

Abstract
Aldosterone-producing adenomas (APAs) vary in phenotype and genotype. Zona glomerulosa (ZG)-like APAs frequently have mutations of an L-type calcium channel (LTCC) CaV1.3. Using a novel antagonist of CaV1.3, compound 8, we investigated the role of CaV1.3 on steroidogenesis in the human adrenocortical cell line, H295R, and in primary human adrenal cells. This investigational drug was compared with the common antihypertensive drug nifedipine, which has 4.5-fold selectivity for the vascular LTCC, CaV1.2, over CaV1.3. In H295R cells transfected with wild-type or mutant CaV1.3 channels, the latter produced more aldosterone than wild-type, which was ameliorated by 100 μM of compound 8. In primary adrenal and non-transfected H295R cells, compound 8 decreased aldosterone production similar to high concentration of nifedipine (100 μM). Selective CaV1.3 blockade may offer a novel way of treating primary hyperaldosteronism, which avoids the vascular side effects of CaV1.2-blockade, and provides targeted treatment for ZG-like APAs with mutations of CaV1.3.
Aldosterone-producing adenomas (APAs), which arise from the adrenal cortex, are one of the most common curable causes of hypertension1,2,3. They account for approximately half of primary aldosteronism, which is estimated to be present in 5–13% of all hypertensive patients, and in at least 20% of patients with resistant hypertension4. However, it is likely that fewer than 10% of APAs are ever diagnosed; and fewer still are removed in time to cure hypertension and prevent resistance to effective drug treatment2,5.
We previously reported somatic gain-of-function mutations in two genes that regulate Na+, K+ and Ca2+ transport in APAs with a zona glomerulosa (ZG)-like phenotype6. Whole exome sequencing of small-cell APAs with a ZG-like gene expression profile found five out of ten to harbour one of four different somatic mutations in the voltage dependent L-type Ca2+ channel, CaV1.3 (encoded by the gene CACNA1D). These four substitution mutations, V259D, G403R, I750M, and P1336R, cluster around the Ca2+ pore between the S5 and S6 domains that line the inner pore surface. The mutations occur in conserved sites within functional domains such as the voltage-sensing domain to the pore (V259D and P1336R) and the channel activation gate (G403R and I750M)6. The G403R and I750M mutations were simultaneously reported as rare de novo germline mutations presenting at birth, together with several patients having somatic mutations of the same residues in sporadic APAs7. Our own replication sequencing revealed three further mutations, and sequencing of APAs in a large European consortium has now identified a total of 19 somatic mutations in or near one of the four Ca2+ channel pore-forming domains 6,8. Patch clamping of HEK293 cells has shown that at least 6 of the 19 mutations affect the CaV1.3 channel function and allow for increased Ca2+ influx through either shifting voltage-dependent activation towards more negative voltages, decelerating inactivation, and/or increasing currents through a higher open channel probability6,9.
The current medical treatment of primary hyperaldosteronism is blockade of the mineralocorticoid receptor, which can lead to an increase in aldosterone secretion10. Therefore, blockade of calcium entry through selective antagonism of CaV1.3 might present a valuable therapeutic target. We therefore aimed to investigate whether CaV1.3 mutations cause the postulated increase in aldosterone secretion from human adrenocortical cells, and whether blockade of calcium entry reverses this. We studied the potential value of this target using 1-(3-chlorophenethyl)-3-cyclopentylpyrimidine-2,4,6-(1H,3H,5H)-trione (compound 8), which was found to be more than 600 times more selective for CaV1.3 than CaV1.211. Nifedipine, a common antihypertensive drug, was used in comparison as a non-selective, or slightly CaV1.2 selective antagonist of L-type calcium channels (IC50 = 0.016 μM)12,13,14. We also undertook immunohistochemistry of normal human adrenals, and APAs, in order to determine whether CaV1.3 is a ZG-selective L-type Ca2+ channel and whether blockade may have greater expected effect on aldosterone secretion from APAs than from normal adrenal.
To study the role of CaV1.3 on aldosterone secretion, we first investigated the substitution mutations near the voltage-sensing domain, P1336R and V259D, on 24-h aldosterone production in transiently transfected H295R cells to find if the different changes seen in our electrophysiology data translated to changes in aldosterone secretion6. We then contrasted the aldosterone secretion of cells transfected with mutant CaV1.3 channels to those transfected with wild-type CaV1.3 channel in the presence of compound 8 or nifedipine to study if blockade of calcium entry affects APAs with a CaV1.3 mutation differently. Transfection of H295R cells with exogenous CaV1.3 was performed with β3 and α2δ accessory subunits, the subunits we used previously to show gain-of function effects of the mutations on Ca2+ currents6. As transfected channels and subunits do not necessarily emulate in vivo expression, we also tested the effect of compound 8 and nifedipine on endogenous CaV1.3 present in H295R cells and primary adrenal cells acquired from adrenals containing an APA (both tumour and adjacent normal adrenal tissues).
Results
CaV1.3 mutations and compound 8 alter aldosterone production
Transfection of H295R cells with CaV1.3 mutants P1336R and V259D caused a 2.4 ± 0.2 (P = 0.0004) and 2.1 ± 0.2 (P = 0.002) fold increase, respectively, in basal aldosterone production compared to wild-type transfected H295R cells (Fig. 1a) and similarly in angiotensin II stimulated aldosterone production (Supplementary Fig. 1).
Figure 1. CaV1.3 mutations and compound 8 alter aldosterone production.
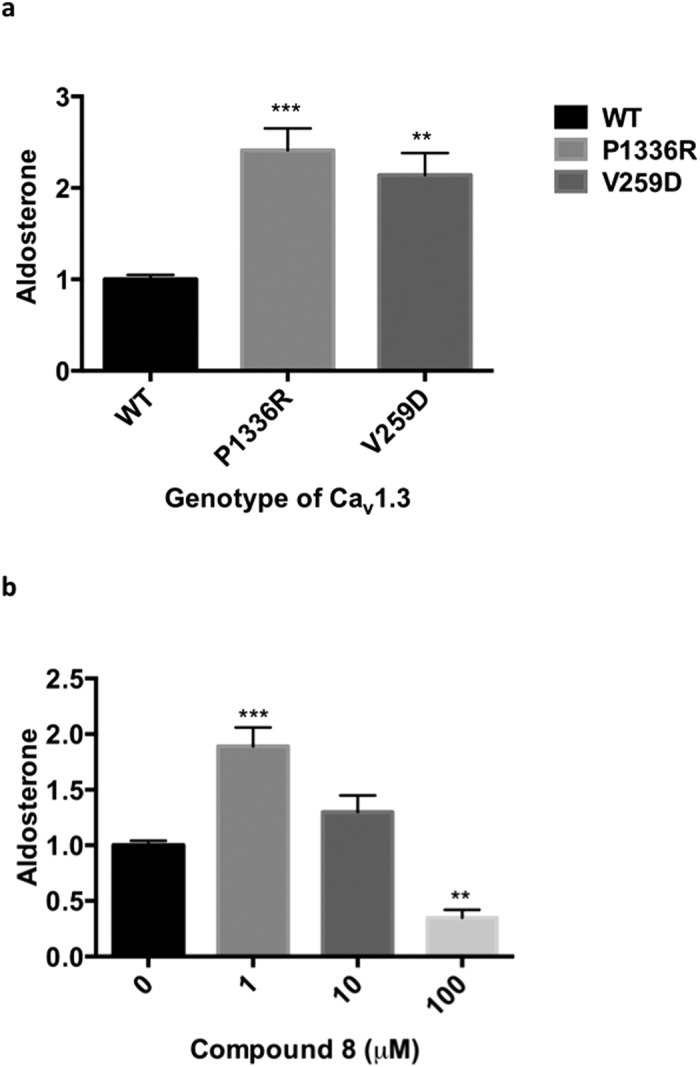
Comparison of stimulated aldosterone production in (a) wild-type (WT), P1336R, and V259D CaV1.3 transfected H295R cells (n = 5) and in (b) different concentration of compound 8 on WT H295R cells (n = 3). Student t-test was used to calculate significance. **P < 0.01 and ***P < 0.001, compared to baseline (Wild-type or 0 M compound 8). The n value represents number of separate experiment/transfection performed. Each experiment/transfection had 6 biological replicates. Aldosterone results shown here were measured by RIA method and are relative to basal level (Wild-type or 0 M compound 8).
Exposure of H295R cells transfected with wild-type CaV1.3 to low concentration (1 μM) of compound 8 almost doubled aldosterone secretion (P = 0.007), whereas high concentration (100 μM) of compound 8 decreased aldosterone production to 35 ± 0.1% of basal level (P = 0.003, Fig. 1b).
Effect of calcium blockade on CaV1.3 genotypes
In cells transiently transfected with wild-type, P1336R, or V259D CaV1.3, there was a similar biphasic effect of compound 8 on aldosterone secretion from the mutant P1336R cells, as that seen in wild-type CaV1.3. In mutant V259D cells, compound 8 was inhibitory only at 100 μM (Fig. 2a). Using the same protocol as for compound 8, the inhibitory effect of nifedipine on aldosterone secretion from CaV1.3 transfected H295R cells was determined. In wild-type CaV1.3 transfected cells, after treatment with 1 μM, 10 μM or 100 μM nifedipine, a 35 ± 12% decrease of aldosterone secretion was observed only at the highest concentration of nifedipine interrogated (100 μM)(P = 0.0001, Fig. 2b) a considerable excess of its Ki for CaV1.2 (IC50 = 0.016 μM)12,13. In P1336R and V259D transfected cells, despite the increased aldosterone secretion compared to that of wild-type cells (as seen at 0 μM), the presence of high concentration of nifedipine (100 μM) decreased aldosterone production similarly across all genotypes (Fig. 2b).
Figure 2.

Effect of compound 8 on aldosterone production of different CaV1.3 genotype (a) Stimulated aldosterone secretion (n = 3) in the presence of compound 8 and (b) stimulated aldosterone secretion in the presence of nifedipine (n = 3) on WT, P1336R and V259D CaV1.3 transfected H295R cells. There was a similar biphasic effect of compound 8 on aldosterone secretion from the mutant P1336R cells (P = 0.02; Student’s t-test), as that seen in wild-type CaV1.3, but not so in mutant V259D cells or when transfected cells were treated with nifedipine. (c) Comparison of basal aldosterone production of non-transfected H295R cells in the presence of 0–100 μM of compound 8 or nifedipine (n = 3). Two-way ANOVA was used to calculate overall significance. Table of P-values shows significance of mutation status (Mutation), concentration of treatment (Concentration), and type of treatment (Drug), on aldosterone production. The n value represents number of separate experiment/transfection performed. Each experiment/transfection had 6 biological replicates. Aldosterone was measured by RIA (a,b) or RIA and HTR-FRET (c) method. Results of both methods are relative to basal level (Wild-type or 0 µM of treatment).
In non-transfected H295R cells (with only endogenous CaV1.3 and endogenous CaV1.3 accessory subunits present), compound 8 and nifedipine, 1–100 μM, decreased basal aldosterone secretion (Fig. 2c).
Compound 8 decreases aldosterone production in primary human adrenal cells
In primary human adrenal cells cultured from the normal adjacent adrenals of patients with an APA, 10 and 100 μM of compound 8 inhibited aldosterone production by 35 ± 10 and 43 ± 11%, respectively (P < 0.05; Fig. 3a). Cortisol secretion was also decreased to 72 ± 1 and 50 ± 4% of basal level, respectively (P < 0.05; Supplementary Fig. 2). As for nifedipine, effect on aldosterone production was varied - not all cell cultures from the different patients showed a reduction, even at the high concentration of 100 μM (Fig. 3b).
Figure 3. Compound 8 decreases aldosterone production in primary human adrenal cells.

Aldosterone secretion of (a,b) normal primary adrenal cells or (c,d) aldosterone-producing adenomas (APAs) in the presence of compound 8 (a,c) or nifedipine (b,d). Dose response curve between 0-100 μM of compound 8 on (a) normal primary adrenal cells (n = 4) and (c) APA cells (n = 3), and nifedipine on (b) normal primary adrenal cells (n = 3) and (d) APA cells (n = 3). Two-way ANOVA was used to calculate overall significance. Table of P-values shows significance of patient differences (Patient variability) and concentration of treatment (Concentration) on aldosterone production. The n value represents number of individual patient samples used for each experiment. Each concentration was replicated 2–12 times within each individual patient samples (which depended on quantity of primary cells available). Aldosterone results shown here are relative to 0 M of treatment. Numbers 181, 182, 184, 187, 196, and 221 represent individual patient ID. Clinical data for these patients is provided in Supplementary Table 1. Primary cell cultures from patients 181, 182, and 184 were performed in the absence of angiotensin II (seen as solid bars) whereas primary cell cultures 187, 196, and 221 were stimulated with 10 nM angiotensin II (seen as hatched bars). Aldosterone was measured by RIA and ELISA method.
In APA cells, with increasing concentrations of 1, 10 and 100 μM, both compound 8 and nifedipine showed a dose dependent decrease in aldosterone production, to a minimum average of 54 ± 2 and 43 ± 13% of basal level, respectively (P < 0.005; Fig. 3c,d).
Localization of CaV1.3 in adrenals containing an APA
In sections of adjacent normal adrenal, that were adjacent to an APA or pheochromocytoma, CaV1.3 was detected in the ZG and the zona reticularis (ZR) (Fig. 4a). Only in ZG were juxtanuclear accumulation seen (as shown in the zoomed image), as ZR staining was mainly cytoplasmic (Fig. 4a). Exogenous CaV1.3 in H295R transfected cells had mainly membranous expression (Supplementary Fig. 3).
Figure 4. Localization of CaV1.3 in human adrenals.

(a) Immunohistochemistry (IHC) of CaV1.3 on formalin-fixed paraffin-embedded (FFPE) adrenal sections localized the channel to the zona glomerulosa (ZG) and zona reticularis (ZR) of the adrenals. In the ZG, cytoplasmic and juxtanuclear accumulation of CaV1.3 was observed whereas in the ZR, staining was mainly cytoplasmic. Picture is representative of 12 normal adjacent adrenal glands, 3 from patients with a phaeochromocytoma and 9 from patients with an aldosterone-producing adenoma (APA). C, capsule; G, zona glomerulosa; F, zona fasciculata; R, zona reticularis; M, adrenal medulla. (b) CaV1.3 expression in APA cells. IHC of CaV1.3 on FFPE adrenal sections were performed on three different types of APAs: (i–iii) ZG-like (low nucleus to cytoplasm ratio) APAs without a CaV1.3 mutation, (iv–vi) APAs with a CaV1.3 mutation, and (vii–ix) APAs with a KCNJ5 mutation. Immunostaining reveals a mixture of cytoplasm and membranous sublocalization in APA cells.
In APAs, different patterns of CaV1.3 expression were observed. CaV1.3 was expressed at the cell membrane, cytoplasmic, at the edge of cell clusters, or sparsely, or not at all (Fig. 4b and Supplementary Fig. 4).
Discussion
We previously reported that somatic mutation of CaV1.3 is present in a subset of APAs, distinguished by several features resembling normal ZG6. In a large multi-centre study of 474 APAs, the frequency of CaV1.3 mutation was estimated to be 9.3%8. Although no particular histological phenotype was found in the multi-centre study8, one centre within the study did subsequently report that of their 71 APAs, CaV1.3 mutant APAs (3 of 71) were composed mainly of ZG-like cells15. Thus the current approximation could be a substantial underestimation since (a) half of our selected ZG-like APAs that were exome sequenced had a CaV1.3 mutation6; and (b) our experience is that such tumours are frequently too small to be detected by conventional adrenal imaging. We therefore wished to show whether the mutations are likely to increase aldosterone production, rather than trigger development of the adenoma, and whether this increase could be reversed by blockade of calcium entry. As few APAs are diagnosed in time to offer high likelihood of surgical cure from hypertension, and the increased recognition of aldosterone morbidity, a need arises for novel therapies that suppress aldosterone production, and lack the adverse effects of aldosterone receptor blockade and other less specific therapies for hypertension.
Herein we report that the two CaV1.3 mutations studied, selected for having different electrophysiological effects6, do increase aldosterone secretion of transfected human adrenocortical cells (Fig. 1). Furthermore, calcium blockade using compound 8, an investigational CaV1.3 inhibitor, and nifedipine, a non-selective L-type calcium channel inhibitor, reversed the increase (Fig. 2). The inhibition of aldosterone secretion was seen in the presence of the highest concentration of compound 8 interrogated in this study (100 μM) in H295R cells transfected with CaV1.3 mutants (Fig. 2); whereas in non-transfected H295R cells and primary adrenal cells, inhibition of aldosterone secretion could be seen at lower concentrations (1 and 10 μM) (Figs 2c and 3d). We also postulate that regardless of whether a given APA has a somatic mutation of CaV1.3, the channel is often more active than in normal ZG cells, where immunohistochemistry suggests CaV1.3 is mainly internalised (Fig. 4).
Compound 8 was interrogated in this study as it was found to be the most selective CaV1.3 antagonist among 60,480 commercial compounds and a few hundred non-commercial compounds (Silverman lab) tested for efficacy in blocking CaV1.3 or CaV1.2 in stably transfected HEK293 cells. Compound 8 was reported to inhibit CaV1.3 > 600-fold more potently than CaV1.211. Subsequent studies have questioned this degree of selectivity, and even whether compound 8 is an agonist or antagonist16,17. Nevertheless, it is well known that the effects of L-type Ca2+ channel blockade can differ among tissues depending on factors such as resting membrane potential18. Consequently, the hyperpolarisation of adrenocortical cells may have enhanced our ability to detect an antagonist effect of compound 8. Further, we may have fortuitously selected the CACNB isoform which maximises compound 8 selectivity, namely CACNB3 (encoding for the β3 subunit). In subsequent analysis, however, we found CACNB2 to be the predominant isoform in human adrenal, indeed being one of the genes most up-regulated in ZG compared to zona fasciculata (ZF)19. Thus, for the pharmacological responses of the different mutations to be legitimately compared, a better CaV1.3 antagonist than Compound 8 is needed. Future antagonists should be developed not only based on its selectivity for CaV1.3 but also on its functionality with the prevalent accessory subunits in the human adrenal.
In our cells transfected with exogenous CaV1.3, the stimulatory effect of apparent low dose calcium blockade on aldosterone secretion was observed only for Compound 8, but not nifedipine. This increase in aldosterone secretion could have been due to low dose compound 8 behaving as a channel activator16; but toxicity (and hence leakage of aldosterone) due to high calcium influx in transfected H295R cells cannot be dismissed, since no stimulation of aldosterone secretion was seen in untransfected cells (Fig. 2c). The limitation of our expression CaV1.3 model, however, was that the cell line we used, H295R cells, express a mixture of endogenous CaV1.2 and CaV1.3 whereas primary human ZG cells express mainly CaV1.319,20. Moreover, the immortalised H295R cells were not a perfect model for primary aldosteronism as other adrenal corticosteroids are secreted21. This cell line was used mainly due to the ease of transfecting exogenous mutant CaV1.3. Hence, to supplement our transfection experiments, not only was compound 8 also studied in un-transfected H295R cells, but also in primary adrenal cells (of which we have a limited supply), to support endogenous CaV1.3 role in aldosterone regulation. To note, as we did not find a linear relationship between increase in aldosterone production and amount of transfected constructs, no correction for transfection efficiency whether by Western blots or qPCR was performed. Transfection rates of exogenous CaV1.3 were confirmed as similar visually, using its GFP-tag.
Previous studies have shown a number of dihydropyridines to reduce aldosterone secretion from adrenocortical cells22. We chose nifedipine as a comparator because of experience with its use in patients, in whom it was the first dihydropyridine to be used23,24,25, and also because of its modest CaV1.2 selectivity. Nifedipine is expected to exert its CaV1.2 blockade at concentrations around 4.45 nM14. At the lowest concentration of nifedipine that we had interrogated (1 μM), a concentration which should have easily blocked CaV1.2, only some inhibition of aldosterone could be seen in non-transfected H295R and primary adrenal cells and none at all in H295R cells transfected with CaV1.3 mutants (Figs 2 and 3). The shallow concentration-response curves are consistent with blockade of different sites at low and high concentrations (Figs 2 and 3). Dihydropyridines sometimes cause substantial reductions in plasma aldosterone in patients with primary aldosteronism26. However this is not the predominant response at usual clinical doses, and increasing the dose to the presumed CaV1.3-blocking range is precluded by the vascular side effects, particularly peripheral edema25,27.
The potential attraction of selective CaV1.3 blockade is that such a drug can be used at a dose which achieves substantial suppression of aldosterone secretion, without the vascular side effects of currently used L-type Ca2+ blockers25,27. Previously, a T-type Ca2+ channel blocker, mibefradil, was introduced whose reduction in aldosterone secretion was among the theoretical advantages over L-type Ca2+ blockade28; however the drug was withdrawn due to reports of dangerous and even fatal interactions with other drugs and was later found to cause serious effects on QTc29. In vitro studies, have shown that single blockade of either L-type or T-type Ca2+ channels can decrease aldosterone production, even though the influx of Ca2+ in the ZG is thought to be mediated by both channels28,30,31,32. While there has also been considerable attempt to develop inhibitors of aldosterone synthase as a therapeutic class33, these have foundered on the challenge of developing a drug, which inhibits aldosterone synthase, without effect on the 95% homologous enzyme catalysing cortisol synthesis (encoded by the gene CYP11B1). By contrast, the homology between CaV1.2 and CaV1.3 is only 75%34. Thus, even though compound 8 itself may not be the ideal drug candidate to progress for treatment of hyperaldosteronism, there are a number of sites outside the dihydropyridine-binding site where CaV1.2 and CaV1.3 differ sufficiently to suggest that selective blockade is achievable.
Three drugs do have clinical efficacy in patients with primary aldosteronism: spironolactone, eplerenone and amiloride35,36. However, the efficacy of the latter two is modest, and the use of spironolactone is limited in men by the anti-androgenic effects of higher doses37,38. All three drugs cause substantial increases in plasma aldosterone secretion, probably secondary to the rise in plasma K+, and there is some concern whether aldosterone could have adverse vascular effects through a non-canonical aldosterone receptor39,40. Although no evidence exists in humans, there is an additional theoretical benefit from blocking aldosterone synthesis rather than response – that such a drug could cause involution of aldosterone-producing cells. This is suggested by the observation that genetic deletion of the enzyme aldosterone synthase leads to apoptosis of the normal ZG cells41.
In summary, we previously reported ZG-like APAs to have CaV1.3 mutations. In this study, we confirmed that CaV1.3 is localized to the human adrenal ZG. By blocking endogenous CaV1.3 in primary human adrenal and transfecting mutant CaV1.3 in the human adrenocortical cell line, H295R, we have also confirmed that CaV1.3 plays a role in human adrenal steroidogenesis. Taken together, these discoveries suggest that CaV1.3 can provide a novel mechanism and target for regulating excess aldosterone secretion and may be a novel way of treating hyperaldosteronism, especially those caused by ZG-like APAs with a CaV1.3 mutation. Since non-selective or CaV1.2 selective dihydropyridines are dose-limited clinically by vascular effects, a selective CaV1.3 antagonist may be valuable for suppressing aldosterone secretion in some patients with aldosterone-dependent hypertension.
Methods
Cell culture experimentation
H295R cells, were cultured in growth medium consisting of DMEM/Nutrient F-12 Ham supplemented with 10% foetal bovine serum, 100 U of penicillin, 0.1 mg/mL streptomycin, 0.4 mM L-glutamine and insulin-transferrin-sodium selenite medium (ITS) at 37 °C in 5% CO2.
For transient transfection, wild-type or mutant P1336R or V259D CaV1.3 GFP-tagged constructs were co-transfected together with constructs for β3 and α2δ auxiliary subunits of CaV1.3 into H295R cells using Amaxa Nucleofector kit R (Lonza, Germany) with electroporation program P20. The GFP-CaV1.3 wild-type and mutant vectors were obtained from our collaborators; Dr. Jöerg Striessnig’s group at University of Innsbruck Center for Chemistry and Biomedicine, Austria. These constructs were derived from the ‘long’ isoform of the CaV1.3 α1 pore-forming subunit, with isoform A of the alternatively spliced exon 8. Transfected cells were seeded into 24-well plates at 100, 000 cells per well in 0.5 mL of growth medium. At 24-h post-transfection, H295R cells were serum deprived in un-supplemented DMEM/Nutrient F-12 Ham medium for 24-h. At 48-h post-transfection, the transfection efficiency was visualised and qualitatively quantified by fluorescence microscopy. Further experiments were performed on cells with 60–80% transfection efficiency.
For primary cell culture, adrenocortical cells were obtained from the adrenals of patients with Conn’s syndrome that had undergone adrenalectomy at Addenbrooke’s Hospital, Cambridge, UK (Supplementary Table 1). Local ethical approval and informed consent were obtained for each patient and the procedures followed were in accordance with institutional guidelines. After macroscopic identification of APA and adjacent normal adrenal by a trained histopathologist, tissue samples were placed in growth medium within 45 minutes of surgical excision. The APA and adjacent normal adrenal was then digested separately in 3.3 mg/ml collagenase at 37 °C for 2-h. Within a week of procurement, the primary human adrenocortical cells were then randomly seeded into 24-well plates at 50, 000 cells per well in 0.5 mL of growth medium and allowed to settle for a further 48-h before drug treatments were performed.
Drug treatments with CaV1.3 selective antagonist, compound 8, and L-type calcium blocker, nifedipine
Compound 8 and nifedipine (Sigma-Aldrich, UK) were reconstituted in DMSO to stock concentrations of 1, 10, and 100 mM. Stock concentrations were further diluted (1:1000) in sterile un-supplemented DMEM/Nutrient F-12 Ham for treatments.
Transfected H295R cells were treated at 48-h post transfection (after 24-h of serum deprivation) with either vehicle or compound 8 or nifedipine in un-supplemented DMEM/Nutrient F-12 Ham medium in the presence of 10 nM angiotensin II. Supernatant and cells were harvested after 24-h incubation at 37 °C.
For non-transfected H295R cells, cells were seeded into 24-well plates at 100 000 cells per well in 0.5 mL of growth medium, serum deprived for 24-h, and treated with either vehicle, compound 8 or nifedipine in un-supplemented DMEM/Nutrient F-12 Ham medium. Supernatant and H295R cells were harvested after 24-h incubation at 37 °C.
For primary human adrenal cells, APA and adjacent normal adrenal cells were serum deprived for 24-h, and treated with either vehicle or compound 8 or nifedipine in un-supplemented DMEM/Nutrient F-12 Ham medium in the presence or absence of 10 nM angiotensin II. Supernatant and H295R cells were harvested after 24-h incubation at 37 °C.
Immunohistochemistry
Immunohistochemistry was performed on formalin-fixed, paraffin-embedded adrenal sections (4 μm) using an automated immunostainer with cover tile technology (Bond-III system, Leica Biosystems). A commercial antibody of CaV1.3, clone N38/8 (UC Davis/NIH NeuroMab Facility; 1:500 dilution), was used as the primary antibody. Negative control experiments, in which the primary antibody was omitted, resulted in a complete absence of staining. Images were captured using a standard bright-field microscope, a U-TV1-X digital camera and CellD software (Olympus UK).
Confocal Imaging
H295R cells were cultured in complete media on sterilised and poly L-lysine coated cover-slips at the density of 105 cells/well in 12-well cell-culture plate for 24-h. Cells were serum-starved overnight before transfection. Serum-free media was replaced with antibiotic-free serum-containing media at the time of transfection with Lipofectamine 3000 transfection reagent (Life Technologies). Cells were co-transfected with GFP-tagged CaV1.3 WT, β3 & α2δ constructs according to manufacturer’s instructions. 48-h later plasma membranes of cells were stained with 2ug/ml Wheat Germ Agglutinin, Alexa Fluor® 633 Conjugate (W21404, Life Technologies) in complete media for 10 min at 37 °C. Cells were washed twice with PBS (5 min each), followed by fixing with 4% paraformaldehyde and permeabilisation with 1% trition-X100 (PBST), 10 min each at room temperature. Cells were incubated with blocking buffer (3% BSA in PBS) for 1-h at room temperature and overnight with the CaV1.3 antibody, clone N38/8 (UC Davis/NIH NeuroMab Facility; 1:500 dilution) in 3% BSA-PBST. Goat anti-mouse IgG, Alexa Fluor® 568 Conjugate (A11004, Life Technologies) was used as secondary antibody at 1:1000 dilution in 3%BSA-PBST for 1-h at room temperature. Finally cells were washed thrice in PBST and cover-slips were mounted on slides using VECTASHIELD Antifade Mounting Medium with DAPI (H-1200, Vector Laboratories). Confocal images were taken using Zeiss LSM510 Meta confocal microscope and analysed using Zen 2011 software.
Aldosterone concentration measurements
Aldosterone concentration was quantitatively measured using three methods due to availability of the kits; Coat-A-Count® Aldosterone (Siemens Medical Solutions, USA), a125I solid-phase radioimmunoassay and after the discontinuation of this kit, an ELISA method adapted from researchers in Gomez-Sanchez’s group and finally a commercially available Homogenous Time Resolved Fluorescence Resonance Energy Transfer (HTR-FRET) assay from Cisbio Bioassays, France (used according to manufacturer’s instructions). ELISA was carried out using a selective validated aldosterone monoclonal antibody gifted to us and produced by Gomez-Sanchez’s lab, USA42. The aldosterone concentrations from transfected H295R cells were normalized to total cell protein, which was determined by performing the bicinchoninic acid (BCA) protein assay (Pierce Biotechnology, USA).
Statistical analysis
Experiments were performed with vehicle/plasmid controls where appropriate. Each experiment was performed with biological replicates and the averages were calculated. Aldosterone measurements are expressed as a ratio of basal (control) for each experiment. Results are shown as mean values ± SEM of separate experiments/transfections unless stated otherwise. Statistical analysis, two-tailed Student’s t-tests or analysis of variance, was performed as indicated using the standard statistical software, Prism 6 (GraphPad Software, Inc).
Additional Information
How to cite this article: Xie, C. B. et al. Regulation of aldosterone secretion by CaV1.3. Sci. Rep. 6, 24697; doi: 10.1038/srep24697 (2016).
Supplementary Material
Acknowledgments
This work is supported by NIHR Senior Investigator grant NF-SI-0512–10052 awarded to M.J.B.; the Austin Doyle Award (Servier Australia) and the Tunku Abdul Rahman Centenary Fund (St Catharine’s College, Cambridge, UK) awarded to E.A.B.A.; Gates Cambridge Scholarship awarded to C.B.X.; L.H.S., S.G. and C.M. are supported by the British Heart Foundation PhD studentship FS/11/35/28871, FS/14/75/31134 and FS/14/12/30540 respectively; J.Z. was supported by the Cambridge Overseas Trust Scholarship and the Sun Hung Kai Properties-Kwoks’ Foundation; A.E.D.T. is funded by the Agency for Science, Technology & Research (A*STAR) Singapore and Wellcome Trust Award 085686/Z/08/A; LHS, JZ and EABA were further supported by the NIHR Cambridge Biomedical Research Centre; the Human Research Tissue Bank is supported by the NIHR Cambridge Biomedical Research Centre. The CaV1.3 constructs were kindly gifted by Dr. Jöerg Striessnig and Dr Petronel Tuluc.
Footnotes
Author Contributions C.B.X. and L.H.S. designed and performed the experiments on CaV1.3 transfected H295R cells with the help of S.G. E.A.B.A. and L.H.S. designed and performed experiments on non-transfected H295R cells and primary adrenal cells with the help of A.E.D.T. and J.Z. W.Z. performed the immunohistochemistry and E.A.B.A. documented the results. S.K. and R.B.S. designed and provided compound 8. G.T. performed the confocal microscopy. L.H.S. performed the statistical analysis on the data generated. C.M. provided the clinical information of patients. C.B.X., L.H.S., E.A.B.A. and M.J.B. wrote the manuscript.
References
- Funder J. W. et al. Case detection, diagnosis, and treatment of patients with primary aldosteronism: an endocrine society clinical practice guideline. J Clin Endocrinol Metab 93, 3266–3281, doi: 10.1210/jc.2008-0104 (2008). [DOI] [PubMed] [Google Scholar]
- Rossi G. P. et al. A prospective study of the prevalence of primary aldosteronism in 1,125 hypertensive patients. J Am Coll Cardiol 48, 2293–2300, doi: 10.1016/j.jacc.2006.07.059 (2006). [DOI] [PubMed] [Google Scholar]
- Rossi G. P. A comprehensive review of the clinical aspects of primary aldosteronism. Nat Rev Endocrinol 7, 485–495, doi: 10.1038/nrendo.2011.76 (2011). [DOI] [PubMed] [Google Scholar]
- Young W. F. Jr. Minireview: primary aldosteronism–changing concepts in diagnosis and treatment. Endocrinology 144, 2208–2213, doi: 10.1210/en.2003-0279 (2003). [DOI] [PubMed] [Google Scholar]
- Hannemann A. et al. Screening for primary aldosteronism in hypertensive subjects: results from two German epidemiological studies. Eur J Endocrinol 167, 7–15, doi: 10.1530/EJE-11-1013 (2012). [DOI] [PubMed] [Google Scholar]
- Azizan E. A. et al. Somatic mutations in ATP1A1 and CACNA1D underlie a common subtype of adrenal hypertension. Nat Genet 45, 1055–1060, doi: 10.1038/ng.2716 (2013). [DOI] [PubMed] [Google Scholar]
- Scholl U. I. et al. Somatic and germline CACNA1D calcium channel mutations in aldosterone-producing adenomas and primary aldosteronism. Nat Genet 45, 1050–1054, doi: 10.1038/ng.2695 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandes-Rosa F. L. et al. Genetic spectrum and clinical correlates of somatic mutations in aldosterone-producing adenoma. Hypertension 64, 354–361, doi: 10.1161/HYPERTENSIONAHA.114.03419 (2014). [DOI] [PubMed] [Google Scholar]
- Pinggera A. et al. Human CaV1.3 (CACNA1D) calcium channel mutations associated with hyperaldosteronism or autism risk. Program No. 2012.12 2014 Neuroscience Meeting Planner. Washington, DC: Society for Neuroscience. 16th Nov 2014. Available at: http://www.abstractsonline.com/Plan/ViewAbstract.aspx?sKey=5fc23105-4242-4a6b-b284-f1838cc2427e&cKey=4b73446b-97ac-4a41-bd40-78f28c79bbe3&mKey=%7b54C85D94-6D69-4B09-AFAA-502C0E680CA7%7d (Accessed: 7th March 2016).
- Hood S. J., Taylor K. P., Ashby M. J. & Brown M. J. The spironolactone, amiloride, losartan, and thiazide (SALT) double-blind crossover trial in patients with low-renin hypertension and elevated aldosterone-renin ratio. Circulation 116, 268–275, doi: 10.1161/CIRCULATIONAHA.107.690396 (2007). [DOI] [PubMed] [Google Scholar]
- Kang S. et al. CaV1.3-selective L-type calcium channel antagonists as potential new therapeutics for Parkinson’s disease. Nat Commun 3, 1146, doi: 10.1038/ncomms2149 (2012). [DOI] [PubMed] [Google Scholar]
- Kuryshev Y. A., Brown A. M., Duzic E. & Kirsch G. E. Evaluating state dependence and subtype selectivity of calcium channel modulators in automated electrophysiology assays. Assay Drug Dev Technol 12, 110–119, doi: 10.1089/adt.2013.552 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balasubramanian B. et al. Optimization of Ca(v)1.2 screening with an automated planar patch clamp platform. J Pharmacol Toxicol Methods 59, 62–72 (2009). [DOI] [PubMed] [Google Scholar]
- Sinnegger-Brauns M. J. et al. Expression and 1,4-dihydropyridine-binding properties of brain L-type calcium channel isoforms. Mol Pharmacol 75, 407–414, doi: 10.1124/mol.108.049981 (2009). [DOI] [PubMed] [Google Scholar]
- Monticone S. et al. Immunohistochemical, genetic and clinical characterization of sporadic aldosterone-producing adenomas. Mol Cell Endocrinol 411, 146–154, doi: 10.1016/j.mce.2015.04.022 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ortner N. J. et al. Pyrimidine-2,4,6-triones are a new class of voltage-gated L-type Ca2+channel activators. Nat Commun 5, 3897, doi: 10.1038/ncomms4897 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang H. et al. Modest CaV1.342-selective inhibition by compound 8 is beta-subunit dependent. Nat Commun 5, 4481, doi: 10.1038/ncomms5481 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Triggle D. J. Calcium-channel antagonists: mechanisms of action, vascular selectivities, and clinical relevance. Cleve Clin J Med 59, 617–627 (1992). [DOI] [PubMed] [Google Scholar]
- Zhou J. et al. DACH1, a Zona Glomerulosa Selective Gene in the Human Adrenal, Activates Transforming Growth Factor-β Signaling and Suppresses Aldosterone Secretion. Hypertension, doi: 10.1161/hyp.0000000000000025 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaikh L. H. et al. LGR5 activates non-canonical Wnt-signaling and inhibits aldosterone production in the human adrenal. J Clin Endocrinol Metab, jc20151734, doi: 10.1210/jc.2015-1734 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gazdar A. F. et al. Establishment and characterization of a human adrenocortical carcinoma cell line that expresses multiple pathways of steroid biosynthesis. Cancer Res 50, 5488–5496 (1990). [PubMed] [Google Scholar]
- Kojima K., Kojima I. & Rasmussen H. Dihydropyridine calcium agonist and antagonist effects on aldosterone secretion. Am J Physiol 247, E645–650 (1984). [DOI] [PubMed] [Google Scholar]
- Clark R. E. et al. Laboratory and initial clinical studies of nifedipine, a calcium antagonist for improved myocardial preservation. Ann Surg 193, 719–732 (1981). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickerson J. E., Hingorani A. D., Ashby M. J., Palmer C. R. & Brown M. J. Optimisation of antihypertensive treatment by crossover rotation of four major classes. Lancet 353, 2008–2013 (1999). [DOI] [PubMed] [Google Scholar]
- Brown M. J. et al. Morbidity and mortality in patients randomised to double-blind treatment with a long-acting calcium-channel blocker or diuretic in the International Nifedipine GITS study: Intervention as a Goal in Hypertension Treatment (INSIGHT). Lancet 356, 366–372, doi: 10.1016/S0140-6736(00)02527-7 (2000). [DOI] [PubMed] [Google Scholar]
- Brown M. J. & Hopper R. V. Calcium-channel blockade can mask the diagnosis of Conn’s syndrome. Postgrad Med J 75, 235–236 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown M. J., McInnes G. T., Papst C. C., Zhang J. & MacDonald T. M. Aliskiren and the calcium channel blocker amlodipine combination as an initial treatment strategy for hypertension control (ACCELERATE): a randomised, parallel-group trial. Lancet 377, 312–320, doi: 10.1016/S0140-6736(10)62003-X (2011). [DOI] [PubMed] [Google Scholar]
- Rossier M. F., Ertel E. A., Vallotton M. B. & Capponi A. M. Inhibitory action of mibefradil on calcium signaling and aldosterone synthesis in bovine adrenal glomerulosa cells. J Pharmacol Exp Ther 287, 824–831 (1998). [PubMed] [Google Scholar]
- Glaser S., Steinbach M., Opitz C., Wruck U. & Kleber F. X. Torsades de pointes caused by Mibefradil. Eur J Heart Fail 3, 627–630 (2001). [DOI] [PubMed] [Google Scholar]
- Uebele V. N., Nuss C. E., Renger J. J. & Connolly T. M. Role of voltage-gated calcium channels in potassium-stimulated aldosterone secretion from rat adrenal zona glomerulosa cells. J Steroid Biochem Mol Biol 92, 209–218, doi: 10.1016/j.jsbmb.2004.04.012 (2004). [DOI] [PubMed] [Google Scholar]
- Lotshaw D. P. Role of membrane depolarization and T-type Ca2+channels in angiotensin II and K+stimulated aldosterone secretion. Mol Cell Endocrinol 175, 157–171 (2001). [DOI] [PubMed] [Google Scholar]
- Rossier M. F. et al. Blocking T-type calcium channels with tetrandrine inhibits steroidogenesis in bovine adrenal glomerulosa cells. Endocrinology 132, 1035–1043, doi: 10.1210/endo.132.3.8382595 (1993). [DOI] [PubMed] [Google Scholar]
- Amar L. et al. Aldosterone synthase inhibition with LCI699: a proof-of-concept study in patients with primary aldosteronism. Hypertension 56, 831–838, doi: 10.1161/HYPERTENSIONAHA.110.157271 (2010). [DOI] [PubMed] [Google Scholar]
- Zuccotti A. et al. Structural and functional differences between L-type calcium channels: crucial issues for future selective targeting. Trends Pharmacol Sci 32, 366–375, doi: 10.1016/j.tips.2011.02.012 (2011). [DOI] [PubMed] [Google Scholar]
- Parthasarathy H. K. et al. A double-blind, randomized study comparing the antihypertensive effect of eplerenone and spironolactone in patients with hypertension and evidence of primary aldosteronism. J Hypertens 29, 980–990, doi: 10.1097/HJH.0b013e3283455ca5 (2011). [DOI] [PubMed] [Google Scholar]
- Kremer D. et al. Amiloride in the treatment of primary hyperaldosteronism and essential hypertension. Clin Endocrinol (Oxf) 7, 151–157 (1977). [DOI] [PubMed] [Google Scholar]
- Clark E. Spironolactone Therapy and Gynecomastia. JAMA 193, 163–164 (1965). [DOI] [PubMed] [Google Scholar]
- Sussman R. M. Spironolactone and gynaecomastia. Lancet 1, 58 (1963). [DOI] [PubMed] [Google Scholar]
- Wehling M. et al. Rapid cardiovascular action of aldosterone in man. J Clin Endocrinol Metab 83, 3517–3522, doi: 10.1210/jcem.83.10.5203 (1998). [DOI] [PubMed] [Google Scholar]
- Funder J. W. Minireview: aldosterone and the cardiovascular system: genomic and nongenomic effects. Endocrinology 147, 5564–5567, doi: 10.1210/en.2006-0826 (2006). [DOI] [PubMed] [Google Scholar]
- Lee G. et al. Homeostatic responses in the adrenal cortex to the absence of aldosterone in mice. Endocrinology 146, 2650–2656, doi: 10.1210/en.2004-1102 (2005). [DOI] [PubMed] [Google Scholar]
- Gomez-Sanchez C. E. et al. Development of monoclonal antibodies against human CYP11B1 and CYP11B2. Mol Cell Endocrinol 383, 111–117, doi: 10.1016/j.mce.2013.11.022 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.