarkers to Predict Risk of Frailty and Its Consequences in Older Individuals: The FRAILOMIC Initiative. Gerontology 2016;62:182-90. 10.1159/000435853 [DOI] [PubMed] [Google Scholar]
- 13.Lippi G, Simundic AM. Laboratory networking and sample quality: a still relevant issue for patient safety. Clin Chem Lab Med 2012;50:1703-5. 10.1515/cclm-2012-0245 [DOI] [PubMed] [Google Scholar]
- 14.Lippi G, Salvagno GL, Montagnana M, et al. Influence of hemolysis on routine clinical chemistry testing. Clin Chem Lab Med 2006;44:311-6. 10.1515/CCLM.2006.054 [DOI] [PubMed] [Google Scholar]
- 15.Agarwal S, Vargas G, Nordstrom C, et al. Effect of interference from hemolysis, icterus and lipemia on routine pediatric clinical chemistry assays. Clin Chim Acta 2015;438:241-5. 10.1016/j.cca.2014.08.008 [DOI] [PubMed] [Google Scholar]
- 16.Simundic AM, Topic E, Nikolac N, Lippi G. Hemolysis detection and management of hemolysed specimens. Biochem Med. 2010;20:154–9. 10.11613/BM.2010.018 [DOI] [Google Scholar]
- 17.Lippi G. Interference studies: focus on blood cell lysates preparation and testing. Clin Lab 2012;58:351-5. [PubMed] [Google Scholar]
- 18.Di Martino G, Stefani AL, Lippi G, et al. The degree of acceptability of swine blood values at increasing levels of hemolysis evaluated through visual inspection versus automated quantification. J Vet Diagn Invest 2015;27:306-12. 10.1177/1040638715585155 [DOI] [PubMed] [Google Scholar]
- 19.Simundic AM, Cornes M, Grankvist K, et al. Standardization of collection requirements for fasting samples: for the Working Group on Preanalytical Phase (WG-PA) of the European Federation of Clinical Chemistry and Laboratory Medicine (EFLM). Clin Chim Acta 2014;432:33-7. 10.1016/j.cca.2013.11.008 [DOI] [PubMed] [Google Scholar]
- 20.Lippi G, Salvagno GL, Montagnana M, et al. Influence of short-term venous stasis on clinical chemistry testing. Clin Chem Lab Med 2005;43:869-75. 10.1515/CCLM.2005.146 [DOI] [PubMed] [Google Scholar]
- 21.Lippi G, Salvagno GL, Montagnana M, et al. Quality standards for sample collection in coagulation testing. Semin Thromb Hemost 2012;38:565-75. 10.1055/s-0032-1315961 [DOI] [PubMed] [Google Scholar]
- 22.Lippi G, Salvagno GL, Lima-Oliveira G, et al. Postural change during venous blood collection is a major source of bias in clinical chemistry testing. Clin Chim Acta 2015;440:164-8. 10.1016/j.cca.2014.11.024 [DOI] [PubMed] [Google Scholar]
- 23.Bowen RA, Remaley AT. Interferences from blood collection tube components on clinical chemistry assays. Biochem Med (Zagreb) 2014;24:31-44. 10.11613/BM.2014.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lippi G, Cornes MP, Grankvist K, et al. EFLM WG-Preanalytical phase opinion paper: local validation of blood collection tubes in clinical laboratories. Clin Chem Lab Med 2016;54:755-60. 10.1515/cclm-2015-1274 [DOI] [PubMed] [Google Scholar]
- 25.EudraCT. EudraCT statistics. Available online: https://eudract.ema.europa.eu/statistics.html, access 5 April 2016.
- 26.Simundic AM, Cornes M, Grankvist K, et al. Survey of national guidelines, education and training on phlebotomy in 28 European countries: an original report by the European Federation of Clinical Chemistry and Laboratory Medicine (EFLM) working group for the preanalytical phase (WG-PA). Clin Chem Lab Med 2013;51:1585-93. [DOI] [PubMed] [Google Scholar]
- 27.Groenen PJ, Blokx WA, Diepenbroek C, et al. Preparing pathology for personalized medicine: possibilities for improvement of the pre-analytical phase. Histopathology 2011;59:1-7. 10.1111/j.1365-2559.2010.03711.x [DOI] [PubMed] [Google Scholar]
- 28.Christenson RH, Duh SH. Methodological and analytic considerations for blood biomarkers. Prog Cardiovasc Dis 2012;55:25-33. 10.1016/j.pcad.2012.05.001 [DOI] [PubMed] [Google Scholar]
- 29.Kellogg MD, Ellervik C, Morrow D, et al. Preanalytical considerations in the design of clinical trials and epidemiological studies. Clin Chem 2015;61:797-803. 10.1373/clinchem.2014.226118 [DOI] [PubMed] [Google Scholar]
- 30.Lippi G, Plebani M, Guidi GC. The paradox in translational medicine. Clin Chem 2007;53:1553. 10.1373/clinchem.2007.087288 [DOI] [PubMed] [Google Scholar]