

Abstract
The NIH-funded LINCS program has been initiated to generate a library of integrated, network-based, cellular signatures (LINCS). A novel high-throughput gene-expression profiling assay known as L1000 was the main technology used to generate more than a million transcriptional profiles. The profiles are based on the treatment of 14 cell lines with one of many perturbation agents of interest at a single concentration for 6 and 24 hours duration. In this study, we focus on the chemical compound treatments within the LINCS data set. The experimental variables available include number of replicates, cell lines, and time points. Our study reveals that compound characterization based on three cell lines at two time points results in more genes being affected than six cell lines at a single time point. Based on the available LINCS data, we conclude that the most optimal experimental design to characterize a large set of compounds is to test them in duplicate in three different cell lines. Our conclusions are constrained by the fact that the compounds were profiled at a single, relative high concentration, and the longer time point is likely to result in phenotypic rather than mechanistic effects being recorded.
Introduction
Chemical compounds can be characterized by their chemical structure and the associated physiochemical properties as well as by the effects they induce in one or more experimental settings. High throughput screen (HTS) is traditionally used to identify lead compounds revealing activity in a biological assay for a single therapeutic target or pathway of interest.1,2 These assays test for activity on the protein level (e.g., binding assays), the biochemical level (e.g., enzyme activity assays), or the cellular level (e.g., cell viability assays). Characterization of biological effects induced by treating cellular systems with small molecules using transcriptional profiling is currently being explored as a means to add compound annotation beyond information, which can be derived from HTS (i.e., chemical structures and measured biological activity). Indeed, compound-induced transcriptional effects can be translated into “gene signatures” (i.e., set of differentially expressed genes characterizing the activity of a given compound, pathway, or disease), which can be used to discover new connections among compounds, pathways, and diseases.2–8
To this end, Peck et al.9 developed a cost-effective transcriptional profiling methodology, in which up to 100 transcripts can be measured in HTS mode, combining multiplex ligation-mediated amplification with the Luminex FlexMap (Luminex, Austin, TX) optically addressed and barcoded microsphere, and flow cytometric detection technology. The combination of ligated-mediated amplification and an optically addressed microsphere and flow cytometric detection has since been extended to 978 gene transcripts. This technology, known as L1000 (developed at the Broad Institute and commercialized by Genometry, Inc., Cambridge, MA), can now be used to screen and measure the transcriptionally induced effect(s) of thousands of compounds per day at a cost far below conventional transcriptomic techniques like microarrays. The 978 genes that are measured using the L1000 platform have been identified to capture most of the information contained within the entire transcriptome (www.lincscloud.org/l1000). The rest of the transcriptome can then be estimated by a model built from computational processing of thousands of gene expression data sets from GEO. Within this study, such extrapolation is not considered, focusing instead on only the 978 directly measured genes.
When exploring compound-induced transcriptional effects, one must, however, find a balance between the number of compounds and the number of conditions that can be tested. Conditions may include the number of (1) biological replicates, needed for sound statistical testing, (2) cellular backgrounds, which are likely to reveal different biological effects, (3) compound concentrations, with low concentrations likely to expose highly potent interactions between a small molecule and its respective target and high concentrations tending to show less specific or even toxic effects, and (4) time points, with earlier time points revealing more primary versus downstream biological effects.
With the availability of the public library of integrated network-based cellular signatures (LINCS) data set, which covers multiple cellular contexts, and time points across multiple compounds, it becomes possible to explore these experimental variables and propose some level of guidance on how to strike the right balance for genome-wide HTS transcriptional profiling.
Materials and Methods
General
Raw L1000 data were downloaded from the Broad Institute's FTP server (www.broadinstitute.org/LINCS/) and processed by Genometry, Inc. using their proprietary methods. A total of 162,499 gene-expression profiles with a “CPC” designation satisfied Genometry's well- and plate-based quality thresholds. These gene-expression profiles covered 14,199 compounds that were repeatedly measured (one to five replicates) at two time points (6 and 24 h), and in subsets of cell lines selected from 14 available lines (PC3, VCAP, A549, HT29, HEPG2, HCC515, HA1E, MCF7, ASC, SKB, NPC, NEU, PHH, and A375; www.lincscloud.org/cell_types/). In total there were 63,119 treatments (i.e., any combination of compound, time, and cell line) as shown in Table 1. These treatments came from 221 of the original plate designs (with one to five replicate plates passing quality control), where a plate design included up to 362 treatment wells and up to 29 DMSO wells on a 384 multiwell plate. There were a minimum of four DMSO wells passing quality control on plates included in this study.
Table 1.
Summary of Tests for Differential Expression Between Treatments and DMSO
| Treatments | Compounds | |
|---|---|---|
| Total | 63,119 | 14,199 |
| >1 significant gene | 50,595 | 13,856 |
| >1 significant gene, FC >2 | 24,306 | 11,392 |
| >10 significant genes, FC >2 | 2,844 | 2,746 |
FC, fold change.
Compound Diversity
A differential gene expression analysis was run for each of the 221 plate designs, using the limma R package.10 In this analysis, the logged gene expression intensities between compound treatment and DMSO are compared.
A linear model was estimated for each of the 978 investigated genes, with a coefficient for the effect of each treatment versus DMSO.11 Based on this model, t-statistics and associated P values were obtained to test separately whether the absolute fold change (FC) is (1) different from 1 (test of any difference) and (2) greater than 2 (test of substantial difference).12 The t-statistics were moderated using an empirical Bayes method to shrink the variance estimates obtained for each gene toward a common value.11 For each test, the P values are adjusted to control the false discovery rate across genes.13 Where there were replicates for a given plate design, the within-plate correlation was estimated and incorporated in the model using generalized least squares to account for potential plate effects.14
To separate the differences between compounds from the differences between cell lines and time points within compounds, a between-compound principal component analysis (PCA) was applied to the moderated t-statistics for the test of any difference from DMSO. In this analysis, genes are weighted by the loadings from a standard PCA of the 63,119 treatments by 978 genes, compounds are weighted by the relative frequency of treatments per compound, and principal components are obtained for the variance between the mean profile per compound.15 Unfortunately, the number of principal components required to adequately represent the variance between compounds, that is, the bioactivity space, was too large to summarize the compound diversity by observing projections in low dimensions. As a result, the compound diversity was summarized by a measure based on the distances between compounds in the bioactivity space. Lacevic and Amaldi16 considered a number of diversity measures for points in Euclidean space and recommended using the sum of Euclidean distances over the minimum spanning tree. Thus compounds were projected onto a certain number of principal components from the between-compound PCA, the minimum spanning tree connecting the compounds was found, and the sum of distances on this tree represented the diversity, that is, how spread out the compounds were in the bioactivity space. Adding a compound in a new area of the bioactivity space adds a large amount to the diversity, whereas adding a compound near to other compounds in the same space adds a small amount and adding a compound that produces the same response as another compound adds nothing at all.
The redundancy in the current set of compounds was explored by how well a subset of the compounds captures the diversity of the full set of compounds. Two methods for selecting subsets were applied. The first method obtained a subset of size k by selecting the medoids of k clusters obtained from a hierarchical clustering, where the medoid is the compound with minimum total distance from other members in the cluster. The second method generated subsets iteratively, starting with a randomly selected compound and then adding the compound with maximum distance to its nearest neighbor in the subset.17 Both methods were applied to diversity based on the full set of between-compound principal components (i.e., 978 components) and to a reduced set, covering 70% of the between-compound variability (i.e., 222 components).
Benefit of Additional Time Points
From the available 14,199 compounds, only 9,236 were tested at both 6 and 24 h. The number of cell lines per compound represented in these treatments ranged from one to six (Table 2). As a result, the analysis was split up into six groups. In each group, the percentage of compounds that revealed no significant genes, at least one significant gene, at least one significant gene at FC >2, and at least 10 significant genes at FC >2, compared with DMSO, was plotted for both time points and graphically compared.
Table 2.
Number of Cell Lines per Compound Represented in Treatments at Both Time Points
| Number of Cell Lines | ||||||
|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 6 | |
| Compounds (N) | 7,541 | 278 | 329 | 574 | 92 | 422 |
Benefit of Different Cellular Backgrounds
Since all cell lines are not always used to measure the transcriptional effect of compounds at the same time point, there is a need to split up the treatments according to time. A total of nine cell lines have 744 compounds in common, which were run at 6 h (i.e., group 1: PC3, VCAP, A549, HT29, HEPG2, HCC515, HA1E, MCF7, and A375). At 24 h, there is less overlap between the treatments run for different cell lines. Four groups can be identified, based on the treatments run for NEU, PHH, A375, and HA1E, respectively. In each case the number of cell lines was chosen to maximize the number of common compounds. These four groups (i.e., group 2: NEU, NPC, MCF7, ASC, A549, and SKB; group 3: A549, ASC, SKB, MCF7, NPC, and PHH; group 4: A549, HT29, MCF7, PC3, VCAP, and A375; and group 5: HCC515, HA1E, PC3, VCAP, SKB, NPC, MCF7, ASC, and A549) cover all cell lines apart from HEPG2, for which no treatments were run at 24 h.
A linear discriminant analysis (LDA) was performed for each of the five cell line groups. In addition, the percentage of compounds that revealed no significant genes, at least one significant gene, at least one significant gene at FC >2, and at least 10 significant genes at FC >2, compared with DMSO, was plotted for each of the cell lines within each of the five cell line groups and was graphically compared.
Benefit of Replicates
The addition of cell lines or time points implies collating results from different differential expression analyses, whereas additional replicates of the same plate design are analyzed together. The benefit of replicate plates on the L1000 differential expression analysis can thus be investigated through a simulation experiment. The log2 expression of gene i on plate j in the kth well is assumed to be
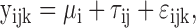 |
where μi is the true gene expression and
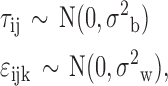 |
in which σ2b is the between-plate variance and σ2w is the within-plate variance. Without loss of generality, the respective DMSO means are assumed to be 0. In each simulation, data are simulated for 978 genes, 24 DMSO, and 1 treatment sample. The true treatment mean is always assumed to be one, representing a twofold change on the log scale. For multiple plates, this requires an estimate of the intra-plate well correlation. Since the same treatment effect is assumed for all wells, this correlation is not estimated in the simulation experiment, but included as one of the simulation parameters. The simulation is run over a grid of parameters for the within-group variance, between-group variance, within-plate correlation, and the number of plates. With the exception of plate numbers, the values for all remaining parameters are set to the lower quartile, median, and upper quartile of the corresponding statistics, taken from the 6,458 DMSO samples. For each set of parameters, the significance of the logged gene expression intensity between the treatment and the DMSO is determined for each of the 978 genes. The proportion of genes significant after correction for multiple testing shows the power to detect a twofold change.
All analyses were run using R version 3.1.118 and interpreted at a 5% significance level, with Bonferroni correction for multiple testing when needed.
Results
Compound Diversity
The number-of-genes-changing is a crude measure for activity and the adopted FC thresholds are high and arbitrarily defined. The current analysis does not capture coordinated (i.e., signature-type) effects that have been shown to be robust biological effects, encoded in and detected from gene-expression data. Against this background, 13,856 compounds from 14,119 compounds that were run in 63,119 treatments yield at least one gene with a significant difference from the corresponding DMSO sample. Within this group, 11,392 compounds reveal a twofold change difference for at least one gene, and 2,746 compounds have 10 or more of suchlike genes (Table 1).
The between-compound PCA reveals that 28% of the total variance is due to differences between the 14,119 compounds. The first two and three principal components (cumulative) only describe 18% and 21%, respectively, of the between-compound variance; therefore, it is impossible to adequately capture the bioactivity space in low dimensions. In fact, up to 222 dimensions are required to explain 70% of the variance. Compound diversity is consequently summarized by means of the sum of the Euclidean distances over the minimum spanning tree in full (i.e., 978) and reduced (i.e., 222) dimensions, for selected subsets of compounds. Figure 1 shows the diversity against the proportion of compounds selected, where the diversity is given as a proportion of the diversity of the full set of 14,119 compounds. If selected compounds were evenly spread throughout the bioactivity space, then the diversity would be proportional to the number of compounds selected, that is, the diversity would fall on the diagonal as shown in Figure 1. This would be true regardless of the selection method. If, in contrast, there are clusters of compounds in the bioactivity space, then it is possible to select subsets that represent disproportionally high amounts of the diversity. Different selection methods will be more or less effective at identifying subsets that efficiently cover the bioactivity space: the closer the curve is to the top left corner as shown in Figure 1, the more effective the method at optimizing diversity and the greater redundancy revealed in the data. Both compound selection methodologies (i.e., cluster medoid and maximum distance to nearest neighbor) are able to find subsets with more efficient coverage, so there is clearly some redundancy in the data (Fig. 1). The cluster medoid method is virtually unaffected by whether the distances are based on all 978 or only 222 of the principal components. This suggests that this method is robust to noise, selecting compounds by the dominant features of bioactivity. Selection of cluster medoids may thus be more stable and reflect true diversity. In contrast, the maximum distance method improves with using distances based on the full dimensionality, suggesting that small differences between compounds allow the method to differentiate between similar compounds and make the best selection to optimize diversity. Thus the maximum distance methodology based on 222 principal components seems to be a good compromise between robustness and coverage. The resulting diversity curve suggests that 50% of the compounds can capture 68% of the diversity, 75% can capture 87% of the diversity, whereas the full diversity is approximately reached when 98% of the compounds are included (Fig. 1).
Fig. 1.

Compound diversity (i.e., sum of Euclidean distances over minimum spanning tree) by proportion of compounds selected, using different selection methods with distances based on all between-compound principal components (i.e., 978 components) or a reduced set explaining 70% of the between-compounds variance (i.e., 222 components).
Benefit of Additional Time Points
The compounds that were treated at both 6 and 24 h are classified by the corresponding number of cell lines in Table 2. The cumulative percentage, across time, of compounds that induce significant differential gene expression in at least one cell line, split up by the number of cell lines, is shown in Figure 2. Around 75% of the compounds affect at least one gene significantly in a single cell line after 6 h of incubation (Fig. 2). This value increases to nearly 100% when an additional time point is considered (Fig. 2). When there are two or more cell lines, the percentage of compounds significantly affecting at least one gene is already nearly 100% (Fig. 2). However, an additional time point approximately doubles the number of compounds where there are at least 10 genes with significant FCs greater than 2 in at least 1 cell line, regardless of the number of cell lines (Fig. 2). Hence, different compounds have large FCs at different points in time.
Fig. 2.

Cumulative significance across time points for the 9,236 compounds with treatments run at both time points. The categories show the number of genes significant in at least one of the seven cell lines represented in the treatments.
Benefit of Different Cellular Backgrounds
At 6 h, differential gene expression was measured in 9 cell lines for 744 compounds. Based on this compound subset, the nine cell lines are ordinated in two dimensions according to LDA (Fig. 3). The first two linear discriminant axes explain 32% of the variance and discriminate PC3 and VCAP from each other as well as all others. The remaining cell lines can, to some extent, be discriminated from each other using a third linear discriminant dimension (Fig. 3). Figure 4A reveals that the highest percentage of compounds that induce at least one differentially expressed gene can be found in VCAP, whereas PC3 has the least. Even though PC3 is less sensitive than VCAP, it still identifies compounds that do not affect VCAP (Fig. 4B). Each additional cell line increases the number of compounds that reveal at least one gene with a significant FC greater than 2, although the additional benefit becomes small after three cell lines (Fig. 4B).
Fig. 3.

Results of an LDA, discriminating PC3, VCAP, A549, HT29, HEPG2, HCC515, HA1E, MCF7, and A375 on the basis of t-statistics summarizing the differential expression of 978 genes, for 744 common compounds after 6 h of incubation. The common compounds are projected for each cell line onto (A) the first and second, (B) the first and third, and (C) the second and third discriminant axes. LDA, linear discriminant analysis.
Fig. 4.

All 744 compounds, common in PC3, VCAP, A549, HT29, HEPG2, HCC515, HA1E, MCF7, and A375, classified according to the number of differentially expressed genes at 6 h of incubation. (A) Significance based on individual cell lines; (B) cumulative significance across all nine cell lines ordered by distance from the other cell lines based on all linear discriminant axes.
At 24 h, differential gene expression was measured in four groups of cell lines, for 197, 445, 569, and 445 common compounds, respectively (Fig. 5). Figure 5 summarizes the number of compounds that affect the different cell lines in the four different cell line groups at 24 h of treatment. Similar to the analysis at 6 h, the effect of adding more cell lines after the first three, LDA based, most distant located cell lines, is marginal.
Fig. 5.

For cell line groups 2 (i.e., NEU, NPC, MCF7, ASC, A549, SKB), 3 (i.e., A549, ASC, SKB, MCF7, NPC, PHH), 4 (i.e., A549, HT29, MCF7, PC3, VCAP, A375), and 5 (i.e., HCC515, HA1E, PC3, VCAP, SKB, NPC, MCF7, ASC, A549); common compounds classified according to the number of differentially expressed genes at 24 h of incubation. (A, C, E, G) Significance based on individual cell lines in the respective group; (B, D, F, H) cumulative significance across the cell line group ordered by distance from the other cell lines based on all linear discriminant axes.
Based on the 6 and 24 h LDAs, the cell lines can be ranked in terms of their dissimilarity from other cell lines, with higher weight given to the analyses that are based on a higher number of compounds (i.e., from highest to lowest rank: PC3, VCAP, A549, HT29, HEPG2, HCC515, HA1E, MCF7, ASC, SKB, NPC, NEU, PHH, and A375). In addition, LDA suggests that at least three cell lines should be considered and that the additional benefit is likely to be marginal after six to seven cell lines. It must be stated, however, that there is no measure of the relative diversity in the chosen LINCS cell line panel and that the cell lines could be highly redundant.
Benefit of Replicates
The benefit of using replicates has been investigated in a simulation-based power analysis. This analysis reveals that when the between-plate variance is at its highest, increasing the number of replicates will adversely affect the power of the test (Fig. 6). In all other cases, the power of the test increases with increasing number of replicates. When the within-plate variance is at its median value or lower, three replicates are sufficient to obtain a power of 50% or more. In particular, with all settings at their median value, three replicates result in a power of around 80%, whereas only one replicate results in a power of 50% (Fig. 6).
Fig. 6.

Power to detect a twofold change from DMSO based on simulations varying the within-plate variance, the between-plate variance, and the within-plate correlation assumed in the joint model of expression over replicate plates. With the exception of the number of replicate plates, parameter settings were determined by the lower quartile, median, and upper quartile of values observed in the DMSO samples. (A) Lower quartile, (B) median, and (C) upper quartile setting for the within-plate correlation.
Trade-Off Between Factors
Finally, the relative importance of time points, cell lines, and replicates is investigated on a reduced data set, representing 3 replicates of 298 compounds at 2 time points for the top 3 most informative cell lines taken from the LDA (i.e., PC3, VCAP, and A549). The differential gene expression analysis is run on the full data set, and then repeated twice. In a first iteration, two of the three replicates in each {cell line, time} combination are taken at random, followed by a second iteration in which just one replicate is taken at random. The percentage of compounds identified as having at least one gene with a significant FC greater than 2 in at least one {cell line, time} combination when varying the number of time points, number of cell lines, and number of replicates is summarized in Figure 7. Starting from PC3 at 6 h, there is more benefit in adding an extra cell line (i.e., VCAP) than an extra time point, irrespective of the number of replicates that are considered. However, the effect of adding an extra cell line is most clearly noted in the case where there is only one replicate (Fig. 7). Having added VCAP at 6 h, there is relatively more benefit in adding A549 than adding an additional time point, given the number of additional plates that would be required for an additional time point. In the three replicate cases, adding an additional cell line would for instance add an extra 20% for three extra plates, whereas adding an additional time point would add an extra 26% for double the amount of extra plates (Fig. 7C).
Fig. 7.

Compounds identified in (A) one replicate, (B) two replicates, and (C) three replicates analyses, as having at least one gene with significant fold change greater than 2 in at least one cell line: time combination when varying the number of time points and/or the number of cell lines, as a percentage of those identified with all three cell lines and both time points.
Discussion
A between-compound PCA cannot adequately reduce the total compound bioactive space to lower dimensions. Indeed, 222 dimensions would be required to capture only 70% of the total variance. It is, however, not surprising that the dimensionality in the LINCS data set cannot be reduced to a small set of principal components, since the genes that were measured on the L1000 platform were deliberately selected to be nonredundant. Nonetheless, the sum of Euclidean distances over the minimum spanning tree, using the maximum distance-based methodology on a reduced set of 222 principal components, represents an elegant and robust way to summarize the compound diversity. As such, only 2% of the compounds (i.e., 282 compounds) can be regarded as totally redundant, as the diversity reaches 100% when 98% of the compounds are considered in the LINCS data set. It should, however, be mentioned that this analysis does not take the coordinated gene expression effects among the compounds into account. Gene signatures, preferentially cross validated, will do so, and will be able to pick up common patterns between compounds that are transcriptionally diverse.5,19–23 Gene signatures were not used in this study, because the transcriptional diversity of the entire LINCS data set needed to be captured, instead of focusing on a set of signatures that could not be cross validated.
In planning transcriptional compound profiling studies, priority should clearly be given to additional replicates to ensure the analysis is not underpowered, so that true FCs of at least twofold can be frequently detected and so that t-statistics are not inflated when true FC is less than twofold. The simulation-based power analysis indicates that three replicates would give power of around 80% on average, whereas six replicates would give a power greater than 50% in nearly all of the simulated tests. Despite the decrease in accuracy, fewer replicates may well result in similar top compound rankings, which will ultimately be the compounds of interest. However, in the study of trade-off between factors, the correspondence between the one replicate analysis and either the two or three replicates analysis is only around 25% for the top 50 compounds, which is little more than the 17% that would be expected by chance alone. Hence, a single replicate will not only lead to poorly estimated statistics but will also result in unreliable compound rankings. In contrast, the correspondence between the ranked lists of the two and three replicate analyses is nearly always above 75%, with 19 common compounds in the top 20. Thus the two-replicate condition is a good approximation for the three-replicate condition, when the top ranked compounds are considered.
Adding extra cell lines will add more information about the compound-induced biological effects as illustrated by the LDA at 6 and 24 h. The combination of PC3, VCAP, and A549 will cover most of the transcriptional effects within the LINCS data set. The benefit of multiple cell lines is slightly greater than adding an extra time point. However, this cell line effect decreases after three cell lines and becomes marginal at six cell lines. In contrast, the benefit of an additional time point remains fairly constant up to six cell lines. Therefore, it would probably be better to run three cell lines at two time points than to run six cell lines at one time point. Obviously, the choice of PC3, VCAP, and A549 should be viewed in the context of this particular data set. Important cell lines may be missing, whereas others may be overrepresented in the current data. PC3 and VCAP are, for example, two cell lines originating from prostate tissue. In addition, the compound groups that were used to assess the effect of cell lines may hold a bias toward a particular biological effect. Finally, as pointed out by Iorio et al.,22 if a compound shows inconsistent transcriptional effects across different cell lines, its biological effect may be diluted when merging gene expression values from these different cell lines.
The current findings must, however, be interpreted with care and viewed against the context of the current LINCS data set and all of its limitations: (1) the LINCS data were not explicitly collected to address which experimental variables are needed in a genome-wide HTS transcriptional profiling study, (2) all compounds have been profiled at the same high single dose, (3) concentration is not considered as a variable, (4) there is no measure of the relative diversity in the chosen cell line panel and could be highly redundant, (5) the compounds used are almost certainly not typical of those in primary-screening libraries nor the molecularly targeted and (allegedly) highly selective agents that pharma companies chose. Finally, the current analysis does not capture coordinated (i.e., signature-type) effects such as those used by other authors,3–5,19–23 but rather uses the number-of-genes-changing as a crude measure for activity.
Conclusions
In this article, a publicly available LINCS data set from the NIH-funded LINCS program and processed by Genometry, Inc. was used to explore different experimental conditions that can be used to assess compound-induced transcriptional effects. Given the limitations, already outlined, we suggest that two replicates from three cell lines at one time point seem to provide the best experimental conditions for genome-wide HTS transcriptional profiling of thousands of compounds, yielding highest power to do correct transcriptional characterization of the compound-induced transcriptional effects.
Abbreviations Used
- A375
malignant melanoma cell line
- A549
lung carcinoma cell line
- ASC
adipocyte cell line
- DMSO
dimethyl sulfoxide
- FC
fold change
- FTP
file transfer protocol
- GEO
Gene Expression Omnibus
- HA1E
immortalized kidney cell line
- HCC515
lung carcinoma cell line
- HEPG2
hepatocellular carcinoma cell line
- HT29
colon adenocarcinoma cell line
- HTS
high throughput screen
- LDA
linear discriminant analysis
- LINCS
library of integrated network-based cellular signatures
- MCF7
breast adenocarcinoma cell line
- NEU
primary terminally differentiated neuron cells
- NIH
National Institute of Health
- NPC
primary iPS-derived neural progenitor cells
- PC3
prostate adenocarcinoma cell line
- PCA
principal component analysis
- PHH
primary hepatocyte cells
- SKB
skeletal myoblast cells
- VCAP
metastatic prostate cancer cell line
Disclosure Statement
No competing financial interests exist.
References
- 1.Drews J: Drug discovery: A historical perspective. Science 2000;287:1960–1964 [DOI] [PubMed] [Google Scholar]
- 2.Iorio F, Rittman T, Ge H, et al. : Transcriptional data: A new gateway to drug repositioning? Drug Discov Today 2013;18: 350–357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lamb J, Crawford ED, Peck D, et al. : The connectivity map: Using gene-expression signatures to connect small molecules, genes and diseases. Science 2006;313:1929–1935 [DOI] [PubMed] [Google Scholar]
- 4.Lamb J: The connectivity map: A new tool for biomedical research. Nat Rev Cancer 2007;7:54–60 [DOI] [PubMed] [Google Scholar]
- 5.D'Arcy P, Brnjic S, Olofsson MH, et al. : Inhibition of proteasome deubiquitinating activity as a new cancer therapy. Nat Med 2011;17:1636–1640 [DOI] [PubMed] [Google Scholar]
- 6.Finley SD, Chu L-H, Popel AS: Computational systems biology approaches to anti-angiogenic cancer therapeutics. Drug Discov Today 2015;20:187–197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Verbist B, Verheyen GE, Vervoort L, et al. : Integrating high-dimensional transcriptomics and image analysis tools into early safety screening: A proof-of-concept for a new early drug development strategy. Chem Res Toxicol 2015;28:1914–1925 [DOI] [PubMed] [Google Scholar]
- 8.Liu C, Su J, Yang F, Wei K, Ma J, Zhou X: Compound signature detection on LINCS L1000 big data. Mol Biosyst 2015;11:714–722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Peck D, Crawford ED, Ross KN, Stegmaier K, Golub TR, Lamb J: A method for high-throughput gene expression signature analysis. Genome Biol 2006;7:R61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ritchie ME, Phipson B, Wu D, et al. : Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res 2015;43:7:e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Smyth GK: Linear models and empirical Bayes methods for assessing differential expression in microarray experiments. Stat Appl Genet Mol Biol 2004;3:Article3 [DOI] [PubMed] [Google Scholar]
- 12.McCarthy DJ, Smyth GK: Testing significance relative to a fold-change threshold is a TREAT. Bioinformatics 2004;25:765–771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Benjamini Y, Hochberg Y: Controlling the false discovery rate: A practical and powerful approach to multiple testing. JRSS B 1995;57:289–300 [Google Scholar]
- 14.Smyth GK, Michaud J, Scott H: The use of within-array replicate spots for assessing differential expression in microarray experiments. Bioinformatics 2005;21:2067–2075 [DOI] [PubMed] [Google Scholar]
- 15.Within PCA and Between PCA. Available at: https://pbil.univ-lyon1.fr/R/pdf/course4.pdf (Last accessed April17, 2016)
- 16.Lacevic B, Amaldi E: Ectropy of diversity measures for populations in Euclidean space. Inf Sci 2011;181:2316–2339 [Google Scholar]
- 17.Wawer MJ, Li K, Gustafsdottir SM, et al. : Toward performance diverse small-molecule libraries for cell-based phenotypic screening using multiplexed high-dimensional profiling. Proc Natl Acad Sci 2014;111:10911–10916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.R: A language and environment for statistical computing. Available at: www.R-project.org (Last accessed July10, 2014)
- 19.Peng T, Golub RT, Sabatini MD: The immunosuppressant rapamycin mimics a starvation-like signal distinct from amino acid and glucose deprivation. Mol Cell Biol 2002;22:5575–5584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fournier MV, Martin KJ, Kenny PA, et al. : Gene expression signature in organized and growth-arrested mammary acini predicts good outcome in breast cancer. Cancer Res 2006;66:7095–7102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ciuffreda L, Del Bufalo D, Desideri M, et al. : Growth-inhibitory and antiangiogenic activity of the MEK inhibitor PD0325901 in malignant melanoma with or without BRAF mutations. Neoplasia 2009;11:720–731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Iorio F, Bosotti R, Scacheri E, et al. : Discovery of drug mode of action and drug repositioning from transcriptional responses. Proc Natl Acad Sci U S A 2010;107:14621–16626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nigsch F, Hutz J, Cornett B, et al. : Determination of minimal transcriptional signatures of compounds for target prediction. EURASIP J Bioinform Syst Biol 2012;2012:2. [DOI] [PMC free article] [PubMed] [Google Scholar]