

Abstract
Fatty acid amide hydrolase (FAAH) is one of the principle enzymes for metabolizing endogenous cannabinoid neurotransmitters such as anandamide, and thus regulates endocannabinoid (eCB) signaling. Selective pharmacological blockade of FAAH has emerged as a potential therapy to discern the endogenous functions of anandamide-mediated eCB pathways in anxiety, pain and addiction. Quantification of FAAH in the living brain by positron emission tomography (PET) would help our understanding of the endocannabinoid system in these conditions. While most FAAH radiotracers operate by an irreversible (‘suicide’) binding mechanism, a FAAH tracer with reversibility would facilitate quantitative analysis. We have identified and radiolabeled a reversible FAAH inhibitor, 7-(2-[11C]methoxyphenyl)-1-(5-(pyridin-2-yl)oxazol-2-yl)heptan-1-one ([11C]MPPO) in 13% radiochemical yield (non-decay corrected) with >99% radiochemical purity and 2 Ci/μmol (74 GBq/μmol) specific activity. The tracer showed moderate brain uptake (0.8 SUV) with heterogeneous brain distribution. However, blocking studies with a potent FAAH inhibitor URB597 demonstrated a low to modest specificity to the target. Measurement of lipophilicity, metabolite and efflux pathway analysis were also performed to study the pharmacokinetic profile of [11C]MPPO. In all, we reported an efficient radiolabeling and preliminary evaluation of the first-in-class FAAH inhibitor [11C]MPPO with α-ketoheterocyclic scaffold.
Keywords: PET, fatty acid amide hydrolase, FAAH, [11C]MPPO, radiotracer
Graphical abstract
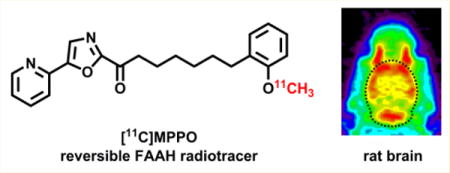
INTRODUCTION
Endocannabinoid signaling systems is a neuromodulatory network which regulates mammalian neurophysiology, including cognition and memory, motor function, anxiety, pain perception, addiction and reward behaviors.1 Endocannabinoids are endogenous small molecules (chemical messengers), and owing to their hydrophobic character which preclude storage into synaptic vesicles, they are biosynthesized and released ‘on request’ in vivo. The signaling network is modulated mainly via two G-protein-coupled receptors, namely, cannabinoid receptors CB1 and CB2. The corresponding endocannabinoid ligands of CB1/2 in mammals have been identified as anandamide (AEA)2 and 2-arachidonoylglycerol (2-AG).3,4 Early efforts have been focused on the direct pharmacological intervention of endocannabinoid system by its natural agonist Δ9-tetrahydrocannabinol (the psychoactive component of Cannabis sativa) and other synthetic cannabinoids; however, the concomitant detrimental effects on cognition and motor control limit their use as therapeutic agents. In order to circumvent this problem, targeting enzyme degradation that controls endocannabinoid (AEA and 2-AG) levels has emerged as a potential strategy to identify drug candidates for medicinal uses.5–9 Two enzymes, fatty acid amide hydrolase (FAAH) and monoacylglycerol lipase (MAGL) are primarily responsible for the degradation of AEA and 2-AG, respectively. In particular, FAAH is a ~60 kDa integral membrane enzyme that is highly expressed in the mammalian brain along with liver and kidney,10,11 and functions as a serine hydrolase equipped with an unusual serine-serine-lysine (Ser241-Ser217-Lys142) catalytic triad.12 Inhibition of FAAH increases the levels of AEA and has been found to reduce anxiety, pain, addiction and inflammation, as well as show anti-depressant and analgesic effects in preclinical models13–15 without adverse effects in motility and behavior change observed with direct CB1 interventions.16,17 Several reversible and irreversible FAAH inhibitors including URB59716 and PF-04457845,18–21 JNJ-4216527922 and V15886623 have advanced to clinical trials for release of osteoarthritis pain, cannabis withdrawal, anxiety and schizophrenia.
A positron emission tomography (PET) tracer for FAAH is desirable to allow quantitative enzyme mapping, target engagement and dose selection in clinical studies with high sensitivity under minimal perturbation of the biological state. As shown in Figure 1, there are continued efforts in the search for suitable FAAH PET tracers, including anadamide analogs,24 [11C-methyl]URB597 analog,25 [11C-carbonyl]URB694 ([11C]CURB; 1),26,27 [18F]DOPP (2),28,29 [11C]PF-04457845 (4),30,31 [18F]PF-9811 (5),32 MK-3168 (6),30 and most recently [11C]MFTC (3).33 To date, only two PET ligands have been reported in human studies, namely [11C]CURB (1)34,35 and [11C]MK-3168 (6).36 Although considerable advances have been made in the development of FAAH tracers, the majority of the reported radioligands are limited to suicide37 inhibitors (Figure 1A–B), which feature a covalent irreversible binding mechanism. For instance, [11C]CURB (1; Scheme 1A) binds to FAAH in vivo by acylation of the Ser241 sidechain and concomitant exclusion of unlabeled O-phenoxy fragment (7) to yield 11C-labeled FAAH (8). As the first radiotracer for FAAH in humans, [11C]CURB has been successfully utilized to map FAAH in the living brain using an irreversible two tissue compartment model.34 However, based on the binding mechanism, [11C]CURB only provides the measurement of FAAH activity, as opposed to FAAH availability,29 which could be extracted from a reversible tracer as used in a ligand-receptor scenario in PET studies.38 A FAAH tracer with reversibility would allow us to access key information from PET quantifications, including volumes of distribution, binding potentials, and monitor neurological response to therapeutics (occupancy and displacement studies).39,40 To date, [11C]MK-3168 (Figure 1C) is the only reversible PET ligand for this target and has advanced to human PET studies,31 but only preliminary proceedings have been reported since 2012.22,36 Therefore there is an urgent need for reversible FAAH radiotracers to address these questions and provide a full quantitative assessment of FAAH distribution, concentration, and activities in brain, to establish it as a viable biomarker for the diagnosis and prognosis of patients with associated disorders in endocannabinoid signaling system.
Figure 1.
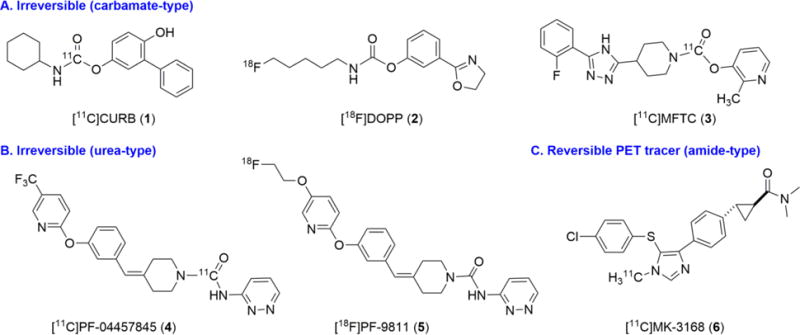
Representative PET radiotracers for imaging FAAH.
Scheme 1.
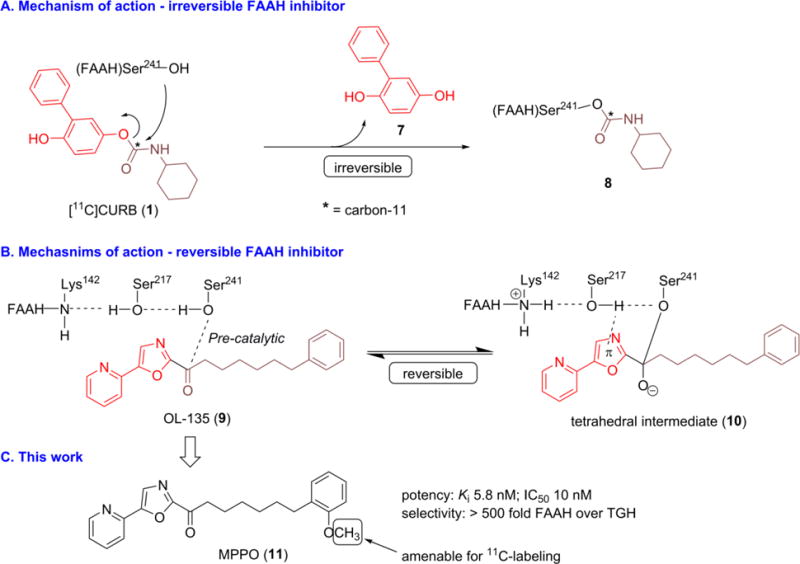
Mechanism of action and this work.
With the goal to develop a novel and reversible FAAH radiotracer, we aimed to design a 11C-labeled molecule based on a potent and selective FAAH inhibitor, 7-phenyl-1-(5-(pyridin-2-yl)oxazol-2-yl)heptan-1-one (OL-135; 9) with an established reversible binding mechanism (Scheme 1B).41,42 Briefly, the hydroxyl group in Ser241 residue of FAAH is added to the carbonyl moiety of OL-135 to form an enzyme-bound intermediate (10), which is further stabilized by hydrogen bonding between FAAH(Ser217) and the oxazole ring. The tetrahedral intermediate 10 is covalently bound to FAAH(Ser241), but in a reversible manner, because the reaction cannot proceed further to release a leaving group. On the basis of these findings, we speculated that a positron-emitting analog of OL-135 would provide a potential reversible PET ligand for FAAH based on the existing mechanism of action. Therefore, we selected a recently reported close analog of OL-135, 7-(2-methoxyphenyl)-1-(5-(pyridin-2-yl)oxazol-2-yl)heptan-1-one (MPPO; 11, Scheme 1C), which is amenable for 11C-labeling and exhibited an excellent binding profile43 to FAAH in vitro with IC50 value of 10 nM and greater than 500 fold selectivity over triacylglycerol hydrolase (TGH) as well as > 105 fold selectivity over hydrolytic enzyme KIAA1363. Herein we describe our synthesis, radiolabeling and preliminary evaluation of [11C]MPPO, the first-in-kind α-ketoheterocyclic PET ligand targeting FAAH.
RESULTS AND DISCUSSION
Chemistry
With MPPO 11 as the target, we designed a synthetic strategy for the synthesis (for detailed retrosynthetic analysis, see Scheme S1 in Supporting Information). As summarized in Scheme 2A, 5-(pyridin-2-yl)oxazole (14) was constructed in a quantitative yield according to the literature procedure.43–45 Protection of alcohol 15 with 3,4-dihydro-2H-pyran in the presence of a catalytic amount of PPTS afforded bromide 16 in 85% yield. Condensation of 2-methoxybenzaldehyde with Wittig salt 17 afforded the mixture of Z/E olefins 18 in a yield of 56% over two steps. It was worthy of note that the synthesis of Wittig salt 17 in organic solvents, such as THF, toluene or CH3CN, led to low to modest yields of 18 (0–21%). The optimized reaction parameters were identified as solvent-free conditions in neat PPh3 at 120 °C for 4 h to generate 17, which was utilized in the subsequent Wittig olefination immediately. Compound 19 was obtained in 99% yield on exposure to a Pd/C catalyzed hydrogenation of 18 and the subsequent deprotection with TsOH yielded 20. Swern oxidation afforded aldehyde 21 in 83% yield. The synthesis of key intermediate 22 was not straightforward since base-mediated condensation using KHMDS, LHMDS, nBuLi, nBuLi/Bu3SnCl, nBuLi/ZnCl2/CuI or MeMgBr failed to produce the desired product. A notable improvement was achieved when iPrMgCl in THF was used, leading to the secondary alcohol 22 in almost quantitative conversion. Compound 22 was used without further purification in the following Dess-Martin oxidation to generate MPPO (11) in 51% yield. Demethylation of 11 with BBr3 in CH2Cl2 gave the radiolabeling precursor phenol (7-(2-hydroxyphenyl)-1-(5-(pyridin-2-yl)oxazol-2-yl)heptan-1-one; 23) in 58% yield after recrystallization.
Scheme 2.
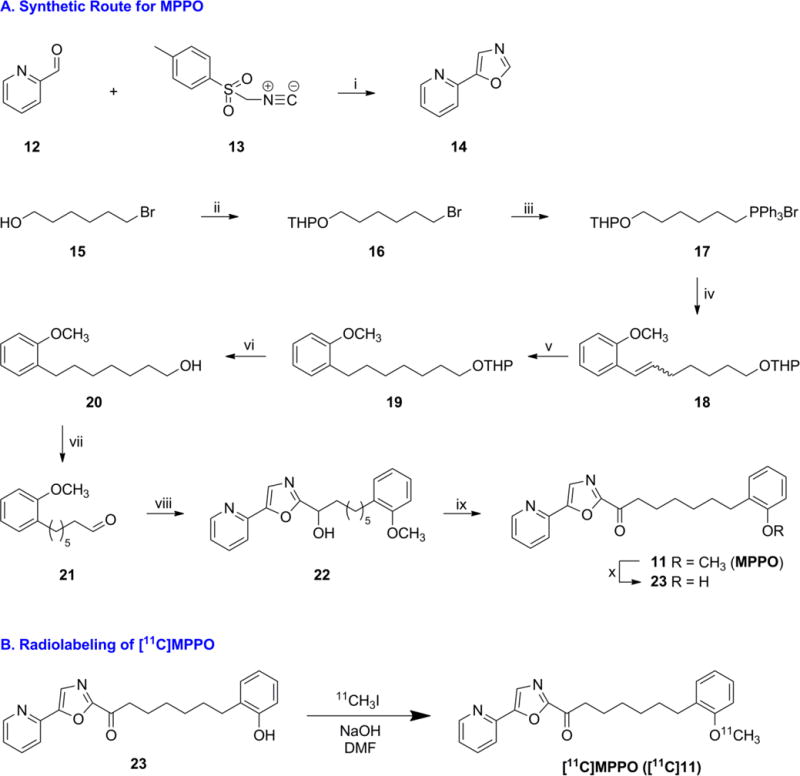
Syntheses of standard and precursor, and radiolabeling of [11C]MPPO
aReagents and conditions: (i) K2CO3, MeOH, 96%. (ii) 3,4-dihydro-2H-pyran, PPTS, CH2Cl2, 85%. (iii) PPh3, 120 °C. (iv) 2-methoxybenzaldehyde, LiHMDS, THF, 56% over two steps. (v) Pd/C, H2, MeOH, 99%. (vi) p-Toluenesulfonic acid monohydrate, MeOH, 89%. (vii) (COCl)2, DMSO, Et3N, CH2Cl2, 83%. (viii) 14, iPrMgCl, THF. (ix) Dess-Martin periodinane, CH2Cl2, 51% over two steps. (x) BBr3, CH2Cl2, 58%.
Radiolabeling
As shown in Scheme 2B, the methyl ether was identified as the most convenient labeling site for MPPO with carbon-11. The automated radiosynthesis of [11C]MPPO was performed by the reaction of a phenolic precursor 23 with [11C]CH3I in the presence of NaOH. [11C]CH3I was bubbled through the reaction vial and after 5 min of reaction time at 80 °C, we observed highly selective conversion to [11C]MPPO. The reaction mixture was purified by reverse phase HPLC, and reformulated in saline for intravenous injection. [11C]MPPO was synthesized in 13 ± 3% radiochemical yields based on [11C]CO2 (non-decay corrected) for the subsequent preclinical PET imaging studies. Specifically, starting from 350 – 530 mCi (13.0 – 19.6 GBq) of [11C]CO2, [11C]MPPO was obtained in 58 ± 17 mCi (2.15 ± 0.65 GBq) at end-of-synthesis (~29 min synthesis time from end-of-bombardment) with > 99% radiochemical purity (n = 10; for semi-preparative and analytical HPLC results, see Figure S1 in Supporting Information). The specific activity was greater than 2 Ci/μmol (74 GBq/μmol) and no radiolysis was observed within 90 min after formulation.
In a parallel approach, we also prepared an aliphatic analog of OL-135 for radiolabeling. Swern oxidation of alcohol 24 gave the corresponding aldehyde 25 in 70% yield. After a sequential iPrMgCl-mediated condensation and Dess-Martin oxidation, radiolabeling precursor bromide 26 was obtained in a yield of 72% over two steps. The resulting bromide 26 was subjected to nucleophilic displacement with KF and 18-crown-6 in CH3CN to give the corresponding fluoro analog 27 in 30% yield after recrystallization. The radiofluorination proceeded to generate [18F]27 in 3% isolated radiochemical yield with greater than 1 Ci/μmol specific activity; however, further evaluation was not pursued due to a rapid defluorination in vivo (Scheme S2 in Supporting Information).
Lipophilicity
Lipophilicity of radiolabeled tracer has predictive utility for assessing blood-brain barrier permeability, with an optimum logP range of 2.0–3.5.46–49
Using liquid-liquid partition between n-octanol and PBS buffer (pH 7.4),47 logD7.4 of [11C]MPPO was determined to be 3.43 (n = 3), which is comparable with several brain penetrant FAAH tracers, including [11C]CURB (1; logD7.4 2.8),26 [11C]PF-04457845 (4; logD7.4 3.48).30
Whole body biodistribution studies in mice
The kinetics and tissue distribution of [11C]MPPO was studied in mice at several experimental time points (1, 5, 15, 30 and 60 min) post-tracer injection. The results are expressed as the percentage of injected dose per wet tissue (%ID/g) in Table 1 (for biodistribution expressed as SUV unit, see Table S1 in Supporting Information). At 1 min post injection, a high uptake (> 3 %ID/g) was observed in the heart, lungs, liver, kidneys and small intestine. After the initial phase the radioactivity levels in most tissues decreased rapidly, while the signals in the liver and small intestine continually increased until 15 min and then decreased slowly. The radiotracer was efficiently cleared from blood (1 min/60 min ratio of 6.1) and high uptake of [11C]MPPO in the liver, kidney, and small intestine suggests that hepatobiliary and urinary excretion, as well as the intestinal reuptake pathway, may dominate the whole body distribution of radioactivity. The present result indicates that the distribution of [11C]MPPO was in agreement with the distribution of FAAH in mice as reported previously,50–52 with high expression in the liver, brain, testes, kidneys and spleen. In addition, rapid clearance of radioactivity from lungs, heart and muscle was observed, which is consistent with low FAAH expression in these organs in mice.50
Table 1.
Distribution of radioactivity in mice after injection of [11C]MPPO. Data are %ID/g (mean ± SD, n = 3)
| tissue | 1 min | 5 min | 15 min | 30 min | 60 min |
|---|---|---|---|---|---|
| blood | 1.83 ± 0.15 | 0.75 ± 0.07 | 0.38 ± 0.03 | 0.34 ± 0.05 | 0.30 + 0.01 |
| heart | 6.89 ± 0.60 | 1.65 ± 0.07 | 0.55 ± 0.01 | 0.51 ± 0.03 | 0.32 ± 0.03 |
| lungs | 17.47 ± 3.88 | 4.88 ± 0.41 | 1.20 ± 0.04 | 1.19 ± 0.18 | 0.64 ± 0.08 |
| liver | 9.78 ± 1.10 | 18.61 ± 2.31 | 20.05 ± 1.43 | 17.47 ± 1.10 | 13.69 ±0.63 |
| spleen | 2.28 ± 0.50 | 1.29 ± 0.22 | 0.60 ± 0.03 | 0.49 ± 0.03 | 0.37 ±0.01 |
| kidneys | 12.64 ± 0.56 | 17.17 ± 1.67 | 13.70 ± 0.69 | 9.55 ± 0.65 | 6.20 ± 0.70 |
| small intestine | 3.08 ± 0.29 | 7.22 ± 0.89 | 18.54 + 4.00 | 15.72 + 4.58 | 13.75 ±3.44 |
| testes | 0.44 ± 0.05 | 0.67 ± 0.02 | 0.59 ± 0.00 | 0.48 ± 0.02 | 0.30 ± 0.03 |
| muscle | 1.78 ±0.16 | 1.03 ± 0.09 | 0.58 ± 0.17 | 0.50 ± 0.17 | 0.24 ± 0.04 |
| brain | 1.87 ± 0.20 | 2.27 ± 0.16 | 1.81 ± 0.09 | 1.40 ± 0.15 | 0.88 + 0.09 |
The total level of initial brain uptake of [11C]MPPO was moderate to high with 1.87 %ID/g and 2.27 %ID/g at 1 min and 5 min post-tracer injection, respectively. The radioactivity washout from the brain was rapid with 0.88 %ID/g at 60 min time point (5 min/60 min ratio of 2.6). These results indicate moderate brain uptake (0.8 SUV) of [11C]MPPO. Therefore we further evaluated [11C]MPPO as a suitable reversible PET tracer for FAAH neuroimaging in PET imaging studies in Sprague-Dawley rats.
PET imaging studies in rats
Representative PET images of rat brain after injection of [11C]MPPO are shown in Figure 2A. PET images in normal rats showed moderate brain penetration and accumulation of radioactivity in the brain. The highest radioactivity was seen in the cerebellar nuclei (0.87 SUV), followed by cerebral cortex, hippocampus, thalamus, striatum while the lowest uptake was observed in the pons. As shown in the time-activity curves of different brain regions, radioactivity in brain tissues increased rapidly after the injection of [11C]MPPO, peaked at 1.5 min (0.87 SUV in cerebellar nuclei), and gradually washed out over 60 min. The distribution pattern of [11C]MPPO was similar to that for [11C]CURB26 or [11C]MFTC33 and the distribution of FAAH in the rat brain.51–54
Figure 2.
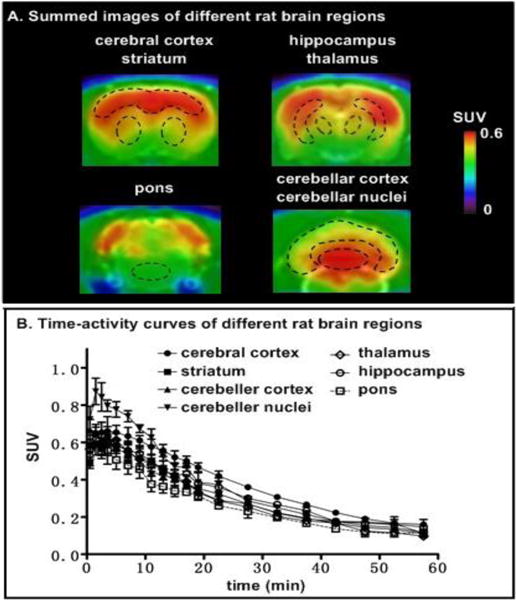
Representative coronal PET images (summed 0–30 min) and time-activity curves of [11C]MPPO in different rat brain regions (n = 3, mean ± SEM).
As shown in Figure 3, pretreatment with a potent FAAH inhibitor, URB597 (3 mg/kg, 30 min iv before injection), successfully abolished the difference of radioactivity uptakes in different regions of interest. The radioactivity distribution became fairly uniform in all brain regions, including cerebral cortex, cerebellar cortex, cerebellar nuclei and pons, indicating certain specificity of [11C]MPPO, albeit modest, to FAAH in the rat brains, although there was only a marginal difference of regional brain uptake between baseline and blocking.
Figure 3.
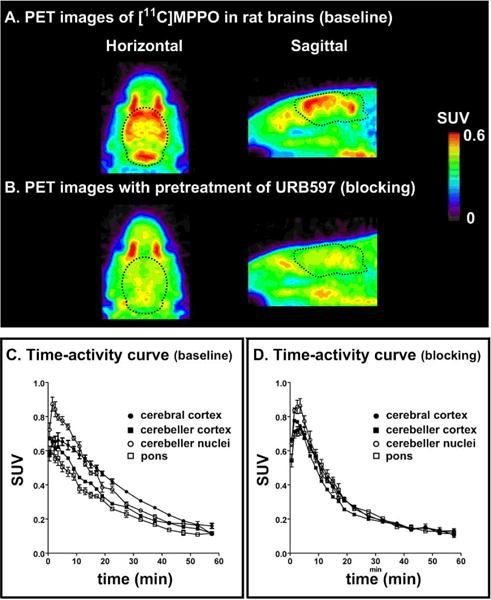
Representative PET images (0–30 min) and time-activity curves (baseline and blocking) of [11C]MPPO in rat brains. (n = 3; mean ± SEM).
[11C]MPPO, as hypothesized in Scheme 1B, may show a reversible binding profile to FAAH (Figure 4). The radioactivity reached the maximum uptake of 0.65 SUV, follow by a steady washout in rat brains (5 min/60 min ratio of 5.1). Compared with an irreversible FAAH radiotracer [11C]MFTC,33 within 60 min post injection, no significant decrease of [11C]MFTC in rat brain uptake was observed (5 min/60 min ratio of 0.9).
Figure 4.
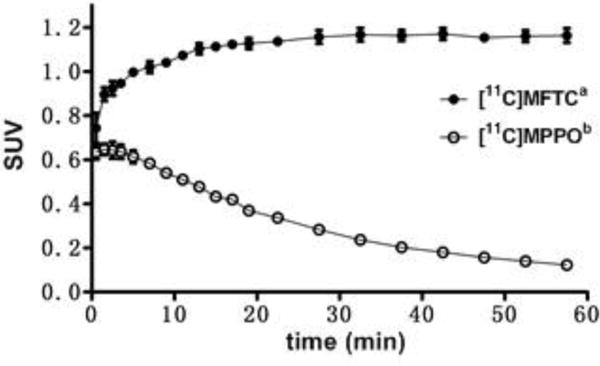
Reversible binding of [11C]MPPO in the brain. (n = 3; mean ± SD). a. SUV data from PET imaging of [11C]MFTC in rat whole brain; b. SUV data from PET imaging of [11C]MPPO in rat whole brain.
Metabolite analysis
To evaluate the in vivo stability of [11C]MPPO, the fraction of radiometabolites in the plasma and brain homogenate of Sprague-Dawley rats was evaluated post-tracer injection. The percentages of unchanged [11C]MPPO and the corresponding radiometabolites, as determined by radio-HPLC, are shown in Figure 5. Analysis of rat brain homogenates and plasma 15 min post-tracer injection showed that 75% and 90% metabolism occurred with the radioactivity associated with hydrophilic metabolites. These results indicates a rapid in vivo metabolism of [11C]MPPO, which may contribute to the low specific binding and/or rapid washout in PET imaging studies.
Figure 5.
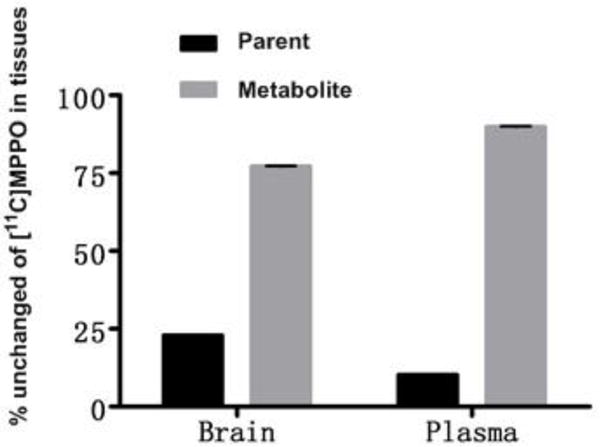
Radiometabolites of [11C]MPPO in rats. (n = 3; mean ± SD)
PET imaging studies in PgP/Bcrp knockout mice
Another possible reason for relatively a low-to-moderate brain uptake and/or specific binding of [11C]MPPO in the brain could be insufficient brain penetration due to ATP-binding cassette (ABC) efflux transporters, including P-glycoprotein (PgP) and breast cancer resistance protein (Bcrp) at the blood-brain barrier.40,55
In order to evaluate the interaction between [11C]MPPO and ABC transporters, we carried out PET imaging studies and compared radiotracer behavior, including brain uptake and washout on wild-type and PgP/Bcrp knockout mice (ABCB1a/1b−/−ABCG2−/−), in cerebral cortex and cerebellum regions. As shown in Figure 6, peak brain uptake in cerebral cortex and cerebellum was 0.95 SUV and 1.04 SUV in PgP/Bcrp KO mice, respectively, representing a marginal difference in comparison to the uptakes of cerebral cortex (0.74 SUV) and cerebellum (0.78 SUV) in wild type controls. In addition, brain uptakes in cerebral cortex and cerebellum of [11C]MPPO was not noticeably increased in PgP/Bcrp KO mice compared with that of wild type mice (ratio of KO/WT in AUC[cerebral cortex]= 1.2 and ratio of KO/WT in AUC[cerebellum]= 1.2); therefore these results indicates [11C]MPPO lacks intensive interactions with PgP/Bcrp efflux pump on the murine blood-brain barrier and is not likely a substrate for Pgp/Bcrp in mice.
Figure 6.
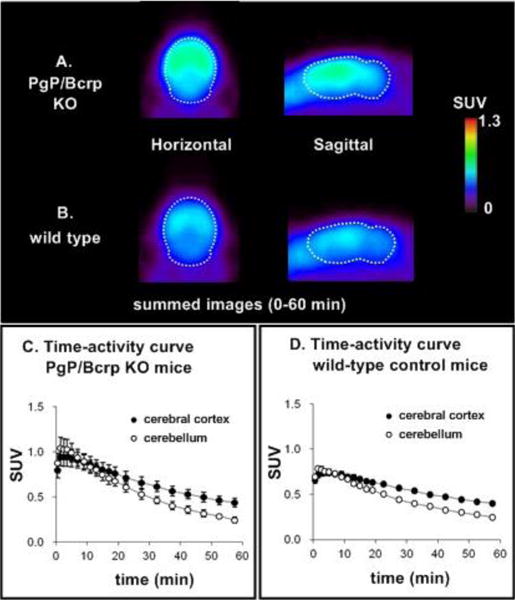
PET images (0–60 min) and time-activity curves in wild-type and PgP/BCRP KO mice (n = 3; mean ± SEM).
CONCLUSION
We have efficiently synthesized a new α-ketoheterocyclic FAAH radioligand, [11C]MPPO, in good radiochemical yield, high radiochemical purity and high specific activity. The lipophilicity, whole body distribution, brain penetration, efflux pump and metabolism studies were evaluated to determine the suitability of [11C]MPPO as a FAAH radiotracer. Preliminary PET imaging studies showed low to moderate level of specific binding to FAAH in murine brains. Although [11C]MPPO is less likely pursued for in vivo mapping of FAAH in the brain, the radiosynthesis and preliminary evaluation by PET imaging studies offers a roadmap for the investigation of other FAAH radiotracer candidates based on the α-ketoheterocyclic drug scaffolds with reversible binding profiles. In the future, radioligands with higher affinity and stability with minimal off-target binding will need to be developed to allow in vivo imaging of FAAH.
METHODS
Materials and Methods
Chemistry
General Conditions
All the chemicals employed in the syntheses were purchased from commercial vendors and used without further purification. Thin-layer chromatography (TLC) was conducted with 0.25 mm silica gel plates (60F-254) and visualized by exposure to UV light (254 nm) or stained with potassium permanganate. Flash column chromatography was performed using silica gel (particle size 0.040–0.063 mm). H-Nuclear magnetic resonance (NMR) spectra were obtained at 400 or 500 MHz on Bruker spectrometers in CDCl3 solutions at room temperature with tetramethylsilane (TMS, δ = 0) as an internal standard. 13C NMR spectra were obtained at 100 or 125 MHz and 19F-NMR spectra were obtained at 376 MHz. Chemical shifts (δ) are reported in ppm and coupling constants are reported in Hertz. The multiplicities are abbreviated as follows: s = singlet, d = doublet, t = triplet, m = multiplet, br = broad signal, dd = doublet of doublets, etc. For all the HRMS measurements, the ionization method is ESI and the mass analyzer type is TOF.
2-((7-(2-methoxyphenyl)hept-6-en-1-yl)oxy)tetrahydro-2H-pyran (18)
Compound 16 (3.9 g, 14.7 mmol) and PPh3 (5.8 g, 22.3 mmol) were placed in an oven-dried reaction vessel charged with a stir bar, which was filled with argon. The mixture was heated at 120 °C for 4 h, then cooled down to ambient temperature, followed by addition of THF (40 mL). The resulting mixture was stirred at room temperature for 10 min, then LiHMDS (1.0 M in THF, 18.4 mL, 18.4 mmol) was added slowly. The reaction was stirred for 1 h and cooled down to 0 °C. A solution of 2-methoxybenzaldehyde (1.8 g, 13.5 mmol) in THF (20 mL) was added into the reaction. The mixture was stirred overnight at room temperature, then diluted with EtOAc (70 mL), and washed with H2O (30 mL × 1) and brine (30 mL × 1), dried over anhydrous Na2SO4, and concentrated. The residue was purified by flash chromatography on silica gel (hexanes to ethyl acetate gradient column) to yield the compound 18 as a yellow oil (2.3 g, 56% over two steps). Rf = 0.5 (Hexanes/EtOAc = 50/1); 1H NMR showed the compound exists as a 1:0.6 mixture of Z- and E- products, which are used for the next step without further separation; HRMS calcd for C19H29O3 (M + H)+ 305.2117, found 305.2121.
7-(2-methoxyphenyl)-1-(5-(pyridin-2-yl)oxazol-2-yl)heptan-1-one (11)
To a solution of the compound 14 (219 mg, 1.5 mmol) in anhydrous THF (7 mL) at 0 °C was added iPrMgCl (2 M in THF, 0.9 mL, 1.8 mmol), the mixture was stirred at 0 °C for 1 h. A solution of compound 21 (330 mg, 1.5 mmol) in THF (4 mL) was added into the reaction, and the mixture was allowed to room temperature and stirred for 3 h and quenched with saturated aqueous NH4Cl (10 mL) and extracted with EtOAc (3 × 10 mL). The organic layers were combined, washed with water (1 × 10 mL) and brine (1 × 10 mL), dried over Na2SO4, filtered and evaporated under reduced pressure. The residue was dissolved immediately into CH2Cl2 (10 mL). Dess-Martin periodinane (1.1 g, 2.6 mmol) was added to a solution and the resultant suspension was stirred at rt for 2 h. Then the reaction was quenched with saturated aqueous Na2SO3 solution (5 mL) and NaHCO3 solution (5 mL), and the mixture was stirred for another 30 min. Then the aqueous layer was extracted with CH2Cl2 (3 × 7 mL). The combined organic portion was washed with brine (5 mL), dried over Na2SO4, filtered, and concentrated in vacuo. The residue was purified by flash chromatography on silica gel (hexanes to ethyl acetate gradient column) to yield the compound 11 as a white solid (278 mg, 51% over two steps). Rf = 0.7 (Hexanes/EtOAc = 2/1); 1H NMR (400 MHz, CDCl3) δ 8.68 (d, J = 4.8 Hz, 1H), 7.90–7.87 (m, 2H), 7.82 (td, J = 8.0, 1.6 Hz, 1H), 7.35–7.31 (m, 1H), 7.20–7.13 (m, 2H), 6.91–6.84 (m, 2H), 3.83 (s, 3H), 3.13 (t, J = 7.2 Hz, 2H), 2.62 (t, J = 7.6 Hz, 2H), 1.85–1.77 (m, 2H), 1.66–1.58 (m, 2H), 1.50–1.40 (m, 4H); 13C NMR (100 MHz, CDCl3) δ 188.5, 157.4, 153.2, 150.1, 146.3, 137.1, 131.0, 129.7, 126.8, 124.1, 120.3, 120.2, 110.1, 55.2, 39.1, 30.0, 29.6, 29.2, 29.0, 23.9; HRMS calcd for C22H25N2O3 (M + H)+ 365.1865; found 365.1859.
7-(2-hydroxyphenyl)-1-(5-(pyridin-2-yl)oxazol-2-yl)heptan-1-one (23)
To a solution of the compound 11 (428 mg, 1.17 mmol) in anhydrous CH2Cl2 (15 mL) at −78 °C was added BBr3 (0.35 mL, 3.53 mmol), the mixture was allowed to room temperature and stirred for 1 h, and quenched with saturated aqueous NaHCO3 (7 mL) and 10% NaOH (7 mL). The aqueous layer was extracted with CH2Cl2 (3 × 7 mL). The combined organic portion was washed with brine (10 mL), dried over Na2SO4, filtered, and concentrated in vacuo. The residue was purified by flash chromatography on silica gel (hexanes to ethyl acetate gradient column) to yield the crude product, which was recrystallized using 10% CH2Cl2/Hexanes to yield pure compound 23 as a white solid (238 mg, 58%). Rf = 0.4 (Hexanes/EtOAc = 2/1); 1H NMR (500 MHz, CDCl3) δ 8.68 (d, J = 4.8 Hz, 1H), 7.89–7.86 (m, 2H), 7.82 (td, J = 8.0, 1.6 Hz, 1H), 7.34–7.31 (m, 1H), 7.12–7.10 (m, 1H), 7.10–7.04 (m, 1H), 6.85 (t, J = 7.5 Hz, 1H), 6.76 (dd, J = 8.0, 0.5 Hz, 1H), 5.35–5.30 (m, 1H), 3.11 (t, J = 7.0 Hz, 2H), 2.62 (t, J = 7.5 Hz, 2H), 1.81–1.77 (m, 2H), 1.68–1.63 (m, 2H), 1.45–1.42 (m, 4H); 13C NMR (125 MHz, CDCl3) δ 188.5, 157.3, 153.5, 153.1, 150.0, 146.2, 137.1, 130.2, 128.5, 126.9, 126.8, 124.1, 120.6, 120.4, 115.1, 39.0, 29.7, 29.4, 28.9, 28.8, 23.8; HRMS calcd for C21H23N2O3 (M + H)+ 351.1709; found 351.1702.
12-bromo-1-(5-(pyridin-2-yl)oxazol-2-yl)dodecan-1-one (26)
To a solution of the compound 14 (500 mg, 3.42 mmol) in anhydrous THF (15 mL) at 0 °C was added iPrMgCl (2 M in THF, 2.1 mL, 4.2 mmol), the mixture was stirred at 0 °C for 1 h. A solution of aldehyde 25 (1.1 g, 4.2 mmol) in THF (10 mL) was added into the reaction, and the mixture was allowed to room temperature and stirred for 3 h and quenched with saturated aqueous NH4Cl (7 mL) and extracted with EtOAc (3 × 10 mL). The organic layers were combined, washed with water (1 × 10 mL) and brine (1 × 10 mL), dried over Na2SO4, filtered and evaporated under reduced pressure. The residue was dissolved immediately into CH2Cl2 (30 mL). Dess-Martin periodinane (1.77 g, 4.18 mmol) was added to a solution and the resultant suspension was stirred at rt for 2 h. Then the reaction was quenched with saturated aqueous Na2SO3 solution (7 mL) and NaHCO3 solution (7 mL), and the mixture was stirred for another 30 min. Then the aqueous layer was extracted with CH2Cl2 (3 × 15 mL). The combined organic portion was washed with brine (15 mL), dried over Na2SO4, filtered, and concentrated in vacuo. The residue was purified by flash chromatography on silica gel (hexanes to ethyl acetate gradient column) to yield the compound 26 as a white solid (997 mg, 72% over two steps). Rf = 0.25 (Hexanes/EtOAc = 10/1); 1H NMR (400 MHz, CDCl3) δ 8.67–8.65 (m, 1H), 7.87–7.85 (m, 2H), 7.80 (td, J = 7.6, 1.6 Hz, 1H), 7.32–7.29 (m, 1H), 3.39 (t, J = 6.8 Hz, 2H), 3.10 (t, J = 7.6 Hz, 2H), 1.88–1.80 (m, 2H), 1.79–1.73 (m, 2H), 1.42–1.27 (m, 14H); 13C NMR (100 MHz, CDCl3) δ 188.6, 157.4, 153.3, 150.1, 146.3, 137.1, 126.8, 124.1, 120.4, 39.1, 34.1, 32.8, 29.4, 29.3, 29.2, 29.1, 28.7, 28.1, 24.0; HRMS calcd for C20H28BrN2O2 (M + H)+ 407.1334; found 407.1331.
12-fluoro-1-(5-(pyridin-2-yl)oxazol-2-yl)dodecan-1-one (27)
A solution of 18-crown-6 (486 mg, 1.84 mmol) in anhydrous CH3CN (5.5 mL) was heated at 85 °C for 10 min, then KF (110 mg, 1.88 mmol) was added into the hot solution. And the mixture was stirred at 85 °C for 20 min. After that, compound 26 (150 mg, 0.37 mmol) was added, and the vessel was sealed tightly. The mixture was stirred at 95 °C for 42 h, then quenched with saturated aqueous NH4Cl (10 mL) and extracted with Et2O (3 × 10 mL). The combined organic portion was washed with brine (2 × 10 mL), dried over Na2SO4, filtered, and concentrated in vacuo. The residue was purified by flash chromatography on silica gel (hexanes to ethyl acetate gradient column) to yield the crude compound, which was recrystallized using 10% CH2Cl2/pentane to yield pure compound 27 as a white solid (38 mg, 30%). Rf = 0.25 (Hexanes/EtOAc = 10/1); 1H NMR (400 MHz, CDCl3) δ 8.67–8.65 (m, 1H), 7.88–7.85 (m, 2H), 7.80 (td, J = 7.6, 2.0 Hz, 1H), 7.33–7.29 (m, 1H), 4.43 (dt, J = 47.2, 6.4 Hz, 2H), 3.11 (t, J = 7.2 Hz, 2H), 1.79–1.70 (m, 2H), 1.69–1.61 (m, 2H), 1.40–1.24 (m, 14H); 13C NMR (100 MHz, CDCl3) δ 188.6, 157.4, 153.3, 150.1, 146.3, 137.1, 126.8, 124.1, 120.4, 84.2 (d, JC–F = 163.1 Hz), 39.1, 30.5, 30.3, 29.4, 29.3, 29.2, 29.1, 25.1, 25.0, 24.0; 19F NMR (376 MHz, CDCl3) δ −217.9; HRMS calcd for C20H28FN2O2 (M + H)+ 347.2135; found 347.2129.
Radiolabeling of [11C]MPPO
Carbon-11 CO2 was produced by 14N(p, α)11C nuclear reaction using a CYPRIS HM18 cyclotron (Sumitomo Heavy Industry, Tokyo, Japan). [11C]Methyl iodide ([11C]CH3I) was synthesized from cyclotron-produced [11C]CO2. Briefly, [11C]CO2 was bubbled into a solution of LiAlH4 (0.4 M in THF, 300 μL). After evaporation, the remaining reaction mixture was treated with hydroiodic acid (57% aqueous solution, 300 μL). The resulting [11C]CH3I was transferred under helium gas with heating into a pre-cooled (−15 to −20 °C) reaction vessel containing precursor 23 (1 mg), NaOH (6 μL, 0.5 M) and anhydrous DMF (300 μL). After the radioactivity reached a plateau during transfer, the reaction vessel was warmed to 80 °C and maintained for 5 min. CH3CN/0.05M NH4OAc (v/v, 2/8, 1 mL) was added to the reaction mixture, which was then injected to a semi-preparative HPLC system. HPLC purification was completed on a Capcell Pak C18 column (10 mm ID × 250 mm; Shiseido, Tokyo) using a mobile phase of CH3CN/0.05M NH4OAc (v/v, 2/8) at a flowrate of 5.0 mL/min. The retention time for [11C]MPPO was 9.5 min, whereas that for unreacted 23 was 5.1 min. The radioactive fraction corresponding to the desired product was collected in a sterile flask, evaporated to dryness in vacuo, re-dissolved in sterile normal saline (3 mL), and passed through a 0.22 μm Millipore filter. The synthesis time was ca. 27 min from end-of-bombardment. Radiochemical and chemical purity were measured by analytical HPLC (Capcell Pak C18, 4.6 mm ID × 250 mm, UV at 254 nm; CH3CN/0.05M NH4OAc (v/v, 2/8); retention time 8.5 min, 1.0 mL/min). The identity of [11C]MPPO was confirmed by the co-injection with unlabeled MPPO. The specific activity of [11C]MPPO was calculated based on a mass calibration curve at 254 nm. Radiochemical yield was 41% decay-corrected based on [11C]CO2 with >99% radiochemical purity and 2.2 Ci/μmol specific activity.
Measurement of Lipophilicity
The LogD value was measured by mixing [11C]MPPO (radiochemical purity: 100%; approximately 150,000 cpm) with n-octanol (3.0 g) and a sodium phosphate-buffered saline (PBS, 3.0 g; 0.1 M, pH 7.40) in a test tube. The tube was vortexed for 3 min at room temperature, followed by centrifugation at 3500 rpm for 5 min. An aliquot of 0.65 mL PBS and 0.65 mL n-octanol was removed, weighted, and its radioactivity was counted using a 1480 Wizard autogamma counter (PerkinElmer, Waltham, MA), respectively. Each sample from the remaining organic layer were removed and repartitioned until consistent LogD value was obtained. The LogD value was calculated by comparing the ratio of cpm/g of n-octanol to that of PBS and expressed as LogD = Log [cpm/g (n-octanol)/cpm/g (PBS)]. All assays were performed in triplicate.
Animal experiments
DdY mice (male; 7 weeks old; 34–36 g), and Sprague-Dawley (male; 7 weeks old; 210–230 g) rats were purchased from Japan SLC (Shizuoka, Japan). Wild-type (male; 17–18 weeks old; 30–32 g) and Pgp/Bcrp knockout (Abcb1a/1b−/−Abcg2−/−; male; 17–18 weeks old; 31–33 g) FAB mice were purchased from Taconic Farm (Hudson, NY). These animals were housed under a 12 h dark-light cycle and were allowed free access to food pellets and water. The animal experiments were approved by the Animal Ethics Committee of Massachusetts General Hospital and/or the National Institute of Radiological Sciences.
Biodistribution in mice
A saline solution of [11C]MPPO (2 MBq, 0.03 nmol/200 μL) was injected into ddy mice via tail vein. Three mice were sacrificed at 1, 5, 15, 30, and 60 min after injection. Whole brain, heart, lung, liver, spleen, kidneys, small intestine (including contents), muscle, testes, and blood samples were quickly removed and weighed. The radioactivity present in these tissues was measured using a gamma counter, and all radioactivity measurements were decay corrected. The uptake of each organ is expressed as a percentage of the injected dose per gram of wet tissue (% ID/g).
Small-animal PET study in rats
PET scans were performed using a small-animal Inveon PET scanner (Siemens Medical Solutions USA, Knoxville, TN), which provides 159 transaxial slices 0.796 mm (center to center) apart, a 10 cm transaxial field of view (FOV), and a 12.7 cm axial FOV. Prior to imaging studies, Sprague-Dawley rats were anesthetized with 5% (v/v) isoflurane and maintained by 1–2% (v/v) isoflurane. To inject [11C]MPPO, a 24-gauge needle with catheter (Terumo Medical Products, Tokyo) was placed into the rat tail vein. Emission scans were acquired for 90 min in three-dimensional list mode with an energy window of 350–750 keV, immediately after intravenous injection of [11C]MPPO (40–51 MBq, 0.7–1 nmol/200 μL). For the pretreatment studies, unlabeled URB597 (3.0 mg/kg) dissolved in 300 μL saline containing 10% ethanol and 5% Tween® 80) were injected at 30 min before the injection of [11C]MPPO. Three rats were used for each experiment.
PET dynamic images were reconstructed with filtered back-projection using a Hanning’s filter, a Nyquist cutoff of 0.5 cycle/pixel. The PET images were reconstructed using ASIPro VM software (Analysis Tools and System Setup/Diagnostics Tool, Siemens Medical Solutions). All list-mode acquisition data were sorted into three-dimensional sinograms, which were then Fourier rebinned into two-dimensional sinograms (1 min × 4 scans, 2 min × 8 scans, 5 min × 8 scans). Regions of interest were placed on the whole brain, cerebral cortex and cerebellum using ASIPro VM. Regional brain uptake of radioactivity was decay-corrected to the injection time and was expressed as the standardized uptake value (SUV), which was normalized to the injected radioactivity and body weight. SUV = (radioactivity per mL tissue/injected radioactivity) × g body weight.
Small-animal PET study in mice
A wide-type or Pgp/Bcrp knockout FAB mouse was secured in a custom-designed chamber, placed in the Inveon PET scanner, and prepared as described above. A 29-gauge needle with 12–15 cm of polyethylene 10 tube prepared in house was placed into the tail vein of mouse to inject [11C]MPPO (18 MBq, 0.12 nmol/100 μL). A dynamic emission scan in 3D list mode was performed for 60 min (1 min × 4 scans, 2 min × 8 scans, 5 min × 8 scans). Three mice were used for each experiment, respectively. The PET data were treated as above.
Metabolite assay for rat plasma and brain homogenate
Following the intravenous injection of [11C]MPPO (82 MBq, 1.1 nmol/200 μL), Sprague-Dawley rats (n = 3) were sacrificed by cervical dislocation at 15 min. Blood and whole brain samples were quickly removed and the blood samples were centrifuged at 15,000 g for 2 min at 4 °C to separate the plasma. The supernatant (0.5 mL) was then collected in a test tube containing CH3CN (0.5 mL) and the resulting mixture was vortexed for 15 s and centrifuged at 15,000 g for 2 min for deproteinization. The rat brain was homogenized in an ice-cooled CH3CN/H2O (1/1, 1 mL) solution. The homogenate was centrifuged at 150,000 rpm for 2 min at 4 °C and the supernatant was collected. The recovery of radioactivity into the supernatant was > 90% based on the total radioactivity in the brain homogenate.
An aliquot of the supernatant (100–500 μL) obtained from the plasma or brain homogenate was injected into the radio-HPLC system, and analyzed under the same analytical conditions described above, except the flow rate of 2 mL/min. The percentage of [11C]MPPO (retention time: 4.6 min) to total radioactivity (corrected for decay) on the HPLC charts was calculated as (peak area for [11C]MPPO/total peak area) × 100.
Supplementary Material
Acknowledgments
We would like to thank the staff at the radiochemistry program, Nuclear Medicine and Molecular Imaging, Massachusetts General Hospital, MA, USA and National Institute of Radiological Sciences, Chiba, Japan for their support with cyclotron operation, radioisotope production, radiosynthesis and animal experiments. S.H.L is a recipient of an NIH career development award (NIH/NIDA K01DA038000).
ABBREVIATIONS
- PET
positron emission tomography
- AEA
anandamide
- 2-AG
2-arachidonoylglycerl
- FAAH
fatty acid amide hydrolase
- MAGL
monoacylglycerol lipase
- SUV
standardized uptake value
- TAC
time-activity curve
- %ID/g
percentage of injected dose per gram of wet tissue
- SUV
standard uptake value
- KO
knockout
- PgP
P-glucoprotein
- Bcrp
breast cancer resistance protein
- 18-crown-6
1,4,7,10,13,16-hexaoxacyclooctadecane
- PPTS
Pyridinium p-toluenesulfonate
- LiHMDS
Lithium bis(trimethylsilyl)amide
- THF
tetrahydrofuran
- DMF
dimethylformamide
- DMSO
Dimethyl sulfoxide
Footnotes
Supporting Information
Characterization of all new compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
Notes
The authors declare no competing financial interest.
References
- 1.Ahn K, McKinney MK, Cravatt BF. Enzymatic Pathways That Regulate Endocannabinoid Signaling in the Nervous System. Chem Rev. 2008;108:1687–1707. doi: 10.1021/cr0782067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Devane WA, Hanus L, Breuer A, Pertwee RG, Stevenson LA, Griffin G, Gibson D, Mandelbaum A, Etinger A, Mechoulam R. Isolation and structure of a brain constituent that binds to the cannabinoid receptor. Science. 1992;258:1946–1949. doi: 10.1126/science.1470919. [DOI] [PubMed] [Google Scholar]
- 3.Mechoulam R, Ben-Shabat S, Hanus L, Ligumsky M, Kaminski NE, Schatz AR, Gopher A, Almog S, Martin BR, Compton DR, et al. Identification of an endogenous 2-monoglyceride, present in canine gut, that binds to cannabinoid receptors. Biochem Pharmacol. 1995;50:83–90. doi: 10.1016/0006-2952(95)00109-d. [DOI] [PubMed] [Google Scholar]
- 4.Sugiura T, Kondo S, Sukagawa A, Nakane S, Shinoda A, Itoh K, Yamashita A, Waku K. 2-Arachidonoylglycerol: a possible endogenous cannabinoid receptor ligand in brain. Biochem Biophys Res Commun. 1995;215:89–97. doi: 10.1006/bbrc.1995.2437. [DOI] [PubMed] [Google Scholar]
- 5.Ahn K, Johnson DS, Cravatt BF. Fatty acid amide hydrolase as a potential therapeutic target for the treatment of pain and CNS disorders. Expert Opinion on Drug Discovery. 2009;4:763–784. doi: 10.1517/17460440903018857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Minkkila A, Saario S, Nevalainen T. Discovery and development of endocannabinoid-hydrolyzing enzyme inhibitors. Curr Top Med Chem. 2010;10:828–858. doi: 10.2174/156802610791164238. [DOI] [PubMed] [Google Scholar]
- 7.Khanna IK, Alexander CW. Fatty acid amide hydrolase inhibitors–progress and potential. CNS Neurol Disord Drug Targets. 2011;10:545–558. doi: 10.2174/187152711796234989. [DOI] [PubMed] [Google Scholar]
- 8.Otrubova K, Ezzili C, Boger DL. The Discovery and Development of Inhibitors of Fatty Acid Amide Hydrolase (FAAH) Biorg Med Chem Lett. 2011;21:4674–4685. doi: 10.1016/j.bmcl.2011.06.096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Blankman JL, Cravatt BF. Chemical probes of endocannabinoid metabolism. Pharmacol Rev. 2013;65:849–871. doi: 10.1124/pr.112.006387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gulyas AI, Cravatt BF, Bracey MH, Dinh TP, Piomelli D, Boscia F, Freund TF. Segregation of two endocannabinoid-hydrolyzing enzymes into pre- and postsynaptic compartments in the rat hippocampus, cerebellum and amygdala. Eur J Neurosci. 2004;20:441–458. doi: 10.1111/j.1460-9568.2004.03428.x. [DOI] [PubMed] [Google Scholar]
- 11.McKinney MK, Cravatt BF. Structure and function of fatty acid amide hydrolase. Annu Rev Biochem. 2005;74:411–432. doi: 10.1146/annurev.biochem.74.082803.133450. [DOI] [PubMed] [Google Scholar]
- 12.Bracey MH, Hanson MA, Masuda KR, Stevens RC, Cravatt BF. Structural adaptations in a membrane enzyme that terminates endocannabinoid signaling. Science. 2002;298:1793–1796. doi: 10.1126/science.1076535. [DOI] [PubMed] [Google Scholar]
- 13.Lichtman AH, Shelton CC, Advani T, Cravatt BF. Mice lacking fatty acid amide hydrolase exhibit a cannabinoid receptor-mediated phenotypic hypoalgesia. Pain. 2004;109:319–327. doi: 10.1016/j.pain.2004.01.022. [DOI] [PubMed] [Google Scholar]
- 14.Gobbi G, Bambico FR, Mangieri R, Bortolato M, Campolongo P, Solinas M, Cassano T, Morgese MG, Debonnel G, Duranti A, Tontini A, Tarzia G, Mor M, Trezza V, Goldberg SR, Cuomo V, Piomelli D. Antidepressant-like activity and modulation of brain monoaminergic transmission by blockade of anandamide hydrolysis. Proc Natl Acad Sci U S A. 2005;102:18620–18625. doi: 10.1073/pnas.0509591102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gaetani S, Dipasquale P, Romano A, Righetti L, Cassano T, Piomelli D, Cuomo V. The endocannabinoid system as a target for novel anxiolytic and antidepressant drugs. Int Rev Neurobiol. 2009;85:57–72. doi: 10.1016/S0074-7742(09)85005-8. [DOI] [PubMed] [Google Scholar]
- 16.Kathuria S, Gaetani S, Fegley D, Valino F, Duranti A, Tontini A, Mor M, Tarzia G, La Rana G, Calignano A, Giustino A, Tattoli M, Palmery M, Cuomo V, Piomelli D. Modulation of anxiety through blockade of anandamide hydrolysis. Nat Med. 2003;9:76–81. doi: 10.1038/nm803. [DOI] [PubMed] [Google Scholar]
- 17.Piomelli D. The molecular logic of endocannabinoid signalling. Nat Rev Neurosci. 2003;4:873–884. doi: 10.1038/nrn1247. [DOI] [PubMed] [Google Scholar]
- 18.Johnson DS, Stiff C, Lazerwith SE, Kesten SR, Fay LK, Morris M, Beidler D, Liimatta MB, Smith SE, Dudley DT, Sadagopan N, Bhattachar SN, Kesten SJ, Nomanbhoy TK, Cravatt BF, Ahn K. Discovery of PF-04457845: A Highly Potent, Orally Bioavailable, and Selective Urea FAAH Inhibitor. ACS Med Chem Lett. 2011;2:91–96. doi: 10.1021/ml100190t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ahn K, Smith SE, Liimatta MB, Beidler D, Sadagopan N, Dudley DT, Young T, Wren P, Zhang Y, Swaney S, Van Becelaere K, Blankman JL, Nomura DK, Bhattachar SN, Stiff C, Nomanbhoy TK, Weerapana E, Johnson DS, Cravatt BF. Mechanistic and pharmacological characterization of PF-04457845: a highly potent and selective fatty acid amide hydrolase inhibitor that reduces inflammatory and noninflammatory pain. J Pharmacol Exp Ther. 2011;338:114–124. doi: 10.1124/jpet.111.180257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li GL, Winter H, Arends R, Jay GW, Le V, Young T, Huggins JP. Assessment of the pharmacology and tolerability of PF-04457845, an irreversible inhibitor of fatty acid amide hydrolase-1, in healthy subjects. Br J Clin Pharmacol. 2012;73:706–716. doi: 10.1111/j.1365-2125.2011.04137.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Huggins JP, Smart TS, Langman S, Taylor L, Young T. An efficient randomised, placebo-controlled clinical trial with the irreversible fatty acid amide hydrolase-1 inhibitor PF-04457845, which modulates endocannabinoids but fails to induce effective analgesia in patients with pain due to osteoarthritis of the knee. Pain. 2012;153:1837–1846. doi: 10.1016/j.pain.2012.04.020. [DOI] [PubMed] [Google Scholar]
- 22.Postnov A, Schmidt M, Penson J, Van Hecken A, Zannikos P, Pemberton D, Palmer J, de Hoon J, Bormans G, Van Laere K. Kinetic modeling of Fatty Acid Amide Hydrolase (FAAH) enzyme occupancy after JNJ-42165279 inhibition based on [11C]MK-3168 PET imaging of human brain. Journal of Nuclear Medicine Meeting Abstracts. 2015;56:362. [Google Scholar]
- 23.ClinicalTrials. Identifier, NCT01634529
- 24.Wyffels L, Muccioli GG, De Bruyne S, Moerman L, Sambre J, Lambert DM, De Vos F. Synthesis, in vitro and in vivo evaluation, and radiolabeling of aryl anandamide analogues as candidate radioligands for in vivo imaging of fatty acid amide hydrolase in the brain. J Med Chem. 2009;52:4613–4622. doi: 10.1021/jm900324e. [DOI] [PubMed] [Google Scholar]
- 25.Wyffels L, Muccioli GG, Kapanda CN, Labar G, De Bruyne S, De Vos F, Lambert DM. PET imaging of fatty acid amide hydrolase in the brain: synthesis and biological evaluation of an 11C-labelled URB597 analogue. Nucl Med Biol. 2010;37:665–675. doi: 10.1016/j.nucmedbio.2010.03.009. [DOI] [PubMed] [Google Scholar]
- 26.Wilson AA, Garcia A, Parkes J, Houle S, Tong J, Vasdev N. [11C]CURB: Evaluation of a novel radiotracer for imaging fatty acid amide hydrolase by positron emission tomography. Nucl Med Biol. 2011;38:247–253. doi: 10.1016/j.nucmedbio.2010.08.001. [DOI] [PubMed] [Google Scholar]
- 27.Wilson AA, Hicks JW, Sadovski O, Parkes J, Tong J, Houle S, Fowler CJ, Vasdev N. Radiosynthesis and Evaluation of [11C-Carbonyl]-Labeled Carbamates as Fatty Acid Amide Hydrolase Radiotracers for Positron Emission Tomography. J Med Chem. 2013;56:201–209. doi: 10.1021/jm301492y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sadovski O, Hicks JW, Parkes J, Raymond R, Nobrega J, Houle S, Cipriano M, Fowler CJ, Vasdev N, Wilson AA. Development and characterization of a promising fluorine-18 labelled radiopharmaceutical for in vivo imaging of fatty acid amide hydrolase. Bioorg Med Chem. 2013;21:4351–4357. doi: 10.1016/j.bmc.2013.04.077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rotstein BH, Wey HY, Shoup TM, Wilson AA, Liang SH, Hooker JM, Vasdev N. PET imaging of fatty acid amide hydrolase with [18F]DOPP in nonhuman primates. Mol Pharm. 2014;11:3832–3838. doi: 10.1021/mp500316h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hicks JW, Parkes J, Sadovski O, Tong J, Houle S, Vasdev N, Wilson AA. Synthesis and preclinical evaluation of [11C-carbonyl]PF-04457845 for neuroimaging of fatty acid amide hydrolase. Nucl Med Biol. 2013;40:740–746. doi: 10.1016/j.nucmedbio.2013.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liu P, Hamill TG, Chioda M, Chobanian H, Fung S, Guo Y, Chang L, Bakshi R, Hong Q, Dellureficio J, Lin LS, Abbadie C, Alexander J, Jin H, Mandala S, Shiao LL, Li W, Sanabria S, Williams D, Zeng Z, Hajdu R, Jochnowitz N, Rosenbach M, Karanam B, Madeira M, Salituro G, Powell J, Xu L, Terebetski JL, Leone JF, Miller P, Cook J, Holahan M, Joshi A, O’Malley S, Purcell M, Posavec D, Chen TB, Riffel K, Williams M, Hargreaves R, Sullivan KA, Nargund RP, DeVita RJ. Discovery of MK-3168: A PET Tracer for Imaging Brain Fatty Acid Amide Hydrolase. ACS Med Chem Lett. 2013;4:509–513. doi: 10.1021/ml4000996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Skaddan MB, Zhang L, Johnson DS, Zhu A, Zasadny KR, Coelho RV, Kuszpit K, Currier G, Fan KH, Beck EM, Chen L, Drozda SE, Balan G, Niphakis M, Cravatt BF, Ahn K, Bocan T, Villalobos A. The synthesis and in vivo evaluation of [18F]PF-9811: a novel PET ligand for imaging brain fatty acid amide hydrolase (FAAH) Nucl Med Biol. 2012;39:1058–1067. doi: 10.1016/j.nucmedbio.2012.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kumata K, Yui J, Hatori A, Maeda J, Xie L, Ogawa M, Yamasaki T, Nagai Y, Shimoda Y, Fujinaga M, Kawamura K, Zhang MR. Development of [11C]MFTC for PET imaging of fatty acid amide hydrolase in rat and monkey brains. ACS Chem Neurosci. 2015;6:339–346. doi: 10.1021/cn500269g. [DOI] [PubMed] [Google Scholar]
- 34.Rusjan PM, Wilson AA, Mizrahi R, Boileau I, Chavez SE, Lobaugh NJ, Kish SJ, Houle S, Tong J. Mapping human brain fatty acid amide hydrolase activity with PET. J Cereb Blood Flow Metab. 2013;33:407–414. doi: 10.1038/jcbfm.2012.180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Boileau I, Bloomfield PM, Rusjan P, Mizrahi R, Mufti A, Vitcu I, Kish SJ, Houle S, Wilson AA, Tong J. Whole-body radiation dosimetry of 11C-carbonyl-URB694: a PET tracer for fatty acid amide hydrolase. J Nucl Med. 2014;55:1993–1997. doi: 10.2967/jnumed.114.146464. [DOI] [PubMed] [Google Scholar]
- 36.Joshi A, Li W, Sanabria S, Holahan M, Purcell M, Declercq R, Depre M, Bormans G, Van Laere K, Hamill T. Translational studies with [11C]MK-3168, a PET tracer for fatty acid amide hydrolase (FAAH) J Nucl Med Meeting Abstracts. 2012;53:397. [Google Scholar]
- 37.Fowler JS, MacGregor RR, Wolf AP, Arnett CD, Dewey SL, Schlyer D, Christman D, Logan J, Smith M, Sachs H, et al. Mapping human brain monoamine oxidase A and B with 11C-labeled suicide inactivators and PET. Science. 1987;235:481–485. doi: 10.1126/science.3099392. [DOI] [PubMed] [Google Scholar]
- 38.Cumming P, Vasdev N. The Assay of Enzyme Activity by Positron Emission Tomography. In: Gründer G, editor. Molecular Imaging in the Clinical Neurosciences. Vol. 71. Humana Press; 2012. pp. 111–135. [Google Scholar]
- 39.Innis RB, Cunningham VJ, Delforge J, Fujita M, Gjedde A, Gunn RN, Holden J, Houle S, Huang SC, Ichise M, Iida H, Ito H, Kimura Y, Koeppe RA, Knudsen GM, Knuuti J, Lammertsma AA, Laruelle M, Logan J, Maguire RP, Mintun MA, Morris ED, Parsey R, Price JC, Slifstein M, Sossi V, Suhara T, Votaw JR, Wong DF, Carson RE. Consensus nomenclature for in vivo imaging of reversibly binding radioligands. J Cereb Blood Flow Metab. 2007;27:1533–1539. doi: 10.1038/sj.jcbfm.9600493. [DOI] [PubMed] [Google Scholar]
- 40.Pike VW. PET Radiotracers: crossing the blood-brain barrier and surviving metabolism. Trends Pharmacol Sci. 2009;30:431–440. doi: 10.1016/j.tips.2009.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mileni M, Garfunkle J, DeMartino JK, Cravatt BF, Boger DL, Stevens RC. Binding and inactivation mechanism of a humanized fatty acid amide hydrolase by alpha-ketoheterocycle inhibitors revealed from cocrystal structures. J Am Chem Soc. 2009;131:10497–10506. doi: 10.1021/ja902694n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Otrubova K, Boger DL. alpha-Ketoheterocycle-based Inhibitors of Fatty Acid Amide Hydrolase (FAAH) ACS Chem Neurosci. 2012;3:340–348. doi: 10.1021/cn2001206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hardouin C, Kelso MJ, Romero FA, Rayl TJ, Leung D, Hwang I, Cravatt BF, Boger DL. Structure-activity relationships of alpha-ketooxazole inhibitors of fatty acid amide hydrolase. J Med Chem. 2007;50:3359–3368. doi: 10.1021/jm061414r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Georgiades SN, Rizeq N. Synthesis of a ‘Propeller-Like’ Oligoheteroaryl with Alternating Pyridine and Oxazole Motifs. Synlett. 2015;26:489–493. [Google Scholar]
- 45.Boger DL, Miyauchi H, Du W, Hardouin C, Fecik RA, Cheng H, Hwang I, Hedrick MP, Leung D, Acevedo O, Guimaraes CR, Jorgensen WL, Cravatt BF. Discovery of a potent, selective, and efficacious class of reversible alpha-ketoheterocycle inhibitors of fatty acid amide hydrolase effective as analgesics. J Med Chem. 2005;48:1849–1856. doi: 10.1021/jm049614v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dishino DD, Welch MJ, Kilbourn MR, Raichle ME. Relationship between lipophilicity and brain extraction of C-11-labeled radiopharmaceuticals. J Nucl Med. 1983;24:1030–1038. [PubMed] [Google Scholar]
- 47.Wilson AA, Jin L, Garcia A, DaSilva JN, Houle S. An admonition when measuring the lipophilicity of radiotracers using counting techniques. Appl Radiat Isot. 2001;54:203–208. doi: 10.1016/s0969-8043(00)00269-4. [DOI] [PubMed] [Google Scholar]
- 48.Waterhouse RN. Determination of lipophilicity and its use as a predictor of blood-brain barrier penetration of molecular imaging agents. Mol Imaging Biol. 2003;5:376–389. doi: 10.1016/j.mibio.2003.09.014. [DOI] [PubMed] [Google Scholar]
- 49.Zoghbi SS, Anderson KB, Jenko KJ, Luckenbaugh DA, Innis RB, Pike VW. On Quantitative Relationships Between Drug-Like Compound Lipophilicity and Plasma Free Fraction in Monkey and Human. J Pharm Sci. 2012;101:1028–1039. doi: 10.1002/jps.22822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Watanabe K, Ogi H, Nakamura S, Kayano Y, Matsunaga T, Yoshimura H, Yamamoto I. Distribution and characterization of anandamide amidohydrolase in mouse brain and liver. Life Sci. 1998;62:1223–1229. doi: 10.1016/s0024-3205(98)00052-6. [DOI] [PubMed] [Google Scholar]
- 51.Ueda N, Puffenbarger RA, Yamamoto S, Deutsch DG. The fatty acid amide hydrolase (FAAH) Chem Phys Lipids. 2000;108:107–121. doi: 10.1016/s0009-3084(00)00190-0. [DOI] [PubMed] [Google Scholar]
- 52.Fowler CJ, Jonsson KO, Tiger G. Fatty acid amide hydrolase: biochemistry, pharmacology, and therapeutic possibilities for an enzyme hydrolyzing anandamide, 2-arachidonoylglycerol, palmitoylethanolamide, and oleamide. Biochem Pharmacol. 2001;62:517–526. doi: 10.1016/s0006-2952(01)00712-2. [DOI] [PubMed] [Google Scholar]
- 53.Hillard CJ, Wilkison DM, Edgemond WS, Campbell WB. Characterization of the kinetics and distribution of N-arachidonylethanolamine (anandamide) hydrolysis by rat brain. Biochim Biophys Acta. 1995;3:249–256. doi: 10.1016/0005-2760(95)00087-s. [DOI] [PubMed] [Google Scholar]
- 54.Thomas EA, Cravatt BF, Danielson PE, Gilula NB, Sutcliffe JG. Fatty acid amide hydrolase, the degradative enzyme for anandamide and oleamide, has selective distribution in neurons within the rat central nervous system. J Neurosci Res. 1997;50:1047–1052. doi: 10.1002/(SICI)1097-4547(19971215)50:6<1047::AID-JNR16>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- 55.Tatsuta T, Naito M, Oh-hara T, Sugawara I, Tsuruo T. Functional involvement of P-glycoprotein in blood-brain barrier. J Biol Chem. 1992;267:20383–20391. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.