

Abstract
Nearly 350 IgG-based therapeutics are approved for clinical use or are under development for many diseases lacking adequate treatment options. These include molecularly engineered biologicals comprising the IgG Fc-domain fused to various effector molecules (so-called Fc-fusion proteins) that confer the advantages of IgG, including binding to the neonatal Fc receptor (FcRn) to facilitate in vivo stability, and the therapeutic benefit of the specific effector functions. Advances in IgG structure-function relationships and an understanding of FcRn biology have provided therapeutic opportunities for previously unapproachable diseases. This article discusses approved Fc-fusion therapeutics, novel Fc-fusion proteins and FcRn-dependent delivery approaches in development, and how engineering of the FcRn–Fc interaction can generate longer-lasting and more effective therapeutics.
Keywords: Fc-fusion proteins, IgG, mucosal drug delivery, neonatal Fc receptor, therapeutic antibody
Introduction
In the recent past, IgG-based therapeutics and antibodies have gained overwhelming importance for the treatment of a wide range of diseases, including autoimmunity and inflammation (Chan & Carter, 2010), cancer treatment (Weiner et al., 2010), cardiovascular and infectious diseases and transplantation medicine (Reichert, 2010, 2012). The broad foundations for the tremendous success of this extremely versatile class of biologically based therapeutics was laid with the invention of hybridoma technologies by Georges Köhler and Cesar Millstein, which allowed for the rapid generation of monoclonal antibodies (mAbs) (Kohler & Milstein, 1975). Since then, the development of chimeric, humanized and more recently human antibodies together with antibody engineering technologies has substantially improved the pharmacokinetic, pharmacodynamic and immunologic properties of this class of therapeutics, making IgG-based therapeutics nowadays a highly potent, specific and generally well-tolerated therapeutic approach (Jiang et al., 2011; Nelson et al., 2010). IgG-based biologic agents also benefit from widely utilized simplified and scalable purification procedures (Huang, 2009; Jazayeri & Carroll, 2008), and the impact of this class of therapeutics is impressively illustrated by the fact that more than 30 antibodies or IgG-based therapeutics have been approved in the past 25 years (Beck et al., 2010) with annual US sales for mAbs reaching ≈$18.5 billion and a combined sales growth rate for mAbs and fusion proteins that reached 14.3% per year in 2010 (Aggarwal, 2011). Furthermore, with nearly 350 antibody-based therapeutics (including Fc-fusion proteins) in the commercial pipeline and in development, mAbs represent the fastest growing category of antibody-based therapeutics entering clinical studies (Nelson et al., 2010; Reichert, 2008, 2012).
In addition to developing mAbs with new specificities against novel targets by using next-generation hybridoma and antibody engineering strategies such as yeast and phage display (Beck, 2011; Beck et al., 2008, 2010; Lonberg, 2008; Nelson et al., 2010; Weiner et al., 2010), the knowledge associated with understanding the biology of IgG molecules is increasingly allowing for the generation of mechanism-based modifications of existing therapeutics. Most of these antibody designs to develop so-called “bio-better” agents (Beck, 2011) are currently based on the properties and functions of the Fc-domain and its relationship to the neonatal Fc receptor (FcRn). FcRn plays a central role in regulating the catabolism and thus the half-life of IgG as well as its functions in the immune system and in particular presentation of complexed antigens (Baker et al., 2009; 2011, 2012; Qiao et al., 2008; Yoshida et al., 2004, 2006). As such, therapeutic agents that are molecularly engineered to include an Fc-domain (Fc-fusion proteins) or mAbs that are modified to exhibit increased affinity toward FcRn confer upon such agents the benefits of improved pharmacokinetics and potentially pharmacodynamics without compromising the specificity of the therapeutic moiety.
In this article, we review currently approved Fc-fusion therapeutics, novel Fc-fusion proteins and FcRn-dependent delivery approaches in development. We provide insights into how engineering of the FcRn–Fc interaction can generate longer-lasting and more effective therapeutics that may be delivered by unique parenteral routes.
Fundamental biology of FcRn-dependent IgG homeostasis
IgG molecules are approximately 150 kDa and composed of two identical light chains and heavy chains. IgG is the most abundant immunoglobulin in humans and together with albumin accounts for approximately 80% of the protein plasma mass (Schultze & Heremans, 1966).
Remarkably, these two biologically unrelated molecules share an extended plasma half-life of 19–22 d, which by far exceeds the typical half-life of a few minutes to a few days for other known human plasma proteins (Peters, 1977, 1985; Watson, 1965). Intimately related to the extended half-life of both proteins is a common natural pathway that revolves around FcRn. Functioning as a single broadly distributed and, contrary to what is suggested by its name, lifelong-expressed Fc receptor, it serves as the key homeostatic regulator in this process and thereby exhibits a profound relevance for the basic physiologic processes and plasma homeostasis of these two major proteins (Roopenian & Akilesh, 2007).
FcRn and IgG from a historical perspective
The identification of FcRn originated in discoveries that were made half a century ago when Francis William Rogers Brambell drew a functional association between the passive acquisition of antibody-based immunity in neonatal rodents (Brambell, 1963) and the observation that the half-life of infused IgG decreased as the amount of the infused IgG increased (Brambell et al., 1964). This suggested to Brambell the existence of a saturable receptor that was responsible for both this transport process and the salvaging of IgG from catabolism. A major clue for the nature of this process came a decade later when Richard Rodewald's group discovered that the ability of suckling neonatal rats to absorb maternal IgG from breast milk was a pH-dependent process that involved endocytosis and transcytosis across the intestinal barrier (Abrahamson et al., 1979; Rodewald, 1973, 1976, 1980; Rodewald & Kraehenbuhl, 1984). Soon thereafter, rat FcRn was isolated and identified as the molecule responsible for these observations and described as a heterodimer between a 48 kDa and a 12 kDa subunit, which was subsequently cloned in 1989 by Simister & Mostov (Simister & Mostov, 1989; Simister & Rees, 1985). The latter component was identified as β2-microglobulin, which served as a non-covalent binding partner of the FcRn major histocompatibility complex (MHC) class I-like alpha (or heavy) chain, which itself was shown to be differentially glycosylated and downregulated upon weaning, accounting for the neonatal transmission of immunity in rodents (Simister & Mostov, 1989; Simister & Rees, 1985). Human FcRn was cloned in 1994 (Story et al., 1994); soon thereafter, FcRn-mediated maternofetal transport of IgG across the placenta during gestation was shown to be a key mechanism for the acquisition of passive immunity in the fetus (Leach et al., 1996; Simister et al., 1996).
These now classic studies allowed for a rapid acquisition of knowledge about the structure and function of FcRn, which is of fundamental importance to the related activities of IgG and albumin.
FcRn and IgG: structural insights for interaction
FcRn, encoded by the Fcgrt gene, is a MHC class I-like transmembrane protein consisting of a heavy chain containing three extracellular domains (α1, α2 and α3), a single pass transmembrane domain and a short cytoplasmatic tail (Burmeister et al., 1994a,b; Martin et al., 2001) (Figure 1). For proper function, the FcRn heavy chain non-covalently associates with the common β2-microglobulin subunit as a light chain, which interacts with FcRn via residues on the underside of the α1-α2 platform and the side of the α3 domain (West & Bjorkman, 2000). Although the tertiary structure resembles MHC class I molecules with which it shares 22–29% sequence homology (Simister & Mostov, 1989), the mouse and human FcRn genes are located outside the MHC locus, on chromosomes 7 and 19, respectively (Ahouse et al., 1993; Kandil et al., 1996). In further divergence from classical MHC molecules, the sites where peptide residues bind to MHC class I molecules are occluded in FcRn by an arginine side chain and a proline residue, so that FcRn does not directly present peptide antigens to T-cells (Burmeister et al., 1994a,b).
Figure 1.

Crystal structure of FcRn and the FcRn–IgG Fc complex. (a) FcRn is a heterodimeric molecule consisting of a heavy chain with the three extracellular domains (shown in brown) that non-covalently associate with beta-2-microglobulin as a light chain (blue). Although the tertiary structure of FcRn resembles an MHC class I molecule, the binding sites where peptides bind to MHC class I molecules are inaccessible in FcRn. Human FcRn and human β2m are shown. (b) At acidic pH, FcRn (brown) binds to the CH2-CH3 hinge region of IgG (green). Critical amino acids in FcRn for the binding to the Fc fragment are highlighted in yellow. Rat FcRn and a rat Fc fragment are shown. Crystal structures were generated with Cn3d based on the protein data bank (PDB) ID 1EXU (West and Bjorkman, 2000) and 1FRT (Burmeister et al., 1994b).
Structural homologues of FcRn have now been identified in many mammalian species including not only rat, mouse and human as described above but also cow (Kacskovics et al., 2000), pig (Schnulle & Hurley, 2003), sheep (Mayer et al., 2002a,b), camel (Kacskovics et al., 2006), marsupials (Adamski et al., 2000), canine (Dumont et al., 2012) and macaque (Bitonti et al., 2004). Furthermore, the chicken yolk sac receptor FcRY has been described as the functional orthologue of FcRn in birds (He & Bjorkman, 2011; West et al., 2004), which, similar to FcRn, exhibits pH-dependent ligand binding and functions in endocytosis, bidirectional transcytosis and recycling of IgY, the avian and reptile counterpart of IgG (He & Bjorkman, 2011; Tesar et al., 2008). Between different species, FcRn exhibits considerable structural variations, which most likely account for the molecule's different ligand binding specificity and slight variations in its functions. The peptide sequences of rat and mouse FcRn, for example, are 91% homologous (Ahouse et al., 1993), whereas the extracellular region of human FcRn shares only 65% amino acid sequence identity with rat FcRn (Story et al., 1994). Bovine FcRn, on the other hand, displays 77% homology to its human counterpart, but exhibits further divergence from rodent FcRn (Kacskovics et al., 2000). Similarly, although mouse and rat FcRn exhibit promiscuous binding to multiple different species of IgG such as horse, rabbit and human, human FcRn binding is significantly more restricted and limited to itself and rabbit (Ober et al., 2001). Clearly, FcRn is an ancient protein, likely present in all mammalian species.
A major advance in understanding FcRn function was in the elucidation of the crystal structure. Such analyses revealed that two FcRn molecules bind to a single IgG in a 2:1 stoichiometry (Huber et al., 1993; Sanchez et al., 1999; Schuck et al., 1999). Each IgG heavy chain contains three constant regions (Huber et al., 1976) with one of the FcRn molecules binding to the CH2-CH3 interface of the IgG Fc region (Huber et al., 1993; Sanchez et al., 1999; Schuck et al., 1999; West & Bjorkman, 2000) (Figure 1). Such binding between IgG and FcRn occurs in a strictly pH-dependent manner with low micro- to nanomolar affinity at pH ≤6.5 but no binding at pH 7.5 (Raghavan et al., 1995). Several amino acids on both molecules have been identified to be critical for this interaction. Site-directed mutagenesis approaches have revealed that the residues Ile253, His310 and His435 of IgG play a central role in the interaction with FcRn, as shown within different species (mouse, human and rat) as well as for interspecies binding (Firan et al., 2001; Kim et al., 1994, 1999; Martin et al., 2001; Medesan et al., 1997; Raghavan et al., 1995; Shields et al., 2001). Apart from these, the His436 residue in mouse immunoglobulin G1 (IgG1), which corresponds to a Tyr in human IgG1, has been shown to be involved in binding of IgG to FcRn, although to a lesser degree than Ile253, His310 and His435 (Medesan et al., 1997; Shields et al., 2001). Both I253 and H310 are well conserved in all human and murine IgG subclasses, whereas H435 and H436 generally show a lesser degree of conservation, but are still relatively well-conserved across species (Deisenhofer, 1981). The highly conserved His433 of IgG is also thought be involved in the interaction between IgG and FcRn, although this is a controversial observation with varying results in different studies (Kim et al., 1999; Martin et al., 2001; Medesan et al., 1997; Raghavan et al., 1995; Shields et al., 2001). The pKa of His is 6.0–6.5 such that several histidine residues of IgG become protonated below physiologic pH, allowing for the formation of salt bridges with acidic residues on FcRn which in doing so provides the structural basis for the strict pH dependency of IgG–FcRn interactions.
As initially identified in the interaction between rat FcRn and rat IgG2a, residues on FcRn involved in binding IgG include Glu117, Glu118, Glu132, Trp133, Glu135 and Asp137 on the α2 helix (Martin et al., 2001). Although these residues are generally conserved between different species and the main tertiary structure of FcRn with three extracellular ligand-binding domains is preserved, differences between rodent and human FcRn have been described at specific residues and contribute to IgG binding (Vaughn et al., 1997). While human FcRn contains only a single N-glycan moiety in its α2 domain, rat FcRn possesses three additional N-glycan moieties in the α1, α2 and α3 domains (Ahouse et al., 1993; Kuo et al., 2009; Martin et al., 2001; West & Bjorkman, 2000). The Asn128 residue in the α2 domain of rat FcRn, which is lacking in human FcRn, binds to IgG forming a functional “carbohydrate handshake” (Martin et al., 2001; Vaughn & Bjorkman, 1998). In another example, human FcRn displays very limited interspecies IgG binding, extending only to rabbit IgG (Ober et al., 2001), whereas human IgG can bind to cynomolgus FcRn (Bitonti et al., 2004; Dall'Acqua et al., 2006; Zalevsky et al., 2010). In fact, cynomolgus and human IgG have been demonstrated to bind equally well to cynomolgus monkey FcRn (Dall'Acqua et al., 2006), thereby further strengthening the evolutionary significance of the interaction between the Fc region and FcRn. Rodent FcRn, however, is known to be promiscuous by binding to IgG molecules from a variety of species including human, rabbit and bovine IgG as discussed above (Ober et al., 2001). Murinization of human FcRn by mutating the poorly conserved Leu137 residue within the α2 domain of human FcRn to the murine counterpart (glutamic acid) confers binding of human FcRn to mouse IgG1 and IgG2a while reducing binding to human IgG1 twofold (Zhou et al., 2003). The L137E mutation demonstrates that single docking topologies are vitally important in the binding of FcRn to IgG and understanding these interactions is of critical importance for the generation of therapeutic antibodies and the optimization of bioavailability and plasma half-life of these macromolecules. Apart from the residues discussed above, Ile1 on β2m contributes to IgG binding, most likely by interacting with hydrophobic residues at position 309 of the IgG-Fc domain.
FcRn-dependent protection of IgG
One of the most important and best-characterized functions of FcRn is the protection of its two ligands, IgG and albumin, from catabolism. Evidence for these processes results from studies in mice genetically deficient in either FcRn or β2m expression wherein both hypogammaglobulinemia and hypoalbuminemia are observed along with a commensurate decrease in plasma half-life for each of these molecules (Akilesh et al., 2007; Chaudhury et al., 2003; Ghetie et al., 1996; Israel et al., 1996; Junghans & Anderson, 1996; Roopenian et al., 2003). In this respect, FcRn knockout mice have a significantly reduced IgG half-life of 1.4 d compared to 9 d in wild-type animals (Roopenian et al., 2003). Furthermore, the plasma concentrations of all IgG subclasses are reduced approximately fourfold in these mice, and values for β2m-deficient mice appear to be similar or even lower (Kim et al., 2008; Roopenian et al., 2003). The dependence of IgG homeostasis on FcRn has also been demonstrated in humans. The only reported defect in FcRn, a single nucleotide transversion in β2m, was identified in two family members with familial hypercatabolic hypoproteinemia (Waldmann & Terry, 1990; Wani et al., 2006). These individuals had low serum IgG levels, and the half-life of IgG was approximately 3 days; although IgG synthesis was determined to be normal, they each showed hypercatabolism of IgG (Waldmann & Terry, 1990; Wani et al., 2006). Furthermore, the rate of FcRn-mediated recycling has been calculated to be 42% greater than the rate of IgG production in humans, indicating that the recycling of IgG rather than its production rate is the dominant process for maintaining elevated IgG concentration in humans (Kim et al., 2007). Similar concepts apply to FcRn-dependent handling of albumin although in this case production of albumin by the liver is considerable (for a review see (Anderson et al., 2006; Kim et al., 2005, 2007)).
It is now clear that both hematopoietic cells and parenchymal cells are responsible for protecting IgG from degradation. This evidence comes from studies in bone marrow chimeric mice, in which wild-type and Fcgrt−/− mice were reconstituted with bone marrow from Fcgrt−/− or wild-type animals, respectively (Akilesh et al., 2007; Kobayashi et al., 2009; Qiao et al., 2008). The studies to date would indicate that there is an equal contribution from parenchymal and hematopoietic cells in protecting IgG from degradation. Interestingly, unpublished data from our laboratory suggest that the hematopoietic compartment may have a particularly important role to play in IgG protection (Figure 2). However, this requires additional verification including studies with models that contain deletion in specific subsets of cells. Such approaches have recently been applied to understanding this problem. For example, using a floxed Fcgrt mouse that was crossed to a Tie2-Cre expressing model, leading to a conditional deletion of FcRn in the vascular endothelium and, to a lesser extent, hematopoietic cells, Montoyo et al. (2009) were able to pinpoint the vascular endothelium as a major parenchymal cell type contributing to the IgG homeostasis.
Figure 2.
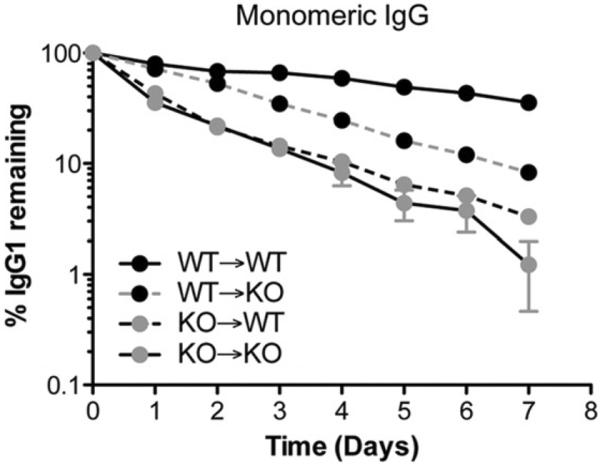
Contribution of the hematopoietic and parenchymal compartment for the protection of monomeric IgG. To assess the contribution of hematopoietic and parenchymal cells to IgG protection, wild-type (WT) and Fcgrt−/− (KO) mice were lethally irradiated and substituted with bone marrow from either WT or Fcgrt−/− mice, and the percentage of a remaining monomeric NIP-OVA mouse IgG was measured over time. Mice in which FcRn was expressed in the hematopoietic compartment (WT→KO) exhibited a higher percentage of remaining IgG compared to mice in which FcRn was limited to the parenchymal compartment (KO→WT), indicating that the contribution of the hematopoietic compartment for IgG protection is greater than that of the parenchymal compartment. Based upon data previously reported by Qiao et al. (2008).
The exact vascular tissue beds within which FcRn operates to regulate IgG levels are unknown but considered to be in skin, muscle, liver and spleen (Akilesh et al., 2007; Montoyo et al., 2009). At these sites, the process of FcRn-mediated IgG recycling from endothelial cells has been deduced from in vitro endothelial cell model systems and consists of the following steps as illustrated in Figure 3(a). Expression of FcRn on the cell surface appears to be limited, with the majority of the receptor located in intracellular compartments in most cell types where FcRn is found. At most cell surfaces, including that associated with endothelial cells, the pH is not acidic and thereby not favorable for FcRn–IgG interactions, requiring naturally occurring IgG to be internalized by fluid-phase processes such as pinocytosis. Following their entry into the cell, IgG molecules then accumulate within early endosomes, where FcRn predominantly resides and wherein the acidic pH is permissive for FcRn binding to IgG. FcRn is observed to co-localize with Rab4+ and Rab11+ vesicles associated with sorting endosomes (Ward et al., 2005). IgG molecules leave the sorting endosome in FcRn+ tubules and vesicles that are also involved in recycling of transferrin (Ober et al., 2004b). Exocytosis of FcRn–IgG complexes is finally associated with a compartment positive for Rab11, but not Rab4 (Ward et al., 2005), and occurs via both classical exocytosis, in which the exocytic vesicle fuses completely with the cell membrane, and a “prolonged-release” exocytosis where the vesicle only partially fuses with the cell membrane but does so in repetitive cycles (Ober et al., 2004a). This slower “kiss and run” exocytosis release mode is characterized by periodic, stepwise release of IgG, rather than the rapid burst that is observed for complete-fusion events. In both processes, the neutral pH of the extracellular milieu associated with the bloodstream facilitates dissociation of IgG from FcRn and its release into the systemic circulation (Ober et al., 2004a).
Figure 3.

FcRn-dependent recycling and protection of monomeric IgG versus degradation and potentially presentation of complexed IgG. (a) In endothelial cells, IgG molecules or IgG Fc-fusion proteins can be internalized by fluid-phase processes such as pinocytosis. FcRn is present within EEA-1 positive early endosomes (Ober et al., 2004b), where acidic pH allows interaction between FcRn and the Fc region of IgG molecules. In nonpolarized endothelial cells, FcRn co-localizes with Rab4+ and Rab11+ vesicles in both fusion and fission events with the sorting endosome (Ward et al., 2005). Exocytosis of FcRn–IgG complexes is associated with a compartment positive for Rab11, but not Rab4 (Ward et al., 2005) and occurs via classical exocytosis, in which the exocytic vesicle fuses completely with the cell membrane and a “prolonged-release” where the vesicle only partially fuses with the cell membrane in repetitive cycles (Ober et al., 2004a). The slower-release mode is characterized by periodic, stepwise release of IgG, rather than the rapid burst that is observed for complete fusion events. In both processes, neutral pH of the bloodstream facilitates dissociation of IgG from FcRn and its release into the systemic circulation. In contrast, proteins not bound to IgG or unbound effector molecules are directed into LAMP-1+ lysosomes and subsequent undergo degradation. (b) FcRn-dependent degradation of IgG-immune complexes. Classical FcγRs on the surface of APCs can bind IgG containing immune complexes, leading to their internalization. In the acidic environment of the endosome, generated, at least in part, by the actions of V-ATPase, the immune complex is released from the internalizing FcγR and subsequently bound by FcRn. FcRn then directs the antigen within the immune complex into antigen processing pathways conducive to the generation of epitopes for loading onto both MHC class I and MHC class II molecules. Thereby, the same APC can effectively prime both CD4+ and CD8+ T cells in FcRn-dependent pathways.
An important consideration to note is that these aforementioned mechanisms apply to single, monomeric IgG molecules (Baker et al., 2011, 2012; Ober et al., 2004b; Qiao et al., 2008; Ward et al., 2003, 2005). As shown in Figure 3(b), after uptake by classical FcγRs, multimeric IgG-containing immune complexes, in contrast, are diverted to intracellular organelles in antigen-presenting cells (APCs) that are involved in FcRn-dependent antigen processing and presentation and ultimately degradation (Baker et al., 2011; Qiao et al., 2008). Consistent with this, whereas the in vivo half-life of monomeric IgG or monomeric IgG-antigen complexes (single antibody and cognate antigen) is prolonged in mice and dependent upon the presence of FcRn, IgG-containing immune complexes are rapidly cleared leading to an extremely short half-life (Qiao et al., 2008). Hence, the intracellular itinerary of the FcRn–IgG complexes is fundamentally different between monomeric IgGs or for Fc-fusion proteins, which structurally resemble single IgG molecules, which FcRn salvages from degradation, and multimeric IgGs, which are degraded and processed in MHC class II and, as recently shown, MHC class I antigen presentation pathways to elicit appropriate T-cell responses as shown in Figure 3(b) (Baker et al., 2011; Qiao et al., 2008).
Rationale for engineering Fc-fusion proteins and general aspects of Fc-fusion proteins
During the past 20 years, advances in molecular cloning techniques and antibody engineering have increasingly allowed for the generation of chimeric molecules in which an effector domain of a short-lived macromolecule is coupled to the Fc region of an IgG molecule.
Among the several advantages that Fc-fusion proteins exhibit, one of the most striking is the ability of these to interact with FcRn allowing for considerable improvement in therapeutic half-life and thus the need for less frequent parenteral administration. Fc-fusion proteins contain the Fc region of human IgG1 that binds to FcRn, which is part of a naturally occurring pathway that delays lysosomal degradation of immunoglobulins by cycling them back into circulation and prolonging their plasma half-life as described above. Moreover, given the vast amount of accrued knowledge on FcRn–IgG interactions, the presence of an IgG Fc domain affords considerable opportunities for antibody engineering to further enable additional beneficial properties to the chimeric molecule as will be discussed further below.
Fc-fusion protein technology can also facilitate the manufacturing process, offering relatively high mammalian cell culture protein expression. As with antibodies, purification of the therapeutic compound may also leverage the Fc region, which binds reversibly and with high affinity to staphylococcal protein A or streptococcal protein G. Thus, fusion proteins containing an Fc fragment with human γ1, γ2 or γ4 heavy chains can easily be purified by affinity chromatography techniques from conditioned medium utilizing protein A or protein G conjugated to resins, or stabilized variants thereof (Ghose et al., 2006).
A final benefit is related to the immunomodulatory properties of IgG molecules and, in particular, the Fc domain. This property derives from original observations that coupling of haptens to IgG can induce antigen-specific tolerance (Borel, 1980), as can IgG antibodies engineered to contain specific epitopes (Zambidis & Scott, 1996). This is further supported by the induction of regulatory T cells (Treg) by intravenously administered immunoglobulin (Ephrem et al., 2008; Kessel et al., 2007). Such approaches have been directly applied as shown by the ability of IgG antibodies engineered to carry potentially immunogenic regions of factor VIII to induce tolerance (Lei & Scott, 2005). Although the precise mechanism remains to be identified, it has been proposed to include the presence of peptide epitopes (Tregitopes) within the Fc region of IgG that are capable of inducing Treg cells (De Groot et al., 2008), among other potential mechanisms (Kaneko et al., 2006). Such properties raise the possibility that Fc-fusion proteins may confer tolerogenic properties on potentially immunogenic therapeutic proteins as has been suggested for etanercept (Enbrel, 2011), an Fc-fusion protein containing the type II tumor necrosis factor (TNF) receptor (De Groot & Scott, 2007).
Generation of IgG Fc-fusion proteins and posttranslational processing
Although Fc-fusion proteins can be expressed in a variety of different expression systems, mammalian systems are often preferred because they enable the correct conformation, proper folding and posttranslational modifications of the protein (Jazayeri & Carroll, 2008). As an expression system, established cell culture lines such as Chinese hamster ovary (CHO) cells, NSO hybridoma cells, human embryonic kidney (HEK) cells or Sp2/0 cells are commonly used and offer the advantage that the IgG-Fc glycoform profile is less heterogeneous (Walsh & Jefferis, 2006). In fact, appropriate Fc glycosylation is an absolute requirement for the elicitation of certain types of cellular effector functions by IgG. In this respect, it has been long recognized that the oligosaccharide moiety covalently attached to Asparagine 297 (Asn297) in the CH2 domain is an essential part of the IgG-Fc structure (Deisenhofer, 1981; Sutton & Phillips, 1983) and that binding to and activation of FcγRs is ablated in antibodies deglycosylated at Asn297 (Tao & Morrison, 1989). However, depending on the clone of the producing cells and the culture conditions, abnormal glycosylation patterns can result in antibodies that are less potent or even immunogenic (Patel et al., 1992; Walsh & Jefferis, 2006). CHO and other murine cell lines are known, for example, to be able to add non-human and thereby strongly immunogenic sugar residues to therapeutic antibodies. One such example of an immunogenic oligosaccharide epitope is the non-human N-glycolylneuraminic acid (Neu5Gc) against which 85% of all untreated humans have been found to possess anti-Neu5Gc antibodies, sometimes at high titers (Padler-Karavani et al., 2008; Tangvoranuntakul et al., 2003; Zhu & Hurst, 2002). It has been shown that these anti-Neu5Gc antibodies from healthy individuals can generate Neu5Gc containing immune complexes in vitro and thus may be predicted to enter the FcRn-driven intracellular pathway leading to the generation of T-cell responses. As such, biopharmaceutical companies as well as academic researchers are actively exploring steps that would reduce levels of Neu5Gc in the engineering process (Borys et al., 2010; Ghaderi et al., 2010). Alternatively, these proteins can be produced in human cell lines, such as HEK293, HT-1080 and PER.C6, which only produce human glycosylation patterns thus obviating this problem (Ghaderi et al., 2010, 2012).
Alternatively, certain types of carbohydrate side-chain modifications of IgG ameliorate immunogenicity and may in fact induce bystander tolerance. The Asn297 residue on human IgG contains many different glycoforms (Arnold et al., 2007; Nimmerjahn & Ravetch, 2007). One group of these isoforms, which is a rare component of IgG, are those that contain terminal α2,6 sialic acid residues, which induce an inhibitory response and are thus tolerogenic (Kaneko et al., 2006). The mechanism for this inhibition is the binding to specific ICAM-3 grabbing non-integrin-related-1 protein (SIGN-R1) on macrophages (Anthony et al., 2008). This is in turn associated with induction of interleukin-33 (IL-33), which promotes the regulatory function of macrophages through upregulation of FcγRIIb, an inhibitory Fcγ receptor, in a pathway that depends upon IL-4 producing basophils (Anthony et al., 2011). This complex pathway dampens immune responses in a potentially broad manner and has been proposed to account for some of the therapeutic properties of IVIG. Whether such modifications confer tolerance to antibody-based therapeutics and Fc-fusion proteins remains to be defined but indeed seems possible. While these studies only briefly outline the importance of glycosylation as the major posttranslational modification of engineered antibodies relevant for both immunogenicity, tolerance and effector functions, detailed and thorough descriptions of posttranslational modifications in protein pharmaceuticals are available elsewhere (Walsh, 2010; Walsh & Jefferis, 2006).
The first successful generation and utilization of a Fc-fusion protein for clinical purposes was in 1989 and involved fusion of the ligand-binding domains of CD4 to the Fc fragment of an IgG1 molecule to generate a chimeric molecule, which was shown to specifically inhibit CD4 activity (Byrn et al., 1990; Capon et al., 1989). Since this first foray into Fc-fusion protein creation, several variations of the general structure of receptor-Fc fusion, in which the C-terminus of the extracellular ligand-binding domain (ECD) of a receptor is genetically fused to the N-terminus of the hinge region, followed by the CH2 and CH3 domains of an IgG, have been designed and engineered (Figure 4).
Figure 4.

Structure of IgG Fc-fusion proteins and mutations in the Fc region to modify FcRn binding. In typical IgG fusion proteins, the C-terminus of the effector molecule is fused to the N-terminus of the hinge region, followed by the CH2 and CH3 domain of human IgG. The effector molecule can be fused as a monomer (i) or a dimer (ii). Apart from full receptors or ligands as the effector molecules, bioactive peptide sequences (iii) have been successfully generated as Fc-fusion proteins. In order to generate improved therapeutics, various amino acid mutations, singly or in combination, have been introduced into the Fc region of human IgG that allow for enhanced binding to FcRn, thereby extending the therapeutic half-life of the fusion proteins. Black labeled residues: mutations with enhanced FcRn binding, grey labeled residues: mutations with decreased FcRn binding. For details see Supplementary Table 2.
The first Fc-fusion proteins consisted of a single or multiple ligand-binding domains or peptide chains that were linked as a homo- or hetero-dimers to a dimeric Fc fragment in a structure reminiscent of a native IgG. However, depending on the nature of the therapeutic protein fused to the Fc dimer, this approach can result in very large molecules in which steric hindrance between the two effector domains and/or the Fc region might impede physiological uptake and function of or binding of the effector molecule to other molecules or receptors. Furthermore, glycosylation might result in heavily charged molecules, thereby additionally hindering therapeutic activity by affecting endocytosis or transport. Nevertheless, these potential limitations can be avoided by molecular engineering as shown by the generation of a monomeric Fc-fusion which consists of only one effector molecule fused to a dimer of Fc, thereby reducing molecular size (Bitonti et al., 2004). As discussed below, this modification of the typical dimeric Fc-fusion protein has proven to increase physiologic uptake, bioavailability and serum half-life of the effector molecule and therefore represents a powerful modification of the conventional dimeric Fc-fusion protein form (Dumont et al., 2006).
Among the many hurdles and challenges in engineering of Fc-fusion proteins and the benefits derived from this combination, one of the most difficult has been the generation of suitable linker regions for conjugating the effector molecule to the Fc region (Kemshead & Hopkins, 1993). Ideally, the linker region should be stable within the systemic circulation and allow efficient interactions of the effector molecule with the target cell. Currently, three types of linkers are mainly used for Fc-fusion proteins and antibody based conjugates: disulfide-based linkers, acide labile hydrazone linkers and peptide-based linkers (Iyer & Kadambi, 2011). Importantly, the linker technologies are not only important for drug stability, but also for efficacy and toxicity of the conjugates (Doronina et al., 2006), and details on the importance of the various linkers can be found elsewhere (Senter, 2009; Wu & Senter, 2005).
Efficacy and safety of approved Fc-fusion proteins
Etanercept (Enbrel®), consisting of the ECD of the human p75 (type II) TNF receptor fused to the Fc fragment of human IgG1, inhibits the binding of TNFα and TNFβ to cell surface TNF receptors and was the first Fc-fusion protein to obtain Food and Drug Administration (FDA) approval for the treatment of rheumatoid arthritis (RA) as well as other forms of arthritis. Since etanercept was approved more than a decade ago in 1998, its safety and immunogenicity has been intensively assessed. Anti-etanercept antibodies have been reported with frequencies of 3–5.6% in RA (Dore et al., 2007; Keystone et al., 2004), 0% in ankylosing spondylitis (de Vries et al., 2009), 8% in children with juvenile idiopathic arthritis (Lovell et al., 2000) and 18% in psoriasis (Tyring et al., 2007). However, no study, to date, has shown a clear association between the presence of these antibodies and lower clinical response suggesting that they are non-neutralizing.
Alefacept (Amevive®, 2011) is a dimeric fusion protein composed of the extracellular CD2-binding portion of human lymphocyte function-associated antigen 3, the primary co-receptor for CD2 on T cells, fused to the CH2 and CH3 domains of human IgG1. Thus, by binding to CD2 on T cells, alefacept inhibits the crosstalk of APCs and T cells thereby inhibiting T-cell activation and inducing apoptosis of pathogenic CD45R0+ memory effector T cells (Strober & Menon, 2007). As the large majority of T lymphocytes in psoriatic lesions are CD45R0+, alefacept has been shown to be beneficial for the treatment of psoriasis and thus was the first Fc-fusion protein compound that gained FDA approval for the treatment of patients with moderate-to-severe plaque psoriasis in 2003 (Strober & Menon, 2007). Subsequently, it was shown that alefacept also depletes CD83+ and CD11c+ DCs and decreases the expression of proinflammatory genes within psoriatric skin lesions (Chamian et al., 2005). Non-neutralizing anti-alefacept antibodies have been detected in <1–6% of the patients having received up to five retreatment cycles with no significant increase in antibodies reported with subsequent retreatment (Gordon & Langley, 2003; Krueger et al., 2002; Lebwohl et al., 2003; Lowe et al., 2003; Roberts et al., 2010). Although generally well-tolerated, production and marketing of alefacept was discontinued in 2011, a decision which was, according to the manufacturer, neither a result of safety concerns nor due to an FDA-mandated or voluntary recall.
Abatacept (Orencia®, 2011), engineered as fusion between the ECD of human cytotoxic T lymphocyte-associated molecule-4 (CTLA-4) and the Fc fragment of human IgG1, was the first costimulation modulator approved by the FDA in 2005 for the treatment of otherwise therapy-refractory RA. As a mechanism of action, CTLA-4 binds to CD80 and CD86 on APCs with 100-fold higher avidity than the activating CD28 ligand and thereby out-competes CD28 mediated costimulation of T cells (Moreland et al., 2006). The efficacy of Abatacept has also been assessed in a variety of diseases besides RA (Kremer et al., 2003) including psoriatic arthritis (Mease et al., 2011), multiple sclerosis (Viglietta et al., 2008), diabetes (Orban et al., 2011), lupus erythematosus (Merrill et al., 2010), alopecia totalis (registered under clinical trials: NCT01314495) and most recently inflammatory bowel diseases (Sandborn et al., 2012). Belatacept (Nulojix®, 2011), which was FDA approved in 2011 for the treatment of kidney transplant rejection, was engineered as a modified form of abatacept in which the introduction of two amino acid alterations (L104E and A29Y) gave rise to slower dissociation of the compound from both CD80 and CD86. With this increased avidity, a 10-fold higher in vitro potency of belatacept has been documented (Larsen et al., 2005). Data from an integrated analysis of phase II and III trials on the immunogenicity of Abatacept report an incidence of anti-abatacept antibodies between 2.8% and 3% (Haggerty et al., 2007), some of which were found to possess neutralizing abilities toward the drug based upon in vitro assays (Haggerty et al., 2007). However, the available evidence indicates that immunogenicity of abatacept does not seem to affect therapeutic efficacy or drug safety (Haggerty et al., 2007). In a long-term extension of a phase II study, with a median follow-up of 5 years, anti-belatacept antibodies were reported with a frequency of 12% and 23% in patients receiving a 4-week and 8-week dosing, respectively. However, no seropositive patients, including two with belatacept neutralizing antibodies, experienced graft loss or serious infusion related side effects or autoimmune adverse events (Vincenti et al., 2010), indicating that these antibodies did not negatively impact treatment. Because both belatacept and abatacept potentially potently inhibit Treg function, which are highly dependent upon CD28 signalling, the relationship that this property has on immunogenicity remains to be understood (Mayer et al., 2012).
Rilonacept (Arcalyst®, 2010) is a so-called cytokine trap Fc-fusion protein, which incorporates the extracellular domains of both IL-1 receptor components, the C-terminal ligand binding region of the IL-1 receptor accessory protein (IL-1RAcP) and the N terminus of the IL-1 receptor type I extracellular region (IL-1RI), which are simultaneously linked to the Fc region of human IgG1 (Economides et al., 2003). Rilonacept functions as a soluble decoy receptor especially for IL-1β, to which it binds with 100-times greater affinity than the native receptor. Furthermore, it binds to IL-1RA and IL-1α, although with lower affinity compared to IL-1β. Rilonacept has orphan drug status and is approved for the treatment of cryopyrin-associated periodic syndromes, including familial cold autoimmflammatory syndrome and Muckle–Wells syndrome. Apart from these, rilonacept is currently being evaluated for the treatment of other IL-1β-driven diseases such as gout (Stahl et al., 2009). Anti-rilonacept antibodies have been reported with frequencies between 28% and 43%. However, most of the study subjects who tested positive for anti-rilonacept antibodies were negative at the study end, and the presence of antibodies had no observable adverse effects on drug activity or safety (Hoffman et al., 2008; Rilonacept SMPC, 2009).
Aflibercept (Eylea®, Zaltrap®) is a vascular endothelium growth factor (VEGF) trap that was engineered as a further evolution of a previous generation of vascular endothelial receptor 1 fused to Fc. Although the original VEGF trap was created by fusing the first three domains of VEGFR1 to the Fc of human IgG1, aflibercept was engineered as a fusion between the second Ig domain of human VEGFR1 and the third Ig domain of human VEGFR2 (Chu, 2009). This modification circumvented the poor pharmacokinetic profile and the non-specific interactions of the original VEGF trap. Aflibercept has thus proven to exhibit markedly improved pharmacokinetics (Holash et al., 2002) and has been shown to effectively inhibit angiogenesis by acting as a decoy receptor that binds VEGF-A and VEGF-B as well as placental growth factor (Papadopoulos et al., 2012). In 2011, aflibercept received approval for the treatment of neovascular age-related macular degeneration. For this indication, aflibercept is prepared as an ultra-purified and iso-osmotic drug specifically designed for intravitreal injection. Alternatively, formulations for intravenous administration of aflibercept are also available and are mainly utilized in oncologic medicine (Wang & Lockhart, 2012). In this respect, aflibercept has recently been approved for use in combination with a folinic acid, fluorouracil and irinotecan chemotherapy regimen to treat patients with metastatic colorectal cancer (Ciombor et al., 2013). After intravenous administration, various phase I–II oncology trials have reported the presence of anti-aflibercept antibodies in 0–7% of patients (Coleman et al., 2011; Diaz-Padilla et al., 2012; Freyer et al., 2012; Isambert et al., 2012). Similarly, aflibercept treatment of macular degeneration is associated with anti-aflibercept antibodies in 1–3% of patients, although no relationship to therapeutic efficacy or safety has been described (Eylea, 2011).
Romiplostim (Nplate®, 2011) is a thrombopoiesis-stimulating peptibody in which four identical peptides mimicking thrombopoietin have been linked to the Fc fragment of human IgG1 (two repeats to each chain of the Fc). However in contrast to the other Fc-fusions, the fusion partner, in this case thrombopoietin mimetic peptides, are linked to the C-terminus of the Fc region, and the protein is produced in Escherichia coli (Molineux & Newland, 2010). Importantly, through phage display screening, the peptide was chosen to have no homology to human thrombopoietin, but rather to possess a tertiary structure that would allow binding and activation of the human thrombopoietin receptor (Cwirla et al., 1997). Upon binding the thrombopoietin receptor, romiplostim stimulates platelet production in a dose-dependent manner and was designated as an orphan drug by the FDA in 2003. In 2008 romiplostim gained FDA approval for the long-term treatment of patients with chronic idiopathic thrombocytopenic purpura. No neutralizing antibodies against romiplostim or thrombopoietin were detected in phase I and II trials nor in two 24-week phase III studies (Molineux & Newland, 2010). In a long-term extension study, one patient developed neutralizing antibodies against romiplostim, but not against thrombopoietin. However, this patient exhibited no thrombocytopenia related to the presence of antibodies and anti-romiplostim antibodies were undetectable again 4 months after treatment discontinuation (Bussel et al., 2009).
Fc-fusion proteins and delivery approaches in clinical development
Apart from the above outlined approved therapeutics, a multitude of other Fc-fusion proteins are in clinical development with several of them having already reached advanced clinical trials (Supplementary Table 1). In the field of autoimmune diseases, atacicept was designed as a fusion protein between the extracellular domain of transmembrane activator and calcium-modulating ligand interactor and human IgG1. As such, atacicept neutralizes B lymphocyte stimulator (BLys) and a proliferation-inducing ligand (APRIL), both of which are critical in the maturation and development of B cells (Bracewell et al., 2009). In several phase Ia/Ib trials, atacicept exhibited a favorable safety and tolerability profile (Dall'Era et al., 2007; Munafo et al., 2007; Pena-Rossi et al., 2009; Tak et al., 2008). Despite the potent biological activity observed in these studies, including reduction of both B cells and immunoglobulin levels, atacicept failed to demonstrate clear clinical efficacy in two phase II/III trials in patients with RA (Genovese et al., 2011; van Vollenhoven et al., 2011). Furthermore, one of the most recent phase II/III trials on atacicept in patients with lupus nephritis who were concomitantly receiving newly initiated corticosteroids (CS) and mycophenolate mofetil (MMF) treatment was terminated prematurely after the enrollment of six patients due to an unexpected decline in serum IgG levels and the occurrence of serious infections (Ginzler et al., 2012). However, it is important to note that Atacicept itself was not identified as the direct cause of these large decreases in IgG levels, and that immunosuppression associated with the combination of CS and MMF administered to these patients is itself known to suppress immunoglobulin levels. In light of this, it is clear that the careful balancing of concurrent immunosuppressants is critical to preventing susceptibility toward serious infections and also that atacicept might represent a powerful agent, which might be especially beneficial in the treatment of IgG-mediated diseases, such as active nephritis.
Blisibimod (AMG-623) is a BLys antagonist currently in development. Unlike Atacicept, Blisibimod is a peptidobody, in which a peptide selected for its ability to bind with high affinity only to BLyS (but not to APRIL) has been fused to an IgG Fc fragment (Stohl, 2012). In a double-blind and placebo-controlled phase I trial among systemic lupus erythematosus (SLE) patients, blisibimod led to a dose-independent decrease in naive and total peripheral blood B cells as well as an increase in memory B cells (Stohl & Hilbert, 2012). In a Phase IIb dose-finding study known as the PEARL SC trial (registered under NCT01162681), a strong trend toward improved clinical response at week 20 in blisibimod-treated SLE patients was observed, with the most favorable effects observed in patients with the most severe disease activity. Based on these results, a phase III trial has recently been initiated in patients with severe SLE (registered under NCT01395745). Furthermore, phase II/III trials are currently evaluating blisibimod for the treatment of immune thrombocytopenia (registered under NCT01609452) and, in combination with methotrexate for the treatment of anti-neutrophil cytoplasmic antibody-associated vasculitis (registered under NCT01598857).
Two therapeutics that are currently in clinical development belong to the class of small modular immunopharmaceutical proteins (SMIPs). SMIPs represent a novel proprietary substance class, consisting of a single chain variable fragment linked via a hinge region to an effector domain, usually a reduced Fc fragment (Zhao et al., 2007). The molecular mass is thus only half to one-third that of mAbs, while Fc-mediated effector functions are retained. The two SMIPs in clinical development are both engineered to specifically bind CD20 on pre-B and mature B lymphocytes and thereby induce B-cell lysis. TRU-015, engineered as a fusion of a single-chain Fv specific for CD20 to an Fc fragment devoid of CH1 and CL has, by design, a decreased ability to engage complement components and complement-dependent cytotoxicity but retains strong antibody-dependent cytotoxicity (ADCC) and potent direct signaling activity in vivo (Burge et al., 2008). It has been shown that it can effectively deplete B lymphocytes in a dose-dependent manner in non-human primates and improve survival in mouse xenograft tumor models (Hayden-Ledbetter et al., 2009). In an open-label dose escalation phase I trial in patients with RA, TRU-015 was associated with dose-dependent B-cell depletion and found to have an acceptable tolerability profile (Burge et al., 2008). SBI-087 is a further SMIP directed against CD20 on B cells, which is currently being evaluated in phase I studies in patients with SLE (registered under clinical trials NCT00714116) and in phase II trials in seropositive patients with RA (registered under clinical trials NCT01008852). Initial results suggest that SBI-087 is not only relatively well-tolerated and safe but also induces robust B-cell depletion upon subcutaneous administration (Fleischmann et al., 2010).
In the field of hematologic diseases, recombinant FVIII and FIX have been generated as the first clinical examples of Fc-fusion proteins engineered as monomers. In this case, either one effector molecule of recombinant FVIII or recombinant FIX has been covalently linked through the carboxy-terminus to the N-terminus of an Fc dimer to form recombinant FVIII Fc fusion protein (rFVIIIFc) and recombinant FIX Fc fusion protein (rFIXFc), respectively (Figure 4). Importantly, when compared to similar doses of the rFIXFc dimer, the monomer was more efficiently retained in the circulation and exhibited a longer terminal half-life in rodents (Dumont et al., 2009). These markedly improved pharmacokinetics of the rFIXFc monomer are likely due to changes in the overall properties of the molecule (i.e. reduced size and reduced negative charge) leading to more efficient transport through the recycling pathway. In addition, it has been shown that the half-life of the rFIXFc monomer is approximately three- to fourfold longer than that of non-fused human rFIX when evaluated across different species including non-human primates (Peters et al., 2010). Importantly, in FcRn knock-out mice, the half-life of rFIXFc was found to be as short as that of rFIX, thereby confirming that the interaction between FcRn and the Fc fragment is responsible for conferring protection of the Fc-fusion proteins from degradation (Peters et al., 2010). Based on these observations, a multicenter phase I/IIa clinical trial on rFIXFc was conducted and the results have recently been published (Shapiro et al., 2012). In 14 patients with hemophilia B, rFIXFc monomer was well tolerated, and no FIX inhibitors or anti-rFIXFc antibodies were detected in any subject. Furthermore, rFIXFc monomer exhibited a markedly longer half-life and mean residence time than that reported for current rFIX products, resulting in rapid and prolonged FIX activity provided by the fusion protein (Shapiro et al., 2012). A multicenter global phase III clinical trial assessing clinical efficacy of different extended interval dosing regimens has recently been completed (Powell et al., 2013).
A similar rescue by creation of a Fc-fusion has been shown for rFVIII. Fusing a B-domain deleted rFVIII with an IgG1 Fc fragment as a monomer provided an approximate twofold longer half-life in hemophilia A mice and dogs, which was shown to require FcRn, while the specific FVIII activity was not altered by the fusion (Dumont et al., 2012). Furthermore, consistent with prolonged half-life, rFVIIIFc monomer exhibited an ~twofold longer prophylactic efficacy in mice with hemophilia A and a sustained protection from venous bleeding in a tail injury model twofold longer than non-fused rFVIII (Dumont et al., 2012). In a multicenter phase I/IIa trial, safety and pharmacokinetics were assessed (Powell et al., 2012). Apart from being well-tolerated in 16 subjects with severe hemophilia A, rFVIIIFc monomer possessed a considerably longer half-life and a lower clearance rate compared to rFVIII (Powell et al., 2012). Based on these results, a phase III trial has been recently completed, which identified and established extended interval prophylaxis dosing regimens (Mahlangu et al., 2013).
Although the above-mentioned rFIX- and rFVIII-fusion proteins consist of a monomeric effector molecule linked to dimeric Fc fragment of IgG1, results of a recent study indicate that a monomeric Fc region can retain the ability to bind to FcRn (Ying et al., 2012). By combining structure-based rational protein design with multiple screening strategies, three human monomeric IgG1 Fc fragments were produced that were highly soluble and exhibited pH-dependent FcRn binding in a manner akin to Fc dimers (Ying et al., 2012). This approach thus offers the possibility of engineering even smaller, and therefore potentially more tissue-penetrant, therapeutics that will still possess enhanced circulating half-life due to interaction with FcRn. Moreover, it has been shown recently in in vivo experiments that a short FcRn-binding peptide consisting of a 16 amino acid sequence conferred pH-dependent binding and FcRn-dependent recycling and transcytosis to a model fluorescent protein when genetically linked to this protein, thereby potentially representing a further strategy in engineering therapeutic proteins with FcRn-dependent half-life extension and low molecular weight (Sockolosky et al., 2012).
Mucosal delivery of therapeutic Fc-fusion proteins
In addition to the aforementioned improvements in pharmacokinetics and pharmacodynamics conferred by IgG-FcRn, interactions following administration of Fc-fusion proteins systemically, clinical exploitation of this receptor–ligand interaction has already been successfully explored as a noninvasive delivery route for therapeutics. This capitalizes upon the knowledge that FcRn is functionally expressed throughout adult life in the upper and central airways as well as intestines of humans and non-human primates (Bitonti et al., 2004; Dickinson et al., 1999; Israel et al., 1996; Spiekermann et al., 2002) and raises the possibility that therapeutic proteins when linked to Fc can be delivered noninvasively across mucosal tissues by a process called transcytosis (Figure 5) (Dumont et al., 2006, 2009). FcRn, in fact, is one of the few molecules known to move from luminal to serosal membranes of the polarized cells that form the epithelial barriers of the lung and intestine (reviewed in (Baker et al., 2009)); thus providing a unique opportunity for study of apical to basolateral transcytosis and a potent opportunity for the non-invasive delivery of protein therapeutics.
Figure 5.

FcRn-mediated transcytosis across polarized epithelial monolayers. In polarized epithelia such as intestinal epithelial cells, IgG molecules and Fc-fusion proteins can be recycled from the basolateral and apical membrane, from which FcRn rapidly directs IgG molecules into the apical and basal early endosomes (AEE and BEE) and the common recycling endosome (CRE). As such, FcRn is a receptor capable of bidirectional transcytosis. From the CRE, the actin motor myosin Vb and the GTPase Rab25 regulate a sorting step that specifies transcytosis in both directions without affecting recycling (Tzaban et al., 2009). Rab11a as a further regulatory component of the RE regulates a sorting step for recycling FcRn to the basolateral membrane, but it is dispensable for transcytosis (Tzaban et al., 2009). Thus the recycling and transcytosis pathway for IgG are distinct in polarized cells and display an inherent polarity.
The first study demonstrating FcRn-dependent absorption of therapeutic molecules across mucosal tissues was performed in mice using a fusion of erythropoietin (Epo) with the human Fc-domain (Spiekermann et al., 2002). In subsequent studies using cynomolgus monkeys, pulmonary delivery of a human Epo Fc-fusion protein (EpoFc) as a dimer or as a monomer was successfully accomplished when the respective fusion proteins were aerosolized with a particle size of 4–6 μm to target the upper airways (Bitonti et al., 2004). In both mice and monkeys, the FcRn-dependence of this transepithelial transport was confirmed by demonstrating that an EpoFc-fusion, which was mutated in three amino acids in the Fc domain critical for FcRn binding (I253A, H310A and H435A), was only poorly absorbed when administered either to the upper airways or deep lung (Bitonti et al., 2004). Also consistent with this FcRn-dependence, the wild-type protein was poorly absorbed when delivered to the deep lung, reflecting that the majority of the FcRn expression is in the upper and central airways and required for transport (Bitonti et al., 2004). Furthermore, similar to the observations made for rFIXFc-fusion protein, reducing the size and charge of the EpoFc by generating a monomeric fusion protein with a single Epo conjoined to a dimeric Fc subunit resulted in a striking increase in transport of the molecule across the lung epithelium of the monkey (Bitonti et al., 2004). Confirmation that this pathway functions similarly in humans was obtained from the successful completion of a phase I clinical trial assessing the efficacy of delivery of an EpoFc-fusion protein into the bloodstream of normal healthy volunteers in which transport of the EpoFc-fusion proteins in the upper airways was shown to occur in a dose-dependent fashion and induce a reticulocytosis (Dumont et al., 2005). These studies have laid the foundation for the broader possibility of transepithelial delivery of different types of macromolecular cargo when fused to the Fc fragment of IgG. The feasibility of this has indeed been confirmed for Fc-fusion proteins associated with not only Epo but also interferon-α, interferon-β and follicle-stimulating hormone (Bitonti & Dumont, 2006; Low et al., 2005; Vallee et al., 2012). Globally, these studies demonstrate that an FcRn-dependent alternative to invasive drug delivery is both potent and clinically feasible.
As has been shown recently, the interaction between IgG and FcRn can not only be utilized for drug delivery but also for the acquisition of humoral immunity at mucosal sites. Based on FcRn's ability to bi-directionally transport IgG molecules across the epithelium lining mucosal barriers such as the intestines, genitourinary tract, the stomach and the lung, it has been shown that FcRn can confer passive immunity against viral or bacterial infections at the site of pathogen entry when neutralizing antibodies were administered to the systemic circulation before the infection (Bai et al., 2011; Ben Suleiman et al., 2012; Li et al., 2011). In this manner, it is expected that such antibodies will be actively transported from a basal-to-apical direction across the polarized epithelium into the lumen as has been demonstrated in animal model systems (Yoshida et al., 2004, 2006). Beyond passive immunity, FcRn can also promote active immunity when pathogens are presented as Fc-fusion proteins together with potent adjuvants to artificially force an immune response. Intranasal immunization of mice with a fusion protein containing the extracellular domain of herpes-simplex virus (HSV) and the Fc fragment of IgG2a in the presence of an adjuvant to disrupt tolerance was found to induce cellular and humoral responses that protected against intravaginal HSV challenge, even 6 months after immunization (Ye et al., 2011). In a similar manner, intranasal vaccination with a fusion between HIV Gag protein p24 and an IgG heavy chain together with an adjuvant led to local and systemic immunity, including durable B- and T-cell memory and Gag-specific immunity, which was sufficiently potent to protect against an intravaginal challenge with recombinant vaccinia virus expressing the HIV Gag protein (Lu et al., 2011). Of note, mice lacking FcRn or those immunized with free antigen or an antigen fused to an Fc fragment disabled in FcRn binding were not protected against HSV or HIV infection, thereby highlighting the central role of FcRn–IgG interactions for efficient mucosal transport across an epithelial barrier (Lu et al., 2011; Ye et al., 2011).
Modulating FcRn–IgG binding to increase half-life of antibodies and Fc-fusion proteins
Based on these novel FcRn-dependent approaches for drug delivery and mucosal vaccination, it is evident that modulating FcRn–IgG interactions is a highly tractable and promising engineering strategy for the generation of optimized and application-specific therapeutics and vaccines. Increasing the FcRn–IgG interactions would help to increase the therapeutic half-life and plasma levels of an Fc-containing compound, thereby potentially reducing the frequency of administration, whereas decreasing the affinity to FcRn would reduce circulating half-life, a circumstance especially desirable for generating diagnostic molecules with reduced toxicity. Following internalization from the bloodstream by fluid-phase pinocytosis, IgG binds to FcRn in acidic endosomal compartments where FcRn predominantly resides (see Figure 3a). This results in the trafficking of this complex to the cell surface where the neutral pH during exocytosis facilitates the release of IgG back into the bloodstream as summarized elsewhere (Ober et al., 2004b; Roopenian & Akilesh, 2007; Roopenian et al., 2003; Ward & Ober, 2009; Ward et al., 2003). In the absence of FcRn rescue, soluble molecules taken up by pinocytosis are instead directed into an active lysosomal degradation pathway and permanently lost from circulation as discussed above.
Importantly, when considering the physiological constraints of FcRn-mediated IgG recycling, it has become clear that Fc mutants created with increased binding to FcRn at both acidic and neutral pH do not exhibit a prolonged circulating half-life (Dall'Acqua et al., 2002; Gurbaxani et al., 2006). In fact, increased IgG–FcRn binding at pH 7.4 prevents IgG dissociation from FcRn and its release into the bloodstream, rather directing IgG into a degradation pathway. Such engineered antibodies (Abdeg's or antibodies that enhance degradation) have shown utility in the treatment of antibody-mediated autoimmune diseases in animal models (Patel et al., 2011). As such, the rate of dissociation at pH 7.4 is considered to be at least equally important in determining serum half-life as the binding at acidic pH (Wang et al., 2011). Therefore, for the engineering of IgG-based therapeutics designed to have improved binding to FcRn and longer half-life, it is of critical importance to improve binding at acidic, but not neutral, pH.
To this end, several different mutations in the IgG Fc domain that selectively modify and increase binding at pH 6.0 without altering binding and dissociation rates at pH 7.4 have been described and are summarized in Figure 4 and Supplementary Table 2.
M252/S254/T256 mutations
Introducing the triple substitution YTE (M252Y/S254T/T256E) in the CH2 domain of the humanized anti-respiratory syncytial virus antibody palivizumab (MEDI-493) has been shown to increase the binding to human FcRn by about 10-fold at pH 6.0 while still allowing efficient release at pH 7.4 (Dall'Acqua et al., 2002). In vivo studies have shown that this YTE substitution in MEDI-524, an affinity matured variant of palivizumab, resulted in a fourfold increase in serum half-life in non-human primates (Dall'Acqua et al., 2006). Importantly, the increased serum persistence of YTE in serum was accompanied by a fourfold higher antibody concentration in broncho-alveolar fluid, the expected active site of the therapeutic and consistent with enhanced FcRn-mediated transcytosis in a basal-to-apical direction in vivo (Dall'Acqua et al., 2006). Apart from enhanced FcRn-dependent binding through the introduction of the YTE triple mutation, crystal structure analysis has confirmed the formation of favorable surface contact and hydrogen bonding surface between FcRn and palivizumab (Oganesyan et al., 2009). Furthermore, the YTE mutation has been described to significantly reduce the binding to human FcγRIIIA as well as concomitant ADCC activity (Dall'Acqua et al., 2006), an observation that is in accordance with evidence that amino acid residues 254 and 256 are implicated in IgG binding to FcγRIIIA (Shields et al., 2001).
T250/M428 mutations
The amino acids T250 and M428 are conserved among all human IgG subclasses. When mutated to T250Q and M428L (QL), initial studies reported markedly increased binding of an anti-hepatitis B virus IgG2-QL mutant to human FcRn at pH 6.0 with retained pH-dependent release that resulted in an approximately twofold increase in serum half-life in rhesus monkeys (Hinton et al., 2004). In subsequent studies, similar results were obtained when the QL mutation was engineered into human IgG1 variants. Here, the antibodies with the QL mutation exhibited an approximately 29-fold increase in FcRn binding and a 2.5-fold increase in serum half-life in rhesus monkeys without alterations in antigen binding or cellular effector functions (Hinton et al., 2006). Compared to human IgG1 and IgG2, the introduction of the QL double mutation into IgG4 resulted in an even more pronounced improvement in binding to human FcRn (Hinton et al., 2006). Surprisingly, when the same mutation was engineered into an anti-TNF antibody, leading to increased and pH-dependent binding to murine and cynomolgus FcRn by factor 500 and 40, respectively, serum half-life of the IgG1 QL mutant was not found to be extended in cynomolgus monkeys compared to the wild-type anti-TNF counterparts (Datta-Mannan et al., 2007). This observation remains as a conundrum but raises the possibility of increased secretion via transcytosis into mucosal secretions as a potential explanation.
H433/N434 mutations
The two residues, His433 and Asn434, are in direct proximity to amino acids such as His435 that play a central role in human FcRn–IgG interactions and have thus been intensively used in engineering Fc regions with high FcRn affinity. In an initial report where His433 and Asn434 were mutated to lysine and phenylalanine, respectively (H433K/N434F), the mutation conferred an approximately 16-fold increase in binding to human FcRn at pH 6.0 while maintaining a very low affinity at pH 7.2 (Vaccaro et al., 2006) and importantly, preserved efficient recycling and transcytosis in human systems. In contrast, the same mutant exhibited increased binding affinities at both pH 6.0 and pH 7.2 to mouse FcRn and was thus not efficiently recycled and had a significantly shorter half-life than that of wild-type IgG in murine systems (Vaccaro et al., 2006). These observations thereby substantiate that species-specific differences need to be taken into account when assessing binding to FcRn and further that an IgG engineered with strong FcRn binding at all pH levels functions rather as an FcRn blocker that enhances IgG degradation. The latter notion is supported by a report directly comparing two different Fc mutations at N434 in a fusion protein consisting of anti-human epidermal growth factor receptor 2 and IgG1 (Trastuzumab). The mutation N434A, possessing a fourfold increased binding to both human and non-human primate FcRn without binding at pH 7.4, exhibited twofold increased serum half-life in cynomolgus monkeys compared to native IgG whereas N434W, with 80-fold increased binding at pH 6 and significant binding also at pH 7.4, did not show any prolongation in serum half-life (Yeung et al., 2009). Variants of a TNF-IgG1 fusion with N434A and N434H mutations further substantiate this concept. Specifically, N434H, with the highest binding affinity to murine FcRn at pH 7.4, exhibited a faster clearance and a lower bioavailability compared to native and N434A TNF–IgG1 (Deng et al., 2010). In contrast when translated into a primate system, these variants both exhibited enhanced binding affinity at pH 6.0, but not pH 7.4, to monkey FcRn and were observed to have significantly higher areas under the serum concentration-time curve in monkeys than did native TNF-IgG1 (Deng et al., 2010).
T307/E380/N434 mutations
Simultaneous mutation of the residues T307, E380 and N434 to alanines (AAA) has been shown to increase binding to human FcRn at pH 6.0 while leaving binding at pH 7.4 unaltered (Petkova et al., 2006, Shields et al., 2001). When the combined AAA set of mutations were engineered into a trastuzumab IgG1 fusion protein (Hu4D5–IgG1) and comparatively analyzed in humanized mice transgenic for human FcRn (hFcRn Tg) along with N343A and a mutation disabling FcRn binding (I253A), the N434A and AAA mutants displayed a two- to threefold extended serum half-life in vivo compared with native antibody, whereas the I253A mutated variant exhibited a very short in vivo half-life consistent with disruption of FcRn binding (Petkova et al., 2006). Furthermore, in an antibody-based arthritis model in which arthritis was induced by passive transfer of plasma from a patient with active RA into hFcRn Tg mice, which were also deficient in FcγRIIB, only the AAA variant engineered into IgG was able to ameliorate experimental arthritis (Petkova et al., 2006). Thus, strengthening the interaction between FcRn and IgG molecules can be exploited as an anti-autoimmune therapy to promote the clearance of endogenous pathogenic IgG.
V259/V308F/M428/N434 mutations
In a recent report, improved binding of Fc-fusion proteins to FcRn has been linked for the first time to enhanced therapeutic efficacy (Zalevsky et al., 2010). In this report, a series of Fc variants with greater affinity for human FcRn were engineered into an anti-VEGF IgG1 fusion, which is currently approved for the treatment of colorectal, lung, breast and renal cancers (Bevacizumab, Avastin®). A fusion protein variant with the M428L/N434S mutations (LS) conferred 11-fold improvement in FcRn binding affinity at pH 6.0 and an approximately threefold improvement in serum half-life relative to native IgG1 in cynomolgus monkeys. Similar results were also obtained by incorporating the V259I/V308F/M428L mutations (IFL), which resulted in a 20-fold increase in FcRn binding affinity at pH 6.0 and a similar threefold increase in half-life (Zalevsky et al., 2010). When the same LS mutation was engineered into an anti-EGFR IgG1 fusion (Cetuximab, Erbitux®), similar results for enhanced FcRn binding and serum half-life extension were observed while binding to the target, human EGFR antigen, was unperturbed (Zalevsky et al., 2010). Importantly, when these variants were introduced to a model of SKOV-3 or A431 tumors in humanized FcRn transgenic mouse on a Rag−/− background, a significant reduction in tumor volume was observed upon administration of both LS variants compared to their wild-type therapeutic counterparts (Zalevsky et al., 2010). Hence, this study illustrates, for the first time, a positive correlation between pharmacokinetic enhancement and in vivo efficacy and provides a forward look on how powerful FcRn-enabling Fc engineering can be for the generation of more effective therapeutics (Zalevsky et al., 2010).
One note of caution should be taken in broadly concluding that these high affinity, pH-dependent variants can extend the half-life of all antibodies and Fc-fusion proteins, however, as the effect of these variants also depends upon the context of the particular protein. A recent report (Igawa et al., 2010) demonstrated that the half-life of the antibody tocilizumab was limited by the fact that once bound to its membrane-associated IL-6R target, the antibody/IL-6R complex would be degraded in the lysosome, allowing only one antigen-binding event in the antibody's lifetime (Igawa et al., 2010). However, by introducing mutations into the antigen-binding region to cause the release under the acidic conditions of the endosome, the antibody could then be recycled back to the cell surface and thus the half-life extended, as well as allowing the antibody to now bind multiple IL-6R targets in its circulating lifetime. Interestingly, and of particular relevance to the current topic, incorporating the N434A described above did not improve the half-life of an IgG with the native tocilizumab antigen binding region but only for the IgG with the pH-dependent binding region (Igawa et al., 2010). From this, one can see that depending on the specificity of the antibody and characteristics of its interaction with its target, these high affinity, pH-dependent variants may not enable half-life extension for all antibodies. In a similar fashion, the effect in the context of an Fc-fusion protein can depend on the nature of the clearance receptor interactions for the particular protein. In fact, incorporation of various high affinity variants into the context of rFIXFc and rFVIIaFc has failed to show any significant prolongation of half-life when examined in transgenic mice or cynomolgus monkeys, while successfully extending the half-life of a control antibody. This lack of effect could be due to the particular pathways that contribute to the clearance of these two clotting factors, which are likely different from those pathways responsible for antibody clearance.
Immunoregulatory properties and immunogenicity of Fc-fusion proteins with respect to FcRn binding
Advances in antibody engineering from chimeric to humanized and finally fully human antibodies have led to major improvements in the generation of safer and less immunogenic therapeutics. In this respect, it is generally acknowledged that the elimination of rodent sequences from antibody-based therapeutics reduces the frequency of anti-antibody responses (AAR) (Hwang & Foote, 2005). Thus, replacement of mouse IgG Fc regions with those of human origin leads to the largest immunogenicity reduction (Hwang & Foote, 2005). As discussed above, approved and in-development Fc-fusion proteins can evoke AARs in relatively low numbers of subjects and are generally well-tolerated. Due to their human Fc regions and therefore the minimal presence of foreign antigens, it would be tempting to assume that these compounds might escape immune recognition and thus have led to the call that Fc-fusion proteins are a relatively “safe” therapeutic substance class (De Groot & Scott, 2007). But can this hold true? The process of eliciting an immune response and generating immunogenicity is rather complex and not limited to the simplified division into “foreign” and “self”. In this respect, factors intrinsic to the therapeutic substance such as presence and number of T-cell epitopes or regulatory epitopes, the chemical formulation and aggregation, the above discussed post-translational modifications and the localization of the therapeutic target within the body (soluble versus membrane-bound) and extrinsic factors such as the HLA status, the route of administration or the concomitant use of (immunosuppressive) medications are all known key factors that influence immunogenicity of a therapeutic substance (De Groot & Scott, 2007; Scott & De Groot, 2010). For the elicitation of a classical adaptive immune response, the immunogenic sequence or protein is taken up by APCs, which upon processing of the protein, load some of the resulting peptides onto MHC molecules for presentation to T cells, which may, in the presence of appropriate co-stimulatory signals, become activated and subsequently interact with B-cells. Despite the fact that many Fc-fusion proteins can be considered autologous, many nonetheless have certain unique sequences in either the Fc fragment due to allo- or isotypic variation (Magdelaine-Beuzelin et al., 2009), at the junction of the therapeutic agent and Fc domain or within the therapeutic domain that is linked to the Fc fragment again due to allelic variation or mutations/ deletions in the case of genetic replacement therapies (Hwang & Foote, 2005; Oldenburg & Pavlova, 2006). Therefore, such proteins may contain T-cell epitopes not presented during thymic maturation and thereby have the potential to generate reactive T-cells. In the light of these considerations, aspects of immunogenicity in fusion proteins that enable FcRn binding require particular attention. Recently, comprehension of the breadth of FcRn's functions has dramatically changed: away from a simply recycling and transcytosis receptor to a molecule that is critically involved in generating immunological responses by shuttling immune complexes across epithelial barriers as well as toward MHC-I and MHC-II presentation pathways in professional APCs such as dendritic cells (Baker et al., 2011, 2012; Qiao et al., 2008; Yoshida et al., 2004, 2006). Therefore, it will be critical to determine whether therapeutics engineered with high affinity for FcRn, which will exhibit the desired extension of circulating half-life, will also elicit stronger immunogenicity, a side effect that might result from the concomitant enabling of more efficient antigen presentation of the high-affinity FcRn therapeutics if the antigen to which the therapeutic binds is repetitive as in the case of a tumor- or infection-associated target molecule. While this concern is being addressed, it is critical to continue efforts to identify immunogenic and tolerogenic properties of IgG-based therapeutics (De Groot & Martin, 2009; De Groot & Scott, 2007; Koren et al., 2007; Scott & De Groot, 2010), to modify or deimmunize critical immunogenic residues (Collen et al., 1996a,b; De Groot & Martin, 2009; Scott & De Groot, 2010; Tangri et al., 2002) and to explore novel engineering pathways enabling the design of therapeutics, which can drive tolerogenic immune responses. Indeed, this has recently been described for the expansion of regulatory T-cell populations by certain residues within the IgG Fc fragments as discussed previously in this review (De Groot et al., 2008). These so-called “Tregitopes” have been shown to activate regulatory FoxP3+ cells and to suppress effector T cells in vitro and in vivo and thus represent a prime example of how IgG Fc engineering can not only be utilized to optimize pharmacokinetics but also to reduce side effects and immunogenicity. In therapeutics developed and utilized clinically to date, all with native Fc, immunogenicity appears to be minimal, as discussed, making them a highly desirable class of therapeutic agents.
Concluding remarks
IgG-based therapeutics and antibodies represent one of the most rapidly evolving substance classes. Following the success of first-generation antibodies, engineering efforts have resulted in the generation of Fc-fusion proteins in which one or more effector molecules are linked to an IgG-Fc region. As such, Fc-fusion proteins combine the benefits of a broader repertoire of therapeutic targets with considerable improvements in therapeutic half-life and in vivo stability provided by the interaction with FcRn. Most recently, in the search for improved peptides, proteins and antibodies, engineering strategies have recognized that the IgG–FcRn interaction is as highly tractable path for the generation of optimized and application-specific therapeutics with improved properties. In this respect, selective mutations within the Fc region of IgG-based therapeutics can not only enhance circulating half-life of therapeutics through increased binding to FcRn but also confer superior therapeutic efficacy in vivo in specific cases. Furthermore, recent studies have demonstrated that the interaction between IgG and FcRn can be successfully co-opted for the non-invasive, potent and clinically feasible delivery of drugs and antibodies for passive acquisition of humoral immunity at mucosal sites. Finally, after a review of the clinical experience with Fc-fusion proteins together with the known potential immunoregulatory properties of IgG, Fc-fusion proteins are an increasingly recognized class of safe and useful therapeutic proteins.
Supplementary Material
Acknowledgments
T. R. receives support from the German Research Foundation Grant no. RA 2040/1-1. K. B. was supported by The Canadian Institutes for Health Research. J. A. D., R. T. P., H. J. and G. F. P. are employees of, and hold equity in Biogen Idec S-W. Q. has no conflict of interest to disclose. W. I. L. is supported by NIH DK48106, DK084424, DK090603 and Harvard Digestive Diseases Center (NIH P30DK034854) and receives licensing payments from Biogen Idec. R. S. B. is supported by NIH DK44319, DK51362, DK53056, DK88199 and Harvard Digestive Diseases Center (NIH P30DK034854) and receives licensing payments from and is a consultant to Biogen Idec.
Footnotes
Declaration of interest The authors report no further conflicts of interest. The authors alone are responsible for the content and writing of this article.
References
- Abrahamson DR, Powers A, Rodewald R. Intestinal absorption of immune complexes by neonatal rats: a route of antigen transfer from mother to young. Science. 1979;206:567–9. doi: 10.1126/science.493961. [DOI] [PubMed] [Google Scholar]
- Adamski FM, King AT, Demmer J. Expression of the Fc receptor in the mammary gland during lactation in the marsupial Trichosurus vulpecula (brushtail possum) Mol Immunol. 2000;37:435–44. doi: 10.1016/s0161-5890(00)00065-1. [DOI] [PubMed] [Google Scholar]
- Aggarwal S. What's fueling the biotech engine – 2010 to 2011. Nature Biotechnol. 2011;29:1083–9. doi: 10.1038/nbt.2060. [DOI] [PubMed] [Google Scholar]
- Ahouse JJ, Hagerman CL, Mittal P, et al. Mouse MHC class I-like Fc receptor encoded outside the MHC. J Immunol. 1993;151:6076–88. [PubMed] [Google Scholar]
- Akilesh S, Christianson GJ, Roopenian DC, Shaw AS. Neonatal FcR expression in bone marrow-derived cells functions to protect serum IgG from catabolism. J Immunol. 2007;179:4580–8. doi: 10.4049/jimmunol.179.7.4580. [DOI] [PubMed] [Google Scholar]
- Amevive® [accessed 24 Apr 2012];Prescribing information. 2011 Available from: http://www.astellas.us/docs/amevive.pdf. last.
- Anderson CL, Chaudhury C, Kim J, et al. Perspective – FcRn transports albumin: relevance to immunology and medicine. Trends Immunol. 2006;27:343–8. doi: 10.1016/j.it.2006.05.004. [DOI] [PubMed] [Google Scholar]
- Anthony RM, Kobayashi T, Wermeling F, Ravetch JV. Intravenous gammaglobulin suppresses inflammation through a novel T(H)2 pathway. Nature. 2011;475:110–3. doi: 10.1038/nature10134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anthony RM, Wermeling F, Karlsson MC, Ravetch JV. Identification of a receptor required for the anti-inflammatory activity of IVIG. Proc Natl Acad Sci U S A. 2008;105:19571–8. doi: 10.1073/pnas.0810163105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arcalyst [accessed 24 Apr 2012];Prescribing information. 2010 Available from: http://www.regeneron.com//ArcalystPI-US-Summary-EMEA/ArcalystPI_June_2010r3-1.pdf. last.
- Arnold JN, Wormald MR, Sim RB, et al. The impact of glycosylation on the biological function and structure of human immunoglobulins. Ann Rev Immunol. 2007;25:21–50. doi: 10.1146/annurev.immunol.25.022106.141702. [DOI] [PubMed] [Google Scholar]
- Bai Y, Ye L, Tesar DB, et al. Intracellular neutralization of viral infection in polarized epithelial cells by neonatal Fc receptor (FcRn)-mediated IgG transport. Proc Natl Acad Sci U S A. 2011;108:18406–11. doi: 10.1073/pnas.1115348108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker K, Qiao SW, Kuo T, et al. Immune and non-immune functions of the (not so) neonatal Fc receptor, FcRn. Semin Immunopathol. 2009;31:223–36. doi: 10.1007/s00281-009-0160-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker K, Qiao SW, Kuo TT, et al. Neonatal Fc receptor for IgG (FcRn) regulates cross-presentation of IgG immune complexes by CD8-CD11b+ dendritic cells. Proc Natl Acad Sci U S A. 2011;108:9927–32. doi: 10.1073/pnas.1019037108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker K, Rath T, Lencer WI, et al. Cross-presentation of IgG-containing immune complexes. Cell Mol Life Sci. 2012;70:1319–34. doi: 10.1007/s00018-012-1100-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beck A. Biosimilar, biobetter and next generation therapeutic antibodies. MAbs. 2011;3:107–10. doi: 10.4161/mabs.3.2.14785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beck A, Wurch T, Bailly C, Corvaia N. Strategies and challenges for the next generation of therapeutic antibodies. Nat Rev Immunol. 2010;10:345–52. doi: 10.1038/nri2747. [DOI] [PubMed] [Google Scholar]
- Beck A, Wurch T, Corvaia N. Therapeutic antibodies and derivatives: from the bench to the clinic. Curr Pharm Biotechnol. 2008;9:421–2. doi: 10.2174/138920108786786420. [DOI] [PubMed] [Google Scholar]
- Ben Suleiman Y, Yoshida M, Nishiumi S, et al. Neonatal Fc receptor for IgG (FcRn) expressed in the gastric epithelium regulates bacterial infection in mice. Mucosal Immunol. 2012;5:87–98. doi: 10.1038/mi.2011.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bitonti AJ, Dumont JA. Pulmonary administration of therapeutic proteins using an immunoglobulin transport pathway. Adv Drug Deliv Rev. 2006;58:1106–18. doi: 10.1016/j.addr.2006.07.015. [DOI] [PubMed] [Google Scholar]
- Bitonti AJ, Dumont JA, Low SC, et al. Pulmonary delivery of an erythropoietin Fc fusion protein in non-human primates through an immunoglobulin transport pathway. Proc Natl Acad Sci U S A. 2004;101:9763–8. doi: 10.1073/pnas.0403235101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borel Y. Haptens bound to self IgG induce immunologic tolerance, while when coupled to syngeneic spleen cells they induce immune suppression. Immunol Rev. 1980;50:71–104. doi: 10.1111/j.1600-065x.1980.tb00308.x. [DOI] [PubMed] [Google Scholar]
- Borys MC, Dalal NG, Abu-Absi NR, et al. Effects of culture conditions on N-glycolylneuraminic acid (Neu5Gc) content of a recombinant fusion protein produced in CHO cells. Biotechnol Bioeng. 2010;105:1048–57. doi: 10.1002/bit.22644. [DOI] [PubMed] [Google Scholar]
- Bracewell C, Isaacs JD, Emery P, Ng WF. Atacicept, a novel B cell-targeting biological therapy for the treatment of rheumatoid arthritis. Expert Opin Biol Ther. 2009;9:909–19. doi: 10.1517/14712590903033919. [DOI] [PubMed] [Google Scholar]
- Brambell FW. Resemblances between passive anaphylactic sensitization and transmission of passive immunity. Nature. 1963;199:1164–6. doi: 10.1038/1991164a0. [DOI] [PubMed] [Google Scholar]
- Brambell FW, Hemmings WA, Morris IG. A theoretical model of gamma-globulin catabolism. Nature. 1964;203:1352–4. doi: 10.1038/2031352a0. [DOI] [PubMed] [Google Scholar]
- Burge DJ, Bookbinder SA, Kivitz AJ, et al. Pharmacokinetic and pharmacodynamic properties of TRU-015, a CD20-directed small modular immunopharmaceutical protein therapeutic, in patients with rheumatoid arthritis: a Phase I, open-label, dose-escalation clinical study. Clin Ther. 2008;30:1806–16. doi: 10.1016/j.clinthera.2008.10.017. [DOI] [PubMed] [Google Scholar]
- Burmeister WP, Gastinel LN, Simister NE, et al. Crystal structure at 2.2 A resolution of the MHC-related neonatal Fc receptor. Nature. 1994a;372:336–43. doi: 10.1038/372336a0. [DOI] [PubMed] [Google Scholar]
- Burmeister WP, Huber AH, Bjorkman PJ. Crystal structure of the complex of rat neonatal Fc receptor with Fc. Nature. 1994b;372:379–83. doi: 10.1038/372379a0. [DOI] [PubMed] [Google Scholar]
- Bussel JB, Kuter DJ, Pullarkat V, et al. Safety and efficacy of long-term treatment with romiplostim in thrombocytopenic patients with chronic ITP. Blood. 2009;113:2161–71. doi: 10.1182/blood-2008-04-150078. [DOI] [PubMed] [Google Scholar]
- Byrn RA, Mordenti J, Lucas C, et al. Biological properties of a CD4 immunoadhesin. Nature. 1990;344:667–70. doi: 10.1038/344667a0. [DOI] [PubMed] [Google Scholar]
- Capon DJ, Chamow SM, Mordenti J, et al. Designing CD4 immunoadhesins for AIDS therapy. Nature. 1989;337:525–31. doi: 10.1038/337525a0. [DOI] [PubMed] [Google Scholar]
- Chamian F, Lowes MA, Lin SL, et al. Alefacept reduces infiltrating T cells, activated dendritic cells, and inflammatory genes in psoriasis vulgaris. Proc Natl Acad Sci U S A. 2005;102:2075–80. doi: 10.1073/pnas.0409569102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan AC, Carter PJ. Therapeutic antibodies for autoimmunity and inflammation. Nat Rev Immunol. 2010;10:301–16. doi: 10.1038/nri2761. [DOI] [PubMed] [Google Scholar]
- Chaudhury C, Mehnaz S, Robinson JM, et al. The major histocompatibility complex-related Fc receptor for IgG (FcRn) binds albumin and prolongs its lifespan. J Exp Med. 2003;197:315–22. doi: 10.1084/jem.20021829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu QS. Aflibercept (AVE0005): an alternative strategy for inhibiting tumour angiogenesis by vascular endothelial growth factors. Expert Opin Biol Ther. 2009;9:263–71. doi: 10.1517/14712590802666397. [DOI] [PubMed] [Google Scholar]
- Ciombor KK, Berlin J, Chan E. Aflibercept. Clin Cancer Res. 2013;19:1920–5. doi: 10.1158/1078-0432.CCR-12-2911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coleman RL, Duska LR, Ramirez PT, et al. Phase 1–2 study of docetaxel plus aflibercept in patients with recurrent ovarian, primary peritoneal, or fallopian tube cancer. Lancet Oncol. 2011;12:1109–17. doi: 10.1016/S1470-2045(11)70244-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collen D, Bernaerts R, Declerck P, et al. Recombinant staphylokinase variants with altered immunoreactivity. I: construction and characterization. Circulation. 1996a;94:197–206. doi: 10.1161/01.cir.94.2.197. [DOI] [PubMed] [Google Scholar]
- Collen D, Moreau H, Stockx L, Vanderschueren S. Recombinant staphylokinase variants with altered immunoreactivity. II: thrombolytic properties and antibody induction. Circulation. 1996b;94:207–16. doi: 10.1161/01.cir.94.2.207. [DOI] [PubMed] [Google Scholar]
- Cwirla SE, Balasubramanian P, Duffin DJ, et al. Peptide agonist of the thrombopoietin receptor as potent as the natural cytokine. Science. 1997;276:1696–9. doi: 10.1126/science.276.5319.1696. [DOI] [PubMed] [Google Scholar]
- Dall'acqua WF, Kiener PA, Wu H. Properties of human IgG1s engineered for enhanced binding to the neonatal Fc receptor (FcRn) J Biol Chem. 2006;281:23514–24. doi: 10.1074/jbc.M604292200. [DOI] [PubMed] [Google Scholar]
- Dall'acqua WF, Woods RM, Ward ES, et al. Increasing the affinity of a human IgG1 for the neonatal Fc receptor: biological consequences. J Immunol. 2002;169:5171–80. doi: 10.4049/jimmunol.169.9.5171. [DOI] [PubMed] [Google Scholar]
- Dall'era M, Chakravarty E, Wallace D, et al. Reduced B lymphocyte and immunoglobulin levels after atacicept treatment in patients with systemic lupus erythematosus: results of a multicenter, phase Ib, double-blind, placebo-controlled, dose-escalating trial. Arthritis Rheum. 2007;56:4142–50. doi: 10.1002/art.23047. [DOI] [PubMed] [Google Scholar]
- Datta-Mannan A, Witcher DR, Tang Y, et al. Monoclonal antibody clearance. Impact of modulating the interaction of IgG with the neonatal Fc receptor. J Biol Chem. 2007;282:1709–17. doi: 10.1074/jbc.M607161200. [DOI] [PubMed] [Google Scholar]
- De Groot AS, Martin W. Reducing risk, improving outcomes: bioengineering less immunogenic protein therapeutics. Clin Immunol. 2009;131:189–201. doi: 10.1016/j.clim.2009.01.009. [DOI] [PubMed] [Google Scholar]
- De Groot AS, Moise L, Mcmurry JA, et al. Activation of natural regulatory T cells by IgG Fc-derived peptide “Tregitopes”. Blood. 2008;112:3303–11. doi: 10.1182/blood-2008-02-138073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Groot AS, Scott DW. Immunogenicity of protein therapeutics. Trends Immunol. 2007;28:482–90. doi: 10.1016/j.it.2007.07.011. [DOI] [PubMed] [Google Scholar]
- De Vries MK, van der Horst-Bruinsma IE, Nurmohamed MT, et al. Immunogenicity does not influence treatment with etanercept in patients with ankylosing spondylitis. Ann Rheum Dis. 2009;68:531–5. doi: 10.1136/ard.2008.089979. [DOI] [PubMed] [Google Scholar]
- Deisenhofer J. Crystallographic refinement and atomic models of a human Fc fragment and its complex with fragment B of protein A from Staphylococcus aureus at 2.9- and 2.8-A resolution. Biochemistry. 1981;20:2361–70. [PubMed] [Google Scholar]
- Deng R, Loyet KM, Lien S, et al. Pharmacokinetics of humanized monoclonal anti-tumor necrosis factor-{alpha} antibody and its neonatal Fc receptor variants in mice and cynomolgus monkeys. Drug Metab Dispos. 2010;38:600–5. doi: 10.1124/dmd.109.031310. [DOI] [PubMed] [Google Scholar]
- Diaz-Padilla I, Siu LL, San Pedro-Salcedo M, et al. A phase I dose-escalation study of aflibercept administered in combination with pemetrexed and cisplatin in patients with advanced solid tumours. Br J Cancer. 2012;107:604–11. doi: 10.1038/bjc.2012.319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickinson BL, Badizadegan K, Wu Z, et al. Bidirectional FcRn-dependent IgG transport in a polarized human intestinal epithelial cell line. J Clin Invest. 1999;104:903–11. doi: 10.1172/JCI6968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dore RK, Mathews S, Schechtman J, et al. The immunogenicity, safety, and efficacy of etanercept liquid administered once weekly in patients with rheumatoid arthritis. Clin Exp Rheumatol. 2007;25:40–6. [PubMed] [Google Scholar]
- Doronina SO, Mendelsohn BA, Bovee TD, et al. Enhanced activity of monomethylauristatin F through monoclonal antibody delivery: effects of linker technology on efficacy and toxicity. Bioconjug Chem. 2006;17:114–24. doi: 10.1021/bc0502917. [DOI] [PubMed] [Google Scholar]
- Dumont JA, Bitonti AJ, Clark D, et al. Delivery of an erythropoietin-Fc fusion protein by inhalation in humans through an immunoglobulin transport pathway. J Aerosol Med. 2005;18:294–303. doi: 10.1089/jam.2005.18.294. [DOI] [PubMed] [Google Scholar]
- Dumont JA, Liu T, Low SC, et al. Prolonged activity of a recombinant factor VIII-Fc fusion protein in hemophilia A mice and dogs. Blood. 2012;119:3024–30. doi: 10.1182/blood-2011-08-367813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dumont JA, Low SC, Peters RT, Bitonti AJ. Monomeric Fc fusions: impact on pharmacokinetic and biological activity of protein therapeutics. BioDrugs. 2006;20:151–60. doi: 10.2165/00063030-200620030-00002. [DOI] [PubMed] [Google Scholar]
- Dumont JA, Low SC, Peters RT, Bitonti AJ. therapeutic monoclonal antibodies: from bench to clinic. John Wiley & Sons Inc; Hoboken, NJ: 2009. Monomeric Fc fusion molecules. [Google Scholar]
- Economides AN, Carpenter LR, Rudge JS, et al. Cytokine traps: multi-component, high-affinity blockers of cytokine action. Nat Med. 2003;9:47–52. doi: 10.1038/nm811. [DOI] [PubMed] [Google Scholar]
- Enbrel® [accessed 24 Apr 2012];Prescribing Information. 2011 Available from: http://pi.amgen.com/united_states/enbrel/derm/enbrel_pi.pdf. last.
- Ephrem A, Chamat S, Miquel C, et al. Expansion of CD4+CD25+ regulatory T cells by intravenous immunoglobulin: a critical factor in controlling experimental autoimmune encephalomyelitis. Blood. 2008;111:715–22. doi: 10.1182/blood-2007-03-079947. [DOI] [PubMed] [Google Scholar]
- Eylea [accessed 24 Apr 2012];Prescribing information. 2011 Available from: http://www.regeneron.com/Eylea/eylea-fpi.pdf. last.
- Firan M, Bawdon R, Radu C, et al. The MHC class I-related receptor, FcRn, plays an essential role in the maternofetal transfer of gamma-globulin in humans. Int Immunol. 2001;13:993–1002. doi: 10.1093/intimm/13.8.993. [DOI] [PubMed] [Google Scholar]
- Fleischmann RM, Cohen SB, Stanley B, et al. Evidence of peripheral B cell depletion in subjects with controlled lupus erythematosus (SLE) following subcutaneous administration of SBI-087 (Abstract) Arthritis Rheum. 2010;62:1154. [Google Scholar]
- Freyer G, Isambert N, You B, et al. Phase I dose-escalation study of aflibercept in combination with docetaxel and cisplatin in patients with advanced solid tumours. Br J Cancer. 2012;107:598–603. doi: 10.1038/bjc.2012.304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genovese MC, Kinnman N, De la Bourdonnaye G, et al. Atacicept in patients with rheumatoid arthritis and an inadequate response to tumor necrosis factor antagonist therapy: results of a phase II, randomized, placebo-controlled, dose-finding trial. Arthritis Rheum. 2011;63:1793–803. doi: 10.1002/art.30373. [DOI] [PubMed] [Google Scholar]
- Ghaderi D, Taylor RE, Padler-Karavani V, et al. Implications of the presence of N-glycolylneuraminic acid in recombinant therapeutic glycoproteins. Nature Biotechnol. 2010;28:863–7. doi: 10.1038/nbt.1651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghaderi D, Zhang M, Hurtado-Ziola N, Varki A. Production platforms for biotherapeutic glycoproteins. Occurrence, impact, and challenges of non-human sialylation. Biotechnol Genet Eng Rev. 2012;28:147–75. doi: 10.5661/bger-28-147. [DOI] [PubMed] [Google Scholar]
- Ghetie V, Hubbard JG, Kim JK, et al. Abnormally short serum half-lives of IgG in beta 2-microglobulin-deficient mice. Eur J Immunol. 1996;26:690–6. doi: 10.1002/eji.1830260327. [DOI] [PubMed] [Google Scholar]
- Ghose S, Hubbard B, Cramer SM. Evaluation and comparison of alternatives to protein A chromatography mimetic and hydrophobic charge induction chromatographic stationary phases. J Chromatogr A. 2006;1122:144–52. doi: 10.1016/j.chroma.2006.04.083. [DOI] [PubMed] [Google Scholar]
- Ginzler EM, Wax S, Rajeswaran A, et al. Atacicept in combination with MMF and corticosteroids in lupus nephritis: results of a prematurely terminated trial. Arthritis Res Ther. 2012;14:R33. doi: 10.1186/ar3738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon KB, Langley RG. Remittive effects of intramuscular alefacept in psoriasis. J Drugs Dermatol. 2003;2:624–8. [PubMed] [Google Scholar]
- Gurbaxani B, Dela Cruz LL, Chintalacharuvu K, Morrison SL. Analysis of a family of antibodies with different half-lives in mice fails to find a correlation between affinity for FcRn and serum half-life. Mol Immunol. 2006;43:1462–73. doi: 10.1016/j.molimm.2005.07.032. [DOI] [PubMed] [Google Scholar]
- Haggerty HG, Abbott MA, Reilly TP, et al. Evaluation of immunogenicity of the T cell costimulation modulator abatacept in patients treated for rheumatoid arthritis. J Rheumatol. 2007;34:2365–73. [PubMed] [Google Scholar]
- Hayden-Ledbetter MS, Cerveny CG, Espling E, et al. CD20-directed small modular immunopharmaceutical, TRU-015, depletes normal and malignant B cells. Clin Cancer Res. 2009;15:2739–46. doi: 10.1158/1078-0432.CCR-08-1694. [DOI] [PubMed] [Google Scholar]
- He Y, Bjorkman PJ. Structure of FcRY, an avian immunoglobulin receptor related to mammalian mannose receptors, and its complex with IgY. Proc Natl Acad Sci U S A. 2011;108:12431–6. doi: 10.1073/pnas.1106925108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinton PR, Johlfs MG, Xiong JM, et al. Engineered human IgG antibodies with longer serum half-lives in primates. J Biol Chem. 2004;279:6213–6. doi: 10.1074/jbc.C300470200. [DOI] [PubMed] [Google Scholar]
- Hinton PR, Xiong JM, Johlfs MG, et al. An engineered human IgG1 antibody with longer serum half-life. J Immunol. 2006;176:346–56. doi: 10.4049/jimmunol.176.1.346. [DOI] [PubMed] [Google Scholar]
- Hoffman HM, Throne ML, Amar NJ, et al. Efficacy and safety of rilonacept (interleukin-1 Trap) in patients with cryopyrin-associated periodic syndromes: results from two sequential placebo-controlled studies. Arthritis Rheum. 2008;58:2443–52. doi: 10.1002/art.23687. [DOI] [PubMed] [Google Scholar]
- Holash J, Davis S, Papadopoulos N, et al. VEGF-Trap: a VEGF blocker with potent antitumor effects. Proc Natl Acad Sci U S A. 2002;99:11393–8. doi: 10.1073/pnas.172398299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang C. Receptor-Fc fusion therapeutics, traps, and MIMETIBODY technology. Curr Opin Biotechnol. 2009;20:692–9. doi: 10.1016/j.copbio.2009.10.010. [DOI] [PubMed] [Google Scholar]
- Huber AH, Kelley RF, Gastinel LN, Bjorkman PJ. Crystallization and stoichiometry of binding of a complex between a rat intestinal Fc receptor and Fc. J Mol Biol. 1993;230:1077–83. doi: 10.1006/jmbi.1993.1220. [DOI] [PubMed] [Google Scholar]
- Huber R, Deisenhofer J, Colman PM, et al. Crystallographic structure studies of an IgG molecule and an Fc fragment. Nature. 1976;264:415–20. doi: 10.1038/264415a0. [DOI] [PubMed] [Google Scholar]
- Hwang WY, Foote J. Immunogenicity of engineered antibodies. Methods. 2005;36:3–10. doi: 10.1016/j.ymeth.2005.01.001. [DOI] [PubMed] [Google Scholar]
- Igawa T, Ishii S, Tachibana T, et al. Antibody recycling by engineered pH-dependent antigen binding improves the duration of antigen neutralization. Nature Biotechnol. 2010;28:1203–7. doi: 10.1038/nbt.1691. [DOI] [PubMed] [Google Scholar]
- Isambert N, Freyer G, Zanetta S, et al. Phase I dose-escalation study of intravenous aflibercept in combination with docetaxel in patients with advanced solid tumors. Clin Cancer Res. 2012;18:1743–50. doi: 10.1158/1078-0432.CCR-11-1918. [DOI] [PubMed] [Google Scholar]
- Israel EJ, Wilsker DF, Hayes KC, et al. Increased clearance of IgG in mice that lack beta 2-microglobulin: possible protective role of FcRn. Immunology. 1996;89:573–8. doi: 10.1046/j.1365-2567.1996.d01-775.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iyer U, Kadambi VJ. Antibody drug conjugates – Trojan horses in the war on cancer. J Pharmacol Toxicol Methods. 2011;64:207–12. doi: 10.1016/j.vascn.2011.07.005. [DOI] [PubMed] [Google Scholar]
- Jazayeri JA, Carroll GJ. Fc-based cytokines: prospects for engineering superior therapeutics. BioDrugs. 2008;22:11–26. doi: 10.2165/00063030-200822010-00002. [DOI] [PubMed] [Google Scholar]
- Jiang XR, Song A, Bergelson S, et al. Advances in the assessment and control of the effector functions of therapeutic antibodies. Nat Rev Drug Discov. 2011;10:101–11. doi: 10.1038/nrd3365. [DOI] [PubMed] [Google Scholar]
- Junghans RP, Anderson CL. The protection receptor for IgG catabolism is the beta2-microglobulin-containing neonatal intestinal transport receptor. Proc Natl Acad Sci U S A. 1996;93:5512–6. doi: 10.1073/pnas.93.11.5512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kacskovics I, Mayer B, Kis Z, et al. Cloning and characterization of the dromedary (Camelus dromedarius) neonatal Fc receptor (drFcRn) Dev Comp Immunol. 2006;30:1203–15. doi: 10.1016/j.dci.2006.02.006. [DOI] [PubMed] [Google Scholar]
- Kacskovics I, Wu Z, Simister NE, et al. Cloning and characterization of the bovine MHC class I-like Fc receptor. J Immunol. 2000;164:1889–97. doi: 10.4049/jimmunol.164.4.1889. [DOI] [PubMed] [Google Scholar]
- Kandil E, Egashira M, Miyoshi O, et al. The human gene encoding the heavy chain of the major histocompatibility complex class I-like Fc receptor (FCGRT) maps to 19q13.3. Cytogenet Cell Genet. 1996;73:97–8. doi: 10.1159/000134316. [DOI] [PubMed] [Google Scholar]
- Kaneko Y, Nimmerjahn F, Ravetch JV. Anti-inflammatory activity of immunoglobulin G resulting from Fc sialylation. Science. 2006;313:670–3. doi: 10.1126/science.1129594. [DOI] [PubMed] [Google Scholar]
- Kemshead JT, Hopkins K. Uses and limitations of monoclonal antibodies (MoAbs) in the treatment of malignant disease: a review. J R Soc Med. 1993;86:219–24. doi: 10.1177/014107689308600413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kessel A, Ammuri H, Peri R, et al. Intravenous immunoglobulin therapy affects T regulatory cells by increasing their suppressive function. J Immunol. 2007;179:5571–5. doi: 10.4049/jimmunol.179.8.5571. [DOI] [PubMed] [Google Scholar]
- Keystone EC, Schiff MH, Kremer JM, et al. Once-weekly administration of 50 mg etanercept in patients with active rheumatoid arthritis: results of a multicenter, randomized, double-blind, placebo-controlled trial. Arthritis Rheum. 2004;50:353–63. doi: 10.1002/art.20019. [DOI] [PubMed] [Google Scholar]
- Kim J, Bronson CL, Hayton WL, et al. Albumin turnover: FcRn-mediated recycling saves as much albumin from degradation as the liver produces. Am J Physiol Gastrointest Liver Physiol. 2005;290:G352–60. doi: 10.1152/ajpgi.00286.2005. [DOI] [PubMed] [Google Scholar]
- Kim J, Bronson CL, Wani MA, et al. Beta 2-microglobulin deficient mice catabolize IgG more rapidly than FcRn- alpha-chain deficient mice. Exp Biol Med. 2008;233:603–9. doi: 10.3181/0710-RM-270. [DOI] [PubMed] [Google Scholar]
- Kim J, Hayton WL, Robinson JM, Anderson CL. Kinetics of FcRn-mediated recycling of IgG and albumin in human: pathophysiology and therapeutic implications using a simplified mechanism-based model. Clin Immunol. 2007;122:146–55. doi: 10.1016/j.clim.2006.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JK, Firan M, Radu CG, et al. Mapping the site on human IgG for binding of the MHC class I-related receptor, FcRn. Eur J Immunol. 1999;29:2819–25. doi: 10.1002/(SICI)1521-4141(199909)29:09<2819::AID-IMMU2819>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- Kim JK, Tsen MF, Ghetie V, Ward ES. Localization of the site of the murine IgG1 molecule that is involved in binding to the murine intestinal Fc receptor. Eur J Immunol. 1994;24:2429–34. doi: 10.1002/eji.1830241025. [DOI] [PubMed] [Google Scholar]
- Kobayashi K, Qiao SW, Yoshida M, et al. An FcRn-dependent role for anti-flagellin immunoglobulin G in pathogenesis of colitis in mice. Gastroenterology. 2009;137:1746–56. e1. doi: 10.1053/j.gastro.2009.07.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohler G, Milstein C. Continuous cultures of fused cells secreting antibody of predefined specificity. Nature. 1975;256:495–7. doi: 10.1038/256495a0. [DOI] [PubMed] [Google Scholar]
- Koren E, De Groot AS, Jawa V, et al. Clinical validation of the “in silico” prediction of immunogenicity of a human recombinant therapeutic protein. Clinical Immunol. 2007;124:26–32. doi: 10.1016/j.clim.2007.03.544. [DOI] [PubMed] [Google Scholar]
- Kremer JM, Westhovens R, Leon M, et al. Treatment of rheumatoid arthritis by selective inhibition of T-cell activation with fusion protein CTLA4Ig. New Engl J Med. 2003;349:1907–15. doi: 10.1056/NEJMoa035075. [DOI] [PubMed] [Google Scholar]
- Krueger GG, Papp KA, Stough DB, et al. A randomized, double-blind, placebo-controlled phase III study evaluating efficacy and tolerability of 2 courses of alefacept in patients with chronic plaque psoriasis. J Am Acad Dermatol. 2002;47:821–33. doi: 10.1067/mjd.2002.127247. [DOI] [PubMed] [Google Scholar]
- Kuo TT, De Muinck EJ, Claypool SM, et al. N-glycan moieties in neonatal Fc receptor determine steady-state membrane distribution and directional transport of IgG. J Biol Chem. 2009;284:8292–300. doi: 10.1074/jbc.M805877200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsen CP, Pearson TC, Adams AB, et al. Rational development of LEA29Y (belatacept), a high-affinity variant of CTLA4-Ig with potent immunosuppressive properties. Am J Transplant. 2005;5:443–53. doi: 10.1111/j.1600-6143.2005.00749.x. [DOI] [PubMed] [Google Scholar]
- Leach JL, Sedmak DD, Osborne JM, et al. Isolation from human placenta of the IgG transporter, FcRn, and localization to the syncytiotrophoblast: implications for maternal-fetal antibody transport. J Immunol. 1996;157:3317–22. [PubMed] [Google Scholar]
- Lebwohl M, Christophers E, Langley R, et al. An international, randomized, double-blind, placebo-controlled phase 3 trial of intramuscular alefacept in patients with chronic plaque psoriasis. Arch Dermatol. 2003;139:719–27. doi: 10.1001/archderm.139.6.719. [DOI] [PubMed] [Google Scholar]
- Lei TC, Scott DW. Induction of tolerance to factor VIII inhibitors by gene therapy with immunodominant A2 and C2 domains presented by B cells as Ig fusion proteins. Blood. 2005;105:4865–70. doi: 10.1182/blood-2004-11-4274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Palaniyandi S, Zeng R, et al. Transfer of IgG in the female genital tract by MHC class I-related neonatal Fc receptor (FcRn) confers protective immunity to vaginal infection. Proc Natl Acad Sci U S A. 2011;108:4388–93. doi: 10.1073/pnas.1012861108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lonberg N. Fully human antibodies from transgenic mouse and phage display platforms. Curr Opin Immunol. 2008;20:450–9. doi: 10.1016/j.coi.2008.06.004. [DOI] [PubMed] [Google Scholar]
- Lovell DJ, Giannini EH, Reiff A, et al. Etanercept in children with polyarticular juvenile rheumatoid arthritis. Pediatric Rheumatology Collaborative Study Group. New Engl J Med. 2000;342:763–9. doi: 10.1056/NEJM200003163421103. [DOI] [PubMed] [Google Scholar]
- Low SC, Nunes SL, Bitonti AJ, Dumont JA. Oral and pulmonary delivery of FSH-Fc fusion proteins via neonatal Fc receptor-mediated transcytosis. Hum Reprod. 2005;20:1805–13. doi: 10.1093/humrep/deh896. [DOI] [PubMed] [Google Scholar]
- Lowe NJ, Gonzalez J, Bagel J, et al. Repeat courses of intravenous alefacept in patients with chronic plaque psoriasis provide consistent safety and efficacy. Int J Dermatol. 2003;42:224–30. doi: 10.1046/j.1365-4362.2003.01793.x. [DOI] [PubMed] [Google Scholar]
- Lu L, Palaniyandi S, Zeng R, et al. A neonatal Fc receptor-targeted mucosal vaccine strategy effectively induces HIV-1 antigen-specific immunity to genital infection. J Virol. 2011;85:10542–53. doi: 10.1128/JVI.05441-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magdelaine-Beuzelin C, Vermeire S, Goodall M, et al. IgG1 heavy chain-coding gene polymorphism (G1m allotypes) and development of antibodies-to-infliximab. Pharmacogenet Genomics. 2009;19:383–7. doi: 10.1097/FPC.0b013e32832a06bf. [DOI] [PubMed] [Google Scholar]
- Mahlangu J, Powell J, Ragni M, et al. Phase 3 clinical study of recombinant Fc fusion factor VIII (rFVIIIFc) demonstrated safety, efficacy and improved pharmacokinetics (A-LONG) Haemophilia. 2013;19:70. [Google Scholar]
- Martin WL, West AP, Jr, Gan L, Bjorkman PJ. Crystal structure at 2.8 A of an FcRn/heterodimeric Fc complex: mechanism of pH-dependent binding. Mol Cell. 2001;7:867–77. doi: 10.1016/s1097-2765(01)00230-1. [DOI] [PubMed] [Google Scholar]
- Mayer B, Zolnai A, Frenyo LV, et al. Localization of the sheep FcRn in the mammary gland. Vet Immunol Immunopathol. 2002a;87:327–30. doi: 10.1016/s0165-2427(02)00059-4. [DOI] [PubMed] [Google Scholar]
- Mayer B, Zolnai A, Frenyo LV, et al. Redistribution of the sheep neonatal Fc receptor in the mammary gland around the time of parturition in ewes and its localization in the small intestine of neonatal lambs. Immunology. 2002b;107:288–96. doi: 10.1046/j.1365-2567.2002.01514.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer L, Kaser A, Blumberg RS. Dead on arrival: understanding the failure of CTLA4-immunoglobulin therapy in inflammatory bowel disease. Gastroenterology. 2012;143:13–7. doi: 10.1053/j.gastro.2012.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mease P, Genovese MC, Gladstein G, et al. Abatacept in the treatment of patients with psoriatic arthritis: results of a six-month, multicenter, randomized, double-blind, placebo-controlled, phase II trial. Arthritis Rheum. 2011;63:939–48. doi: 10.1002/art.30176. [DOI] [PubMed] [Google Scholar]
- Medesan C, Matesoi D, Radu C, et al. Delineation of the amino acid residues involved in transcytosis and catabolism of mouse IgG1. J Immunol. 1997;158:2211–7. [PubMed] [Google Scholar]
- Merrill JT, Burgos-Vargas R, Westhovens R, et al. The efficacy and safety of abatacept in patients with non-life-threatening manifestations of systemic lupus erythematosus: results of a twelve-month, multicenter, exploratory, phase IIb, randomized, double-blind, placebo-controlled trial. Arthritis Rheum. 2010;62:3077–87. doi: 10.1002/art.27601. [DOI] [PubMed] [Google Scholar]
- Molineux G, Newland A. Development of romiplostim for the treatment of patients with chronic immune thrombocytopenia: from bench to bedside. Br J Haematol. 2010;150:9–20. doi: 10.1111/j.1365-2141.2010.08140.x. [DOI] [PubMed] [Google Scholar]
- Montoyo HP, Vaccaro C, Hafner M, et al. Conditional deletion of the MHC class I-related receptor FcRn reveals the sites of IgG homeostasis in mice. Proc Natl Acad Sci U S A. 2009;106:2788–93. doi: 10.1073/pnas.0810796106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreland L, Bate G, Kirkpatrick P. Abatacept. Nature Rev Drug Discov. 2006;5:185–6. doi: 10.1038/nrd1989. [DOI] [PubMed] [Google Scholar]
- Munafo A, Priestley A, Nestorov I, et al. Safety, pharmacokinetics and pharmacodynamics of atacicept in healthy volunteers. Eur J Clin Pharmacol. 2007;63:647–56. doi: 10.1007/s00228-007-0311-7. [DOI] [PubMed] [Google Scholar]
- Nelson AL, Dhimolea E, Reichert JM. Development trends for human monoclonal antibody therapeutics. Nat Rev Drug Discov. 2010;9:767–74. doi: 10.1038/nrd3229. [DOI] [PubMed] [Google Scholar]
- Nimmerjahn F, Ravetch JV. The antiinflammatory activity of IgG: the intravenous IgG paradox. J Exp Med. 2007;204:11–15. doi: 10.1084/jem.20061788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nplate [accessed 24 Apr 2012];Prescribing information. 2011 Available from: http://pi.amgen.com/united_states/nplate/nplate_pi_hcp_english.pdf. last.
- Nulojix [accessed 24 Apr 2012];Prescribing Information. 2011 Available from: http://packageinserts.bms.com/pi/pi_nulojix.pdf. last.
- Ober RJ, Martinez C, Lai X, et al. Exocytosis of IgG as mediated by the receptor, FcRn: an analysis at the single-molecule level. Proc Natl Acad Sci U S A. 2004a;101:11076–81. doi: 10.1073/pnas.0402970101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ober RJ, Martinez C, Vaccaro C, et al. Visualizing the site and dynamics of IgG salvage by the MHC class I-related receptor, FcRn. J Immunol. 2004b;172:2021–9. doi: 10.4049/jimmunol.172.4.2021. [DOI] [PubMed] [Google Scholar]
- Ober RJ, Radu CG, Ghetie V, Ward ES. Differences in promiscuity for antibody-FcRn interactions across species: implications for therapeutic antibodies. Int Immunol. 2001;13:1551–9. doi: 10.1093/intimm/13.12.1551. [DOI] [PubMed] [Google Scholar]
- Oganesyan V, Damschroder MM, Woods RM, et al. Structural characterization of a human Fc fragment engineered for extended serum half-life. Mol Immunol. 2009;46:1750–5. doi: 10.1016/j.molimm.2009.01.026. [DOI] [PubMed] [Google Scholar]
- Oldenburg J, Pavlova A. Genetic risk factors for inhibitors to factors VIII and IX. Haemophilia. 2006;12:15–22. doi: 10.1111/j.1365-2516.2006.01361.x. [DOI] [PubMed] [Google Scholar]
- Orban T, Bundy B, Becker DJ, et al. Co-stimulation modulation with abatacept in patients with recent-onset type 1 diabetes: a randomised, double-blind, placebo-controlled trial. Lancet. 2011;378:412–9. doi: 10.1016/S0140-6736(11)60886-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orencia [accessed 24 Apr 2012];Prescribing Information. 2011 Available from: http://packageinserts.bms.com/pi/pi_orencia.pdf. last.
- Padler-Karavani V, Yu H, Cao H, et al. Diversity in specificity, abundance, and composition of anti-Neu5Gc antibodies in normal humans: potential implications for disease. Glycobiology. 2008;18:818–30. doi: 10.1093/glycob/cwn072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papadopoulos N, Martin J, Ruan Q, et al. Binding and neutralization of vascular endothelial growth factor (VEGF) and related ligands by VEGF Trap, ranibizumab and bevacizumab. Angiogenesis. 2012;15:171–85. doi: 10.1007/s10456-011-9249-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel DA, Puig-Canto A, Challa DK, et al. Neonatal Fc receptor blockade by Fc engineering ameliorates arthritis in a murine model. J Immunol. 2011;187:1015–22. doi: 10.4049/jimmunol.1003780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel TP, Parekh RB, Moellering BJ, Prior CP. Different culture methods lead to differences in glycosylation of a murine IgG monoclonal antibody. Biochem J. 1992;285:839–45. doi: 10.1042/bj2850839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pena-Rossi C, Nasonov E, Stanislav M, et al. An exploratory dose-escalating study investigating the safety, tolerability, pharmacokinetics and pharmacodynamics of intravenous atacicept in patients with systemic lupus erythematosus. Lupus. 2009;18:547–55. doi: 10.1177/0961203309102803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters RT, Low SC, Kamphaus GD, et al. Prolonged activity of factor IX as a monomeric Fc fusion protein. Blood. 2010;115:2057–64. doi: 10.1182/blood-2009-08-239665. [DOI] [PubMed] [Google Scholar]
- Peters T., Jr Serum albumin: recent progress in the understanding of its structure and biosynthesis. Clin Chem. 1977;23:5–12. [PubMed] [Google Scholar]
- Peters T., Jr Serum albumin. Adv Protein Chem. 1985;37:161–245. doi: 10.1016/s0065-3233(08)60065-0. [DOI] [PubMed] [Google Scholar]
- Petkova SB, Akilesh S, Sproule TJ, et al. Enhanced half-life of genetically engineered human IgG1 antibodies in a humanized FcRn mouse model: potential application in humorally mediated autoimmune disease. Int Immunol. 2006;18:1759–69. doi: 10.1093/intimm/dxl110. [DOI] [PubMed] [Google Scholar]
- Powell J, Ozelo M, Pasi J, et al. Safety, efficacy, and improved pharmacokinetics (PK) demonstrated in a phase 3 clinical trial of extended half life recombinant Fc fusion factor IX (B-LONG) Haemophilia. 2013;19:76. [Google Scholar]
- Powell JS, Josephson NC, Quon D, et al. Safety and prolonged activity of recombinant factor VIII Fc fusion protein in hemophilia A patients. Blood. 2012;119:3031–7. doi: 10.1182/blood-2011-09-382846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiao SW, Kobayashi K, Johansen FE, et al. Dependence of antibody-mediated presentation of antigen on FcRn. Proc Natl Acad Sci U S A. 2008;105:9337–42. doi: 10.1073/pnas.0801717105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raghavan M, Bonagura VR, Morrison SL, Bjorkman PJ. Analysis of the pH dependence of the neonatal Fc receptor/immunoglobulin G interaction using antibody and receptor variants. Biochemistry. 1995;34:14649–57. doi: 10.1021/bi00045a005. [DOI] [PubMed] [Google Scholar]
- Reichert JM. Monoclonal antibodies as innovative therapeutics. Curr Pharmaceut Biotechnol. 2008;9:423–30. doi: 10.2174/138920108786786358. [DOI] [PubMed] [Google Scholar]
- Reichert JM. Antibodies to watch in 2010. MAbs. 2010;2:84–100. doi: 10.4161/mabs.2.1.10677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reichert JM. Which are the antibodies to watch in 2012? MAbs. 2012;4:1–3. doi: 10.4161/mabs.4.1.18719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rilonacept_Smpc [accessed 18 Apr 2012];Summary of product characteristics. 2009 Available from: http://www.regeneron.com/ArcalystPI-US-Summary-EMEA/emea-combined-_rilonaceptSmPCFeb2012.pdf. last.
- Roberts JL, Ortonne JP, Tan JK, et al. The safety profile and sustained remission associated with response to multiple courses of intramuscular alefacept for treatment of chronic plaque psoriasis. J Am Acad Dermatol. 2010;62:968–78. doi: 10.1016/j.jaad.2009.07.032. [DOI] [PubMed] [Google Scholar]
- Rodewald R. Intestinal transport of antibodies in the newborn rat. J Cell Biol. 1973;58:189–211. doi: 10.1083/jcb.58.1.189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodewald R. pH-dependent binding of immunoglobulins to intestinal cells of the neonatal rat. J Cell Biol. 1976;71:666–9. doi: 10.1083/jcb.71.2.666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodewald R. Distribution of immunoglobulin G receptors in the small intestine of the young rat. J Cell Biol. 1980;85:18–32. doi: 10.1083/jcb.85.1.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodewald R, Kraehenbuhl JP. Receptor-mediated transport of IgG. J Cell Biol. 1984;99:159s–64s. doi: 10.1083/jcb.99.1.159s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roopenian DC, Akilesh S. FcRn: the neonatal Fc receptor comes of age. Nat Rev Immunol. 2007;7:715–25. doi: 10.1038/nri2155. [DOI] [PubMed] [Google Scholar]
- Roopenian DC, Christianson GJ, Sproule TJ, et al. The MHC class I-like IgG receptor controls perinatal IgG transport, IgG homeostasis, and fate of IgG-Fc-coupled drugs. J Immunol. 2003;170:3528–33. doi: 10.4049/jimmunol.170.7.3528. [DOI] [PubMed] [Google Scholar]
- Sanchez LM, Penny DM, Bjorkman PJ. Stoichiometry of the interaction between the major histocompatibility complex-related Fc receptor and its Fc ligand. Biochemistry. 1999;38:9471–6. doi: 10.1021/bi9907330. [DOI] [PubMed] [Google Scholar]
- Sandborn WJ, Colombel JF, Sands BE, et al. Abatacept for Crohn's disease and ulcerative colitis. Gastroenterology. 2012;143:62–69. e4. doi: 10.1053/j.gastro.2012.04.010. [DOI] [PubMed] [Google Scholar]
- Schnulle PM, Hurley WL. Sequence and expression of the FcRn in the porcine mammary gland. Vet Immunol Immunopathol. 2003;91:227–31. doi: 10.1016/s0165-2427(02)00294-5. [DOI] [PubMed] [Google Scholar]
- Schuck P, Radu CG, Ward ES. Sedimentation equilibrium analysis of recombinant mouse FcRn with murine IgG1. Mol Immunol. 1999;36:1117–25. doi: 10.1016/s0161-5890(99)00093-0. [DOI] [PubMed] [Google Scholar]
- Schultze HE, Heremans JF. Nature and metabolism of extracellular proteins. Elsevier; New York: 1966. Molecular biology of human proteins: with special reference to plasma proteins. [Google Scholar]
- Scott DW, De Groot AS. Can we prevent immunogenicity of human protein drugs? Ann Rheum Dis. 2010;69:i72–6. doi: 10.1136/ard.2009.117564. [DOI] [PubMed] [Google Scholar]
- Senter PD. Potent antibody drug conjugates for cancer therapy. Curr Opin Chem Biol. 2009;13:235–44. doi: 10.1016/j.cbpa.2009.03.023. [DOI] [PubMed] [Google Scholar]
- Shapiro AD, Ragni MV, Valentino LA, et al. Recombinant factor IX-Fc fusion protein (rFIXFc) demonstrates safety and prolonged activity in a phase 1/2a study in hemophilia B patients. Blood. 2012;119:666–72. doi: 10.1182/blood-2011-07-367003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shields RL, Namenuk AK, Hong K, et al. High resolution mapping of the binding site on human IgG1 for Fc gamma RI, Fc gamma RII, Fc gamma RIII, and FcRn and design of IgG1 variants with improved binding to the Fc gamma R. J Biol Chem. 2001;276:6591–604. doi: 10.1074/jbc.M009483200. [DOI] [PubMed] [Google Scholar]
- Simister NE, Mostov KE. An Fc receptor structurally related to MHC class I antigens. Nature. 1989;337:184–7. doi: 10.1038/337184a0. [DOI] [PubMed] [Google Scholar]
- Simister NE, Rees AR. Isolation and characterization of an Fc receptor from neonatal rat small intestine. Eur J Immunol. 1985;15:733–8. doi: 10.1002/eji.1830150718. [DOI] [PubMed] [Google Scholar]
- Simister NE, Story CM, Chen HL, Hunt JS. An IgG-transporting Fc receptor expressed in the syncytiotrophoblast of human placenta. Eur J Immunol. 1996;26:1527–31. doi: 10.1002/eji.1830260718. [DOI] [PubMed] [Google Scholar]
- Sockolosky JT, Tiffany MR, Szoka FC. Engineering neonatal Fc receptor-mediated recycling and transcytosis in recombinant proteins by short terminal peptide extensions. Proc Natl Acad Sci U S A. 2012;109:16095–100. doi: 10.1073/pnas.1208857109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spiekermann GM, Finn PW, Ward ES, et al. Receptor-mediated immunoglobulin G transport across mucosal barriers in adult life: functional expression of FcRn in the mammalian lung. J Exp Med. 2002;196:303–10. doi: 10.1084/jem.20020400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stahl N, Radin A, Mellis S. Rilonacept – CAPS and beyond. Ann New York Acad Sci. 2009;1182:124–34. doi: 10.1111/j.1749-6632.2009.05074.x. [DOI] [PubMed] [Google Scholar]
- Stohl W. Biologic differences between various inhibitors of the BLyS/BAFF pathway: should we expect differences between belimumab and other inhibitors in development? Curr Rheumatol Rep. 2012;14:303–9. doi: 10.1007/s11926-012-0254-6. [DOI] [PubMed] [Google Scholar]
- Stohl W, Hilbert DM. The discovery and development of belimumab: the anti-BLyS-lupus connection. Nature Biotechnol. 2012;30:69–77. doi: 10.1038/nbt.2076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Story CM, Mikulska JE, Simister NE. A major histocompatibility complex class I-like Fc receptor cloned from human placenta: possible role in transfer of immunoglobulin G from mother to fetus. J Exp Med. 1994;180:2377–81. doi: 10.1084/jem.180.6.2377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strober BE, Menon K. Alefacept for the treatment of psoriasis and other dermatologic diseases. Dermatol Ther. 2007;20:270–6. doi: 10.1111/j.1529-8019.2007.00140.x. [DOI] [PubMed] [Google Scholar]
- Sutton BJ, Phillips DC. The three-dimensional structure of the carbohydrate within the Fc fragment of immunoglobulin G. Biochem Soc Trans. 1983;11:130–2. [PubMed] [Google Scholar]
- Tak PP, Thurlings RM, Rossier C, et al. Atacicept in patients with rheumatoid arthritis: results of a multicenter, phase Ib, double-blind, placebo-controlled, dose-escalating, single- and repeated-dose study. Arthritis Rheum. 2008;58:61–72. doi: 10.1002/art.23178. [DOI] [PubMed] [Google Scholar]
- Tangri S, Licalsi C, Sidney J, Sette A. Rationally engineered proteins or antibodies with absent or reduced immunogenicity. Curr Med Chem. 2002;9:2191–9. doi: 10.2174/0929867023368647. [DOI] [PubMed] [Google Scholar]
- Tangvoranuntakul P, Gagneux P, Diaz S, et al. Human uptake and incorporation of an immunogenic nonhuman dietary sialic acid. Proc Natl Acad Sci U S A. 2003;100:12045–50. doi: 10.1073/pnas.2131556100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao MH, Morrison SL. Studies of aglycosylated chimeric mouse-human IgG. Role of carbohydrate in the structure and effector functions mediated by the human IgG constant region. J Immunol. 1989;143:2595–601. [PubMed] [Google Scholar]
- Tesar DB, Cheung EJ, Bjorkman PJ. The chicken yolk sac IgY receptor, a mammalian mannose receptor family member, transcytoses IgY across polarized epithelial cells. Mol Biol Cell. 2008;19:1587–93. doi: 10.1091/mbc.E07-09-0972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyring S, Gordon KB, Poulin Y, et al. Long-term safety and efficacy of 50 mg of etanercept twice weekly in patients with psoriasis. Arch Dermatol. 2007;143:719–26. doi: 10.1001/archderm.143.6.719. [DOI] [PubMed] [Google Scholar]
- Tzaban S, Massol RH, Yen E, et al. The recycling and transcytotic pathways for IgG transport by FcRn are distinct and display an inherent polarity. J Cell Biol. 2009;185:673–84. doi: 10.1083/jcb.200809122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaccaro C, Bawdon R, Wanjie S, et al. Divergent activities of an engineered antibody in murine and human systems have implications for therapeutic antibodies. Proc Natl Acad Sci U S A. 2006;103:18709–14. doi: 10.1073/pnas.0606304103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vallee S, Rakhe S, Reidy T, et al. Pulmonary administration of interferon Beta-1a-fc fusion protein in non-human primates using an immunoglobulin transport pathway. J Interferon Cytokine Res. 2012;32:178–84. doi: 10.1089/jir.2011.0048. [DOI] [PubMed] [Google Scholar]
- van Vollenhoven RF, Kinnman N, Vincent E, et al. Atacicept in patients with rheumatoid arthritis and an inadequate response to methotrexate: results of a phase II, randomized, placebo-controlled trial. Arthritis Rheum. 2011;63:1782–92. doi: 10.1002/art.30372. [DOI] [PubMed] [Google Scholar]
- Vaughn DE, Bjorkman PJ. Structural basis of pH-dependent antibody binding by the neonatal Fc receptor. Structure. 1998;6:63–73. doi: 10.1016/s0969-2126(98)00008-2. [DOI] [PubMed] [Google Scholar]
- Vaughn DE, Milburn CM, Penny DM, et al. Identification of critical IgG binding epitopes on the neonatal Fc receptor. J Mol Biol. 1997;274:597–607. doi: 10.1006/jmbi.1997.1388. [DOI] [PubMed] [Google Scholar]
- Viglietta V, Bourcier K, Buckle GJ, et al. CTLA4Ig treatment in patients with multiple sclerosis: an open-label, phase 1 clinical trial. Neurology. 2008;71:917–24. doi: 10.1212/01.wnl.0000325915.00112.61. [DOI] [PubMed] [Google Scholar]
- Vincenti F, Blancho G, Durrbach A, et al. Five-year safety and efficacy of belatacept in renal transplantation. J Am Soc Nephrol. 2010;21:1587–96. doi: 10.1681/ASN.2009111109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waldmann TA, Terry WD. Familial hypercatabolic hypoproteinemia. A disorder of endogenous catabolism of albumin and immunoglobulin. J Clin Investig. 1990;86:2093–8. doi: 10.1172/JCI114947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh G. Post-translational modifications of protein biopharmaceuticals. Drug Discov Today. 2010;15:773–80. doi: 10.1016/j.drudis.2010.06.009. [DOI] [PubMed] [Google Scholar]
- Walsh G, Jefferis R. Post-translational modifications in the context of therapeutic proteins. Nat Biotechnol. 2006;24:1241–52. doi: 10.1038/nbt1252. [DOI] [PubMed] [Google Scholar]
- Wang TF, Lockhart AC. Aflibercept in the treatment of metastatic colorectal cancer. Clin Med Insights Oncol. 2012;6:19–30. doi: 10.4137/CMO.S7432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, Lu P, Fang Y, et al. Monoclonal antibodies with identical Fc sequences can bind to FcRn differentially with pharmacokinetic consequences. Drug Metab Dispos. 2011;39:1469–77. doi: 10.1124/dmd.111.039453. [DOI] [PubMed] [Google Scholar]
- Wani MA, Haynes LD, Kim J, et al. Familial hypercatabolic hypoproteinemia caused by deficiency of the neonatal Fc receptor, FcRn, due to a mutant beta2-microglobulin gene. Proc Natl Acad Sci U S A. 2006;103:5084–9. doi: 10.1073/pnas.0600548103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward ES, Martinez C, Vaccaro C, et al. From sorting endosomes to exocytosis: association of Rab4 and Rab11 GTPases with the Fc receptor, FcRn, during recycling. Mol Biol Cell. 2005;16:2028–38. doi: 10.1091/mbc.E04-08-0735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward ES, Ober RJ. Chapter 4: multitasking by exploitation of intracellular transport functions the many faces of FcRn. Adv Immunol. 2009;103:77–115. doi: 10.1016/S0065-2776(09)03004-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward ES, Zhou J, Ghetie V, Ober RJ. Evidence to support the cellular mechanism involved in serum IgG homeostasis in humans. Int Immunol. 2003;15:187–95. doi: 10.1093/intimm/dxg018. [DOI] [PubMed] [Google Scholar]
- Watson D. Albumin and “total globulin” fractions of blood. Adv Clin Chem. 1965;8:237–303. [PubMed] [Google Scholar]
- Weiner LM, Surana R, Wang S. Monoclonal antibodies: versatile platforms for cancer immunotherapy. Nat Rev Immunol. 2010;10:317–27. doi: 10.1038/nri2744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West AP, Jr, Bjorkman PJ. Crystal structure and immunoglobulin G binding properties of the human major histocompatibility complex-related Fc receptor(,) Biochemistry. 2000;39:9698–708. doi: 10.1021/bi000749m. [DOI] [PubMed] [Google Scholar]
- West AP, Jr, Herr AB, Bjorkman PJ. The chicken yolk sac IgY receptor, a functional equivalent of the mammalian MHC-related Fc receptor, is a phospholipase A2 receptor homolog. Immunity. 2004;20:601–10. doi: 10.1016/s1074-7613(04)00113-x. [DOI] [PubMed] [Google Scholar]
- Wu AM, Senter PD. Arming antibodies: prospects and challenges for immunoconjugates. Nat Biotechnol. 2005;23:1137–46. doi: 10.1038/nbt1141. [DOI] [PubMed] [Google Scholar]
- Ye L, Zeng R, Bai Y, et al. Efficient mucosal vaccination mediated by the neonatal Fc receptor. Nat Biotechnol. 2011;29:158–63. doi: 10.1038/nbt.1742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeung YA, Leabman MK, Marvin JS, et al. Engineering human IgG1 affinity to human neonatal Fc receptor: impact of affinity improvement on pharmacokinetics in primates. J Immunol. 2009;182:7663–71. doi: 10.4049/jimmunol.0804182. [DOI] [PubMed] [Google Scholar]
- Ying T, Chen W, Gong R, et al. Soluble monomeric IgG1 Fc. J Biol Chem. 2012;287:19399–408. doi: 10.1074/jbc.M112.368647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida M, Claypool SM, Wagner JS, et al. Human neonatal Fc receptor mediates transport of IgG into luminal secretions for delivery of antigens to mucosal dendritic cells. Immunity. 2004;20:769–83. doi: 10.1016/j.immuni.2004.05.007. [DOI] [PubMed] [Google Scholar]
- Yoshida M, Kobayashi K, Kuo TT, et al. Neonatal Fc receptor for IgG regulates mucosal immune responses to luminal bacteria. J Clin Invest. 2006;116:2142–51. doi: 10.1172/JCI27821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zalevsky J, Chamberlain AK, Horton HM, et al. Enhanced antibody half-life improves in vivo activity. Nat Biotechnol. 2010;28:157–9. doi: 10.1038/nbt.1601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zambidis ET, Scott DW. Epitope-specific tolerance induction with an engineered immunoglobulin. Proc Natl Acad Sci U S A. 1996;93:5019–24. doi: 10.1073/pnas.93.10.5019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao X, Lapalombella R, Joshi T, et al. Targeting CD37-positive lymphoid malignancies with a novel engineered small modular immunopharmaceutical. Blood. 2007;110:2569–77. doi: 10.1182/blood-2006-12-062927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J, Johnson JE, Ghetie V, et al. Generation of mutated variants of the human form of the MHC class I-related receptor, FcRn, with increased affinity for mouse immunoglobulin G. J Mol Biol. 2003;332:901–13. doi: 10.1016/s0022-2836(03)00952-5. [DOI] [PubMed] [Google Scholar]
- Zhu A, Hurst R. Anti-N-glycolylneuraminic acid antibodies identified in healthy human serum. Xenotransplantation. 2002;9:376–81. doi: 10.1034/j.1399-3089.2002.02138.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.