

Abstract
Atomoxetine is a selective norepinephrine (NE) reuptake inhibitor approved for the treatment of attention-deficit/hyperactivity disorder (ADHD) in children (≥6 years of age), adolescents, and adults. Its metabolism and disposition are fairly complex, and primarily governed by cytochrome P450 (CYP) 2D6 (CYP2D6), whose protein expression varies substantially from person to person, and by race and ethnicity because of genetic polymorphism. These differences can be substantial, resulting in 8–10-fold differences in atomoxetine exposure between CYP2D6 poor metabolizers and extensive metabolizers. In this review, we have attempted to revisit and analyze all published clinical pharmacokinetic data on atomoxetine inclusive of public access documents from the new drug application submitted to the United States Food and Drug Administration (FDA). The present review focuses on atomoxetine metabolism, disposition, and genetic polymorphisms of CYP2D6 as they specifically relate to atomoxetine, and provides an in-depth discussion of the fundamental pharmacokinetics of the drug including its absorption, distribution, metabolism, and excretion in pediatric and adult populations. Further, a summary of relationships between genetic variants of CYP2D6 and to some degree, CYP2C19, are provided with respect to atomoxetine plasma concentrations, central nervous system (CNS) pharmacokinetics, and associated clinical implications for pharmacotherapy. Lastly, dosage adjustments based on pharmacokinetic principles are discussed.
Introduction
Attention-deficit/hyperactivity disorder (ADHD) is a neuropsychiatric disorder characterized by developmentally inappropriate levels of hyperactive, impulsive, and inattentive behaviors (Warikoo and Faraone 2013). ADHD is the most common behavioral disorder of childhood, with a worldwide prevalence estimated to be between 8% and 10% in school-age children (Polanczyk et al. 2007; National Institute of Mental Health 2015). No single etiology has been identified as the cause of ADHD and no reliable biomarkers are known. Multifactorial hypotheses on its etiology have been advanced, which include prenatal and perinatal risk factors, genetics, psychosocial factors, and neurobiological deficits (Polanczyk et al. 2007; Warikoo and Faraone 2013). Although the pathophysiology of ADHD remains incompletely understood, converging evidence supports an underlying dysregulation of noradrenergic and dopaminergic pathways that are associated with the neurocircuitry of higher cortical functions, including attention and executive functioning (Agster et al. 2011; Del Campo et al. 2011). Abnormalities in monoamines dopamine (DA) and norepinephrine (NE), as well as their metabolism and transport – particularly within the frontal cortex and subcortical neural networks – appear to underlie the disorder. Further, at the neuronal level, the medications with the greatest efficacy in ADHD target central DA and/or NE transporters (NETs) (Agster et al. 2011; Del Campo et al. 2011; Warikoo and Faraone 2013). Taken together, these observations suggest that abnormalities in DA and NE metabolism and turnover, as well as alterations in the expression and function of associated transporters and receptors, are the major determinants of the pathophysiology of ADHD (Agster et al. 2011; Del Campo et al. 2011; Warikoo and Faraone 2013).
Although multimodal treatment approaches to ADHD are advocated, the majority of those diagnosed with the disorder will be prescribed medication. When pharmacotherapy is utilized in the treatment of ADHD, the psychostimulants methylphenidate and amphetamine are generally considered as first-line medications. Although psychostimulants have a lengthy and impressive track record of safety and efficacy, up to 30% of patients treated with one or the other of these agents may experience an inadequate clinical response or treatment-limiting side effects including behavioral changes, appetite suppression, and others (Hodgkins et al. 2012; Warikoo and Faraone 2013). Beyond those inadequately responding to psychostimulants, other patient populations/factors may make the use of psychostimulants less desirable. Appropriate candidates for nonstimulant medications include those with a medical condition that may pose a relative contraindication, or those with comorbid disorders such as Tourette syndrome or tic disorders that may be exacerbated by this drug class, or symptoms such as anxiety or insomnia that may be worsened by psychostimulant use (Cortese et al. 2013; Rizzo et al. 2013; Childress and Sallee 2014). Additionally, in some instances, patients may have caregivers who may have a bias against the use of controlled substances, or otherwise valid concerns over potential misuse or drug diversion by the patient or other members of the household (Childress and Sallee 2014).
The first truly novel nonstimulant medication to receive United States Food and Drug Administration (FDA) approval for ADHD, not previously marketed for another indication (i.e., guanfacine, clonidine), was the selective NE reuptake inhibitor atomoxetine (Strattera®). Initially approved in 2002, atomoxetine has a labeled indication for the treatment of ADHD in children ≥6 years of age as well as adults. To date, atomoxetine remains the only FDA- approved selective NE reuptake inhibitor for the treatment of ADHD, and the only nonstimulant approved for use in adult ADHD.
Atomoxetine (previously referred to as R-tomoxetine), is marketed as the R (-) isomer of the racemic mixture of R- and S-isomers. The R- isomer is an approximately nine-fold more potent inhibitor of the NET relative to the S (+) isomer. As a potent and selective inhibitor of the NET, atomoxetine increases the central nervous system (CNS) availability of intrasynaptic NE (Bymaster et al. 2002). This action is believed to underpin the improvements in higher cognitive functions that are typically impaired in patients with ADHD, and improved with drug therapy (Robbins and Arnsten 2009; Childress and Sallee 2014). Further, atomoxetine specifically increases extracellular levels of DA in the prefrontal cortex but not in the nucleus accumbens or striatum. This is believed to be the result of atomoxetine modulation of cortical synaptic DA uptake via the NET (Tatsumi et al. 1997; Bymaster et al. 2002; Swanson et at. 2006; Robbins and Arnsten 2009). Recently, in vivo investigations utilizing positron emission tomography (PET) imaging assessing clinically relevant doses of atomoxetine in rhesus monkeys, reported the surprising finding that the drug occupied not only the NET (as anticipated), but also the serotonin transporter (Ding et al. 2014). These latter results suggest that the mechanism of therapeutic action of atomoxetine may extend beyond what was previously understood. However, these aspects of the drug's neuropharmacology await further study.
Atomoxetine, a widely recognized cytochrome P450 (CYP) 2D6 (CYP2D6) substrate, has accrued more pharmacogenomic data related to its metabolism and disposition than all other FDA-approved medications for ADHD combined. Further, CYP2D6 is the most intensively investigated isozyme with regard to drug disposition and response because of the large geographical and interethnic differences observed in the genetic polymorphism and its involvement in the metabolism of ∼25% of all marketed drugs (Teh and Bertilsson 2012). CYP2D6 genes are highly polymorphic, with numerous allelic variants identified, which may result in varying degrees of loss (or gain) of the catalytic capacity of the CYP2D6 enzyme, and, accordingly, may alter the disposition of drugs that are CYP2D6 substrates. The frequency of CYP2D6 polymorphisms varies considerably within different racial and ethnic groups. It has been estimated that in the general population, up to 10% of Caucasians, 2% of blacks, and 1% of Asians exhibit CYP2D6 polymorphisms (Teh and Bertilsson 2012). Because autosomal chromosomes are paired, every individual has two alleles, with those of the usual or “normal/wild” type having the phenotypic allelic designation of CYP2D6*1/*1 and are referred to as extensive metabolizers (EMs). However, those individuals with variant alleles (e.g., CYP2D6*4) will not be able to metabolize drugs as efficiently as EMs. Therefore, individuals who have dysfunctional CYP2D6 enzyme function are predisposed to the accumulation of any drug highly dependent on the CYP2D6 pathway, and adverse effects/toxicities associated with higher systemic concentrations of the substrate could ensue. Genetic polymorphisms of CYP2D6 result in four primary phenotypes that can have significant clinical implications. These phenotypes include EM (“normal”), intermediate metabolizer (IM), and poor metabolizer (PM) phenotypes. On the other hand, gene duplication and multiplication can result in the “ultrarapid” metabolizer (UM) phenotype. Most literature reports and reference laboratories agree that the term “PM” should be reserved for individuals who have no active CYP. An in-depth review of CYP2D6 pharmacogenomics is beyond the scope of the present review, but excellent reviews are available elsewhere (Teh and Bertilsson 2012). The present review focuses on atomoxetine metabolism and genetic polymorphisms of CYP2D6. In the following sections, the fundamental absorption, distribution, metabolism, and excretion (ADME) characteristics are discussed in detail followed by an extensive discussion and summary of CYP2D6 polymorphisms and atomoxetine clinical pharmacokinetics.
Absorption
Atomoxetine is formulated as a hydrochloride salt and is available in 10, 18, 25, 40, 60, 80, and 100 mg capsules. Results from in vitro solubility studies indicate that the solubility of atomoxetine over a biologically relevant pH range is all well above the maximum dose strength (100 mg)/250 mL, demonstrating the high solubility of atomoxetine hydrochloride (United States Food and Drug Administration 2002). Although results from specific in vitro/in situ experiments concerning intestinal permeability are not available, two crossover pilot bioavailability studies conducted in healthy subjects showed that the absolute bioavailability of atomoxetine (40 mg oral capsule) in CYP2D6 EMs (n = 20) and PMs (n = 9) were 63% (90% confidence interval [CI] 0.59, 0.67) and 94% (90% CI 0.88, 0.99), respectively (United States Food and Drug Administration 2002). The differences in atomoxetine bioavailability between CYP2D6 EMs and PMs are primarily attributed to the different contributions of hepatic first-pass metabolism in CYP2D6 EM and PM subjects rather than intestinal absorption; therefore, intestinal permeability of atomoxetine in either CYP2D6 EMs or PMs is believed to be high. Following the oral administration of atomoxetine, the median time to maximum plasma concentration (Tmax) of atomoxetine in CYP2D6 EMs and PMs were ∼1.0 h and 2.5 h, respectively, suggesting a rapid absorption rate. A relative bioavailability study found essentially identical oral bioavailability between atomoxetine capsule and solution, hence oral absorption and bioavailability appear to be unaffected by the capsule formulation (United States Food and Drug Administration 2002). Atomoxetine does not appear to be a substrate of the P-glycoprotein (ABCB1) transmembrane transporter, although in vitro studies suggest that it is a moderate to potent inhibitor of the transporter (Zhu et al. 2008).
Tables 1 and 2 are compilations of single- and multiple-dose pharmacokinetic parameters of atomoxetine in healthy adults displaying different polymorphisms of CYP2D6 and/or CYP2C19 across various races/ethnicities, which were gleaned from the available published literature. As shown in Table 1, the area under the plasma concentration-time curve (AUC) extrapolated to infinity (AUCinf) and maximum plasma concentration (Cmax) generally increased proportionally to dose in both Japanese and United States EM subjects (Matsui et al. 2012). Based on the results of a power model analysis, similar dose proportionality relationships of atomoxetine AUC and Cmax over a dose range of 10–120 mg were observed in both populations (Matsui et al. 2012). Additionally, no significant differences in atomoxetine exposures were documented in Japanese and United States EM subjects (Matsui et al. 2012).
Table 1.
| Subject (n) | Dose | Ethnicity/race | AUCinf (μg/h/mL) | Cmax (ng/mL) | tmax (h)c | CL/F (L/h/kg) | Vz/F (L/kg) | t1/2 (h)d |
|---|---|---|---|---|---|---|---|---|
| Healthy CYP2D6 EMs (16) | 10 mg | US American | 0.512 (70) | 84.5 (37) | 1.5 | 0.356 (47) | 1.92 (52) | 4.2 (2.2–7.5) |
| Healthy CYP2D6 EMs (10) | 20 mg | Caucasian | 0.706 (68) | 142.2 (36) | 1.0 | 0.506 (54) | 2.66 (41) | 4.3 (2.4–8.0) |
| Healthy CYP2D6 EMs (15) | 90 mg | US American | 5.47 (72) | 812.5 (30) | 1.0 | 0.289 (42) | 2.22 (52) | 5.6 (3.8–8.6) |
| Healthy CYP2D6 EMs (15) | 120 mg | US American | 7.43 (66) | 1053 (31) | 1.5 | 0.278 (40) | 1.99 (45) | 5.2 (3.7–7.6) |
| Healthy CYP2D6 EMs (22) | 10 mg | Japanese | 0.574 (70) | 110.5 (33) | 1.3 | 0.377 (43) | 1.64 (26) | 3.5 (1.9–6.6) |
| Healthy CYP2D6 EMs (22) | 40 mg | Japanese | 2.51 (69) | 478.4 (34) | 1.0 | 0.347 (47) | 1.83 (34) | 4.1 (2.1–7.1) |
| Healthy CYP2D6 EMs (22) | 90 mg | Japanese | 5.30 (54) | 920 (33) | 1.8 | 0.337 (40) | 1.79 (31) | 4.0 (2.2–7.0) |
| Healthy CYP2D6 EMs (22) | 120 mg | Japanese | 6.43 (38) | 1086 (31) | 1.0 | 0.348 (39) | 2.06 (32) | 4.3 (2.9–6.2) |
| Healthy CYP2D6 EMs (16) | 40 mg | Chinese | 3.63 (48) | 449 (32) | 1.3 | 0.241 (63) | NA | 4.2 (2.4–5.9) |
| Healthy (21 CYP2D6 EMs and 1 PM) | 40 mg | Chinese | 2.69 (99) | 415 (41) | 1.1 | NA | NA | 3.9 ± 3.3e |
| Healthy CYP2D6 EMs, CYP2C19 EMs (14) | 40 mg | Korean | 0.910 (13) | 221.5 (19) | NA | 0.669 (18) | NA | 2.7 (2.5–2.9) |
| Healthy CYP2D6 EMs, CYP2C19 IMs (14) | 40 mg | Korean | 1.08 (19) | 269.4 (27) | NA | 0.603 (16) | NA | 2.9 (2.5–3.2) |
| Healthy CYP2D6 EMs, CYP2C19 PMs (12) | 40 mg | Korean | 1.63 (25) | 386.1 (18) | NA | 0.406 (24) | NA | 3.6 (3.0–4.1) |
References: Chalon et al. 2003; Cui et al. 2007; Matsui et al. 2012; Shang et al. 2013; and Choi et al. 2014.
Data are expressed as mean (% coefficient of variance), unless noted.
Median value.
Median (minimum-maximum).
Mean ± standard deviation.
AUCinf, area under the plasma concentration-time curve from zero to infinite; CL/F apparent plasma clearance; Cmax, maximum plasma concentration, CYP, cytochrome P450; EM, extensive metabolizer; IM, intermediate metabolizer; PM, poor metabolizer; t1/2, half-life; tmax, time to maximum plasma concentration; Vz/F, apparent volume of distribution during the terminal phase.
Table 2.
| Subject (n) | Dose & dosing regimen | Ethnicity/race | Cmax,ss (ng/mL) | Cmin,ss (ng/mL) | Cavg,ss (ng/mL) | AUCτ,ss (μg/h/mL) | CLss/F (L/h/kg) | Vz/F (L/kg) | t1/2 (h)c |
|---|---|---|---|---|---|---|---|---|---|
| Healthy CYP2D6 EMs (21) | 20 mg BID | US American | 184 (36) | NA | NA | 0.846 (45) | 0.395 (55) | 2.20 (50) | 4.0 (2.9–7.2) |
| Healthy CYP2D6 EMs (4) | 20 mg BID | US American | 160 (52) | 36.1 (116) | 89.6 (64) | 1.08 (64) | 0.373 (75) | 2.33 (51) | 5.3 (3.7–9.1) |
| Healthy CYP2D6 EMs (6) | 40 mg BID | US American | 552 (45) | 105 (138) | 265 (85) | 3.18 (85) | 0.327 (73) | 1.48 (55) | NA |
| Healthy CYP2D6 EMs (8) | 40 mg BID | US American | 527 (67) | 69.5 (134) | 216 (78) | 2.59 (78) | 0.298 (53) | NA | NA |
| Healthy CYP2D6 EMs (15) | 60 mg BID | US American | 591 (46) | 56.6 (46) | 224 (57) | 2.69 (57) | 0.399 (62) | 1.76 (45) | NA |
| Healthy CYP2D6 EMs (10) | 40 mg BID | Japanese | 605 (35) | 34.6 (95) | 206 (42) | 2.47 (42) | 0.321 (50) | NA | NA |
| Healthy CYP2D6 EMs (9) | 60 mg BID | Japanese | 874 (26) | 59.1 (87) | 311 (41) | 3.73 (42) | 0.292 (41) | NA | NA |
| Healthy CYP2D6 EMs (16) | 80 mg QD | Chinese | 1020 (33) | NA | 297 (48) | 7.12 (48) | 0.242 (59) | NA | 4.3 (2.2–6.4) |
| Healthy CYP2D6 PMs (3) | 20 mg BID | US American | 915 (31) | 503 (30) | 704 (27) | 8.44 (27) | 0.0375 (26) | 1.06 (43) | 20 (17–25) |
| Healthy CYP2D6 PMs (6) | 60 mg BID | US American | 2610 (18) | 1353 (26) | 1946 (19) | 23.4 (19) | 0.042 (45) | NA | NA |
References: Belle et al. 2002; Sauer et al. 2003; Sauer et al. 2004; Cui et al. 2007; and Matsui et al. 2012.
Data are expressed as mean (% coefficient of variance), unless noted.
Median (minimum-maximum).
AUCτ,ss, area under the plasma concentration-time curve during a dosage interval (τ) at steady state; BID, twice daily, CLss/F, apparent plasma clearance at steady-state; Cav,ss, average steady-state plasma concentration; Cmax,ss, maximum steady-state plasma concentration; Cmin,ss, minimum steady-state plasma concentration; CYP, cytochrome P450; EM, extensive metabolizer; IM, intermediate metabolizer; PM, poor metabolizer; QD, once daily; t1/2, half-life; Vz/F, apparent volume of distribution during the terminal phase.
Recognizing the pH-dependent solubility of atomoxetine, pharmacokinetic drug–drug interaction (DDI) studies have been conducted to assess the potential for acid-suppressing agents to influence the disposition of atomoxetine. In theory, gastric acid-associated DDIs could alter oral absorption and exposure of drugs with pH-dependent solubility, resulting in loss of clinical benefit (Yu et al. 2014b; Zhang et al. 2014). Based on the results of a crossover clinical study (United States Food and Drug Administration 2002), the AUCinf and Cmax of atomoxetine were unchanged when coadministered with either the over-the-counter (OTC) antacid Maalox® (aluminum /magnesium hydroxide) or the proton pump inhibitor omeprazole, suggesting that gastric acid-related DDIs are of little concern. Consequently, concurrent administration of drugs that suppress gastric acid secretion (e.g., omeprazole) or gastric acid neutralizers (e.g., aluminum /magnesium hydroxides) and histamine-2 receptor antagonists (e.g., famotidine) is inconsequential to atomoxetine pharmacokinetics. Because such DDIs are mediated via the physiological environment of the gastrointestinal tract, such as gastrointestinal pH (Zhang et al. 2014), the results can reasonably extrapolate to CYP2D6 PM subjects as well. Additionally, in view of the similarities in gastric and small intestinal pH between school-age children and adults from a developmental perspective (Yu et al. 2014a), the recommendations are also suitable for the intended pediatric population.
Distribution and CNS Pharmacokinetics
Knowledge pertinent to distribution of drugs within human tissues and fluids can improve the understanding of pharmacological and toxicological responses relative to exposure. However, there is usually a lack of such information provided for most marketed drugs. In this regard, plasma pharmacokinetic data gained from intravenous (i.v.) dosing studies are highly valuable in separating the processes of absorption from that of tissue distribution and elimination, thereby providing insight into in vivo distribution. The volume of distribution (Vd) of atomoxetine at steady-state in humans following i.v. administration is estimated to be 0.85 L/kg, which is only slightly larger than the volume of total body water (Christman et al. 2004). On the basis of the i.v. data, in vivo distribution of atomoxetine is thought to be principally into total body water.
There has been interest in therapeutic monitoring of ADHD therapeutic agents in saliva as a less invasive means of monitoring drug therapy. A study performed in pediatric patients with ADHD (7–16 years of age) who were treated daily with typical therapeutic doses (0.26–1.79 mg/kg/day) of atomoxetine measured the parent drug and its primary phase I metabolite, 4-hydroxyatomoxetine in collected saliva samples. Atomoxetine concentrations in saliva were significantly lower than those in plasma, and the N-desmethylatomoxetine metabolite was not detected (Papaseit et al. 2013). The saliva-to-plasma concentration ratio of atomoxetine and 4-hydroxyatomoxetine were time dependent, with a range of 0.01–0.12 for atomoxetine and 0.16–1.96 for 4-hydroxyatomoxetine, for which the minimum occurs at 1 and 2 hours postadministration, respectively. Peak concentrations (Cmax) of atomoxetine in saliva occurred 3 hours after oral administration, whereas the Tmax in plasma occurred between 1 and 2 hours postdosing (Papaseit et al. 2013). However, CYP2D6 genotyping was not performed in this study, and the potential influence of CYP2D6 polymorphisms on atomoxetine and 4-hydroxyatomoxetine concentrations in saliva cannot be discounted.
In the case of one subject receiving the lowest oral dose (0.26 mg/kg/day or 18 mg/day), the atomoxetine AUC in both plasma and saliva were greater than those observed in subjects treated with the highest dose (1.42–1.79 mg/kg/day or 60 mg/day) (Papaseit et al. 2013). Additionally, the subject had the lowest AUC and Cmax of 4-hydroxyatomoxetine in plasma and an ∼10-fold higher plasma concentration of N-desmethylatomoxetine than study peers, whereas 4-hydroxyatomoxetine concentrations in saliva fell within the range of those observed in the other individuals (Papaseit et al. 2013). These subject-specific observations suggest that compromised CYP2D6 enzymatic activity may be caused by genetic variation, metabolic inhibition, or both. Although study correlations between plasma and saliva concentrations of atomoxetine and 4-hydroxyatomoxetine were found to be statistically significant, saliva concentrations of atomoxetine and 4-hydroxyatomoxetine appeared to be too low and variable to adequately serve as noninvasive surrogates for plasma concentrations when therapeutic drug monitoring is desired.
Sufficient concentrations of the unbound (free) drug at the target site over a sufficient time are crucial in linking plasma pharmacokinetics to pharmacodynamics (Mouton et al. 2008; Mariappan et al. 2013; Wagner et al. 2015). Because atomoxetine is a potent and selective inhibitor of the presynaptic NET associated with noradrenergic nerve endings within the CNS, adequate penetration of atomoxetine into the CNS is crucial. Cerebrospinal fluid (CSF) is the only accessible fluid in the human CNS, and because of a lack of protein binding, CSF drug concentrations are acknowledged to be the best estimate for unbound concentrations in the brain extracellular fluid (ECF) to assess drug exposure at the site of action (De Lange 2013; De Lange and Hammarlund-Udenaes 2015). The concentrations of atomoxetine in the CSF under steady-state conditions were measured in healthy EM males, together with plasma concentrations (Kielbasa et al. 2015). In the study, atomoxetine was initiated at 40 mg once daily for 3 days, and subsequently increased to 80 mg once daily for another 11 days. The CSF samples obtained at 8 hours (n = 3) and 24 hours (n = 3) postdosing after 14 days of exposure were collected by lumbar puncture to determine atomoxetine concentrations. The mean (coefficient of variation [CV]) plasma AUC and Cmax at steady-state was 5.19 (104%) μg/h/mL and 945 (77%) ng/mL (n = 6). The mean concentration of atomoxetine in the human CSF was 6.6 ng/mL at 8 hours and 1.4 ng/mL at 24 hours, with a large CV of 95% and 107%, respectively (Kielbasa et al. 2015). Interestingly, Kielbasa and colleagues concluded that this high variability in human CSF concentrations was likely related to variable plasma exposure of atomoxetine rather than CYP2D6 genotype, because CYP2D6 PM subjects had been excluded from this study (Kielbasa et al. 2015). Unfortunately, specific information on CYP2D6 genotype was unavailable in this report. Given that CYP2D6 non-PM phenotypes include IM, EM, and UM, the potential influence of CYP2D6 status on the observed variability in the CSF and plasma concentrations cannot be ruled out.
Overall, the study demonstrated that atomoxetine appeared in human CSF, and, therefore, can reach the human CNS directly via the blood–CSF fluid barrier (BCSFB) or/and indirectly via the blood–brain barrier (BBB).
The CNS pharmacokinetics of atomoxetine were also investigated in rats by means of microdialysis (Kielbasa et al. 2009), followed by translational pharmacokinetic modeling (Kielbasa and Stratford 2012). The extent of transport across CNS barriers at steady state is governed by the relative contribution of passive diffusion, and active influx and efflux. The BBB equilibrium can be delineated by the brain ECF to plasma unbound drug concentrations under steady-state conditions (CECF/CuP); similarly, the BCSFB equilibrium can be depicted by the ratio of the steady-state unbound drug concentrations in CSF to those in plasma (CCSF/CuP) (Hammarlund-Udenaes et al. 2008; De Lange and Hammarlund-Udenaes 2015). The steady-state CECF/CuP in rats was estimated to be 0.7 ± 0.4 (n = 18), which was close to unity (Kielbasa et al. 2009), suggesting that the BBB transport of atomoxetine is predominantly passive rather than active. This is consistent with a previous report documenting that atomoxetine is not a substrate of P-glycoprotein, an important efflux transporter localized in the BBB (Zhu et al. 2008). Similarly, the steady-state CCSF/CuP in rats was 1.7 ± 0.8 (n = 25), and also approached unity (Kielbasa et al. 2009), implying that passive diffusion rather than an active processes is also the primary mechanism in atomoxetine transport across the BCSFB. Further, unbound atomoxetine distributing into the rat CNS has been well characterized by a physiologically based pharmacokinetic (PBPK) model (Kielbasa and Stratford 2012). The PBPK model consisted of a systemic (plasma) compartment and three CNS compartments (i.e., the CSF, the brain ECF, and the brain cells). In the CNS compartments, the volume of the CSF, the brain ECF, and the brain cells approximated actual physiological values for these volumes from the published literature, but mass exchanges between these compartments (defined as the intercompartment clearances) were obtained by model fitting using the observed concentration data in individual rats.
The PBPK model was then applied to predict atomoxetine pharmacokinetics in the human CNS, by translating the physiological volumes to human and scaling the clearances utilizing established allometric relationships, and based upon differences in brain weight between rats and humans (Kielbasa and Stratford 2012). It is of note that in the PBPK model for humans, the clinical pharmacokinetic parameters of atomoxetine were also incorporated to simultaneously describe the plasma pharmacokinetics in humans, which were taken from CYP2D6 EM subjects. Ultimately, the simulated pharmacokinetic profile of atomoxetine in the human brain ECF after oral administration of 40 and 80 mg of atomoxetine was compared with the in vitro human NE transporter inhibition constant (hNET Ki = 1.1 ng/mL). For 40 mg of atomoxetine, the predicted median unbound Cmax in the human ECF was approximately two-fold higher than the hNET Ki, and the predicted median ECF concentrations were 1.1 at 16 hours and 0.74 ng/mL at 24 hours postdose. For 80 mg of atomoxetine, the simulated median unbound concentration in the ECF was approximately four-fold higher than the hNET Ki at 1 ∼ 2 hours, and ∼1.5 ng/mL at 24 hours postdose, which was slightly greater than the hNET Ki. The results of these simulations suggested that with clinical dosing regimens of 40 and 80 mg (once daily), unbound atomoxetine in the ECF in a representative CYP2D6 EM population might be expected to achieve or at least approach the requisite concentration to produce sufficient NE transporter inhibition (the hNET Ki) needed for clinical effects. Nonetheless, relevant simulations for CYP2D6 UM subjects are not available, which could potentially address concerns that have been raised that currently recommended dosing regimens may not be adequate for CYP2D6 UM individuals to achieve full therapeutic benefit (see de Leon 2015). Future simulation studies may be of value in assessing the impact of genetic variation on atomoxetine CNS pharmacokinetics and pharmacodynamics.
Unbound Fraction in Plasma
Because of the inherent inaccessibility of physiological fluids surrounding the therapeutic target site in a clinical setting, plasma unbound drug concentrations are commonly used as surrogate measures to gain insight into unbound drug concentrations in the site of action (i.e., the CNS), based on the assumption that only the unbound drug can permeate from the vascular space into the tissue (Mariappan et al. 2013). The unbound fraction of atomoxetine in human plasma can be assessed in vitro via protein binding assay. Binding of atomoxetine to human plasma protein in vitro was 98.7%, being predominantly bound to albumin (97.5%), with lesser binding to α1-acid glycoprotein and immunoglobulin G (Sauer et al. 2003). Human plasma protein binding of the two metabolites, N-desmethylatomoxetine and 4-hydroxyatomoxetine, were 99.1% and 66.6%, respectively (Sauer et al. 2003). Plasma protein binding of atomoxetine and its metabolites has been assessed independently using tested concentrations of 150–3000 ng/mL for atomoxetine and N-desmethylatomoxetine and 15–1500 ng/mL for 4-hydroxyatomoxetine) (United States Food and Drug Administration 2002), indicating that unbound fractions of atomoxetine and its metabolites in human plasma are constant over the likely therapeutic ranges, and that these unbound fractions are reliable for estimating plasma unbound concentrations. High plasma protein binding of atomoxetine has raised some concerns that DDIs could potentially result in increases in unbound concentrations in plasma via protein displacement reactions. In vitro protein binding interaction studies have shown that the plasma protein binding of atomoxetine in humans was only reduced in the presence of acetylsalicylic acid at what are considered toxic concentrations (>300 μg/mL), leading to a nearly three-fold increase in the unbound fraction of atomoxetine (United States Food and Drug Administration 2002). However, given the high concentrations of acetylsalicylic acid that were evaluated, the actual clinical significance of this observation is questionable.
Benet and Hoener (2002) have proposed that for all drugs given orally and eliminated predominately by hepatic metabolism, the plasma unbound AUC is independent of plasma protein binding, hence no dosing modifications will be necessary for actual or anticipated changes in the plasma unbound fraction. Regardless of the presence of CYP2D6 genetic polymorphisms, the elimination of atomoxetine is dominated by hepatic clearance (see Metabolism section), and protein-binding displacement DDIs do not appear to be of clinical significance.
Metabolism and Hepatic Extraction
Hepatic metabolism often plays a central role in drug disposition and might be a key to understanding interindividual variability in pharmacokinetics and pharmacodynamics (Markowitz 2013). The biotransformation of atomoxetine has been studied in humans (Sauer et al. 2003), animal models (Mattiuz et al. 2003), and in vitro human liver systems (Ring et al. 2002), which have been previously reviewed (Christman et al. 2004; Sauer et al. 2005). Aromatic ring hydroxylation, benzylic oxidation, and N-demethylation are the three major phase I metabolic pathways governing the biotransformation of atomoxetine in humans (Sauer et al. 2003). The O-glucuronidation of the hydroxylated metabolites appears to be the only major phase II metabolic pathway involved in atomoxetine biotransformation (Fig. 1) (Sauer et al. 2003). CYP2D6 was identified to be the primary enzyme responsible for the formation of 4-hydroxyatomoxetine, which is the major phase I metabolite not only in CYP2D6 EMs but also in PMs, produced via aromatic ring hydroxylation, whereas a variety of other CYP enzymes are capable of catalyzing the conversion of atomoxetine to 4-hydroxyatomoxetine in the absence of CYP2D6, albeit at a considerably slower rate (Ring et al. 2002). 4-hydroxyatomoxetine is subsequently glucuronidated to form the major urinary metabolite 4-hydroxyatomoxetine-O-glucuronide, irrespective of CYP2D6 polymorphisms (Sauer et al. 2003). N-desmethylatomoxetine is a minor metabolite whose formation is mediated by CYP2C19, and undergoes further hydroxylation via CYP2D6 followed by subsequent O-glucuronidation (Ring et al. 2002; Sauer et al. 2003). Although 4-hydroxyatomoxetine has similar pharmacological activity to the parent compound in terms of NE transporter inhibition, the inhibitory activity of the N-desmethylatomoxetine metabolite is nearly 20-fold less than that of atomoxetine (United States Food and Drug Administration 2002). In addition, on the basis of the qualitative metabolic profiles in CYP2D6 EMs and PMs, the same metabolites were observed regardless of phenotype, indicating that no CYP2D6 phenotype-specific metabolites are formed (Sauer et al. 2003).
FIG. 1.

Metabolism of atomoxetine to its major metabolites in humans.
Although the biotransformation of atomoxetine is generally well described in the available literature, relatively little is known regarding its hepatic clearance and first-pass extraction, which can often be correlated with changes in pathology and physiology and may ultimately determine oral bioavailability (Rowland et al. 1973; Benet 2010). As mentioned, atomoxetine displayed linear pharmacokinetic characteristics at therapeutic doses and the oral bioavailability of atomoxetine in CYP2D6 EMs and PMs were 63% (90% CI: 0.59, 0.67) and 94% (90% CI: 0.88, 0.99), respectively. Oral bioavailability (F), defined as the fraction of an oral dose that reaches the systemic circulation, can be considered as the continuous product of the fraction absorbed (fa), intestinal availability (FG), and hepatic availability (FH) (Fig. 2), that is, fa × FG × FH. Further, the FG, and FH are the fraction of drug not metabolized by the gut wall and liver, respectively, which is directly determined by the extraction ratio of the gut (EG) and liver (EH); that is, FG = (1-EG), and FH = (1-EH) (Fig. 2). In contrast to FG and FH, fa is not associated with drug-metabolizing enzymes (Lin 1995; Lin and Lu 1997; Lin 1998).
FIG. 2.
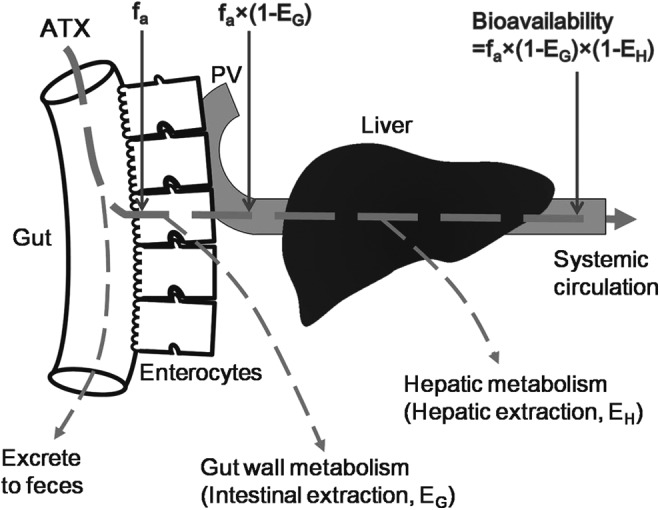
Depiction of the absorption and metabolism of atomoxetine prior to reaching the systemic circulation. ATX, atomoxetine; fa, the fraction of dose absorbed from the intestinal lumen; PV, portal vein.
As shown in the FDA's clinical pharmacology review document (United States Food and Drug Administration 2002), atomoxetine has low to moderate pharmacokinetic variability in both CYP2D6 EMs and PMs. Further, a greater variability in CYP2D6 EMs is observed (relative to PMs), which has primarily been attributed to metabolic differences in EM subpopulations on the basis of the number of wild type alleles: UMs (multiple wild type alleles), homozygous (two wild type alleles), and heterozygous (one wild type allele) (United States Food and Drug Administration 2002). Therefore, it is a reasonable assumption that there is no significant intersubject variability in the fa of atomoxetine, and that the product of intestinal availability and hepatic availability of atomoxetine in CYP2D6 PMs is ∼1.49-fold that observed in EMs; that is,(1-EG,PMs) × (1-EH,PMs)≈1.49 × (1-EG,EMs) × (1-EH,EMs). Unlike the situation with CYP3A, the expression and enzymatic activity of CYP2D6 in the human gut is quite limited (< 8% of median hepatic content and <1% of total immunoquantified P450). Therefore, the relative contribution of the human intestine to the presystemic metabolism of CYP2D6 substrates is substantially lower than that of the human liver (Lin et al. 1999; Lalovic et al. 2004; Paine et al. 2006). Accordingly, as a major substrate of CYP2D6, the intestinal availability of atomoxetine in CYP2D6 PMs and EMs is comparable, and the average ratio of intestinal availability in PMs to EMs approaches 1; that is, (1-EG,PMs)/(1-EG,EMs)≈1, then the average ratio of hepatic availability in PMs to EMs (i.e.,[1-EH,PMs]/[1-EH,EMs]) approaches 1.49. In another words, the differences in oral bioavailability of atomoxetine between PMs and EMs are proportional to the respective contributions of hepatic extraction in PMs and EMs.
On the basis of mass balance relationships, EH can be calculated by dividing hepatic blood clearance (CLH,B) by hepatic blood flow (QH,B); that is, EH = CLH,B/QH,B. A radiolabeled pharmacokinetic study of a 20 mg dose of orally administered 14C-atomoxetine in humans showed that >80% of the radiolabeled dose was recovered in the urine as metabolites, with <3% recovered as unchanged parent compound, demonstrating that atomoxetine is eliminated from the systemic circulation primarily through extensive metabolism, with minimal renal clearance (Sauer et al. 2003). Considering that negligible extrahepatic clearance occurs, the hepatic plasma clearance (CLH,P) of atomoxetine can be approximated by the total plasma clearance (CLiv,P) of atomoxetine after i.v. administration, which were reported to be 0.22 and 0.034 L/h/kg following an i.v. dose of 20 mg in adult CYP2D6 EMs and PMs, respectively, in the FDA clinical pharmacology review (United States Food and Drug Administration 2002). However, if hepatic blood clearance (CLH,B) is to be estimated by CLH,P, then the blood-to-plasma concentration ratio (CB/CP) must be taken into account. Because only very limited amounts of atomoxetine partition into human blood cells (Sauer et al. 2005), the CB/CP of atomoxetine approaches a minimum, that is, (1-hematocrit), which is ∼55% (hematocrit of 45% for a typical healthy adult [Li et al. 2012]). Hence, the average hepatic extraction in CYP2D6 EMs and PMs is estimated to be ∼35% and 5% (assuming hepatic blood flow of 81 L/h for a typical 70 kg human [Rowland and Tozer 1995]), which is in good agreement with the magnitude of the ratio of 1-EH,PMs to 1-EH,EMs and the value of atomoxetine bioavailability in CYP2D6 EMs and PMs.
This also confirms the minimal contribution of intestinal metabolism in the first-pass extraction of atomoxetine. On the other hand, because atomoxetine has an intermediate extraction ratio (between 0.3 and 0.7) in CYP2D6 EMs and a low extraction ratio (< 0.3) in PMs, on the basis of clearance concepts, the hepatic blood clearance in EMs is related to enzyme activity, unbound fraction in blood, and hepatic blood flow. In CYP2D6 PMs, however, hepatic blood clearance depends only on enzyme activity and the unbound fraction in blood. Consequently, genetic polymorphisms of genes encoding for CYP2D6 can influence catalytic activity, and ultimately, atomoxetine metabolism and disposition.
Role of CYP Genetic Polymorphisms
The pivotal role of CYP2D6 and the effect of genetic polymorphisms were observed in both the steady-state and single-dose pharmacokinetics of atomoxetine. A within-study comparison showed that the average steady-state oral clearance (CLss/F) of atomoxetine in CYP2D6 EMs (0.373 L/h/kg) is ∼10-fold greater than that of PMs (0.0357 L/h/kg), confirming the predominant role of CYP2D6 in atomoxetine clearance and first-pass metabolism (Sauer et al. 2003). The significant difference in atomoxetine clearance between CYP2D6 EMs and PMs results in markedly different peak and systemic atomoxetine exposures in these subjects of differing phenotypes. As shown in the same study (Sauer et al. 2003), steady-state AUC (AUCτ,ss), maximal concentration at steady state (Cmax,ss), and average steady-state concentration (Cavg,ss) of atomoxetine in CYP2D6 PMs are approximately eight-fold, six-fold, and eight-fold higher, respectively, than those in EMs. Similarly, the mean Cmax and AUCinf of atomoxetine following single-dose oral administration of a 40 mg capsule were 326 ng/mL and 1.80 μg/h/mL in CYP2D6 EMs, which were both substantially less than those in CYP2D6 PMs (564 ng/mL and 14.5 μg/h/mL) (United States Food and Drug Administration 2002). Additionally, in Chinese subjects, the AUCinf in CYP2D6 PMs was also eight-fold greater than that of EMs (Shang et al. 2013). Importantly, a post-hoc data analysis based on a pooled data set from two phase III trials in children and adolescents with ADHD (6–14 years of age, weight 20–70 kg) found that on average, pediatric CYP2D6 PMs had an 8.3-fold higher systemic exposure (based on predicted AUC) of atomoxetine than EMs between weeks 8 through 10 of atomoxetine treatment (Fig. 3). However, neither the predictive methods nor the actual input data used to generate these predictions were provided in that report (Trzepacz et al. 2008). Several studies have directly compared the total and peak atomoxetine exposures in homozygous CYP2D6*10 subjects with other CYP2D6 EM subjects (Cui et al. 2007; Matsui et al. 2012; Byeon et al. 2015). As presented within Table 3, an approximately two-fold higher AUC was observed in the homozygous CYP2D6*10 Japanese subjects than in the CYP2D6*1/*1, *1/*2, *1/*10 and *2/*10 subjects (Matsui et al. 2012). The oral clearance of the homozygous CYP2D6*10 Japanese subjects were significantly lower than those of the CYP2D6*1/*1, *1/*2, *1/*10 and *2/*10 subjects, and were clearly distinguished from PMs, with the most profound effects noted on clearance in homozygous CYP2D6*10 subjects relative to CYP2D6*1/*1 and *1/*2 genotypes (Matsui et al. 2012). In Chinese subjects, homozygous CYP2D6*10 subjects had, on average, a two-fold higher systemic exposure than the heterozygous CYP2D6*10 and the homozygous CYP2D6*1 subjects (Table 3), as a result of 50% reduction in oral clearance (Cui et al. 2007). Likewise, systemic atomoxetine exposure in Korean CYP2D6*10/*10 individuals was approximately threefold greater than that observed in Korean CYP2D6*wt/*wt (*wt = *1 or *2) and *wt/*10 subjects (Byeon et al. 2015).
FIG. 3.

Predicted plasma atomoxetine exposures in cytochrome P450 2D6 (CYP2D6) extensive and poor metabolizers by week on treatment. Systemic exposure in poor metabolizers was more than eight-fold higher than in extensive metabolizers between weeks 8 and 10. AUC, area under the curve (Trzepacz et al. 2008, reprinted with permission).
Table 3.
Comparison of Atomoxetine Exposures in the Homozygous CYP2D6*10 Subjects with Other CYP2D6 EM Subjects
| AUCinf (μg/h/mL) | Cmax (ng/mL) | ||||||
|---|---|---|---|---|---|---|---|
| Ethnicity | Dose & dosing regimen | Genotype | n | Geometric mean | Ratio (90% CI) | Geometric mean | Ratio (90% CI) |
| Japanese (Matsui et al. 2012) | 10 mg single | *10/*10 | 4 | 0.727 | 125.1 | ||
| *1/*10 or *2/*10 | 5 | 0.406 | 1.79 (1.31, 2.45) | 93.5 | 1.34 (1.03, 1.73) | ||
| *1/*1 or *1/*2 | 5 | 0.331 | 2.20 (1.61, 3.00) | 86.5 | 1.45 (1.12, 1.87) | ||
| 120 mg single | *10/*10 | 4 | 9.83 | 1270.8 | |||
| *1/*10 or *2/*10 | 4 | 5.51 | 1.79 (1.25, 2.56) | 1032.1 | 1.23 (0.909, 1.67) | ||
| *1/*1 or *1/*2 | 5 | 4.69 | 2.10 (1.49, 2.95) | 841.3 | 1.51 (1.13, 2.01) | ||
| Chinese (Cui et al. 2007) | 40 mg single | *10/*10 | 7 | 4.962 | 530 | ||
| other EMs | 9 | 2.242 | 2.21 (1.53, 3.21) | 360 | 1.47 (1.15, 1.88) | ||
| 80 mg QD | *10/*10 | 7 | 9.693a | 1199b | |||
| other EMs | 9 | 4.427a | 2.19 (1.53, 3.13) | 815b | 1.47 (1.13, 1.92) | ||
AUCτ,ss(area under the plasma concentration-time curve during a dosage interval at steady state) instead of AUCinf.
Cmax,ss(maximum steady-state plasma concentration) instead of Cmax.
AUCinf, area under the plasma concentration-time curve from zero to infinite; CI, confidence interval; Cmax, maximum plasma concentration; EM, extensive metabolizer; QD, once daily.
Atomoxetine circulates principally in the plasma of CYP2D6 EMs as the parent drug and 4-hydroxyatomoxetine-O-glucuronide metabolite; in PMs, however, the most abundant species are the parent compound and the N-desmethylatomoxetine metabolite. In adult CYP2D6 EM subjects, after the oral administration of 20 mg twice daily for 5 days, the exposure to 4-hydroxyatomoxetine is only ∼1% (based on Css,max) of that of atomoxetine, whereas the exposure to N-desmethylatomoxetine is ∼5% (based on Css,max and AUCτ) of that of atomoxetine (Sauer et al. 2003). After oral administration of 75 mg of atomoxetine to adult CYP2D6 PMs twice daily over 5 days, the exposure to 4-hydroxyatomoxetine was just 0.1% of that of atomoxetine, whereas the exposure to N-desmethylatomoxetine was ∼45% of that of atomoxetine (United States Food and Drug Administration 2002). Collectively, exposure to 4-hydroxyatomoxetine appears to range from 0.1% of the parent drug in CYP2D6 PMs to 1% of the parent drug in EMs; however, exposure to N-desmethylatomoxetine ranges from 5% of the parent compound in CYP2D6 EMs to 45% of the parent drug in PMs. 4-hydroxyatomoxetine-O-glucuronide plasma concentrations in CYP2D6 EMs are 2.6-fold greater than that of atomoxetine, and >40-fold greater than that of N-desmethylatomoxetine, but the concentrations of the glucuronide metabolite in PMs are substantially less than either the parent or N-desmethylatomoxetine (Sauer et al. 2003).
The contribution of the CYP2C19 metabolic pathway to atomoxetine clearance is relatively minor (Fig. 1), with the CYP2C19 mediated formation of N-desmethylatomoxetine accounting for only 3–6% of the total dose in CYP2D6 EMs and PMs (Sauer et al. 2003). Over the past decade of research in the area, the CYP2C19 metabolic pathway has not been considered to have a remarkable effect on the overall pharmacokinetics of atomoxetine (Sauer et al. 2005). Nevertheless, a recent study suggested the clearance and exposure to atomoxetine may be significantly influenced by the genotype and phenotype of CYP2C19 (Choi et al. 2014). To avoid the influence of CYP2D6 genetic polymorphisms in this investigation, all volunteers who carried the CYP2D6*1/*10 genotype (classified as CYP2D6 EMs) were included for further study of the effect of CYP2C19 status. Forty healthy male Korean subjects carrying the CYP2C19*1/*1 (EM), CYP2C19*1/*2 or *1/*3 (IM), or CYP2C19*2/*2, *2/*3 or *3/*3 (PM) genotype were selected to participate in the study. As summarized in Table 1, CYP2C19 PMs appeared to have statistically significantly higher exposures and lower oral clearance than either EMs or IMs. Further, the exposures to 4-hydroxyatomoxetine in CYP2C19 PMs were significantly greater than those in EMs and IMs, whereas the exposures to N-desmethylatomoxetine were significantly lower in CYP2C19 PMs than in EMs and PMs. These results lend support to the hypothesis that reduced CYP2C19 enzymatic expression in CYP2C19*2 or *3 allele carriers led to impaired metabolic clearance of atomoxetine, thereby increasing overall atomoxetine exposure and decreasing exposure to the CYP2C19 mediated metabolite, N-desmethylatomoxetine. The increases in 4-hydroxyatomoxetine exposures in CYP2C19 PMs are thought to be the consequence of enhanced 4-hydroxyatomoxetine-forming pathways in compensation for the overall reduction in atomoxetine clearance pathways (Choi et al. 2014).
Excretion
Secondary to extensive phase I and phase II metabolism, the largest fraction (84% in CYP2D6 EMs and 31% in PMs) of atomoxetine dose is excreted into the urine principally as 4-hydroxyatomoxetine-O-glucuronide (Sauer et al. 2003). Less than 3% of the administered dose is excreted into the urine as unchanged parent, suggesting a minimal role of renal excretion in the overall clearance of the drug. The cumulative fraction of an orally administered radioactive dose of atomoxetine excreted in urine is ∼96% in EMs and 80% in PMs. The cumulative fraction of radioactive dose excreted via feces is ∼2% in CYP2D6 EMs and 17% in PMs. In CYP2D6 EMs, almost the entire radiolabeled dose was cleared from the body within 48 hours, but in PMs, the entire dose was not eliminated until ∼144 hours postdosing (Sauer et al. 2003). At the 24 hour postdose time point in which almost 90% of the radiolabeled dose had been cleared in CYP2D6 EMs, only 30% of the dose was excreted in PMs (Sauer et al. 2003).
Population Pharmacokinetics
Population pharmacokinetics is the study of the sources and correlates of variability in drug concentrations among individuals who are the actual target patient population receiving the medication of interest (Aarons 1991). Patient specific features including body weight, concomitant use of other medications, and individual metabolic capacity often alter expected dose-concentration relationships. Using the population pharmacokinetic approach also permits the gaining of integrated information on pharmacokinetics from relatively sparse data sets obtained from specific populations (e.g. pediatric patients), as well as more comprehensive data sets, or a combination of both.
Accordingly, a population pharmacokinetic analytic approach has been applied to characterize atomoxetine pharmacokinetics and the effect of intrinsic or/and extrinsic variables on the pharmacokinetics of atomoxetine in pediatric patients with ADHD (United States Food and Drug Administration 2002). Three hundred forty-nine male and 71 female children or/and adolescents with a total of 2354 plasma concentration data points were included in the population pharmacokinetic analysis. A one-compartment model with first-order input and elimination well described the atomoxetine pharmacokinetic profiles in the pediatric populations. The relationships among CYP2D6 phenotype and atomoxetine CL/F, total body weight and atomoxetine CL/F and V/F were retained in the final model. Dose, age, sex, race/ethnicity, hepatic function, albumin, food, and concomitant caffeine use were not identified as significant covariates, and were excluded in the final model.
Based on the population pharmacokinetic analysis, CL/F in pediatric CYP2D6 PMs (n = 25; 6.0%) was approximately nine-fold lower than that observed in EMs (n = 384; 91.4%). Pediatric CYP2D6 UMs (n = 11; 2.6%) appeared to have a two-fold greater CL/F relative to EMs, whereas the values of clearances in UMs were not distinguishable from those in EMs based on the distribution of clearances in these subpopulations. The clearance and volume of distribution of atomoxetine generally increased proportionally with total body weight over the range of 23–125 kg, which is consistent with a comparative pharmacokinetic study in which pharmacokinetic profiles of atomoxetine in CYP2D6 EM pediatric patients with ADHD (7–14 years of age) were similar to those observed in adult EM subjects after normalization by body weight (Witcher et al. 2003). Age (range 7–15 years) and patient sex did not markedly affect atomoxetine disposition. No statistically significant difference in the pharmacokinetics of atomoxetine disposition was identified among individuals of Caucasian (n = 342, 81.4%), Hispanic (n = 23, 5.5%), and African origin (n = 36, 8.6%). Fluctuations in hepatic function (based on total bilirubin and alanine aminotransferase) within the normal range of healthy children did not alter atomoxetine disposition. Slight changes (< 25%) in atomoxetine pharmacokinetics associated with increasing albumin concentrations and food intake are unlikely to be clinically relevant. The disposition of atomoxetine was unchanged in the presence of caffeine.
Dosing Adjustments Based on Pharmacokinetic Principles
Although the manufacturer indicates that atomoxetine, at typical therapeutic doses, appears to be well tolerated in pediatric subjects regardless of CYP2D6 metabolic status (Trzepacz et al. 2008), there appear to have been at least two reasons for the concern over higher plasma concentrations. First, atomoxetine-induced increases in heart rate and diastolic blood pressure appeared to be dose associated (United States Food and Drug Administration 2002; Beasley et al. 2010; Medicines and Healthcare Products Regulatory Agency 2012). Also, compared with CYP2D6 non-PM subjects, PM subjects had statistically significant increases in heart rate and diastolic blood pressure (Fijal et al. 2015). Moreover, increases in standing heart rate were observed in both CYP2D6 EM and PM subjects following a 40 mg dose of atomoxetine given twice daily for 7 days; however, maximum heart rates were ∼10 beats per minute higher in PMs than in EMs (United States Food and Drug Administration 2002). Second, a statistically significant prolongation in the heart rate corrected QT interval was associated with increasing atomoxetine concentrations (Loghin et al. 2013). Additionally, a recent safety analysis by the drug's manufacturer found that CYP2D6 PMs experienced a significantly greater frequency of some treatment-emergent adverse events (such as dry mouth, erectile dysfunction, hyperhidrosis, insomnia, and urinary retention; p < 0.05) than did non-PM subjects (Fijal et al. 2015). Therefore, pharmacokinetic-based dosage adjustment is recommended when patients are expected to be exposed to supratherapeutic atomoxetine concentrations.
Hepatic insufficiency
The systemic clearance of atomoxetine was decreased by nearly 50% and 75% in patients with moderate hepatic impairment (Child–Pugh Class B) and patients with severe hepatic impairment (Child–Pugh Class C), respectively, resulting in a two- to four-fold increase in AUCinf in patients with moderate to severe hepatic insufficiency relative to healthy CYP2D6 EM subjects (Chalon et al. 2003). The increase in atomoxetine exposure is correlated with the progressive severity of hepatic insufficiency (based on the Child–Pugh classification). It is of note that all participants in the study were CYP2D6 EMs. PM patients with hepatic impairment might be anticipated to have greater systemic atomoxetine exposure than normal PM patients (United States Food and Drug Administration 2002). As a result, initial doses of atomoxetine should be decreased to 50% and 25% of the normal dose for patients with moderate and severe hepatic impairment, respectively (Chalon et al. 2003), because a 50% and 75% dose reduction in patients with moderate and severe hepatic impairment, respectively, could provide exposure matching that of healthy CYP2D6 EM subjects receiving atomoxetine at a full dose.
Weight-based dosing regimen
As described, atomoxetine pharmacokinetics have been shown to be proportional to body weight over the range of 23–125 kg. Further, weight-normalized pharmacokinetic parameters were similar among those subjects. On the basis of these findings, the manufacturer proposes a weight-normalized dosing regimen (normalized to a mg/kg basis for children and adolescents up to 70 kg body weight) for atomoxetine in pediatric subjects, to minimize the influence of changes in weight during growth and development on the dose-exposure relationship. Given that atomoxetine exposure in pediatric patients with a 70 kg body weight is two- to three-fold greater than that in children with a 23 kg body weight, weight-normalized dosing regimen for pediatric subjects up to 70 kg body weight has been recommended in both the Prescribing Information (Eli Lilly and Company 2015a) that has been approved by the FDA and the Summary of Product Characteristics (Eli Lilly and Company 2015b) that has been approved by the Medicines and Healthcare Products Regulatory Agency (MHRA) in the United Kingdom.
Genotype
The two- to four-fold increases in atomoxetine exposure that may be observed in hepatic insufficiency have led to the development of consensus guidelines on dosing adjustments for atomoxetine in patients with hepatic insufficiency, which are provided in both the Prescribing Information (Eli Lilly and Company 2015a) and the Summary of Product Characteristics (Eli Lilly and Company 2015b). Similarly, a consensus on weight-based dosing guidelines for atomoxetine in children and adolescents has also been reached because of the two- to three-fold difference in atomoxetine exposure based on differences in body weight (Eli Lilly and Company 2015a,b). However, dosing adjustments for atomoxetine in CYP2D6 PMs in relation to the 8- to 10-fold increases in atomoxetine exposure are only recommended in the Prescribing Information (Eli Lilly and Company 2015a), and do not appear in the Summary of Product Characteristics (Eli Lilly and Company 2015b). Interestingly, even in the Prescribing Information, recommendations for genotyping prior to atomoxetine treatment are absent. An 8- to 10-fold difference in atomoxetine exposure in CYP2D6 PM patients would seem to warrant CYP2D6 genotyping in routine clinical management to avoid excessive dosing early cessation caused by adverse events (ter Laak et al. 2010). Future investigation into the potential benefits of genotyping should include cost–benefit analyses as part of an overall evaluation of the utility of genotyping patients prior to atomoxetine initiation and the development of corresponding genotype-guided dosing guidelines. Another concern related to genotype is the underexposure in CYP2D6 UMs, which may risk therapeutic failure without the employment of necessary dosage modifications (de Leon 2007). On the basis of current evidence, patients who fail to respond to the drug or who show suspected atomoxetine-related toxicities should be considered candidates for CYP2D6 genotyping, and dosing regimens could be further individualized accordingly.
Coadministration with potent inhibitors of CYP2D6
The influence of several known potent inhibitors of CYP2D6 on the steady-state exposure to atomoxetine have been assessed in normal volunteers who were CYP2D6 EMs. When coadministrated with potent CYP2D6 inhibitors such as paroxetine (Belle et al. 2002), fluoxetine, and quinidine (Eli Lilly and Company 2015a), atomoxetine exposure was increased by 500–700% compared with atomoxetine alone, which was similar to what was observed in CYP2D6 PMs. Accordingly, similar dosage adjustments are recommended.
Based upon the reviewed data, clear effects on atomoxetine metabolism and disposition have been observed for various polymorphisms in the two CYP encoding genes CYP2D6, and, to a lesser degree, CYP2C19. The differences in oral bioavailability of atomoxetine between CYP2D6 EMs and PMs are significant and primarily attributed to the contributions of hepatic extraction in CYP2D6 EMs and PMs rather than absorption, transport, or intestinal metabolism.
Limited data pertaining to tissue distribution of atomoxetine are available from CYP2D6 PMs. Concentrations of atomoxetine in saliva are more than one order of magnitude lower than plasma concentrations, and the saliva-to-plasma concentration ratios are time dependent and variable, precluding saliva concentrations from presently serving as noninvasive surrogates for plasma concentration in monitoring atomoxetine levels in pediatric patients. As a drug targeting the CNS, it is not surprising that atomoxetine appears in the CSF in healthy CYP2D6 EMs. The average concentrations of atomoxetine in the human CSF was 6.6 ng/mL at 8 hours and 1.4 ng/mL at 24 hours postdose, which was higher than the in vitro hNET Ki (1.1 ng/mL). However, significant interindividual variability in the CSF and plasma concentrations was observed, which may be related to CYP2D6 genotypes. The BBB and BCSFB transport of atomoxetine have been shown to occur predominately by passive diffusion. Further, pharmacokinetic modeling and simulations revealed that for atomoxetine 40 mg once daily, the predicted median unbound Cmax in the human ECF was almost two-fold higher than the hNET Ki, and the predicted median ECF concentrations were 1.1 at 16 hours and 0.74 ng/mL at 24 hours postdose, indicating that at clinical doses of 40 and 80 mg once daily, unbound atomoxetine in the CYP2D6 EM human ECF may reach or approach the target level of NET inhibition.
Although sufficient unbound atomoxetine exposure in the human CNS is directly related to therapeutic efficacy, little is known about atomoxetine exposure in the CNS in CYP2D6 PMs and UMs, which warrants further investigation. Additionally, unbound fractions of atomoxetine and its metabolites in plasma appear to be constant over the therapeutic range, which enable clinicians to reliably estimate plasma unbound concentrations for therapeutic monitoring when necessary.
Conclusions
Of >120 pharmacogenomics markers for drugs currently described by the FDA, ∼20% are for drugs with neuropsychiatric indications. However, specific guidelines for how to utilize these genetic markers to individualize medication regimens are generally lacking at present. Although specific recommendations for a small number of CYP genotypes have been published, they have not been widely adapted or implemented in mainstream clinical practice (Hamilton 2015). For CYP2D6 and atomoxetine, a clear gene-associated exposure effect is in evidence, which is able to explain, in principle, the bimodal distribution of atomoxetine exposure in two population subgroups. However, for clinicians reviewing the full prescribing literature for Strattera® (atomoxetine), although it provides fairly extensive information on the influence of PM/EM status on the clinical pharmacokinetics of atomoxetine, as well as the greater incidence of many side effects in PMs, they will find no specific language on the need for or value of CYP2D6 genotyping. However, combined under a heading addressing coadministration of atomoxetine with CYP2D6 inhibitors, there are some dosing adjustments recommended for “patients who are known to be CYP2D6 PMs.” The recommendations for both of these clinical scenarios (i.e., DDIs and PM status) are to initiate atomoxetine at 0.5 mg/kg/day and only increase to the otherwise recommended target dose of 1.2 mg/kg/day if symptoms fail to improve after 4 weeks (Eli Lilly and Company 2015a). Recently, Gressier and associates (2015) published a small study (n = 87) and suggested that the development of a “CYP2D6 composite phenotype,” based on genotype as well as the concurrent use of known CYP2D6 metabolic inhibitors, could be of potential clinical utility in attaining antidepressant response in patients treated for major depression. An analogous approach, formalized or otherwise, may be a useful consideration in the individualization of atomoxetine treatment.
Although the Clinical Pharmacogenetics Implementation Consortium (CPIC) has issued a number of guidelines and recommendations for a variety of therapeutic agents including CYP2D6 substrates codeine, tricyclic antidepressants (TCAs), and selective serotonin reuptake inhibitors (SSRIs), no guidelines are presently available for atomoxetine (PharmGKB 2015). This situation may leave the clinician prescriber unsatisfied, as there are no clear guidelines or consensus on this issue. The clinical utility of pharmacogenetic testing, however, continues to be a topic of considerable debate both in psychiatry and general medicine (Gillis and Innocenti 2014).
Clinical Significance
It is widely accepted that only an unbound drug can exert biological effects. Unbound drug exposures at the site of action are believed to determine the clinical consequences and intersubject differences in therapeutic response or adverse responses to a medication. Specific CYP2D6 genetic polymorphisms not only govern atomoxetine metabolism and systemic exposure, but also appear to influence overall CNS exposure as well. Nearly 10-fold increases in systemic atomoxetine exposure as well as higher frequencies of some treatment-emergent adverse events in CYP2D6 PMs have been confirmed, and seem to warrant genotyping guidelines for patients, while ongoing debate continues as to whether CYP2D6 genotyping before treatment with atomoxetine (and most known CYP2D6 substrates) should be implemented. Further investigation into correlations between genetic variation and pharmacokinetic and pharmacodynamic interrelationships in the CNS are also warranted, and will ultimately facilitate the understanding and optimization of atomoxetine therapy under both simple and complex clinical scenarios.
Disclosures
No competing financial interests exist.
References
- Aarons L: Population pharmacokinetics: Theory and practice. Br J Clin Pharmacol 32:669–670, 1991 [PMC free article] [PubMed] [Google Scholar]
- Agster KL, Bates AT, Cain RE, Newman LA, Waterhouse BD, McGaughy JA: The role of cortical norepinephrinein the development of executive function. Neuropsychopharmacology 36:S83, 2011 [Google Scholar]
- Beasley C, Loghin C, Haber H, Kothare P, Kauffman L, April J, Jin L, Allen A, Mitchell M: Effects of atomoxetine on the QT interval, heart rate, and blood pressure in healthy CYP2D6 poor metabolizers. Abstract presented at the International Pharmaceutical Federation (FIP) and American Association of Pharmaceutical Scientists (AAPS), FIP Pharmaceutical Sciences World Congress 2010 in association with the AAPS Annual Meeting and Exposition, New Orleans, 2010 [Google Scholar]
- Belle DJ, Ernest CS, Sauer JM, Smith BP, Thomasson HR, Witcher JW: Effect of potent CYP2D6 inhibition by paroxetine on atomoxetine pharmacokinetics. J Clin Pharmacol 42:1219–1227, 2002 [DOI] [PubMed] [Google Scholar]
- Benet LZ: Clearance concepts: A downdate and an update. J Pharmacokinet Pharmacodyn 37:529–539, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benet LZ, Hoener BA: Changes in plasma protein binding have little clinical relevance. Clin Pharmacol Ther 71:115–121, 2002 [DOI] [PubMed] [Google Scholar]
- Byeon JY, Kim YH, Na HS, Jang JH, Kim SH, Lee YJ, Bae JW, Kim IS, Jang CG, Chung MW, Lee SY: Effects of the CYP2D6* 10 allele on the pharmacokinetics of atomoxetine and its metabolites. Arch Pharm Res 38:2083–2091, 2015 [DOI] [PubMed] [Google Scholar]
- Bymaster FP, Katner JS, Nelson DL, Hemrick–Luecke SK, Threlkeld PG, Heiligenstein JH, Morin SM, Gehlert DR, Perry KW: Atomoxetine increases extracellular levels of norepinephrine and dopamine in prefrontal cortex of rat: a potential mechanism for efficacy in attention deficit/hyperactivity disorder. Neuropsychopharmacology 27:699–711, 2002 [DOI] [PubMed] [Google Scholar]
- Chalon SA, Desager JP, Desante KA, Frye RF, Witcher J, Long AJ, Sauer JM, Golnez JL, Smith BP, Thomasson HR, Horsmans Y: Effect of hepatic impairment on the pharmacokinetics of atomoxetine and its metabolites. Clin Pharmacol Ther 73:178–191, 2003 [DOI] [PubMed] [Google Scholar]
- Childress AC, Sallee FR: Attention-deficit/hyperactivity disorder with inadequate response to stimulants: Approaches to management. CNS Drugs 28:121–129, 2014 [DOI] [PubMed] [Google Scholar]
- Choi CI, Bae JW, Lee YJ, Lee HI, Jang CG, Lee SY: Effects of CYP2C19 genetic polymorphisms on atomoxetine pharmacokinetics. J Clin Psychopharmacol 34:139–142, 2014 [DOI] [PubMed] [Google Scholar]
- Christman AK, Fermo JD, Markowitz JS: Atomoxetine: A novel treatment for attention-deficit/hyperactivity disorder. Pharmacotherapy 24:1020–1036, 2004 [DOI] [PubMed] [Google Scholar]
- Cortese S, Holtmann M, Banaschewski T, Buitelaar J, Coghill D, Danckaerts M, Dittmann RW, Graham J, Taylor E, Sergeant J, European ADHD Guidelines Group: Practitioner review: Current best practice in the management of adverse events during treatment with ADHD medications in children and adolescents. J Child Psychol Psychiatry 54:227–246, 2013 [DOI] [PubMed] [Google Scholar]
- Cui YM, Teng CH, Pan AX, Yuen E, Yeo KP, Zhou Y, Zhao X, Long AJ, Bangs ME, Wise SD: Atomoxetine pharmacokinetics in healthy Chinese subjects and effect of the CYP2D6*10 allele. Br J Clin Pharmacol 64:445–449, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Lange EC: Utility of CSF in translational neuroscience. J Pharmacokinet Pharmacodyn 40:315–326, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Lange EC, Hammarlund–Udenaes M: Translational aspects of blood–brain barrier transport and central nervous system effects of drugs: from discovery to patients. Clin Pharmacol Ther 97:380–394, 2015 [DOI] [PubMed] [Google Scholar]
- de Leon J: The crucial role of the therapeutic window in understanding the clinical relevance of the poor versus the ultrarapid metabolizer phenotypes in subjects taking drugs metabolized by CYP2D6 or CYP2C19. J Clin Psychopharmacol 27:241–245, 2007 [DOI] [PubMed] [Google Scholar]
- de Leon J: Translating pharmacogenetics to clinical practice: Do cytochrome P450 2D6 ultrarapid metabolizers need higher atomoxetine doses?. J Am Acad Child Adolesc Psychiatry 54:532–534, 2015 [DOI] [PubMed] [Google Scholar]
- Del Campo N, Chamberlain SR, Sahakian BJ, Robbins TW: The roles of dopamine and noradrenaline in the pathophysiology and treatment of attention-deficit/hyperactivity disorder. Biol Psychiatry 69:e145–157, 2011 [DOI] [PubMed] [Google Scholar]
- Ding YS, Naganawa M, Gallezot JD, Nabulsi N, Lin SF, Ropchan J, Weinzimmer D, McCarthy TJ, Carson RE, Huang Y, Laruelle M: Clinical doses of atomoxetine significantly occupy both norepinephrine and serotonin transports: implications on treatment of depression and ADHD. Neuroimage 86:164–171, 2014 [DOI] [PubMed] [Google Scholar]
- Eli Lilly and Company: Atomoxetine Full Prescribing Information, Strattera® (atomoxetine), 2015a. Available at http://www.accessdata.fda.gov/drugsatfda_docs/label/2015/021411s046lbl.pdf Accessed 20September2015
- Eli Lilly and Company: Atomoxetine Summary of Product Characteristics, Strattera® (atomoxetine), 2015b. Available at: https://www.medicines.org.uk/emc/medicine/14482 Accessed 20September2015
- Fijal BA, Guo Y, Li SG, Ahl J, Goto T, Tanaka Y, Nisenbaum LK, Upadhyaya HP: CYP2D6 predicted metabolizer status and safety in adult patients with attention-deficit hyperactivity disorder participating in a large placebo-controlled atomoxetine maintenance of response clinical trial. J Clin Pharmacol 55:1167–174, 2015 [DOI] [PubMed] [Google Scholar]
- Gillis NK, Innocenti F: Evidence required to demonstrate clinical utility of pharmacogenetic testing: The debate continues. Clin Pharmacol Ther 96:655–657, 2014 [DOI] [PubMed] [Google Scholar]
- Gressier F, Verstuyft C, Hardy P, Becquemont L, Corruble E: Response to CYP2D6 substrate antidepressants is predicted by a CYP2D6 composite phenotype based on genotype and comedications with CYP2D6 inhibitors. J Neural Transm 122:35–42, 2015 [DOI] [PubMed] [Google Scholar]
- Hamilton SP: The promise of psychiatric pharmacogenomics. Biol Psychiatry 77:29–35, 2015 [DOI] [PubMed] [Google Scholar]
- Hammarlund–Udenaes M, Friden M, Syvanen S, Gupta A: On the rate and extent of drug delivery to the brain. Pharm Res 25, 1737–1750, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodgkins P, Shaw M, Coghill D, Hechtman L: Amfetamine and methylphenidate medications for attention-deficit/hyperactivity disorder: Complementary treatment options. Eur Child Adolesc Psychiatry 21:477–492, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kielbasa W, Pan A, Pereira A: A pharmacokinetic/pharmacodynamic investigation: Assessment of edivoxetine and atomoxetine on systemic and central 3,4-dihydroxyphenylglycol, a biochemical marker for norepinephrine transporter inhibition. Eur Neuropsychopharmacol 25:377–385, 2015 [DOI] [PubMed] [Google Scholar]
- Kielbasa W, Kalvass JC, Stratford R: Microdialysis evaluation of atomoxetine brain penetration and central nervous system pharmacokinetics in rats. Drug Metab Dispos 37:137–142, 2009 [DOI] [PubMed] [Google Scholar]
- Kielbasa W, Stratford RE: Exploratory translational modeling approach in drug development to predict human brain pharmacokinetics and pharmacologically relevant clinical doses. Drug Metab Dispos 40, 877–883, 2012 [DOI] [PubMed] [Google Scholar]
- Lalovic B, Phillips B, Risler LL, Howald W, Shen DD: Quantitative contribution of CYP2D6 and CYP3A to oxycodone metabolism in human liver and intestinal microsomes. Drug Metab Dispos 32:447–454, 2004 [DOI] [PubMed] [Google Scholar]
- Li GF, Wang K, Chen R, Zhao HR, Yang J, Zheng QS: Simulation of the pharmacokinetics of bisoprolol in healthy adults and patients with impaired renal function using whole-body physiologically based pharmacokinetic modeling. Acta Pharmacol Sin 33:1359–1371, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin JH: Applications and limitations of interspecies scaling and in vitro extrapolation in pharmacokinetics. Drug Metab Dispos 26:1202–1212, 1998 [PubMed] [Google Scholar]
- Lin JH: Species similarities and differences in pharmacokinetics. Drug Metab Dispos 23:1008–1021, 1995 [PubMed] [Google Scholar]
- Lin JH, Chiba M, Baillie TA: Is the role of the small intestine in first-pass metabolism overemphasized?. Pharmacol Rev 51:135–158, 1999 [PubMed] [Google Scholar]
- Lin JH, Lu AY: Role of pharmacokinetics and metabolism in drug discovery and development. Pharmacol Rev 49:403–449, 1997 [PubMed] [Google Scholar]
- Loghin C, Haber H, Beasley CM, Jr, Kothare PA, Kauffman L, April J, Jin L, Allen AJ, Mitchell MI: Effects of atomoxetine on the QT interval in healthy CYP2D6 poor metabolizers. Br J Clin Pharmacol 75:538–549, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mariappan TT, Mandlekar S, Marathe P: Insight into tissue free concentration: Utility in drug discovery and development. Curr Drug Metab 14:324–340, 2013 [DOI] [PubMed] [Google Scholar]
- Markowitz JS: Transforming events: Hepatic metabolism and individual variability. Clin Ther 35:201–204, 2013 [DOI] [PubMed] [Google Scholar]
- Matsui A, Azuma J, Witcher JW, Long AJ, Sauer JM, Smith BP, DeSante KA, Read HA, Takahashi M, Nakano M: Pharmacokinetics, safety, and tolerability of atomoxetine and effect of CYP2D6*10/*10 genotype in healthy Japanese men. J Clin Pharmacol 52:388–403, 2012 [DOI] [PubMed] [Google Scholar]
- Mattiuz EL, Ponsler GD, Barbuch RJ, Wood PG, Mullen JH, Shugert RL, Li Q, Wheeler WJ, Kuo F, Conrad PC, Sauer JM: Disposition and metabolic fate of atomoxetine hydrochloride: pharmacokinetics, metabolism, and excretion in the Fischer 344 rat and beagle dog. Drug Metab Dispos 31:88–97, 2003 [DOI] [PubMed] [Google Scholar]
- Medicines and Healthcare Products Regulatory Agency: MHRA UK Public assessment report. Atomoxetine: a review of the effects on heart rate and blood pressure, 2012. Available at http://www.mhra.gov.uk/home/groups/s-par/documents/websiteresources/con152778.pdf Accessed 20September2015
- Mouton JW, Theuretzbacher U, Craig WA, Tulkens PM, Derendorf H, Cars O: Tissue concentrations: Do we ever learn? J Antimicrob Chemother 61:235–237, 2008 [DOI] [PubMed] [Google Scholar]
- National Institute of Mental Health: Attention deficit hyperactivity disorder (ADHD). Available at www.nimh.nih.gov/health/topics/attention-deficit-hyperactivity-disorder-adhd/index.shtml Accessed 10May2015
- Paine MF, Hart HL, Ludington SS, Haining RL, Rettie AE, Zeldin DC: The human intestinal cytochrome P450 “pie”. Drug Metab Dispos 34:880–886, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papaseit E, Marchei E, Farré M, Garcia–Algar O, Pacifici R, Pichini S: Concentrations of atomoxetine and its metabolites in plasma and oral fluid from paediatric patients with attention deficit/hyperactivity disorder. Drug Test Anal 5:446–452, 2013 [DOI] [PubMed] [Google Scholar]
- PharmGKB: Clinical Pharmacogenetics Implementation Consortium, 2015. Available at https://www.pharmgkb.org/page/cpic Accessed June12, 2015
- Polanczyk G, de Lima MS, Horta BL, Biederman J, Rohde LA: The worldwide prevalence of ADHD: A systematic review and meta regression analysis. Am J Psychiatry 164:942–948, 2007 [DOI] [PubMed] [Google Scholar]
- Ring BJ, Gillespie JS, Eckstein JA, Wrighton SA: Identification of the human cytochromes P450 responsible for atomoxetine metabolism. Drug Metab Dispos 30:319–323, 2002 [DOI] [PubMed] [Google Scholar]
- Rizzo R, Gulisano M, Calì PV, Curatolo P: Tourette Syndrome and comorbid ADHD: Current pharmacological treatment options. Eur J Paediatr Neurol 17:421–428, 2013 [DOI] [PubMed] [Google Scholar]
- Robbins TW, Arnsten AF: The neuropsychopharmacology of fronto-executive function:monoaminergic modulation. Annu Rev Neurosci 32:267–287, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowland M, Benet LZ, Graham GG: Clearance concepts in pharmacokinetics. J Pharmacokinet Biopharm 1:123–135, 1973 [DOI] [PubMed] [Google Scholar]
- Rowland M, Tozer TN: Clinical Pharmacokinetics: Concepts and Applications, 3rd ed. Philadelphia: Williams &Wilkins; 1995 [Google Scholar]
- Sauer JM, Long AJ, Ring B, Gillespie JS, Sanburn NP, DeSante KA, Petullo D, VandenBranden MR, Jensen CB, Wrighton SA, Smith BP, Read HA, Witcher JW: Atomoxetine hydrochloride: Clinical drug–drug interaction prediction and outcome. J Pharmacol Exp Ther 308:410–418, 2004 [DOI] [PubMed] [Google Scholar]
- Sauer JM, Ponsler GD, Mattiuz EL, Long AJ, Witcher JW, Thomasson HR, Desante KA: Disposition and metabolic fate of atomoxetine hydrochloride: The role of CYP2D6 in human disposition and metabolism. Drug Metab Dispos 31:98–107, 2003 [DOI] [PubMed] [Google Scholar]
- Sauer JM, Ring BJ, Witcher JW: Clinical pharmacokinetics of atomoxetine. Clin Pharmacokinet 44:571–590, 2005 [DOI] [PubMed] [Google Scholar]
- DW , Guo W, Zhou FC, Wang XP, Li AN, Zhang L, Li WB, Lu W, Wang CY: Relative bioequivalence evaluation of two oral atomoxetine hydrochloride capsules: a single dose, randomized, open-label, 2-period crossover study in healthy Chinese volunteers under fasting conditions. Drug Res (Stuttg) 63:564–567, 2013 [DOI] [PubMed] [Google Scholar]
- Swanson CJ, Perry KW, Koch–Krueger S, Katner J, Svensson KA, Bymaster FP: Effect of the attention deficit/hyperactivity disorder drug atomoxetine on extracellular concentrations of norepinephrine and dopamine in several brain regions of the rat. Neuropharmacology 50:755–760, 2006 [DOI] [PubMed] [Google Scholar]
- Tatsumi M, Groshan K, Blakely RD, Richelson E: Pharmacological profile of antidepressants and related compounds at human monoamine transporters. Eur J Pharmacol 40:249–258, 1997 [DOI] [PubMed] [Google Scholar]
- Teh LK, Bertilsson L: Pharmacogenomics of CYP2D6: Molecular genetics, interethnic differences and clinical importance. Drug Metab Pharmacokinet 27:55–67, 2012 [DOI] [PubMed] [Google Scholar]
- ter Laak MA, Temmink AH, Koeken A, van 't Veer NE, van Hattum PR, Cobbaert CM: Recognition of impaired atomoxetine metabolism because of low CYP2D6 activity. Pediatr Neurol 43:159–162, 2010 [DOI] [PubMed] [Google Scholar]
- Trzepacz PT, Williams DW, Feldman PD, Wrishko RE, Witcher JW, Buitelaar JK: CYP2D6 metabolizer status and atomoxetine dosing in children and adolescents with ADHD. Eur Neuropsychopharmacol 18:79–86, 2008 [DOI] [PubMed] [Google Scholar]
- United States Food and Drug Administration: Atomoxetine approval package 2002. Available at www.accessdata.fda.gov/drugsatfda_docs/nda/2002/21–411_Strattera.cfm Accessed 20March2015
- Wagner C, Zhao P, Pan Y, Hsu V, Grillo J, Huang SM, Sinha V: Application of physiologically based pharmacokinetic (PBPK) modeling to support dose selection: report of an FDA public workshop on PBPK. CPT Pharmacometrics Syst Pharmacol 4:226–230, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warikoo N, Faraone SV: Background, clinical features and treatment of attention deficit hyperactivity disorder in children. Expert Opin Pharmacother 14:1885–906, 2013 [DOI] [PubMed] [Google Scholar]
- Witcher JW, Long A, Smith B, Sauer JM, Heilgenstein J, Wilens T, Spencer T, Biederman J: Atomoxetine pharmacokinetics in children and adolescents with attention deficit hyperactivity disorder. J Child Adolesc Psychopharmacol 13:53–63, 2003 [DOI] [PubMed] [Google Scholar]
- Yu G, Zheng QS, Li GF: Similarities and differences in gastrointestinal physiology between neonates and adults: a physiologically based pharmacokinetic modeling perspective. AAPS J 16:1162–1166, 2014a [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu G, Zheng QS, Wang DX, Zhou HH, Li GF: Drug interactions between tyrosine-kinase inhibitors and acid suppressive agents: More than meets the eye. Lancet Oncol 15:e469–e470, 2014b [DOI] [PubMed] [Google Scholar]
- Zhang L, Wu F, Lee SC, Zhao H, Zhang L: pH–Dependent drug–drug interactions for weak base drugs: Potential implications for new drug development. Clin Pharmacol Ther 96:266–277, 2014 [DOI] [PubMed] [Google Scholar]
- Zhu H-J, Wang J-S, Donovan JL, Jiang Y, Gibson BB, DeVane CL, Markowitz JS: Interactions of attention-deficit/hyperactivity disorder therapeutic agents with the efflux transporter P-glycoprotein. Eur J Pharmacol 578:148–158, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]