

SUMMARY
The fungal protein CBP (calcium binding protein) is a known virulence factor with an unknown virulence mechanism. The protein was identified based on its ability to bind calcium and its prevalence as Histoplasma capsulatum’s most abundant secreted protein. However, CBP has no sequence homology with other calcium binding proteins and contains no known calcium-binding motifs. Here, the NMR structure of CBP reveals a highly intertwined homodimer and represents the first atomic level NMR model of any fungal virulence factor. Each CBP monomer is comprised of four α-helices that adopt the saposin fold, characteristic of a protein family that binds to membranes and lipids. This structural homology suggests that CBP functions as a lipid-binding protein, potentially interacting with host glycolipids in the phagolysosome of host cells.
Keywords: Histoplasma capsulatum, phagolysosome, structural homology, lipid binding
INTRODUCTION
The fungal pathogen Histoplasma capsulatum is uniquely adapted to survive and replicate within the phagolysosome of macrophages (Eissenberg et al., 1988). H. capsulatum secretes its virulence factor CBP, originally designated a calcium-binding protein (Batanghari and Goldman, 1997), into this intracellular compartment, and this process is essential for causing respiratory infections in mammals (Sebghati et al., 2000). CBP is the only detectable calcium-binding macromolecule released by Histoplasma yeasts and is the major component of culture supernatant, indicative of an important function for this protein in sensing, modifying, and/or regulating the environment within the phagolysosome (Batanghari and Goldman, 1997; Batanghari et al., 1998). Equilibrium calcium-binding constants for CBP reveal significant Ca2+- binding affinity (KD = 6.45 ± 0.4 nM), however no structural changes occur upon binding calcium (Beck et al., 2008). These features suggest that the role of CBP may be more complex than simple calcium acquisition, because other calcium binding proteins in this class (i.e. with high affinity for calcium coupled with no conformational change) are involved in deactivation of signal transducers and/or quenching of Ca2+ signals. The ultimate goal of this work was to determine the tertiary structure of CBP to generate hypotheses regarding its specific function and precise role in pathogenesis.
CBP was first identified as a secreted calcium-binding protein produced only in the yeast form, not the mold form, of this dimorphic fungus (Batanghari and Goldman, 1997). Later studies showed that it is the most abundant protein released by H. capsulatum and that its production continues as the yeast multiplies inside host cells (Kugler et al., 2000). Most importantly, CBP has been genetically proven to be indispensable for survival of H. capsulatum in both macrophage and mouse models of pulmonary infection (Sebghati et al., 2000).
Previous characterization of CBP’s structure has yielded information on the oligomeric status, disulfide bond linkages, and overall protein stability (Beck et al., 2008). Biophysical studies revealed that CBP exists as a symmetric homodimer in solution and that the structure is unperturbed by calcium binding. Additionally, three critical disulfide bond connections were determined experimentally (Fig. 1). Characterization of the structural stability and unfolding mechanisms of CBP indicated potential mechanisms of protein regulation and insight into the functional status as an extremely stable, protease-resistant protein.
1.

CBP homology and secondary structure. H. capsulatum strain G186AR CBP1 (accession O42720) sequence alignment with two other H. capsulatum strains CBP1 (G217B), CBP1 (WU24), and CBP1 from P. brasiliensis (Q6TS31). The four helices of CBP are shown as gray bars above the alignment. Conserved cysteine residues are numbered and brackets depict disulfide bond connections determined for G186AR CBP1. The triangle above the sequences indicates the start of the mature, secreted protein. The level of sequence homology is indicated by bar chart below sequences, as well as with color using the ClustalX default parameters (red, basic; blue, hydrophobic; green, uncharged hydrophilic; and orange, polar uncharged).
CBP shares no significant sequence homology with other proteins in NCBI’s GenBank Database, with the exception of orthologs in three different strains of H. capsulatum and the closely related dimorphic fungal pathogen Paracoccidioides brasilensis (Fig. 1). To gain insight into the specific function(s) of CBP, we have determined the three-dimensional solution structure of this homodimeric protein. The resulting global fold of CBP is highly similar to saposin B (SapB) (Ahn et al., 2003), a mammalian sphingolipid activator protein with broad lipid binding specificity and lysosomal localization. Despite lack of primary sequence homology, the structural similarity between the two proteins suggests a common mechanism whereby CBP is involved in lipid binding, lipid metabolism, membrane remodeling, and/or membrane stability.
RESULTS
NMR structure determination
We have recently reported the full backbone and sidechain chemical shift assignments for CBP (Beck et al., 2008) (BRMB accession #15404). From the analysis of short range NOE, CSI, and dihedral angle constraints; we concluded that CBP is composed of four helical segments: α1 (residues 5-26), α2 (residues 32-37), α3 (residues 42-50), and α4 (residues 58-63) (Fig. 1). Here we determined the complete structure of endogenously synthesized CBP using 2,827 experimental restraints, including 2,679 inter-proton distance restraints derived from nuclear Overhauser enhancements (NOEs). Dihedral angle restraints and hydrogen bonds were included from the analysis of chemical shifts using TALOS (Cornilescu et al., 1999) and CSI (Wishart and Sykes, 1994a), respectively. During initial NOE assignment and structure calculations for monomeric CBP (using ARIA 1.2 interfaced to CNS (Brunger et al., 1998; Nilges et al., 1997)), some restraints could be assigned manually to intermonomer NOEs. For example, during the assignment process, intense NOEs were observed between the aromatic protons of F19 and Cβ proton of V5, which are not compatible with an intact α1 helix. NOEs such as these can only be explained in terms of dimer structure where the N-terminus of α1 of one monomer is in close contact with the C-terminus of α1 of the second monomer in an antiparallel coiled-coil structure. However, the symmetry-related degeneracy of the spectra was an obstacle toward the full structural study of the dimer, and the lack of a sufficient number of unambiguous distance restraints initially prevented us from obtaining a high-resolution quaternary structure of CBP.
To further distinguish inter- from intramolecular NOEs, we prepared a heterolabeled dimer by mixing eqiumolar unlabeled and fully 13C, 15N- labeled monomeric CBP obtained after denaturing dimers with 8 M urea. Isotope filtered/edited NOESY experiments (Zwahlen et al., 1997) performed on this heterolabeled dimer allowed for the identification of a set of intermonomer NOE connectivities. The unambiguous distance constraints derived from these NOEs could be used for structure determination using ARIAv2.2β (Rieping et al., 2007), which accommodates restraints for symmetric homodimer structure calculations, and accordingly produced the ensemble of lowest energy NMR structures presented in Figure 2A.
2.

Three-dimensional structure of CBP. (A) Superposition of 20 NMR structures representative of the CBP dimeric fold. The monomers are shown in cyan or magenta with disulfide bonds in orange for both. (B, C) Ribbon drawing of the lowest-energy conformers of monomer (B) and dimer (C). Red, N-terminal helix 1; yellow, helix 2; green, helix 3; and blue, helix 4; with orange disulfide bonds. Homodimer structures (A, C) are in same orientation.
Architecture of the dimer
Each monomer consists of four α-helices arranged in a long hairpin that is bent into a simple V-shape. The outer surface of the monomer is predominantly hydrophilic, whereas the inner surface of the “V” is lined with the side chains from hydrophobic residues. CBP has 12 charged residues (two lysines, one arginine, six aspartate, and three glutamate) resulting in a net negative charge and an estimated pI of 3.93. The charges are not uniformly positioned on the surface (Fig. 3A and B) so that a high density of basic charge is buried by the dimer interface. The fold of each subunit is stabilized by three disulfide bridges, two connecting the extreme C-terminus with the N-terminus of α2 and another between α3 and α4.
3.
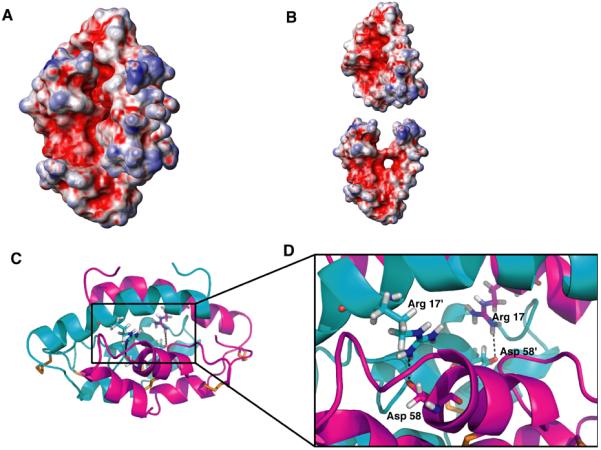
CBP dimer interface features burial of charges and hydrophobicity. (A, B) Surface electrostatic potential of CBP as a dimer (A) and monomer (B) (blue, positive charge; red, negative charge; and white, neutral). Charged surfaces calculated using MOLMOL (Koradi et al., 1996). (C, D) Structures of CBP homodimer (monomers are colored cyan or magenta) showing arrangement of intermolecular Arg 17-Asp 58′ polar interactions. (D) Enlarged detail of salt bridges (indicated by dotted lines) stabilizing the CBP dimer visualized using PyMol(DeLano, 2002).
The homodimer is formed by a head-to-tail interaction of the two monomers involving all four helices. Helix α1 (V5-A25) is especially interesting because of its amphipathic nature and predominance of hydrophobic (V5, F9, Y12, V16, V18, F19, and L22) residues. Several key long-range intermolecular NOEs assigned to these same residues in α1 indicate their participation in the dimer interface. Interestingly, there is little evidence for ion pairing between the subunits. Only one symmetrical pair of intermolecular salt bridges is formed between the side chains of R17 and D58 (Fig. 3C and D). Presumably this interaction further stabilizes the dimer.
In this structure, more than 25% of the surface of each monomer (representing 1122 Å 2) is buried in the dimer interface. Nearly all the hydrophobic residues of the protein are positioned in the cavity at the dimer interface. Disruption of the dimer would expose at least 12 of these residues to the solvent; suggesting that the dimer is the energetically favorable form. This correlates well with previous unfolding data that indicated that an isolated monomeric form of CBP is unstable (Beck et al., 2008).
Backbone disorder and dynamics
{1H} 15N heteronuclear NOE measurements provide a qualitative assessment of the mobility of the N-H bond vectors for individual residues and is sensitive to both the mobility of the overall tumbling time of the protein (τm) and fast internal motions (Palmer, 1993). Although heteronuclear NOE data alone are not sufficient for fully quantifying the dynamic behavior of molecules, they have been used as a probe for assessing the structural flexibility of a protein (Cho et al., 1996; Farrow et al., 1995). Measured NOE values, shown in Figure 4, are all positive and hover near the slow tumbling limit. Areas of intermediate NOEs (such as residues 4, 39-41, and 47) reflect an increase in dynamic flexibility. The {1H} 15N heteronuclear NOE in these distinct areas also correlates with a decrease in the number of long range NOEs and an increase in local rmsd (Fig. 4A and C), indicating a high degree of mobility in these loop regions of the structure. Taken as a whole, these results indicate that CBP is very rigid, with little disorder and dynamic flexibility with respect to the overall tumbling of the protein.
4.

Sequence-dependent NOEs highlight structural and dynamic features of stable CBP homodimer. (a) Distribution of NOE distance restraints, where the intermolecular (light gray), long-range (white), medium-range (dark gray), and short-range (black) NOE distance restraints used for the calculation of the final ensemble of structures of CBP are displayed. (b) Measured heteronuclear {1H}-15N NOE data (±1 std dev) indicate the protein is very rigid, with only discrete region of slightly increased backbone mobility. (c) Local precision of CBP conformation is reflected in rms deviation values plotted against the residue number. Location of secondary structure elements, sequence, and residue numbering are given at top of figure.
Structural homology
Dimerization of proteins via a coiled-coil segment is common, therefore a Dali(Holm and Sander, 1993) search of the Protein Data Bank (PDB) retrieved an abundance of structural homologs for the paired helix α1 of CBP. However, the Dali search did not yield a close match for the overall structure. Subsequently, the FATCAT server (Ye and Godzik, 2004a, 2004b) was used to search the PDB for structural homologs to CBP. The closest match was to the mammalian homodimeric SapB protein (PDB accession number 1N69), which is a member of the saposin-fold family of proteins that bind to membranes and lipids (Ahn et al., 2003; Munford et al., 1995). Despite a lack of sequence homology, the three-dimensional structure of CBP is highly similar (FATCAT p-value = 4.5e-2 and a RMSD of 2.39 Å with 1 twist) to that of SapB as revealed in superposition of two structures in Figure 5A.
5.

CBP is structurally homologous to saposin B. (A) Backbone structural superposition of CBP (red) and SapB (blue; chain A, PDB 1N69) from FATCAT structural alignment (p-value = 4.5 e-2). RMSD for alignment was 2.51 Å with one twist (introduced by FATCAT alignment procedure at hinge region in order to get better alignment of two structures). Side-by-side structural comparison of homodimeric CBP (B) and SapB (C) with similar orientation of N-terminal helices and four helices colored identically with disulfide bonds in orange.
CBP dimers are formed by interlocking two V-shaped monomers in the same manner as the SapB monomers, based on the crystal structure of human saposin B (Ahn et al., 2003) (Fig. 5B and C). Like CBP, the structure of SapB is comprised of two symmetric monomer halves that each contain four amphipathic α-helices folded into a single globular domain. Notably CBP shares the same disulfide linkage pattern (1-6, 2-5, and 3-4), which is also characteristic of the saposin-like protein (SAPLIP) family (Munford et al., 1995). However, the linear spacing of these cysteine residues in CBP is quite different from the consensus spacing in the sequence of the saposins. The SAPLIP family of proteins ranges from 70 to 84 residues in length, analogous to CBP at 78 amino acids in length.
Evidence of bound lipid
Upon further review of the FATCAT structural homology hit to SapB (1N69A), we realized that the highest structural similarity found for CBP (P-value of 4.5e-02; RMSD 2.51 Å) corresponds to one of three distinct monomeric forms of saposin B that were obtained by x-ray crystallography (Ahn et al., 2003). Unintentional co-crystallization of lipids with SapB resulted in the observation of two different conformations of this protein: an asymmetric, closed holo conformation (chain A and B) and a symmetric, more solvent-accessible apo conformation (chain C and C’) (Ahn et al., 2003). CBP shares the greatest structural similarity with chain A of SapB, which corresponds to the monomer conformation with lipid bound.
In light of this finding, we hypothesized that the sample of CBP used for NMR data collection might contain residual lipids much like the SapB crystallization samples. After running a 15N-HSQC on a 15N-labeled sample of CBP used in previous NMR experiments, this protein sample was passed over a lipophillic (“lipidex”) column to remove any bound lipids. After this delipidation procedure, the amide chemical shifts of several residues were perturbed (Fig. 6A), indicating a change in the local environment of these amino acids. Previous unsuccessful attempts to change the solution-state structure of CBP (with addition of calcium or EGTA), as well as the observed extreme stability of NMR samples over five years, indicate the stability and immobility of this protein and highlight the significance of its chemical shift changes upon this delipidation experiment. The putative lipid-binding residues were then mapped onto the protein structure of CBP and overlaid with residues identified in SapB that line the hydrophobic putative ligand-binding cavity (Fig. 6B and C). Most of these residues from CBP are similarly situated to those in the lipid-binding pocket of SapB, consistent with a shared role in lipid binding. Validation that these chemical shift perturbations are the result of lipid removal will require reconstitution of the lipid-bound form of CBP and demonstration that the original structure is restored. Preliminary lipid co-sedimentation experiments have confirmed that CBP is able to bind a variety of lipids, but we have not yet been able to identify the native lipid bound to CBP by mass spectrometry approaches. There are a wide variety of lipid ligands that bind to members of the SAPLIP family, and the structural basis for lipid recognition and specificity by any of the saposins remains unclear, especially with regard to native ligands.
6.

Evidence for lipid binding by CBP. (A) Chemical shift perturbation of CBP after delipidation procedure monitored by HSQC spectra. Ribbon diagram of CBP monomer (B) and dimer (C) colored according to chemical shift changes induced following presumed delipidation of CBP. Residues colored by gradient, with most affected residues colored with darkest blue to those unaffected in white. The residues most affected are also labeled in (B).
DISCUSSION
CBP was first identified ten years ago as the major protein secreted by the yeast form of H. capsulatum (Batanghari and Goldman, 1997), and subsequent studies on CBP focused on gene regulation, protein localization, and its essential role as a virulence factor. However, it has been difficult to predict any precise mechanism for CBP, because the primary sequence lacks homology to any protein with a known function. Even the relevance of in vitro measurements of calcium binding is unclear with respect to CBP function in vivo. Structure determination provided the only viable outlet for uncovering potential functional roles for this protein.
The final NMR ensemble of homodimeric CBP structures exhibits a backbone conformation generally consistent with the dominant α-helical motif described for all members of the saposin-like protein (SAPLIP) family, with significant structural homology to the mammalian saposin SapB (as determined from the FATCAT structural homology database search). CBP and SapB contain identical disulfide linkages (1-6, 2-5, 3-4) and are homodimers, despite the fact that the database search engines did not include these key features as search criteria. Recent homodimeric structures of Sap C and D (Rossmann et al., 2008) which complement earlier SapA and B dimers (Ahn et al., 2003; Ahn et al., 2006), indicate that dimerization is a common feature of saposins. This is a feature also shared by homodimeric CBP. The SAPLIP family members, like CBP, are small proteins ranging in size from 8-11 kDa. A similar three-dimensional fold, conserved disulfide linkages, and shared biochemical property of lipid binding are the only identified common properties of all SAPLIPs (Bruhn, 2005). Besides the disulfide bridges, no other individual conserved residues have been identified that contribute to stability or function throughout the whole SAPLIP family (Bruhn and Leippe, 1999).
Although CBP shares little amino acid sequence homology with the mammalian saposins (A-D), the shared molecular architecture suggests that CBP is the first structurally characterized fungal member of the saposin-fold family. The four mammalian saposins have multiple functions, including that of enhancing lysosomal enzyme-lipid interactions by solubilizing the lipids and/or activating the lipid degrading enzymes (Bruhn, 2005; Kolter and Sandhoff, 2005; Schuette et al., 2001; Soeda et al., 1993). Members of the larger saposin-like family include NK-lysin (Liepinsh et al., 1997), granulysin (Anderson et al., 2003), the pore-forming amoebapores (Hecht et al., 2004), the microbial cyclic polypeptide bacteriocin AS-48 (Gonzalez et al., 2000; Sanchez-Barrena et al., 2003) and the membrane-targeting domain of the aspartic proteinase phytepsin (Kervinen et al., 1999). All these proteins share a common fold and the ability to interact with membranes, but have a wide variety of biological functions.
Based on biochemical and structural information, several mechanisms have been proposed to explain how saposin-like proteins interact with membranes (Alattia et al., 2006; Qi and Grabowski, 2001). For example, amoebapore A has been proposed to act via pore formation upon oligomerization (Hecht et al., 2004). In contrast, it has been suggested that NK-lysin, granulysin and bacteriocin AS-48 destabilize the membrane via binding of positively charged regions, a process termed “molecular electroporation” (Miteva et al., 1999). Yet a general mechanism for saposin-membrane interaction has been recognized whereby positively charged clusters on the surface of saposins interact with negatively charged lipid head groups (Ciaffoni et al., 2001; de Alba et al., 2003; Liu et al., 2005). The recently identified sulfate binding sites of Lys10 and Arg17 in the crystal structure of SapD were linked to biologically relevant interaction of SapD with phosphate groups of lipids (Rossmann et al., 2008). The sidechains of Lys11 and Arg17 in CBP may interact with anionic lipids in a similar fashion. Another congruence is that co-purification with endogenous lipids may influence the folding and structure of saposins, already noted for SapB and C (Ahn et al., 2003; Rossmann et al., 2008) and inferred from structural comparisons of CBP before and after a delipidation procedure.
The common lipid-binding feature among the SAPLIP family of proteins obviously does not limit their functional capabilities, which include antigen presentation and antimicrobial activity (Munford et al., 1995). H. capsulatum could take advantage of a number of lipid-binding mechanisms in order to alter the environment of the phagolysosome for survival and replication of the yeast. Modification of the phagolysosomal lipid content could alter the normal maturation or facilitate expansion of this compartment, thus providing a more suitable environment for H. capsulatum. It is also possible that membrane destabilization and/or pore formation arising from the binding of CBP could facilitate pH modification of the phagolysosome associated with in vivo survival and replication of H. capsulatum (Eissenberg et al., 1993).
Obviously several different models of pathogenesis related to saposin-like membrane/lipid binding by CBP can be hypothesized, but the detailed mechanism of action of CBP during Histoplasma infection still remains to be determined. Intriguingly, CBP shows the closest structural resemblance to a mammalian saposin that is found in lysosomes, which are major components of the intracellular home of H. capsulatum (Eissenberg et al., 1988). It is therefore tempting to speculate that the primary role of CBP is molecular mimicry or direct subversion of host saposin function in the phagolysosomal compartment. Regardless, the quaternary structure of CBP suggests new biological roles for this secreted protein that are distinct from the originally identified correlation with calcium binding.
EXPERIMENTAL PROCEDURES
Sample preparation
Unenriched, 15N- and 13C/ 15N-enriched CBP proteins were expressed in the native organism (H. capsulatum) and purified using a modification of a procedure described earlier (Batanghari and Goldman, 1997) and modified (Beck et al., 2008). Samples used for NMR spectroscopy contained ~1 mM CBP in 10 mM deuterated-HEPES (pH 6.5), 100 mM KCl, 0.02% (w/v) NaN3 and either 5% or 99% D2O.
Heterolabeled dimers were obtained by mixing equimolar unlabeled and fully (13C/15N-) labeled CBP (2 mM total protein concentration). The mixture was denatured to a final concentration of 8 M urea, then refolded by extensively dialyzing against 10 mM ammonium acetate, pH 7.0 for 48 hours, followed by lyophilization and resuspension in NMR buffer (99% D2O).
Delipidation of 15N-labeled CBP was attempted by passing protein over a lipophillic (“lipidex”) column to remove bound lipids (H6258 Hydroxyalkoxypropyl-Dextran; Sigma, St. Louis). Sample was then lyophilized to dryness and resuspended in NMR buffer.
NMR spectroscopy
All NMR data were collected at 25°C on 600- and 700-MHz Varian Unity INOVA (Palo Alto, CA) spectrometers. Pulse sequences were either provided by Varian (Palo Alto, CA) BioPack or were kindly provided by Dr. Lewis Kay (University of Toronto). Data were processed with FELIX 2001 (Accelrys, Inc.).
Distance restraints were obtained from isotope-edited 3D-NOESY experiments: 15N-15N-edited NOESY (τ = 200ms), 15N-edited (τ = 180 ms), 13C-edited (τ = 180 ms), and aromatic 13C-edited (τ = 200 ms) NOESY-HSQC. Inter-monomer distance restraints were obtained from 3D 13C F1-filtered, 12C F3-edited NOESY-HSQC(Zwahlen et al., 1997) (τ = 200 ms) with the heterolabeled dimer. Backbone dihedral angle restraints were obtained from backbone Cα, Cβ, C’, HN and N chemical shifts using TALOS (Cornilescu et al., 1999) and verified by directly measured 3JHNHα coupling constants (Vuister and Bax, 1993). Slowly exchanging amide protons were identified by recording 2D 1H-15N HSQC experiments, 24 and 48 hours after the transfer of lyophilized protein into D2O. Hydrogen bond restraints were obtained from the consensus of a combination of amide hydrogen exchange data, chemical shift index analysis (Wishart and Sykes, 1994b), and 3JHNHα (Vuister and Bax, 1993) experiments.
{1H} 15N heteronuclear NOEs were obtained by recording spectra with and without the use of presaturation applied during the delay between successive transients (Kay et al., 1989). Steady-state {1H}15N NOE values were determined from the ratio of peak intensities in the spectra recorded with and without saturation of protons during the NOE delay period and averaged for the duplicate spectra.
Structure calculations
NOESY spectra were peak picked in FELIX (Accelrys, Inc.) and used as input into ARIA (version 1.2) (Nilges et al., 1997) implemented in CNS (Version 1.1) (Brunger et al., 1998). For the first stage of NOE assignment and structure calculation, an in-house script was used to remove all peaks found in filtered-edited CH-NOESY spectra from the peak lists of the CH-, NH- and aromatic-NOESY spectra. Three pairs of disulfide bonds together with 46 backbone H-bond restraints were also incorporated into the calculations. Backbone H-bond restraints were included in regions of regular secondary structure as defined by both CSI consensus and characteristic NOE patterns. A total of 96 ϕ and ψ angle constraints from TALOS were also used.
The calculation protocol comprised eight iterations of 20 structures each and a final cycle of 100 structures using the default ARIA protocol to determine the structure of the monomeric subunit of homodimeric CBP. Manually assigned 15N-15N NOEs were included; however all remaining NOE restraints were assigned using ARIA’s ADR method for iterative structure calculation and NOE assignment (Nilges, 1995, 1997; Nilges et al., 1997). A total of 1152 unambiguous and 213 ambiguous restraints were identified by ARIA v1.2, and these restraints were manually checked. The 20 lowest energy structures from iteration 8 were refined in a 9-Å shell of water molecules.
The program HADDOCK (High Ambiguity Driven protein-protein DOCKing) (Dominguez et al., 2003) was used to “dock” the 10 lowest energy CBP monomers calculated in the previous stage using ARIA v1.2. The dimer interface was constrained with a set of nine manually assigned intermolecular NOEs from the filtered-edited CH-NOESY experiment. All default parameters of HADDOCK were used.
For the final stage of quaternary structure refinement, a pre-release ARIA version 2.2 (Rieping et al., 2007) structure calculation protocol was obtained from B. Bardiaux and M. Nilges (personal communication). ARIA v2.2β was implemented in order to calculate symmetric homodimers because all distance restraints can be designated as intermonomer, intramonomer, or both. In addition, non-crystallographic symmetry is enforced and a packing force can be used to maintain monomers in close proximity during simulated annealing. Default parameters were used with a few notable exceptions due to the fact that the initial linear chain structure for simulated annealing was replaced with the HADDOCK-derived CBP homodimer. The assignment parameter p was set to a smaller value (e.g. 0.9999 instead of 1) and the violation tolerance to an intermediate value of 5.0 Å (instead of default value of 1000 Å) as suggested for it0 (Linge et al., 2001). The final 20 conformers were refined in a shell of water and the resulting structural ensemble was validated by PROCHECK (Laskowski et al., 1993). The statistics for these are listed in Table 1 (per dimer). The final structural ensemble includes the 20 lowest-energy (total energy) structures with no distance violations >0.5 Å and no angle violations >10.0°. There were 71.1% of residues in the most favored region of the Ramachandran plot of the final 20 structures of CBP. All structural representations of CBP were drawn in MOLMOL (Koradi et al., 1996) or PyMol (DeLano, 2002). The atomic coordinates for CBP have been deposited in the Protein Data Bank, www.pdb.org (PDB ID code 2JV7). The chemical shifts for CBP have previously been deposited in BRMB with accession number 15404.
Table 1.
NMR and refinement statistics for CBP
| NMR distance and dihedral constraints | per monomer |
|---|---|
| Distance constraints | |
| Total NOE | 2679 |
| Intra-residue | 508 |
| Inter-residue | 1475 |
| Sequential (∣i − j∣=1) | 416 |
| Medium-range (∣i −j∣<4) | 297 |
| Long-range (∣i−j∣>5) | 381 |
| Intermolecular | 251 |
| Hydrogen bonds | 55 |
| Dihedral angle restraints | 90 |
| Structure statistics | |
| Violations (mean and s.d.) | |
| Distance constraints (Å) | 0.288 ± 0.002 |
| Dihedral angle constraints (°) | 1.91 ± 0.06 |
| Max. dihedral angle violation (°) | 7.43 |
| Max. distance constraint violation (Å) | 0.43 |
| Deviations from idealized geometry | |
| Bond lengths (Å) | 0.008 ± 0.0001 |
| Bond angles (°) | 1.1 ± 0.008 |
| Impropers (°) | 1.2 ± 0.01 |
| Average pairwise rms deviationa (Å) | |
| Heavy atoms | 1.12 ± 0.01 |
| Backbone | 0.61 ± 0.20 |
| Ramachandran plot (%)b | |
| Most favored regions | 71.1 |
| Additionally allowed regions | 14.8 |
| Generously allowed regions | 8.9 |
| Disallowed regions | 5.2 |
Pairwise r.m.s. deviation was calculated among 20 refined structures using ARIA2.2 (Rieping et al., 2007).
Calculated with PROCHECK-NMR (Laskowski et al., 1993).
NMR chemical shift mapping
Changes in the 2D 1H-15N HSQC spectra of 15N-labeled CBP upon delipidation were monitored and quantified using the length of the vector describing the chemical shift change for each peak: Δδ = [(ΔδHN)2 + (ωΔδN)2]1/2, where the weighting factor (0.15) is determined by the relative magnitudes of the amide nitrogen chemical shift range compared to that for the NH proton chemical shift range (Tugarinov and Kay, 2003).
ACKNOWLEDGMENTS
This work was supported by National Institutes of Health research grants AI25584 to W.E.G., DK48046 to D.P.C., and DK52574 to the Protein Structure Core of the Washington University Digestive Diseases Research Core Center, as well as a St. Louis-Pasteur Collaboration mini-grant to M.R.B from the Institut Pasteur. We thank K.B. Hall for constant support and critical reading of this manuscript, as well as C. Tang for writing scripts used in NMR data manipulation. We acknowledge M. Nilges, B. Bardiaux, M. Delepierre, and I. Guijjaro (Institut Pasteur) for privileged access to ARIA v2.2 during development and help with structure calculations.
References
- Ahn VE, Faull KF, Whitelegge JP, Fluharty AL, Prive GG. Crystal structure of saposin B reveals a dimeric shell for lipid binding. Proc Natl Acad Sci U S A. 2003;100:38–43. doi: 10.1073/pnas.0136947100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahn VE, Leyko P, Alattia JR, Chen L, Prive GG. Crystal structures of saposins A and C. Protein Sci. 2006;15:1849–1857. doi: 10.1110/ps.062256606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alattia JR, Shaw JE, Yip CM, Prive GG. Direct visualization of saposin remodelling of lipid bilayers. J Mol Biol. 2006;362:943–953. doi: 10.1016/j.jmb.2006.08.009. [DOI] [PubMed] [Google Scholar]
- Anderson DH, Sawaya MR, Cascio D, Ernst W, Modlin R, Krensky A, Eisenberg D. Granulysin crystal structure and a structure-derived lytic mechanism. J Mol Biol. 2003;325:355–365. doi: 10.1016/s0022-2836(02)01234-2. [DOI] [PubMed] [Google Scholar]
- Batanghari JW, Goldman WE. Calcium dependence and binding in cultures of Histoplasma capsulatum. Infect Immun. 1997;65:5257–5261. doi: 10.1128/iai.65.12.5257-5261.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batanghari JW, Deepe GS, Jr., Di Cera E, Goldman WE, Bhattacharya A, Murthy MR, Surolia A. Histoplasma acquisition of calcium and expression of CBP1 during intracellular parasitism. Mol Microbiol. 1998;27:531–539. doi: 10.1046/j.1365-2958.1998.00697.x. [DOI] [PubMed] [Google Scholar]
- Beck MR, DeKoster GT, Hambly DM, Gross ML, Cistola DP, Goldman WE. Structural Features Responsible for Biological Stability of Histoplasma’s Virulence Factor CBP. Biochemistry. 2008;47:4427–4438. doi: 10.1021/bi701495v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruhn H, Leippe M. Comparative modeling of amoebapores and granulysin based on the NK-lysin structure-structural and functional implications. Biol Chem. 1999;380:1001–1007. doi: 10.1515/BC.1999.124. [DOI] [PubMed] [Google Scholar]
- Bruhn H. A short guided tour through functional and structural features of saposin-like proteins. Biochem J. 2005;389:249–257. doi: 10.1042/BJ20050051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr D Biol Crystallogr. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- Cho HS, Liu CW, Damberger FF, Pelton JG, Nelson HC, Wemmer DE. Yeast heat shock transcription factor N-terminal activation domains are unstructured as probed by heteronuclear NMR spectroscopy. Protein Sci. 1996;5:262–269. doi: 10.1002/pro.5560050210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciaffoni F, Salvioli R, Tatti M, Arancia G, Crateri P, Vaccaro AM. Saposin D solubilizes anionic phospholipid-containing membranes. J Biol Chem. 2001;276:31583–31589. doi: 10.1074/jbc.M102736200. [DOI] [PubMed] [Google Scholar]
- Cornilescu G, Delaglio F, Bax A. Protein backbone angle restraints from searching a database for chemical shift and sequence homology. J Biomol NMR. 1999;13:289–302. doi: 10.1023/a:1008392405740. [DOI] [PubMed] [Google Scholar]
- de Alba E, Weiler S, Tjandra N. Solution structure of human saposin C: pH-dependent interaction with phospholipid vesicles. Biochemistry. 2003;42:14729–14740. doi: 10.1021/bi0301338. [DOI] [PubMed] [Google Scholar]
- DeLano WL. The PyMOL Molecular Graphics System. Vol. 2007. DeLano Scientific; Palo Alto, CA, USA: 2002. [Google Scholar]
- Dominguez C, Boelens R, Bonvin AM. HADDOCK: a protein-protein docking approach based on biochemical or biophysical information. J Am Chem Soc. 2003;125:1731–1737. doi: 10.1021/ja026939x. [DOI] [PubMed] [Google Scholar]
- Eissenberg LG, Schlesinger PH, Goldman WE. Phagosome-lysosome fusion in P388D1 macrophages infected with Histoplasma capsulatum. J Leukoc Biol. 1988;43:483–491. doi: 10.1002/jlb.43.6.483. [DOI] [PubMed] [Google Scholar]
- Eissenberg LG, Goldman WE, Schlesinger PH. Histoplasma capsulatum modulates the acidification of phagolysosomes. J Exp Med. 1993;177:1605–1611. doi: 10.1084/jem.177.6.1605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farrow NA, Zhang O, Forman-Kay JD, Kay LE. Comparison of the backbone dynamics of a folded and an unfolded SH3 domain existing in equilibrium in aqueous buffer. Biochemistry. 1995;34:868–878. doi: 10.1021/bi00003a021. [DOI] [PubMed] [Google Scholar]
- Gonzalez C, Langdon GM, Bruix M, Galvez A, Valdivia E, Maqueda M, Rico M. Bacteriocin AS-48, a microbial cyclic polypeptide structurally and functionally related to mammalian NK-lysin. Proc Natl Acad Sci U S A. 2000;97:11221–11226. doi: 10.1073/pnas.210301097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hecht O, Van Nuland NA, Schleinkofer K, Dingley AJ, Bruhn H, Leippe M, Grotzinger J. Solution structure of the pore-forming protein of Entamoeba histolytica. J Biol Chem. 2004;279:17834–17841. doi: 10.1074/jbc.M312978200. [DOI] [PubMed] [Google Scholar]
- Holm L, Sander C. Protein structure comparison by alignment of distance matrices. J Mol Biol. 1993;233:123–138. doi: 10.1006/jmbi.1993.1489. [DOI] [PubMed] [Google Scholar]
- Kay LE, Torchia DA, Bax A. Backbone dynamics of proteins as studied by 15N inverse detected heteronuclear NMR spectroscopy: application to staphylococcal nuclease. Biochemistry. 1989;28:8972–8979. doi: 10.1021/bi00449a003. [DOI] [PubMed] [Google Scholar]
- Kervinen J, Tobin GJ, Costa J, Waugh DS, Wlodawer A, Zdanov A. Crystal structure of plant aspartic proteinase prophytepsin: inactivation and vacuolar targeting. Embo J. 1999;18:3947–3955. doi: 10.1093/emboj/18.14.3947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolter T, Sandhoff K. Principles of lysosomal membrane digestion: stimulation of sphingolipid degradation by sphingolipid activator proteins and anionic lysosomal lipids. Annu Rev Cell Dev Biol. 2005;21:81–103. doi: 10.1146/annurev.cellbio.21.122303.120013. [DOI] [PubMed] [Google Scholar]
- Koradi R, Billeter M, Wuthrich K. MOLMOL: a program for display and analysis of macromolecular structures. J Mol Graph. 1996;14:51–55. 29–32. doi: 10.1016/0263-7855(96)00009-4. [DOI] [PubMed] [Google Scholar]
- Kugler S, Young B, Miller VL, Goldman WE, Bhattacharya A, Murthy MR, Surolia A. Monitoring phase-specific gene expression in Histoplasma capsulatum with telomeric GFP fusion plasmids. Cell Microbiol. 2000;2:537–547. doi: 10.1046/j.1462-5822.2000.00078.x. [DOI] [PubMed] [Google Scholar]
- Laskowski RA, MacArthur MW, Moss DS, Thornton JM. PROCHECK: a program to check the stereochemical quality of protein structures. J. Appl. Cryst. 1993;26:283–291. [Google Scholar]
- Liepinsh E, Andersson M, Ruysschaert JM, Otting G. Saposin fold revealed by the NMR structure of NK-lysin. Nat Struct Biol. 1997;4:793–795. doi: 10.1038/nsb1097-793. [DOI] [PubMed] [Google Scholar]
- Linge JP, O’Donoghue SI, Nilges M. Automated assignment of ambiguous nuclear overhauser effects with ARIA. Methods Enzymol. 2001;339:71–90. doi: 10.1016/s0076-6879(01)39310-2. [DOI] [PubMed] [Google Scholar]
- Liu A, Wenzel N, Qi X. Role of lysine residues in membrane anchoring of saposin C. Arch. Biochem. Biophys. 2005;443:101–112. doi: 10.1016/j.abb.2005.09.007. [DOI] [PubMed] [Google Scholar]
- Miteva M, Andersson M, Karshikoff A, Otting G. Molecular electroporation: a unifying concept for the description of membrane pore formation by antibacterial peptides, exemplified with NK-lysin. FEBS Lett. 1999;462:155–158. doi: 10.1016/s0014-5793(99)01520-3. [DOI] [PubMed] [Google Scholar]
- Munford RS, Sheppard PO, O’Hara PJ. Saposin-like proteins (SAPLIP) carry out diverse functions on a common backbone structure. J Lipid Res. 1995;36:1653–1663. [PubMed] [Google Scholar]
- Nilges M. Calculation of protein structures with ambiguous distance restraints. Automated assignment of ambiguous NOE crosspeaks and disulphide connectivities. J Mol Biol. 1995;245:645–660. doi: 10.1006/jmbi.1994.0053. [DOI] [PubMed] [Google Scholar]
- Nilges M. Ambiguous distance data in the calculation of NMR structures. Fold Des. 1997;2:S53–57. doi: 10.1016/s1359-0278(97)00064-3. [DOI] [PubMed] [Google Scholar]
- Nilges M, Macias MJ, O’Donoghue SI, Oschkinat H. Automated NOESY interpretation with ambiguous distance restraints: the refined NMR solution structure of the pleckstrin homology domain from beta-spectrin. J Mol Biol. 1997;269:408–422. doi: 10.1006/jmbi.1997.1044. [DOI] [PubMed] [Google Scholar]
- Palmer AG., 3rd Dynamic properties of proteins from NMR spectroscopy. Curr Opin Biotechnol. 1993;4:385–391. doi: 10.1016/0958-1669(93)90002-e. [DOI] [PubMed] [Google Scholar]
- Qi X, Grabowski GA. Differential membrane interactions of saposins A and C: implications for the functional specificity. J Biol Chem. 2001;276:27010–27017. doi: 10.1074/jbc.M101075200. [DOI] [PubMed] [Google Scholar]
- Rieping W, Habeck M, Bardiaux B, Bernard A, Malliavin T, Nilges M. ARIA2: automated NOE assignment and data integration in NMR structure calculation. Bioinformatics. 2007;23:381–382. doi: 10.1093/bioinformatics/btl589. [DOI] [PubMed] [Google Scholar]
- Rossmann M, Schultz-Heienbrok R, Behlke J, Remmel N, Alings C, Sandhoff K, Saenger W, Maier T. Crystal structures of human saposins C and D: Implications for lipid recognition and membrane interactions. Structure. 2008;16:809–817. doi: 10.1016/j.str.2008.02.016. [DOI] [PubMed] [Google Scholar]
- Sanchez-Barrena MJ, Martinez-Ripoll M, Galvez A, Valdivia E, Maqueda M, Cruz V, Albert A. Structure of bacteriocin AS-48: from soluble state to membrane bound state. J Mol Biol. 2003;334:541–549. doi: 10.1016/j.jmb.2003.09.060. [DOI] [PubMed] [Google Scholar]
- Schuette CG, Pierstorff B, Huettler S, Sandhoff K. Sphingolipid activator proteins: proteins with complex functions in lipid degradation and skin biogenesis. Glycobiology. 2001;11:81R–90R. doi: 10.1093/glycob/11.6.81r. [DOI] [PubMed] [Google Scholar]
- Sebghati TS, Engle JT, Goldman WE. Intracellular parasitism by Histoplasma capsulatum: fungal virulence and calcium dependence. Science. 2000;290:1368–1372. doi: 10.1126/science.290.5495.1368. [DOI] [PubMed] [Google Scholar]
- Soeda S, Hiraiwa M, O’Brien JS, Kishimoto Y. Binding of cerebrosides and sulfatides to saposins A-D. J Biol Chem. 1993;268:18519–18523. [PubMed] [Google Scholar]
- Tugarinov V, Kay LE. Quantitative NMR studies of high molecular weight proteins: application to domain orientation and ligand binding in the 723 residue enzyme malate synthase G. J Mol Biol. 2003;327:1121–1133. doi: 10.1016/s0022-2836(03)00238-9. [DOI] [PubMed] [Google Scholar]
- Vuister GW, Bax A. Quantitative J Correlation: A New Approach for Measuring Homonuclear Three-Bond J(HNHa) Coupling Constants in 15N-Enriched Proteins. J Am Chem Soc. 1993;115:7772–7777. [Google Scholar]
- Wishart DS, Sykes BD. The 13C chemical-shift index: a simple method for the identification of protein secondary structure using 13C chemical-shift data. J Biomol NMR. 1994a;4:171–180. doi: 10.1007/BF00175245. [DOI] [PubMed] [Google Scholar]
- Wishart DS, Sykes BD. Chemical shifts as a tool for structure determination. Methods Enzymol. 1994b;239:363–392. doi: 10.1016/s0076-6879(94)39014-2. [DOI] [PubMed] [Google Scholar]
- Ye Y, Godzik A. Database searching by flexible protein structure alignment. Protein Sci. 2004a;13:1841–1850. doi: 10.1110/ps.03602304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye Y, Godzik A. FATCAT: a web server for flexible structure comparison and structure similarity searching. Nucleic Acids Res. 2004b;32:W582–585. doi: 10.1093/nar/gkh430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zwahlen C, Legault P, Vincent S, Greenblatt J, Konrat R, Kay L. Methods for measurement of intermolecular NOEs by mulitnuclear NMR spectroscopy: application to a bacteriophage N-peptide/boxB RNA complex. J Am Chem Soc. 1997;119:6711–6721. [Google Scholar]