

Case Report
A 58-year-old woman was evaluated at our institution for management of treatment-refractory breast cancer. She had a 5-year history of an initially estrogen receptor (ER), progesterone receptor (PR)–positive and human epidermal growth factor receptor 2 (HER2)–amplified pT1a, pN0, pMx invasive ductal breast carcinoma treated with standard local therapies and without any adjuvant systemic treatment. Subsequently she developed what appeared to be a primary triple-negative (ER/PR/HER2) disease with features of inflammatory breast cancer. Physical exam demonstrated erythema, warmth, and induration extending cephalic from her medial left breast to her lateral left neck (Figs 1A and 2A). She was experiencing dysphagia, attributed to infiltration of the tumor into the infrahyoid muscles. The disease proved to be refractory to multiple systemic therapies, including combination regimens with taxanes (Taxotere, sanofi-aventis, Bridgewater, NJ), bevacizumab (Avastin, Genentech, South San Francisco, CA), dasatinib (Sprycel, Bristol-Myers Squibb, Princeton, NJ), ixabepilone (Ixempra, Bristol-Myers Squibb) and gemcitabine (Gemzar, Eli Lilly, Indianapolis, IN). This lack of response to cytotoxic chemotherapy is typical of inflammatory breast cancer.1
Fig 1.

Fig 2.

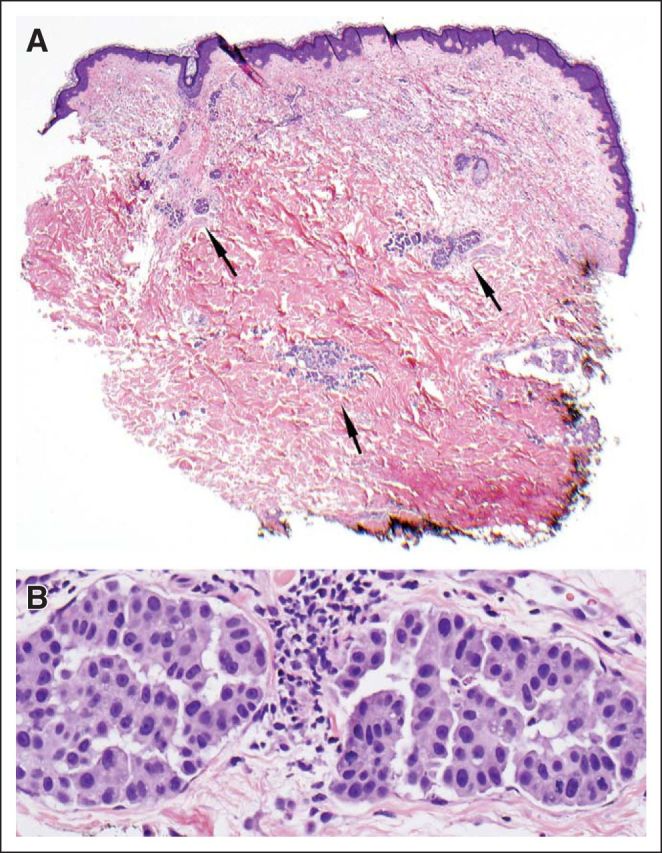
After progressive disease, a new diagnostic skin biopsy was performed to re-evaluate standard biomarkers and to permit sequencing of a broad range of genes known to be associated with cancer within a single integrated assay. Pathologic examination revealed breast carcinoma in the dermal tissue with associated lymphovascular invasion (Figs 3A [low magnification] and 3B [higher magnification]), and these findings along with the clinical presentation fulfilled the diagnostic criteria for inflammatory breast carcinoma.2 Biomarker testing was performed, and the tumor was again negative for ER/PR staining by immunohistochemistry and negative for ERBB2 amplification by fluorescent in situ hybridization (FISH). Accordingly, the patient was considered ineligible for HER2-targeted therapy. At this time, this cutaneous sample of recurrent tumor was submitted for diagnostic next generation sequencing (NGS) by a Clinical Laboratories Improvement Amendments–certified commercial laboratory, where the entire coding regions of 182 cancer-related genes were sequenced, along with 36 introns of 14 genes frequently involved in gene fusions. Consistent with the prior FISH-based test for the recurrent disease, NGS demonstrated no evidence of an ERBB2 copy number gain (amplification), instead revealing two distinct ERBB2 mutations (V777L and S310F, both at 47% allele frequency). Both mutations have previously been described in breast cancer in Catalog of Somatic Mutations in Cancer (COSMIC; December 2012). Mutations in PIK3CA (K111E at 30%) and TP53 (C229fs*10 at 38%) were also identified. The percent of tumor in this sample was estimated to be 50%, and the consistency of allele frequency of the ERBB2 mutations with this histologic assessment of purity suggests these mutations are ubiquitous within the tumor. In vitro studies using established cell lines suggested that both of the identified ERBB2 mutations activate HER2 by either affecting HER2 autophosphorylation or phosphorylation of downstream substrates.3 The V777L mutation is in the kinase domain of HER2 and is associated with aberrant and enhanced activation of downstream signaling pathways in breast cancer cells. The S310F mutation is in the extracellular domain and has also recently been demonstrated to be activating in lung cancer cells.4 It has also been demonstrated that HER2-targeted therapy has a dramatic inhibitory effect on breast cancer cells with these and other ERBB2 mutations.3 On the basis of these data, these mutations were deemed to be activating base substitutions, which suggested that the ERRB2 pathways could be drivers of the patient's disease, even in the absence of either ERBB2 amplification or protein overexpression. In contrast, the PIK3CA mutation seen here (K111E) has not previously been seen in breast carcinoma but has been identified within endometrial endometrioid carcinoma,5 The functional significance of this mutation is therefore unclear, but it suggests some modulation of PIK3CA function.
Fig 3.
The patient began treatment with lapatinib (Tykerb, GlaxoSmithKline, Philadelphia, PA) and capecitabine (Xeloda, Genentech) and experienced initial symptomatic and clinical improvement visible on physical exam (Fig 1B). However, continuing dysphagia limited the possibility of continuing this oral combination. Capecitabine was discontinued, and albumin-bound paclitaxel and trastuzumab (Herceptin, Genentech) were added to the maintained treatment with lapatinib. After 4 weeks, the patient showed significant symptomatic improvement with a lessening of her dysphagia, but her neuropathy worsened and the albumin-bound paclitaxel was replaced with vinorelbine (Navelbine, Pierre Fabre Pharmaceuticals, Parsippany, NJ). The patient tolerated the three-drug regimen and demonstrated a response in the skin and nodes, which was confirmed by positron emission tomography/computed tomography scan (Figs 4A and 4B; standardized uptake value maximum 13.3 v 3.0, respectively). Although this response cannot formally be characterized under RECIST criteria because of lack of other imaging studies, the patient nonetheless experienced great symptomatic improvement, which corresponded to dramatic changes on pharmacodynamic imaging. But the disease progressed after 6 months of treatment, and the patient died shortly thereafter.
Fig 4.

Discussion
The introduction of ER testing in the early 1970s combined with US Food and Drug Administration approval of HER2 companion diagnostics and targeted HER2 therapy in HER2-amplified disease has significantly contributed to the improvement in overall survival of patients with invasive breast cancer.6 However, despite the use of available biomarker-based hormonal and anti-HER2 therapy selection, inflammatory breast cancer, a distinct but poorly understood type of breast cancer, has a lower rate of survival than other breast cancers.2 Patients with inflammatory breast cancer have a survival rate of only 40% after 5 years compared with greater than 80% for all other types of invasive breast cancer.1 There are currently no chemotherapy regimens specific to inflammatory breast cancer, despite the severity of this disease, and patients are treated with general combination chemotherapy regimens when biomarkers do not indicate that either endocrine or HER2-targeted therapy is appropriate.
The efficacy of HER2-targeted therapy is most pronounced in patients with breast cancer with ERBB2 amplification, as identified by FISH.7 This overexpression of the HER2 protein resulting from ERBB2 amplification ectopically activates downstream signaling pathways necessary for oncogenesis. An alternative mechanism of activation of the HER2 pathway also exists given that mutation of HER2 drives oncogenesis in a variety of malignant neoplasms as first identified in non–small-cell lung cancer8 and then later described in GI, genitourinary, and gynecologic carcinomas and some sarcomas.4 Bose et al3 recently described an analogous subset of patients with breast cancer who carry a tumoral HER2 mutation. This is the first case report, to our knowledge, of a HER2 mutation within an inflammatory breast cancer, suggesting the existence of a subset of patients with inflammatory breast cancer who would be newly eligible for HER2-targeted therapy as a result of carrying this mutation. Moreover, our report is the first description, to our knowledge, of a response to HER2-targeted therapy in a patient with breast cancer with tumoral mutations in ERBB2, but whose tumor is simultaneously negative for ERBB2 amplification by FISH or protein overexpression by immunohistochemistry.
Of note, the inflammatory type of breast cancer seen in this patient, although notoriously resistant to chemotherapy, demonstrated a dramatic clinical and symptomatic response to HER2-targeted therapy combined with baseline chemotherapy. The detection of the two separate tumoral ERBB2 driver mutations and with allele frequencies consistent with a ubiquitous presence in the tumor suggest that they are likely to cause an “addiction” of the patient's cancer cells to the “hyperactivated” HER2 pathway and may explain, the dramatic clinical response to HER2-targeted therapy.9 This is also the first case of a breast cancer containing two HER2 mutations within the same tumor, and identifying future similar cases and their response to HER2-targeted therapy will help assess whether more mutations confer great therapeutic sensitivity. Although chemotherapy was coadministered with HER2-targeted therapy, the previous progression on multiple lines of cytotoxic chemotherapy suggests that the HER2-targeted therapy was the critical element in inducing a response in the patient's inflammatory breast cancer.
This observation precedes any report of a clinical trial of HER2-targeted therapy in HER2-mutated breast cancer but demonstrates both the utility of NGS as a clinical diagnostic assay and the potential efficacy of HER2-targeted therapy in breast cancers with HER2 mutations. A recent report from Bose et al3 suggests that the overall incidence of HER2 somatic mutation in breast cancer is around 1.6%, which could translate into 4,000 women per year in the United States. This incidence and the ability to routinely test for these alterations in clinical care should mandate prospective clinical trials of single agents or combinations of HER2-targeted therapies in this subset of patients. Table 1 includes a current list of known HER2 mutations in breast cancer, deriving from publications by other groups and identified by the authors of the current study. It is anticipated that this list will grow over time as more breast cancers are evaluated by deep sequencing technologies and the full spectrum of HER2 mutations in this disease, including patients with inflammatory breast cancer, can be characterized and aligned with the potential benefit from HER2-targeted therapy.
Table 1.
Mutations in ERBB2 Identified in Breast Carcinoma
| Genomic Alteration | Reference |
|---|---|
| G309A | Bose et al 20123 |
| S310F | FMI 2012 (previously unpublished data) |
| R678Q+L755W | Bose et al 20123 |
| del.755-759 | Bose et al 20123 |
| L755S | Bose et al 2012,3 FMI 2012 |
| D769Y | Bose et al 2012,3 FMI 2012 |
| D769H | Bose et al 20123 |
| A775-G776insYVMA | FMI 2012 |
| P780-Y781insGDP | Bose et al 2012,3 FMI 2012 |
| V777L | Bose et al 2012,3 FMI 2012 |
| V842I | Bose et al 20123 |
| R896C | Bose et al 20123 |
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
Although all authors completed the disclosure declaration, the following author(s) and/or an author's immediate family member(s) indicated a financial or other interest that is relevant to the subject matter under consideration in this article. Certain relationships marked with a “U” are those for which no compensation was received; those relationships marked with a “C” were compensated. For a detailed description of the disclosure categories, or for more information about ASCO's conflict of interest policy, please refer to the Author Disclosure Declaration and the Disclosures of Potential Conflicts of Interest section in Information for Contributors.
Employment or Leadership Position: Siraj M. Ali, Foundation Medicine (C); Sean R. Downing, Foundation Medicine (C); Philip J. Stephens, Foundation Medicine (C); Jamie K. Buell, Foundation Medicine (C); Vincent A. Miller, Foundation Medicine (C); Doron Lipson, Foundation Medicine (C); Gary A. Palmer, Foundation Medicine (C); Jeffrey S. Ross, Foundation Medicine (C) Consultant or Advisory Role: None Stock Ownership: Siraj M. Ali, Foundation Medicine; Sean R. Downing, Foundation Medicine; Philip J. Stephens, Foundation Medicine; Jamie K. Buell, Foundation Medicine; Vincent A. Miller, Foundation Medicine; Doron Lipson, Foundation Medicine; Gary A. Palmer, Foundation Medicine; Jeffrey S. Ross, Foundation Medicine Honoraria: Massimo Cristofanilli, Foundation Medicine Research Funding: Jeffrey S. Ross, Foundation Medicine Expert Testimony: None Patents: None Other Remuneration: None
REFERENCES
- 1.Dawood S, Merajver SD, Viens P, et al. International expert panel on inflammatory breast cancer: Consensus statement for standardized diagnosis and treatment. Ann Oncol. 2010;22:515–523. doi: 10.1093/annonc/mdq345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Robertson FM, Bondy M, Yang W, et al. Inflammatory breast cancer: The disease, the biology, the treatment. CA Cancer J Clin. 2010;60:351–375. doi: 10.3322/caac.20082. [DOI] [PubMed] [Google Scholar]
- 3.Bose R, Kavuri SM, Searleman AC, et al. Activating HER2 mutations in HER2 gene amplification negative breast cancer. Cancer Discovery. 2013;3:224–237. doi: 10.1158/2159-8290.CD-12-0349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Greulich H, Kaplan B, Mertins P, et al. Functional analysis of receptor tyrosine kinase mutations in lung cancer identifies oncogenic extracellular domain mutations of ERBB2. Proc Natl Acad Sci U S A. 2012;109:14476–14481. doi: 10.1073/pnas.1203201109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rudd ML, Price JC, Fogoros S, et al. A unique spectrum of somatic PIK3CA (p110alpha) mutations within primary endometrial carcinomas. Clin Cancer Res. 2011;17:1331–1340. doi: 10.1158/1078-0432.CCR-10-0540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ross JS, Slodkowska EA, Symmans WF, et al. The HER-2 receptor and breast cancer: Ten years of targeted anti-HER-2 therapy and personalized medicine. Oncologist. 2009;14:320–368. doi: 10.1634/theoncologist.2008-0230. [DOI] [PubMed] [Google Scholar]
- 7.Piccart-Gebhart MJ, Procter M, Leyland-Jones B, et al. Trastuzumab after adjuvant chemotherapy in HER2-positive breast cancer. N Engl J Med. 2005;353:1659–1672. doi: 10.1056/NEJMoa052306. [DOI] [PubMed] [Google Scholar]
- 8.Stephens P, Hunter C, Bignell G, et al. Lung cancer: Intragenic ERBB2 kinase mutations in tumours. Nature. 2004;431:525–526. doi: 10.1038/431525b. [DOI] [PubMed] [Google Scholar]
- 9.Torti D, Trusolino L. Oncogene addiction as a foundational rationale for targeted anti-cancer therapy: Promises and perils. EMBO Mol Med. 2011;3:623–636. doi: 10.1002/emmm.201100176. [DOI] [PMC free article] [PubMed] [Google Scholar]