

Abstract
Two isoforms of the transmembrane protein tyrosine phosphatase PTPα, which differ by nine amino acids in their extracellular regions, are expressed in a tissue-specific manner. Over-expression of the shorter isoform transforms rodent cells, and it has previously been reasonable to assume that this was a direct consequence of its dephosphorylation and activation of Src. Transformation by the longer wild-type isoform has not previously been studied. We tested the activities of both isoforms in NIH3T3 cells and found that, while both dephosphorylated and activated Src similarly, only the shorter isoform induced focus formation or anchorage-independent growth. Differences in phosphorylation of PTPα at its known regulatory sites, Grb2 binding to PTPα, phosphorylation level of focal adhesion kinase by PTPα, or overall localization were excluded as possible explanations for the differences in transforming activities. The results suggest that transformation by PTPα involves at least one function other than, or in addition to, its activation of Src and that this depends on PTPα’s extracellular domain. Previous studies have suggested that PTPα might be a useful target in breast and colon cancer therapy, and the results presented here suggest that it may be advantageous to develop isoform-specific therapeutic reagents.
Introduction
Protein tyrosine phosphatase (PTP)α, a transmembrane tyrosine phosphatase, transduces cellular signals by dephosphorylating the Src family and insulin receptor tyrosine kinases and possibly other signaling proteins (Pallen 2003). Like other receptor PTPs, it consists of extracellular and membrane spanning-regions and tandem intracellular phosphatase domains, but its extracellular region is much shorter and more glycosylated than that of other receptor PTPs (Tonks 2006). Whether the PTPα extracellular region binds to extracellular ligands that regulate its phosphatase activity remains to be determined. However, it has been shown that the neural cell adhesion molecule contactin forms a complex with the PTPα extracellular region, and this might play a role in the established role of PTPα in regulating neural outgrowth (Zeng et al. 1999).
Two isoforms of PTPα (Fig. 1), differing only in their extracellular regions, arise by alternative splicing and are expressed in a tissue-specific manner (Kaplan et al. 1990; Krueger et al. 1990; Matthews et al. 1990; Sap et al. 1990; Daum et al. 1994). The shorter, 793 amino acid form of the protein (which we will call PTPα793) has 123 extracellular amino acids and is expressed in most tissues; the longer form, PTPα802, which has an additional mini-exon that introduces nine extra amino acids immediately preceding the transmembrane region, is expressed only in a few tissues such as brain and fat (Sap et al. 1990; Norris et al. 1997; Kapp et al. 2007). The interaction between PTPα and contactin was observed with PTPα793 (Zeng et al. 1999), and it remains to be determined whether PTPα802 interacts in the same manner.
Figure 1.
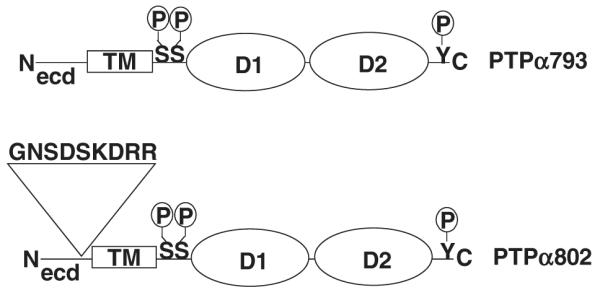
Schematic representation of the two isoforms of PTPα. The long form, PTPα802, includes an additional nine amino acids encoded by exon 3 located in the extracellular domain. The locations of the phosphorylation sites Ser180, Ser204 and Tyr789 are indicated. (D1 and D2, catalytic domains; ecd, extracellular domain; TM, transmembrane domain.)
PTPα activates Src family kinases (SFKs) by dephosphorylating the regulatory phosphotyrosine (pTyr)527 (chicken gene numbering for historical reasons) found in their C-terminal regions (Pallen 2003). Src family dephosphorylation is regulated both by protein–protein interactions and phosphorylations within PTPα. Phosphorylation of Ser180 and Ser204 (for simplicity, we will often use PTPα793 numbering for phosphorylation sites, even when referring to PTPα802), which lie within protein kinase C consensus sequences located between the transmembrane region and first catalytic domain of the protein (Tracy et al. 1995), stimulates PTPα tyrosine phosphatase activity about twofold (den Hertog et al. 1995; Zheng & Shalloway 2001; Zheng et al. 2002). In addition, approximately 20% of PTPα molecules are phosphorylated at Tyr789 (den Hertog et al. 1994; Su et al. 1994). This phosphorylation does not directly modulate catalytic activity, but specifically facilitates dephosphorylation of pTyr527 in Src and the homologous residue in at least one other SFK (Zheng et al. 2000; Maksumova et al. 2007): While wild-type (wt) PTPα793 and the Tyr789 → Phe mutant PTPα793(Y789F) have the same ability to dephosphorylate accessible phosphotyrosines in peptides and generic, nonspecific protein substrates, the Y789F mutant does not dephosphorylate pTyr527, which is protected by intramolecular binding to the Src SH2 domain, nor activate Src in vitro or in vivo in NIH3T3 cells (Zheng et al. 2000). However, it can still dephosphorylate pTyr416, which is autophosphorylated (but not protected by SH2 binding) in activated mutant Src(Y527F) (Kmiecik & Shalloway 1987; Piwnica-Worms et al. 1987), as well as unprotected pTyrs in other substrates [e.g. myelin basic protein (MBP)] (Zheng et al. 2000).
These and related experimental results are explained by a phosphotyrosine-displacement model, which describes pTyr789 transiently binding to the Src SH2 domain, competitively displacing pTyr527 from its intramolecular interaction with Src SH2 and thereby making pTyr527 available for dephosphorylation by PTPα (Zheng et al. 2000). Most tyrosine-phosphorylated PTPα molecules are bound to Grb2 by an interaction between pTyr789 and the Grb2 SH2 domain (den Hertog et al. 1994; Su et al. 1994). Because this sequesters pTyr789, Grb2 binding competitively inhibits dephosphorylation and activation of Src by PTPα (Zheng et al. 2000; Zheng & Shalloway 2001). Thus, Src-directed PTPα activity can be sensitively regulated in fibroblasts by modulating the size of the small fraction of PTPα molecules containing pTyr789 that are not bound by Grb2. The model furthermore predicts that such modulation of the amount of free versus bound pTyr789 will specifically regulate dephosphorylation of Src but not of substrates in which the target phosphotyrosine is not protected by SH2 domain binding.
PTyr789-independent activation of Src can occur in special circumstances and in other cell types, for instance during integrin-mediated cell adhesion (Pallen 2003; Chen et al. 2006) or in PC12 cells (Yang et al. 2002; Chen et al. 2006). It has been suggested that this may result from displacement of pTyr527 from the SH2 domain by phosphotyrosines in co-associated proteins; e.g. by focal adhesion kinase (FAK) within a focal adhesion plaque complex containing it, Src, and PTPα (Pallen 2003; Chen et al. 2006).
Over-expression of PTPα793 transforms rat embryo fibroblasts and mouse NIH3T3 fibroblasts, as measured by focus formation in monolayer culture and anchorage-independent colony formation in soft agarose (Zheng et al. 1992, 2000, 2002). This depends critically on PTPα phosphorylation status, and mutation of Tyr789, Ser180 or Ser204 drastically reduces transforming potential, suggesting that this reflects the observed lack of Src activation by these mutants (Zheng et al. 2000, 2002). However, there is no direct evidence indicating that Src or other SFKs are required for PTPα-induced transformation, and the requirement for Tyr789 phosphorylation may result from the interaction of this phosphorylated residue with Grb2 (den Hertog & Hunter 1996; Su et al. 1996) or other binding partners (Pallen 2003).
In contrast with the effects of PTPα phosphorylation, little is known about the role of the extracellular domain of PTPα in its biological activity, so we were interested in determining whether the additional sequence in the longer form, PTPα802, might impact cell transformation potential. Kapp et al. (2007) showed that transient expression of either PTPα793 or PTPα802 in human embryonic kidney 293 cells caused similar decreases in in vivo tyrosine phosphorylation of overexpressed Fyn and caused similar increases in the abilities of lysates from such cells to dephosphorylate para-nitrophenylphosphate. However, they were unable to obtain stable expression of wt PTPα793 or PTPα802 in untransformed cells (Lammers et al. 2000; Kapp et al. 2007), precluding comparison of their transforming abilities. We therefore used our established expression model to compare the transforming activities of the two splice variants. We found that PTPα793 and PTPα802 activated Src to the same extent, but surprisingly found that PTPα802 was transformation-defective. This suggests that transformation by PTPα involves at least one function other than, or in addition to, its activation of Src and that this depends on PTPα’s extracellular domain.
Results
We have previously described the creation of NIH3T3-derived mouse cells for tet-off inducible expression of hemagglutinin (HA) epitope-tagged human PTPα793 and a phosphatase-defective variant PTPα793(CCSS), which contains inactivating Cys433 → Ser and Cys723 → Ser substitutions within the D1 and D2 catalytic domains, respectively (Zheng et al. 2000). Using a homologous inducible expression plasmid, which differed only in the insertion of the additional nine codons found in the extracellular coding domain of PTPα802 (Fig. 1), we similarly created NIH3T3-derived cell lines that inducibly expressed HA-tagged PTPα802. Lines were maintained in media containing 10 ng/mL doxycycline, which inhibited detectable exogenous PTPα expression, to prevent any potential toxicity effects or other selective pressures resulting from long-term culture with PTPα over-expression. Immunoblotting with polyclonal anti-PTPα antibody showed that exogenous PTPα expression levels increased to half-maximum by 6–8 h after removal of doxycycline and by 16–18 h reached a maximum of ~17–24 times the endogenous level of mouse PTPα (Fig. 3A and data not shown). Therefore, in subsequent experiments, cells were typically induced by removal of doxycycline for 20 h. Previously described cell lines transfected only with G418 antibiotic-resistance and inducible transactivator plasmids (Neo) (Zheng et al. 2000) or transformed by transfection with Src(Y527F) (Kmiecik & Shalloway 1987) were used as negative and positive controls, respectively.
Figure 3.

Effect of PTPα over-expression on Src in vivo tyrosine phosphorylation and kinase activity. (A) Neo, PTPα793, PTPα802 and PTPα793(CCSS) overexpressor cells were induced (by removal of doxycycline for 20 h), and lysates were immunoblotted with anti-PTPα polyclonal antibody (panel a) or Src was immunoprecipitated from the lysates and aliquots were immunoblotted with either anti-Src monoclonal antibody (panel b) or anti-pTyr mAb (panel c). Over-expression of PTPα793, PTPα802 or PTPα793(CCSS) decreased tyrosine phosphorylation of Src by 76 ± 7% (n = 4), 79 ± 5% (n = 4) or 19 ± 10% (n = 3), respectively. (B) Lysates from the induced cells were immunoblotted with anti-PTPα polyclonal antibody (panel a) or Src was immunoprecipitated from the lysates and portions were immunoblotted with anti-Src monoclonal antibody (panel b), immunoblotted using anti-dephospho-Y527 Src monoclonal antibody, which reacts only with the activated, Tyr527-dephosphorylated form of Src (panel c), or were subjected to in vitro kinase assay in buffer containing [γ-32P]ATP and acid-denatured enolase (panel d). Over-expression of PTPα793, PTPα802 or PTPα793(CCSS) increased the amount of dephospho-Y527 by factors of 3.8 ± 1.0, 3.9 ± 1.1 or 1.6 ± 0.5 (n = 5) and increased Src kinase activity by factors of 4.9 ± 2.4, 5.9 ± 2.9 or 1.7 ± 0.8 (n = 3), respectively. The positions of molecular weight markers (in kDa) are indicated.
In vitro phosphatase activities of PTPα variants
We examined the phosphatase activities of the PTPα variants toward nonspecific substrates by incubating anti-HA immunoprecipitates of PTPα793, PTPα802 and PTPα793(CCSS) in phosphatase buffer with [32P]tyrosine-phosphorylated MBP. Relative specific activities (Fig. 2A) were determined after measuring the amounts of [32P]phosphate released and the (approximately equal, data not shown) amounts of PTPα in the reactions. Reactions were carried out for 5 and 10 min to verify linearity. As expected, PTPα793(CCSS) had no detectable activity, whereas PTPα793 and PTPα802 dephosphorylated MBP at the same rate.
Figure 2.

In vitro dephosphorylation of nonspecific and Src substrates by PTPα. (A) Anti-HA immunoprecipitates from lysates from PTPα793, PTPα802 or PTPα793(CCSS) overexpressor cells (induced by removal of doxycycline for 20 h) were incubated with [32P]phosphotyrosine-containing MBP and incubated for 5 or 10 min (in separate experiments to verify reaction linearity) at 30 °C. The amount of [32P] phosphate released was determined by scintillation counting. Immunoblots were used to determine the amount of PTPα present in each reaction and specific activities (relative to that of PTPα793) and standard errors of the mean (n = 3) were computed. (B) Wild-type Src was immunoprecipitated from Src overexpressor cells and subjected to in vitro dephosphorylation by control (Neo) or PTPα proteins that had been immunopurified from induced overexpressor cells. [Equality of the amounts of immunopurified PTPα proteins added to the phosphatase reactions was verified by immunoblotting with anti-PTPα polyclonal antibody (panel a).] The reaction products were divided into three portions that were immunoblotted with either anti-Src (panel b) or anti-phosphotyrosine (panel c) monoclonal antibodies, or used in an in vitro Src kinase assay with [γ-32P]ATP and acid-denatured enolase as substrate (panel d). Src tyrosine phosphorylation was reduced 51 ± 10%, 56 ± 12% or 9 ± 6% (n = 3) by over-expression of PTPα793, PTPα802 or PTPα793(CCSS), respectively; Src kinase activity was increased by factors of 3.4 ± 0.8, 3.6 ± 1.2 or 1.1 ± 0.1 (n = 3; errors are SEMs) by PTPα793, PTPα802 or PTPα793(CCSS), respectively. The positions of molecular weight markers (in kDa) are indicated.
As discussed in the Introduction, PTPα-catalyzed dephosphorylation of Src can be regulated independently of its activity on generic substrates such as MBP. Thus, we also compared the abilities of PTPα793 and PTPα802 to dephosphorylate Src itself: PTPα793, PTPα802 and PTPα793(CCSS) were immunopurified from the induced overexpressor lines with anti-HA polyclonal antibody (using the Neo line as a negative control) and were incubated in dephosphorylation buffer with Src immunoprecipitated from NIH3T3-derived Src overexpressor cells. Tyrosine dephosphorylation of Src was measured by immuno-precipitating Src from the reaction mixtures and immunoblotting with either anti-Src or anti-phosphotyrosine antibodies (Fig. 2B). In concordance with the MBP dephosphorylation results, PTPα793 and PTPα802 both dephosphorylated Src to the same extent (~50%). As expected, PTPα793(CCSS) did not significantly dephosphorylate Src.
To directly test the ability of the PTPα variants to activate Src, a portion of the immunoprecipitated Src that had been subjected to dephosphorylation by the immunopurified PTPα variants was used in an in vitro kinase assay with [γ32P]ATP and enolase as substrate. Consistent with the pTyr dephosphorylation results, PTPα793 and PTPα802 both increased Src specific activity 3–4 times whereas PTPα793(CCSS) had no significant effect.
In vivo dephosphorylation and activation of Src by PTPα variants
Although the in vitro activities of the variants appeared to be identical, it was possible that differences in localization or association might affect their abilities to dephosphorylate and activate Src in vivo. To test this, the phosphorylation state and kinase activity of endogenous Src immunoprecipitated from induced control (Neo) cells and lines overexpressing the different PTPα variants were examined. Over-expression of the PTPα variants had no effect on the level of Src expression (data not shown). To measure the level of Src tyrosine phosphorylation, aliquots of Src immunoprecipitates were immunoblotted with either anti-Src monoclonal antibody or anti-phosphotyrosine antibody (Fig. 3A). Consistent with the in vitro results, over-expression of either PTPα793 or PTPα802 reduced tyrosine phosphorylation by the same amount, approximately 75%. The fact that almost all tyrosine phosphorylation of overexpressed Src in unstimulated NIH3T3 cells is at Tyr 527 (Kmiecik & Shalloway 1987) suggested (but did not prove) that this reflected reduced Tyr527 phosphorylation. [While Src activated by Tyr527 → Phe mutation has increased autophosphorylation at Tyr416 (Kmiecik & Shalloway 1987; Piwnica-Worms et al. 1987), PTPα can dephosphorylate pTyr416 as well as pTyr527 (Zheng et al. 2000). Dephosphorylation of both residues in this experiment probably accounts for the very low level of Src phosphotyrosine observed in the PTPα overexpressor cells.] Over-expression of PTPα793(CCSS) had little or no effect.
In addition (Fig. 3B), Src was immunoprecipitated from induced control and PTPα overexpressor cells and was then either immunoblotted with anti-dephospho-Tyr527 antibody, which only recognizes Src that is dephosphorylated at Tyr527 (i.e. the active form) (Kawakatsu et al. 1996) (panel c), or subjected to kinase assay with [γ-32P]ATP using enolase as substrate (panel d). In agreement with the in vitro results and in vivo anti-pTyr immunoblotting, both PTPα793 and PTPα802 increased dephospho-Tyr527 (~3–4×) and increased Src specific activity (~5–6×) to the same extent. The use of a Tyr527-specific antibody in this experiment proved that the activities of the two isoforms on this residue are the same. PTPα793(CCSS) had no significant effect.
PTPα variants differ in transforming ability
Neo, Src(Y527F), PTPα793 and PTPα802 overexpressor cells were tested for their abilities to induce foci in monolayer culture when mixed with normal NIH3T3 cells in the absence of doxycycline (Fig. 4A). As expected, Neo cells failed to form foci after 16 days, while cells overexpressing Src(Y527F) or PTPα793 formed foci in <14 days. Surprisingly, the PTPα802 overexpressing cells formed almost no foci, even though three separate lines, each expressing approximately the same amount of PTPα802 as the PTPα793-expressor cells, were tested.
Figure 4.

Focus formation and colony formation by PTPα overexpressor cells. (A) Neo (negative control), PTPα793 and PTPα802 NIH3T3-derived overexpressor cells and cSrc(Y527F) overexpressor cells (positive control) were separately (500 cells each) mixed with 2 × 105 NIH3T3 cells and cultured in monolayer without doxycycline for 14 days. The percentages of cells forming foci and standard errors of the mean are shown [two independent experiments each, except six for PTPα802]. [The focus-forming activities of PTPα793 and PTPα802 were statistically different (two-sided t-test) at α = 10−4.] (B) The same cell lines [plus PTPα793(Y789F) and PTPα793(CCSS), not shown] were cultured in 0.3% agarose without doxycycline, and colonies were photographed after 16–18 days. (C) Percentages of cells forming colonies in soft agarose with standard errors of the mean (two independent experiments each, except six for PTPα802). (The colony-forming activities of PTPα793 and PTPα802 were statistically different at α = 10−8).
To extend this finding, anchorage-independent growth of these cell lines plus PTPα793(Y789F) and PTPα793(CCSS) overexpressor cells was assayed by suspending them in semi-solid medium containing 0.3% soft agarose without doxycycline. Consistent with the results above, PTPα793 overexpressor cells formed colonies in soft agarose (~40%), while three different lines of PTPα802 overexpressor cells failed to form any discernible colonies (Fig. 4B,C). As expected, no colonies were formed by the Neo control cells, PTPα793(Y789F)-expressing cells, or PTPα793(CCSS)-expressing cells, while ~75% of fully transformed Src(Y527F)-expressing cells formed colonies. Taken together, these data indicate that PTPα802 has a much lower ability than PTPα793 to transform NIH3T3 cells.
Phosphorylation of PTPα variants
The equality of their phosphatase activities suggested that phosphorylation of the regulatory sites in the PTPα variants was probably the same, but this was examined directly to be certain. Phosphospecific antibodies that recognize pTyr789, pSer204 or pSer180/pSer204 were used to measure their phosphorylation levels. Immunoblots of anti-HA immunoprecipitates from the PTPα793 and PTPα802 overexpressor cells detected no differences in Tyr789, Ser204, or Ser180/Ser204 (Fig. 5). Thus, differences in the phosphorylation levels at these residues do not account for the different transforming activities.
Figure 5.
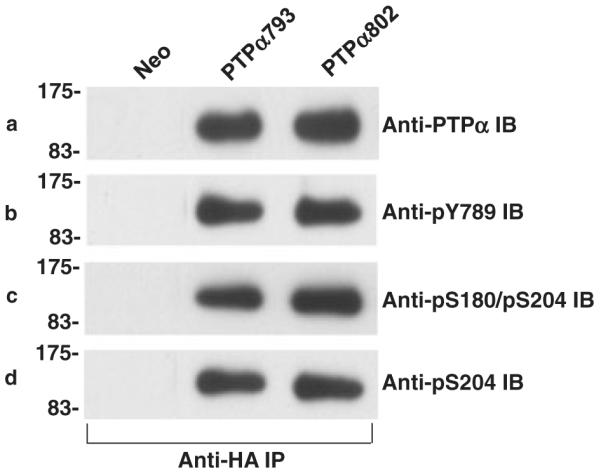
Phosphorylation status of PTPα proteins. Anti-HA immunoprecipitates from lysates from Neo (control) or PTPα793 or PTPα802 overexpressor cells (induced for 20 h) were divided into four aliquots and immunoblotted with anti-PTPα antibody (panel a), anti-phosphoY789 antibody (panel b), anti-phosphoS180/phosphoS204 antibody (panel c) or anti-phosphoS204 antibody (panel d). No signals were detected with control anti-HA immunoprecipitates from Neo control cells. The ratios between phosphorylation of PTPα793 and PTPα802 at pY789, pS180/pS204 and pS204 were 1.1 ± 0.1, 1.0 ± 0.1 and 1.1 ± 0.1, respectively (n = 4; errors are SEMs). The positions of molecular weight markers (in kDa) are indicated.
In vivo association of PTPα variants with Grb2
The equality of the Src-directed phosphatase activities and Tyr789 phosphorylation of PTPα793 and PTPα802 suggested that there was no difference in their Grb2 binding, which has a negative regulatory effect. To confirm, this was tested using co-immunoprecipitation experiments. Anti-Grb2 immunoprecipitates were prepared and aliquots were immunoblotted with either anti-Grb2 or anti-HA (to detect PTPα) antibody (Fig. 6). For comparison, portions of the whole cell lysates were directly immunoblotted with anti-HA antibody. To within statistical error, the same fractions of cellular PTPα793 and PTPα802 were bound by Grb2. As expected (Zheng et al. 2000), no Grb2 was bound by a PTPα793(Y789F) control (not shown).
Figure 6.
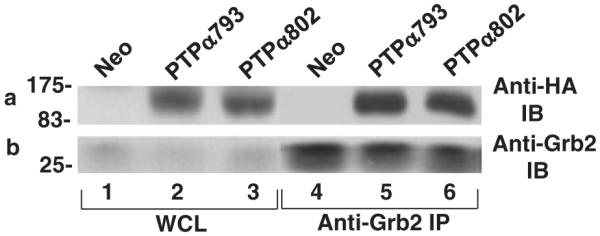
Co-immunoprecipitation of PTPα and Grb2. (a) Lysates from induced Neo (control), PTPα793 or PTPα802 overexpressor cells were immunoprecipitated with anti-Grb2 antibody, and the immunoprecipitates were immunoblotted with either anti-HA (panel a) or Grb2 (panel b) antibodies. The positions of molecular weight markers (in kDa) are indicated. The average over six experiments of the ratio of PTPα793-Grb2 binding to PTPα802-Grb2 binding, normalized by the total amounts of PTPα in the cells, was 1.1 ± 0.1.
Activation of FAK by PTPα variants
PTPα, probably via its activation of SFKs, stimulates phosphorylation of tyrosines within FAK that are known to stimulate its activity and may be required for PTPα-induced transformation (Su et al. 1999; Zeng et al. 2003; Cohen & Guan 2005). Colocalization of PTPα, Src and FAK is probably important for these interactions (Pallen 2003; Chen et al. 2006), and it might depend on the PTPα extracellular domain, thus explaining the different transforming activities of the variants. We examined this possibility by determining whether PTPα793 and PTPα802 differed in their ability to stimulate FAK tyrosine phosphorylation. FAK immunoprecipitates from induced control, PTPα793 and PTPα802 expressor cell lines were immunoblotted with anti-pTyr antibody (Fig. 7). FAK tyrosine phosphorylation levels were the same, suggesting that the difference in PTPα transformation is not explained by differential activation of FAK. However, as there are at least six sites of tyrosine phosphorylation in FAK (Cox et al. 2006), the possibility that there are site-specific differences that cause differences in downstream signaling cannot be excluded.
Figure 7.
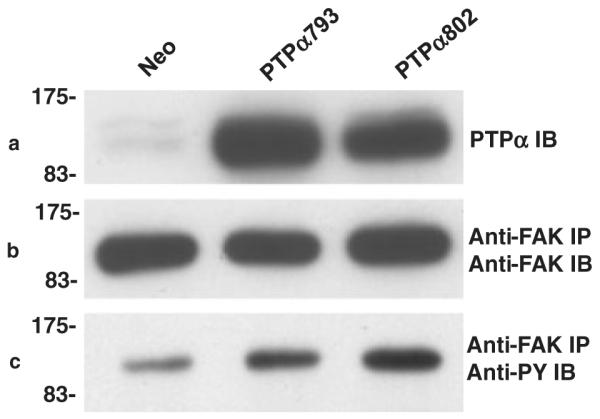
Regulation of focal adhesion kinase (FAK) phosphorylation by PTPα. Lysates from induced Neo (control), PTPα793, PTPα802 or PTPα793(CCSS) overexpressor cells were immunoblotted with anti-PTPα antibody (panel a) or immunoprecipitated with anti-FAK antibody (panels b and c). Aliquots of the immunoprecipitates were immunoblotted with anti-FAK monoclonal antibody (panel b) or anti-pTyr monoclonal antibody (panel c). FAK tyrosine phosphorylation was increased to roughly equal extents by PTPα793 (4.8 ± 2.6) and PTPα802 (4.65 ± 0.8) in two experiments (errors are SEMs). The positions of molecular weight markers (in kDa) are indicated.
Localization
PTPα793 and PTPα802 are localized to membranes by identical signal sequences and juxtamembrane regions (Kaplan et al. 1990; Krueger et al. 1990; Matthews et al. 1990; Sap et al. 1990; Daum et al. 1994). Moreover, the fact that they induce the phosphorylation of FAK, which is primarily localized to focal adhesions in attached cells (such as those used in this study) (Cohen & Guan 2005), to the same extent suggests similarity of distribution to this location as well. Immunofluorescence studies were consistent with this; we detected no obvious differences between the plasma membrane localization of the two isoforms (Fig. 8). This does not exclude the possibility that there are localization differences (e.g. in lipid rafts) that are too subtle to be detected by this technique.
Figure 8.

Immunofluorescent localization of overexpressed PTPα793 and PTPα802. PTPα793 and PTPα802 NIH3T3-derived overexpressor cells were induced for 20 h and the subcellular localizations of the HA-tagged PTPα proteins were determined by confocal microscopy as described in Experimental procedures. Ten micron reference bars are shown.
Discussion
Two PTPα splice variants, PTPα793 and PTPα802, which differ only in their extracellular domains, were similar in all tested biochemical parameters, namely dephosphorylation and activation of Src, phosphorylation at Ser 180, Ser 204 and Tyr 789, association with Grb2, and overall increase in tyrosine phosphorylation of FAK. Nonetheless, while NIH3T3 cells stably expressing PTPα793 readily formed foci in monolayer culture and colonies in soft agarose, cells expressing PTPα802 did not. Thus, at least one PTPα action that stimulates or suppresses transformation other than, or in addition to, its dephosphorylation of Src must be involved.
Quantitative considerations suggest that transformation by PTPα793 may require a function other than its activation of Src: Transforming Tyr527 → Phe Src mutants have ~15-fold increased activity (Kmiecik & Shalloway 1987) and NIH3T3 cells transformed by over-expression of murine Src typically express at least 18 times the endogenous level of Src (Lin et al. 1995). Thus, it is possible that lower levels of activation, such as the five to sixfold increase observed in cells transformed by PTPα793 (Fig. 3B), may not be adequate, by itself, for transformation. Indeed, there is no evidence that activation of SFKs is even required for transformation by PTPα. (We have attempted to test this hypothesis using Src/Fyn/Yes−/− mouse cells, but for unknown reasons have been unable to overexpress PTPα in them.)
However, even if another function is required for transformation, PTPα802 may also possess it, and isoform-dependent transformation may result from greater transforming activity of PTPα793 or greater transformation-suppressing activity of PTPα802. [The observations that PTPα793 over-expression suppresses the growth rate of MCF-7 cells and the tumorigenicity of a mouse line transformed by HER2/neu over-expression (Ardini et al. 2000) provide evidence that it can suppress transformation under some circumstances. PTPα802 could have even higher suppressive activity.] The isoform-specific difference could reflect differential activation of other fibroblast SFKs (i.e. Fyn and Yes), differential substrate-specific activation of SFKs, or differential dephosphorylation of non-SFK targets. Such differential activities could be mediated by interactions of the PTPα extracellular domains with localizing factors, dephosphorylation targets, or SFK phosphorylation targets (which altered activated SFK substrate specificity).
Differential action in focal adhesion plaques is one possibility: In addition to stimulating Src phosphorylation of pTyrs 576 and 577 in FAK, PTPα is required for integrin-stimulated FAK autophosphorylation of Tyr397 (Zeng et al. 2003). Although we did not observe an isoform-specific difference in the overall tyrosine phosphorylation of FAK, a difference in phosphorylation at just one site could have been masked by phosphorylation at the others. Phosphospecific antibodies could be used to test such possibilities. This possibility that this and/or other activities of PTPα in focal adhesions involves non-SFK targets is supported by studies in fibronectin-stimulated fibroblasts (Chen et al. 2006): Reintroducing either wild-type PTPα793 or PTPα793(Y789F) into PTPα−/− cells restored integrin-induced SFK, FAK and paxillin phosphorylation, but only wt PTPα793, and not PTPα793(Y789F), rescued physiological integrin-induced processes including stress fiber assembly, focal adhesion formation and cell spreading. Chen et al. (2006) explained this by suggesting that pTyrs in FAK may replace PTPα’s pTyr789 in phosphotyrosine displacement but that there are non-SFK PTPα targets that also require that it be phosphorylated at Tyr789. In this context, it will be interesting to test whether the ability of PTPα to dephosphorylate the focal adhesion protein Cas (Brunton et al. 1997; Buist et al. 2000) or its role in recruiting Rac1 to focal adhesions (Herrera Abreu et al. 2008) is isoform specific.
Vacaresse et al. (2008) have shown that one-fourth of PTPα (presumably PTPα793) is found in fibroblast lipid rafts, where it can act on colocalized targets, and that growth-factor stimulation induces further recruitment. Moreover, they find that the activation of Src and Fyn in rafts by epidermal growth factor (EGF) or platelet-derived growth factor (PDGF) absolutely requires PTPα (in contrast with Fyn outside rafts, which does not require PTPα for PDGF-induced activation). Thus, isoform-specific localization to and/or activity of PTPα in lipid rafts is another interesting possibility.
There are many other potential explanations. For example, the possibility that differences in PTPα phosphorylation are involved has not been excluded: while we have excluded differences at the primary phosphorylation sites, Ser 180, Ser 204 and Tyr 789, PTPα can also be phosphorylated at Ser 202 and Thr 205 (Yang et al. 2006), and differences in phosphorylation at these or other unidentified phosphorylation sites might be involved. Communication between the extracellular and cytoplasmic domains of PTPα (van der Wijk et al. 2003) might also play a role. Alternatively, the finding that PTPα can direct Src and Fyn substrate specificity differently in response to EGF or PDGF (Vacaresse et al. 2008) opens the interesting possibility that the extracellular domains differentially modulate PTPα’s ability to affect SFK substrate specificity. Determining how the extracellular domain of PTPα governs its biological activity remains an important, open question.
Our results are reminiscent of those with the PTP CD45, which dephosphorylates the SFKs Lck and Fyn in T cells (Hermiston et al. 2003). CD45 also has tandem PTPase domains and a carboxy-proximal phosphotyrosine that has been suggested to participate in phosphotyrosine displacement (Autero et al. 1994). Moreover, CD45 has multiple extracellular domain isoforms having similar in vitro PTPase activities but different biological activities (Ostergaard et al. 1989). The differences could be explained by the proposal of Thomas & Brown (1999) that binding of the CD45 extracellular domain to macrophage surface proteins directs SFK activity to these specific targets if such binding was isoform specific. A similar mechanism could explain the substrate-specific targeting observed for PTPα by Vacaresse et al. (2008). Alternatively, the differences may involve the isoform-specific ability of the extracellular domain to modulate CD45 homodimerization (Hermiston et al. 2003), which reduces CD45 PTPase (Majeti et al. 1998; Takeda et al. 2004) and affects CD45 association with other cell surface proteins (Dornan et al. 2002). In addition, the different isoforms have different glycosylation patterns, which affect homodimerization (Xu & Weiss 2002) and possibly interactions with other ligands and proteins. Similar mechanisms might modulate differential activities of the PTPα isoforms. In particular, PTPα793 activity is reduced by homodimerization (Jiang et al. 1999; Blanchetot et al. 2002), but this has not been tested for PTPα802.
It is interesting to compare our results with those of Lammers’ group (Lammers et al. 2000; Kapp et al. 2007), who studied PTPα793 and PTPα802 mutants that contained Tyr → Phe mutations at their homologous carboxy-proximal phosphorylation sites, Tyr789 (in PTPα793) and Tyr798 (in PTPα802): They found that only phosphatase-defective or Tyr789/798 → Phe PTPα mutants, but not wt PTPαs, could be overexpressed in NIH3T3 cells using a retroviral expression system and suggested ‘growth-inhibiting or toxic effects of PTPα which are not exerted by the mutant PTPs’ (Lammers et al. 2000). This is plausible because over-expression of PTPα at very high levels by a retroviral system could hyperactivate Src leading to cytotoxicity (Tarpley & Temin 1984; Wu & Hackett 1995). This would also explain why the Tyr789 → Phe mutants [that have little ability to directly activate Src in NIH3T3 cells (Zheng et al. 2000)] were not cytotoxic and thus could be overexpressed. Lammers’ group was therefore not able to compare the biological activities of the wt PTPα splice variants, and instead studied the transforming activities of PTPα793(Y789F) and PTPα802(Y798F). In agreement with previous results with PTPα793(Y789F) (Zheng et al. 2000), they found that overexpressed Tyr789 → Phe mutants alone did not transform normal NIH3T3 cells (Lammers et al. 2000; Kapp et al. 2007). However, they found that both PTPα793(Y789F) and PTPα802 (Y798F) did induce foci in NIH3T3 cells that had been primed by over-expression of Src (Kapp et al. 2007). This is not surprising, because the reduced ability of the Tyr789 → Phe mutants to activate Src (Zheng et al. 2000) was compensated by the over-expression of Src itself.
However, in apparent contrast with our results showing that wt PTPα793, but not wt PTPα802, induced focus formation and anchorage-independent growth, they found that the combination of PTPα802(Y798F) and Src over-expression induced 2–4× more foci than the combination of PTPα793(Y789F) and Src over-expression (Kapp et al. 2007). (Anchorage-independent growth was not tested.) The reversal of the relative transforming activities of the long and short isoforms when they are concomitantly mutated and co-overexpressed with Src may result from differences in the non-Src substrates of the Tyr → Phe mutants and wt PTPαs. For example, Tyr798 → Phe mutation of PTPα802 blocks its localization to focal adhesion plaques (Lammers et al. 2000), and this is likely to be true for PTPα793(Y789F) as well. Thus, PTPα activities at focal adhesion plaques will not be observed with the Tyr → Phe mutants. Indeed, wt PTPα793, but not PTPα793(Y789F), affects focal adhesion formation and remodeling and transformation-related phenotypes such as cell adhesion and cell spreading (Chen et al. 2006; Herrera Abreu et al. 2008); potential isoform-dependent differences in these activities would not be observed with the Tyr → Phe mutants.
Conversely, there appear to be proteins that are affected more by the Tyr789/798 → Phe mutants than by the wt PTPαs (Lammers et al. 1998; Maksumova et al. 2007), and these might have different isoform sensitivities. Alternatively, the fact that only the wt PTPαs bind Grb2 (den Hertog et al. 1994; Su et al. 1994) might be explanatory if the extracellular domains and Grb2 acted cooperatively to mediate associations. In summary, there are multiple differences between the activities of the wt and Tyr789/798 → Phe PTPα mutants (particularly because concurrent Src over-expression is required the latter case) that could explain the different relative transforming activities of their isoforms.
We have shown that siRNA-mediated silencing of PTPα in estrogen receptor-negative breast carcinoma and colon cancer cell lines results in apoptosis, suggesting it has a survival role in these cancer cell types (Zheng et al. 2008). Yet, in contrast with SFK knockout, complete PTPα knockout causes only subtle central nervous system defects in mice (Petrone et al. 2003; Skelton et al. 2003), so PTPα may be an attractive therapeutic target. Consistent with the results presented here, as well as the limited tissue-specific expression of PTPα802, we have only detected expression of PTPα793, and not PTPα802, in human breast, colon, prostate and cervical cancer cell lines (Zheng et al. 2008) and in human breast, colon, liver, lung or thyroid tumors (unpublished results). This suggests that the use of small molecule inhibitors or antibodies directed specifically against the PTPα793’s extracellular region so as to spare PTPα802 could be a useful strategy for reducing side effects in these tumor types. Because PTPα802 is preferentially expressed in the brain, this strategy might minimize unwanted central nervous system side effects.
Experimental procedures
Antibodies
Rabbit anti-PTPα antibody 7-091 (anti-intracellular domain) and anti-Src monoclonal antibody mAb 327 (Lipsich et al. 1983) have been described. Rabbit anti-Grb2 (SC-255) and rabbit anti-HA (SC-805) were from Santa Cruz Biotechnology (Santa Cruz, CA, USA), mouse anti-Grb2 (#610111) was from BD Biosciences (San Diego, CA, USA), mouse anti-FAK (F15020) was from Transduction Labs (San Diego, CA, USA), mouse anti-phosphotyrosine PY100 (#9411) and rabbit anti-PTPαpY789 (#4481) were from Cell Signaling Technology (Danvers, MA, USA) and mouse anti-Y527 Src (Clone 28; #AHO0051) was from Invitrogen (Carlsbad, CA, USA). horseradish peroxidase-linked secondary antibodies for immunoblots and goat anti-Rabbit IgG conjugated to Texas Red for Immunofluorescence were from Jackson ImmunoResearch Laboratories (Westgrove, PA, USA). Protein A-sepharose beads and GammaBind sepharose beads were from Amersham Biosciences (Piscataway, NJ, USA).
Anti-phosphoS180/phosphoS204 and anti-phosphoS204 antibodies were commercially generated in rabbits against peptide antigens (QAGSHSNpSFRLSNGRTEC and CPLLARSPpSTNRKYPP, respectively) and then purified using nonphospho- and phospho-peptide affinity columns. Antibody purity was verified by mass spectroscopy or HPLC, and specificity was verified by immunoblotting against wt PTPα793 protein (in which Ser180 and Ser204 are partially phosphorylated), PTPα793 proteins containing either Ser180 → Ala or Ser204 → Ala mutations, or PTPα793 that had been dephosphorylated with the serine/threonine phosphatase PP2A. Although the antigen used to generate the pS180/pS204 antibody only contained pSer at position 180, tests with PTPα793(S180A), PTPα793(S204A) and PTPα793 (S180A/S204A) showed that this antibody reacted with both pSer180 and pSer204 [but not with other phosphoserines (Zheng et al. 2002) in PTPα]. Thus, it was used to measure the combined level of phosphorylation at the two sites.
Cell lines
The c-Src(Y527F) overexpressor cell line NIH[pcsrc527/foc/ep]B1 has been described.(Kmiecik & Shalloway 1987). NIH3T3-derived tet-off inducible cell lines NIH(pTPTPα/cos/)1, NIH(pTPTPαCCSS/cos)1, NIH(pTPTPα789F/cos)1 expressing HA-tagged wt PTPα793, PTPα793(CCSS) and PTPα793(Y789F) and NIH(pTet-splice/cos)1, a control line containing the tet expression vector but expressing no exogenous protein (Neo control), have been described (Zheng et al. 2000). (References to PTPα in our prior papers should be understood as references to PTPα793.)
To generate homologous cell lines expressing HA-tagged PTPα802, we first constructed a tet-off inducible PTPα802-expressing plasmid that was identical to the inducible HA-tagged PTPα793-expressing plasmid pTPTPα except for the insertion of 27 bp encoding the nine amino acid PTPα802 insert: The 824-bp ClaI-ClaI restriction fragment from plasmid pPTPα802 (that contains the N-proximal PTPα802 coding sequence and 17 bp upstream from the coding ATG) was gel purified and used to replace the homologous 797-bp ClaI-ClaI fragment in pTPTPα. The resulting plasmid, pTPTPα802, was sequenced to verify that it was identical to pTPTPα except for the 27-bp insertion.
pTPTPα802 was then cotransfected into NIH3T3 cells along with the transactivator pTet-tTAK (Life Technologies) and the G418-resistance plasmid pSV2neo (Southern & Berg 1982) in the presence of 10 ng/mL doxycycline (Sigma). G418-resistant colonies were selected and screened by immunoblotting with anti-PTPα and anti-HA antibodies for inducible expression of PTPα802 and subsequently endpoint cloned to generate lines NIH (pTPTPα802/cos/ep) 1A, 1B and 2B.
Cell culture
Inducible PTPα cell lines and the NIH(pTet-splice/cos)1 (Neo) control line were grown in monolayer culture in complete Dulbecco’s modified Eagle medium (DMEM) containing 4.5 g/L glucose and 2 mM glutamine (Mediatech; Manassas, VA, USA) plus 10% calf serum (Invitrogen), 3.7 g/L NaHCO3, penicillin/streptomycin [(100 units/mL)/(100 μg/mL)] and 10 ng/mL doxycycline (for suppression of PTPα expression). NIH3T3 and NIH (pcsrc527/foc/ep)B1 cells were grown in the same medium but without doxycycline. All cells were maintained at 37 °C, 10% CO2, 90% humidity.
Transformation assays
Cells were assayed (in the absence of doxycycline) for focus-forming ability by mixing 500 cells of each test line with 2 × 105 normal NIH 3T3 cells in 60-mm tissue culture dishes in DMEM containing 5% calf serum or assayed for colony formation in 0.3% soft agarose containing 10% calf serum as described (Zheng et al. 2000).
Immunoprecipitation and immunoblotting
Cells were plated into medium lacking doxycycline and grown for 12–24 h (80–90% confluence) before harvesting. Alternatively, cells were plated in the presence of doxycycline for approximately 12–24 h, washed, refed with medium without doxycycline and incubated for an additional 12–24 h. Total cell protein was prepared, and immunoprecipitations were carried out as described (Zheng et al. 2000), except that the lysis buffer was 50 mM HEPES (pH 7.2), 150 mM NaCl, 2 mm EDTA, 50 mm NaF, 1% NP-40, 1 mm Na3VO4, 10 μg/mL aprotinin, 10 μg/mL leupeptin and 1 mm phenylmethylsulfonyl fluoride.
Immunoblotting was carried out as described (Zheng et al. 2000) with minor variations: Blots were blocked with phosphate-buffered saline (PBS) supplemented with 0.05% Tween 20 and 5% nonfat milk for PTPα antibody, with Tris-buffered saline supplemented with 0.1% Tween 20 and 5% nonfat milk (TBST) for anti-HA, anti-Grb2 and anti-FAK antibodies, or with TBST supplemented with 2.5% bovine serum albumin for mAb 327, anti-Y527 Src and all phosphospecific antibodies. Membranes were incubated with primary antibodies for 14–18 h at 4 °C at the following dilutions: anti-PTPα (1 : 2500), mAb327 (1 : 10 000), anti-Y527 Src (Clone 28, 1 : 50 000), anti-PY 100 (1 : 2000); anti-HA (1 : 2000), mouse anti-Grb2 (1 : 5000), anti-FAK (1 : 2500), anti-pY789 (1 : 1000), anti-pS180 (1 : 1000) and anti-pS204 (1 : 1000) followed by either peroxidase-conjugated anti-rabbit or anti-mouse-IgG (1 : 10 000) for 1–2 h at room temperature. Proteins were visualized by enhanced chemiluminescence (Perkin Elmer; Boston, MA, USA).
Phosphatase assays
Preparation and dephosphorylation of [32P]tyrosine-phosphorylated MBP by PTPα was carried out as described (Zheng et al. 2000). Incubations were for 5 or 10 min at 30 °C, and released radioactive phosphate was measured by scintillation counting. Separate aliquots were analyzed by immunoblotting to determine PTPα levels. Dephosphorylation of Src [immunoprecipitated from NIH([pMcsrc/foc)B cells (Kmiecik & Shalloway 1987)] by immunopurified PTPα was analyzed as described (Zheng et al. 2000).
Src kinase assay
Immune-complex kinase assays using acid-denatured enolase as a target were carried out as described (Zheng et al. 2000) except that cells were lysed directly on the dish in RIPA buffer.
PTPα phosphorylation
PTPα overexpressor or control cells were induced by removal of doxycycline for 20 h, PTPα proteins were immunoprecipitated from cell lysates containing 2 mg total cell protein (2.5 mg/mL) with 4 μg anti-HA antibody, and equal aliquots of the immunoprecipitates were immunoblotted with the specified antibodies.
Grb2 binding assay
PTPα overexpressor or control cells were induced by removal of doxycycline for 12 h, and lysates (0.5 mg total cell protein, 2.5 mg/mL) prepared with lysis buffer supplemented with 10% glycerol were incubated with 2 μg rabbit anti-Grb2. Aliquots of the total cell lysates and immunoprecipitates were immunoblotted with anti-PTPα and anti-Grb2 antibodies as indicated.
Focal adhesion kinase phosphorylation
PTPα overexpressor cells were induced and PTPα proteins were immunoprecipitated from 1 mg total cell protein (3 mg/mL final concentration) with 1 μg anti-FAK antibody. Equal aliquots of the immunoprecipitates were analyzed by immunoblotting for FAK protein and tyrosine-phosphorylated FAK, and total cell protein was analyzed for PTPα expression.
Immunofluorescence microscopy
Cells were plated on glass coverslips, grown for 24 h in media with doxycycline, washed 3x with media to remove doxycycline and grown for an additional 20 h to induce PTPα expression. Cells were fixed for 10 min with PBS containing 4% paraformaldehyde, washed 3x with PBS, and then permeabilized for 15 min with PBS containing 0.1% Triton X-100. HA-tagged PTPα proteins were sequentially stained for 1 h each with rabbit anti-HA IgG and goat anti-rabbit IgG conjugated to Texas Red in PBS containing 3% bovine serum albumin. Cells were washed 3x with PBS, drained and mounted on glass slides using Vectashield (Vector Laboratories, Burnlingame, CA, USA) containing 4′,6′-diamidino-2-phenylindole dihydrochloride. Images were collected on a Leica SP2 Laser scanning confocal microscope using a 40×/1.25 oil objective lens and 543 nm laser excitation. Emission was collected from 575–715 nm.
Acknowledgements
Confocal microscopy was performed at the Microscopy and Imaging Facility in the Life Sciences Core Laboratory Center at Cornell University with the assistance of Carol Bayles. We are indebted to Steve Taylor for his expert assistance in preparing the manuscript and to the National Cancer Institute (RO1 CA32317) for supporting this research.
References
- Ardini E, Agresti R, Tagliabue E, Greco M, Aiello P, Yang LT, Menard S, Sap J. Expression of protein tyrosine phosphatase α (RPTPα) in human breast cancer correlates with low tumor grade, and inhibits tumor cell growth in vitro and in vivo. Oncogene. 2000;19:4979–4987. doi: 10.1038/sj.onc.1203869. [DOI] [PubMed] [Google Scholar]
- Autero M, Saharinen J, Pessa-Morikawa T, Soula-Rothhut M, Oetken C, Gassmann M, Bergman M, Alitalo K, Burn P, Gahmberg CG, Mustelin T. Tyrosine phosphorylation of CD45 phosphotyrosine phosphatase by p50csk kinase creates a binding site for p56lck tyrosine kinase and activates the phosphatase. Mol. Cell. Biol. 1994;14:1308–1321. doi: 10.1128/mcb.14.2.1308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanchetot C, Tertoolen LG, den Hertog J. Regulation of receptor protein-tyrosine phosphatase alpha by oxidative stress. EMBO J. 2002;21:493–503. doi: 10.1093/emboj/21.4.493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunton VG, Ozanne BW, Paraskeva C, Frame MC. A role for epidermal growth factor receptor, c-Src and focal adhesion kinase in an in vitro model for the progression of colon cancer. Oncogene. 1997;14:283–293. doi: 10.1038/sj.onc.1200827. [DOI] [PubMed] [Google Scholar]
- Buist A, Blanchetot C, Tertoolen LG, den Hertog J. Identification of p130cas as an in vivo substrate of receptor protein-tyrosine phosphatase α. J. Biol. Chem. 2000;275:20754–20761. doi: 10.1074/jbc.M001626200. [DOI] [PubMed] [Google Scholar]
- Chen M, Chen SC, Pallen CJ. Integrin-induced tyrosine phosphorylation of protein-tyrosine phosphatase-α is required for cytoskeletal reorganization and cell migration. J. Biol. Chem. 2006;281:11972–11980. doi: 10.1074/jbc.M600561200. [DOI] [PubMed] [Google Scholar]
- Cohen LA, Guan JL. Mechanisms of focal adhesion kinase regulation. Curr. Cancer Drug Targets. 2005;5:629–643. doi: 10.2174/156800905774932798. [DOI] [PubMed] [Google Scholar]
- Cox BD, Natarajan M, Stettner MR, Gladson CL. New concepts regarding focal adhesion kinase promotion of cell migration and proliferation. J. Cell. Biochem. 2006;99:35–52. doi: 10.1002/jcb.20956. [DOI] [PubMed] [Google Scholar]
- Daum G, Regenass S, Sap J, Schlessinger J, Fischer EH. Multiple forms of the human tyrosine phosphatase RPTPα. Isozymes and differences in glycosylation. J. Biol. Chem. 1994;269:10524–10528. [PubMed] [Google Scholar]
- Dornan S, Sebestyen Z, Gamble J, Nagy P, Bodnar A, Alldridge L, Doe S, Holmes N, Goff LK, Beverley P, Szollosi J, Alexander DR. Differential association of CD45 isoforms with CD4 and CD8 regulates the actions of specific pools of p56lck tyrosine kinase in T cell antigen receptor signal transduction. J. Biol. Chem. 2002;277:1912–1918. doi: 10.1074/jbc.M108386200. [DOI] [PubMed] [Google Scholar]
- Hermiston ML, Xu Z, Weiss A. CD45: a critical regulator of signaling thresholds in immune cells. Annu. Rev. Immunol. 2003;21:107–137. doi: 10.1146/annurev.immunol.21.120601.140946. [DOI] [PubMed] [Google Scholar]
- Herrera Abreu MT, Castellanos Penton P, Kwok V, Vachon E, Shalloway D, Vidali L, Lee W, McCulloch CA, Downey GP. Tyrosine Phosphatase PTPα regulates focal adhesion remodeling through Rac1 Activation. Am. J. Physiol. Cell Physiol. 2008;294:C931–C944. doi: 10.1152/ajpcell.00359.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- den Hertog J, Hunter T. Tight association of GRB2 with receptor protein-tyrosine phosphatase alpha is mediated by the SH2 and C-terminal SH3 domains. EMBO J. 1996;15:3016–3027. [PMC free article] [PubMed] [Google Scholar]
- den Hertog J, Sap J, Pals CE, Schlessinger J, Kruijer W. Stimulation of receptor protein-tyrosine phosphatase α activity and phosphorylation by phorbol ester. Cell Growth Differ. 1995;6:303–307. [PubMed] [Google Scholar]
- den Hertog J, Tracy S, Hunter T. Phosphorylation of receptor protein-tyrosine phosphatase α on Tyr789, a binding site for the SH3-SH2-SH3 adaptor protein GRB-2 in vivo. EMBO J. 1994;13:3020–3032. doi: 10.1002/j.1460-2075.1994.tb06601.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang G, den Hertog J, Su J, Noel J, Sap J, Hunter T. Dimerization inhibits the activity of receptor-like protein-tyrosine phosphatase-α. Nature. 1999;401:606–610. doi: 10.1038/44170. [DOI] [PubMed] [Google Scholar]
- Kaplan R, Morse B, Huebner K, Croce C, Howk R, Ravera M, Ricca G, Jaye M, Schlessinger J. Cloning of three human tyrosine phosphatases reveals a multigene family of receptor-linked protein-tyrosine-phosphatases expressed in brain. Proc. Natl Acad. Sci. USA. 1990;87:7000–7004. doi: 10.1073/pnas.87.18.7000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapp K, Siemens J, Weyrich P, Schulz JB, Haring HU, Lammers R. Extracellular domain splice variants of a transforming protein tyrosine phosphatase α mutant differentially activate Src-kinase dependent focus formation. Genes Cells. 2007;12:63–73. doi: 10.1111/j.1365-2443.2006.01034.x. [DOI] [PubMed] [Google Scholar]
- Kawakatsu H, Sakai T, Takagaki Y, Shinoda Y, Saito M, Owada MK, Yano J. A new monoclonal antibody which selectively recognizes the active form of Src tyrosine kinase. J. Biol. Chem. 1996;271:5680–5685. doi: 10.1074/jbc.271.10.5680. [DOI] [PubMed] [Google Scholar]
- Kmiecik TE, Shalloway D. Activation and suppression of pp60c-src transforming ability by mutation of its primary sites of tyrosine phosphorylation. Cell. 1987;49:65–73. doi: 10.1016/0092-8674(87)90756-2. [DOI] [PubMed] [Google Scholar]
- Krueger NX, Streuli M, Saito H. Structural diversity and evolution of human receptor-like protein tyrosine phosphatases. EMBO J. 1990;9:3241–3252. doi: 10.1002/j.1460-2075.1990.tb07523.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lammers R, Lerch MM, Ullrich A. The carboxyl-terminal tyrosine residue of protein-tyrosine phosphatase α mediates association with focal adhesion plaques. J. Biol. Chem. 2000;275:3391–3396. doi: 10.1074/jbc.275.5.3391. [DOI] [PubMed] [Google Scholar]
- Lammers R, Moller NP, Ullrich A. Mutant forms of the protein tyrosine phosphatase α show differential activities towards intracellular substrates. Biochem. Biophys. Res. Commun. 1998;242:32–38. doi: 10.1006/bbrc.1997.7906. [DOI] [PubMed] [Google Scholar]
- Lin PH, Shenoy S, Galitski T, Shalloway D. Transformation of mouse cells by wild-type mouse c-Src. Oncogene. 1995;10:401–405. [PubMed] [Google Scholar]
- Lipsich LA, Lewis AJ, Brugge JS. Isolation of monoclonal antibodies that recognize the transforming proteins of avian sarcoma viruses. J. Virol. 1983;48:352–360. doi: 10.1128/jvi.48.2.352-360.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majeti R, Bilwes AM, Noel JP, Hunter T, Weiss A. Dimerization-induced inhibition of receptor protein tyrosine phosphatase function through an inhibitory wedge. Science. 1998;279:88–91. doi: 10.1126/science.279.5347.88. [DOI] [PubMed] [Google Scholar]
- Maksumova L, Wang Y, Wong NK, Le HT, Pallen CJ, Johnson P. Differential function of PTPα and PTPα Y789F in T cells and regulation of PTPα phosphorylation at Tyr-789 by CD45. J. Biol. Chem. 2007;282:20925–20932. doi: 10.1074/jbc.M703157200. [DOI] [PubMed] [Google Scholar]
- Matthews RJ, Cahir ED, Thomas ML. Identification of an additional member of the protein-tyrosine-phosphatase family: evidence for alternative splicing in the tyrosine phosphatase domain. Proc. Natl Acad. Sci. USA. 1990;87:4444–4448. doi: 10.1073/pnas.87.12.4444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norris K, Norris F, Kono DH, Vestergaard H, Pedersen O, Theofilopoulos AN, Moller NP. Expression of protein-tyrosine phosphatases in the major insulin target tissues. FEBS Lett. 1997;415:243–248. doi: 10.1016/s0014-5793(97)01133-2. [DOI] [PubMed] [Google Scholar]
- Ostergaard HL, Shackelford DA, Hurley TR, Johnson P, Hyman R, Sefton BM, Trowbridge IS. Expression of CD45 alters phosphorylation of the lck- encoded tyrosine protein kinase in murine lymphoma T-cell lines. Proc. Natl Acad. Sci. USA. 1989;86:8959–8963. doi: 10.1073/pnas.86.22.8959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pallen CJ. Protein tyrosine phosphatase α (PTPα): a Src family kinase activator and mediator of multiple biological effects. Curr. Top Med. Chem. 2003;3:821–835. doi: 10.2174/1568026033452320. [DOI] [PubMed] [Google Scholar]
- Petrone A, Battaglia F, Wang C, Dusa A, Su J, Zagzag D, Bianchi R, Casaccia-Bonnefil P, Arancio O, Sap J. Receptor protein tyrosine phosphatase alpha is essential for hippocampal neuronal migration and long-term potentiation. EMBO J. 2003;22:4121–4131. doi: 10.1093/emboj/cdg399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piwnica-Worms H, Saunders KB, Roberts TM, Smith AE, Cheng SH. Tyrosine phosphorylation regulates the biochemical and biological properties of pp60c-src. Cell. 1987;49:75–82. doi: 10.1016/0092-8674(87)90757-4. [DOI] [PubMed] [Google Scholar]
- Sap J, D’Eustachio P, Givol D, Schlessinger J. Cloning and expression of a widely expressed receptor tyrosine phosphatase. Proc. Natl Acad. Sci. USA. 1990;87:6112–6116. doi: 10.1073/pnas.87.16.6112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skelton MR, Ponniah S, Wang DZ, Doetschman T, Vorhees CV, Pallen CJ. Protein tyrosine phosphatase alpha (PTP alpha) knockout mice show deficits in Morris water maze learning, decreased locomotor activity, and decreases in anxiety. Brain Res. 2003;984:1–10. doi: 10.1016/s0006-8993(03)02839-7. [DOI] [PubMed] [Google Scholar]
- Southern PJ, Berg P. Transformation of mammalian cells to antibiotic resistance with a bacterial gene under control of the SV40 early region promoter. J. Mol. Appl. Genet. 1982;1:327–341. [PubMed] [Google Scholar]
- Su J, Batzer A, Sap J. Receptor tyrosine phosphatase R-PTP-α is tyrosine-phosphorylated and associated with the adaptor protein Grb2. J. Biol. Chem. 1994;269:18731–18734. [PubMed] [Google Scholar]
- Su J, Muranjan M, Sap J. Receptor protein tyrosine phosphatase α activates Src-family kinases and controls integrin-mediated responses in fibroblasts. Curr. Biol. 1999;9:505–511. doi: 10.1016/s0960-9822(99)80234-6. [DOI] [PubMed] [Google Scholar]
- Su J, Yang LT, Sap J. Association between receptor protein-tyrosine phosphatase RPTPα and the Grb2 adaptor. Dual Src homology (SH) 2/SH3 domain requirement and functional consequences. J. Biol. Chem. 1996;271:28086–28096. doi: 10.1074/jbc.271.45.28086. [DOI] [PubMed] [Google Scholar]
- Takeda A, Matsuda A, Paul RM, Yaseen NR. CD45-associated protein inhibits CD45 dimerization and up-regulates its protein tyrosine phosphatase activity. Blood. 2004;103:3440–3447. doi: 10.1182/blood-2003-06-2083. [DOI] [PubMed] [Google Scholar]
- Tarpley WG, Temin HM. The location of v-src in a retrovirus vector determines whether the virus is toxic or transforming. Mol. Cell. Biol. 1984;4:2653–2660. doi: 10.1128/mcb.4.12.2653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas ML, Brown EJ. Positive and negative regulation of Src-family membrane kinases by CD45. Immunol. Today. 1999;20:406–411. doi: 10.1016/s0167-5699(99)01506-6. [DOI] [PubMed] [Google Scholar]
- Tonks NK. Protein tyrosine phosphatases: from genes, to function, to disease. Nat. Rev. Mol. Cell Biol. 2006;7:833–846. doi: 10.1038/nrm2039. [DOI] [PubMed] [Google Scholar]
- Tracy S, van der Geer P, Hunter T. The receptor-like protein-tyrosine phosphatase, RPTPα, is phosphorylated by protein kinase C on two serines close to the inner face of the plasma membrane. J. Biol. Chem. 1995;270:10587–10594. doi: 10.1074/jbc.270.18.10587. [DOI] [PubMed] [Google Scholar]
- Vacaresse N, Moller B, Danielsen EM, Okada M, Sap J. Activation of c-Src and Fyn kinases by protein-tyrosine phosphatase RPTPalpha is substrate-specific and compatible with lipid raft localization. J. Biol. Chem. 2008;283:35815–35824. doi: 10.1074/jbc.M807964200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Wijk T, Blanchetot C, Overvoorde J, den Hertog J. Redox-regulated rotational coupling of receptor protein-tyrosine phosphatase alpha dimers. J. Biol. Chem. 2003;278:13968–13974. doi: 10.1074/jbc.M300632200. [DOI] [PubMed] [Google Scholar]
- Wu LW, Hackett PB. Development of cellular resistance to pp60v-src kinase-induced cell death. Oncogene. 1995;11:1459–1468. [PubMed] [Google Scholar]
- Xu Z, Weiss A. Negative regulation of CD45 by differential homodimerization of the alternatively spliced isoforms. Nat. Immunol. 2002;3:764–771. doi: 10.1038/ni822. [DOI] [PubMed] [Google Scholar]
- Yang F, Stenoien DL, Strittmatter EF, Wang J, Ding L, Lipton MS, Monroe ME, Nicora CD, Gristenko MA, Tang K, Fang R, Adkins JN, Camp DG, II, Chen DJ, Smith RD. Phosphoproteome profiling of human skin fibroblast cells in response to low- and high-dose irradiation. J. Proteome Res. 2006;5:1252–1260. doi: 10.1021/pr060028v. [DOI] [PubMed] [Google Scholar]
- Yang LT, Alexandropoulos K, Sap J. c-SRC mediates neurite outgrowth through recruitment of Crk to the scaffolding protein Sin/Efs without altering the kinetics of ERK activation. J. Biol. Chem. 2002;277:17406–17414. doi: 10.1074/jbc.M111902200. [DOI] [PubMed] [Google Scholar]
- Zeng L, D’Alessandri L, Kalousek MB, Vaughan L, Pallen CJ. Protein tyrosine phosphatase α (PTPα) and contactin form a novel neuronal receptor complex linked to the intracellular tyrosine kinase fyn. J. Cell Biol. 1999;147:707–714. doi: 10.1083/jcb.147.4.707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng L, Si X, Yu WP, Le HT, Ng KP, Teng RM, Ryan K, Wang DZ, Ponniah S, Pallen CJ. PTPα regulates integrin-stimulated FAK autophosphorylation and cytoskeletal rearrangement in cell spreading and migration. J. Cell Biol. 2003;160:137–146. doi: 10.1083/jcb.200206049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng XM, Resnick RJ, Shalloway D. A phosphotyrosine displacement mechanism for activation of Src by PTPα. EMBO J. 2000;19:964–978. doi: 10.1093/emboj/19.5.964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng XM, Resnick RJ, Shalloway D. Mitotic activation of protein-tyrosine phosphatase α and regulation of its Src-mediated transforming activity by its sites of protein kinase C phosphorylation. J. Biol. Chem. 2002;277:21922–21929. doi: 10.1074/jbc.M201394200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng XM, Resnick RJ, Shalloway D. Apoptosis of estrogen-receptor negative breast cancer and colon cancer cell lines by PTP and Src RNAi. Int. J. Cancer. 2008;122:1999–2007. doi: 10.1002/ijc.23321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng XM, Shalloway D. Two mechanisms activate PTP during mitosis. EMBO J. 2001;20:6037–6049. doi: 10.1093/emboj/20.21.6037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng XM, Wang Y, Pallen CJ. Cell transformation and activation of pp60c-src by overexpression of a protein tyrosine phosphatase. Nature. 1992;359:336–339. doi: 10.1038/359336a0. [DOI] [PubMed] [Google Scholar]