

Abstract
Background
Cardiovascular risk and plasma total homocysteine (tHcy) are high in patients with renal failure. High tHcy may account for a substantial part of the increased risk. We assessed mediation by tHcy of the association of estimated glomerular filtration rate (eGFR CKD/EPI) with carotid total plaque area (TPA) and carotid stenosis.
Methods
TPA and carotid stenosis were measured by ultrasound. Multiple linear regression was used to assess the effects of eGFR and/or tHcy after adjustment for age, sex, systolic blood pressure (SBP), smoking, LDL, HDL and weight.
Results
Complete data were available for 1967 patients. eGFR decreased, and TPA and total stenosis increased linearly with age. After adjustment [age, sex, SBP, smoking (in pack years), low-density lipoprotein (LDL), high-density lipoprotein (HDL) and weight], eGFR and tHcy were independently associated with TPA (P < 0.01), but when both were added to the model, their significance was attenuated (P = 0.06 for eGFR, 0.03 for tHcy). Mediation analysis showed that tHcy seems to contribute to a significant mediation of the association of eGFR with TPA but not stenosis; after adjustment for the set of risk factors listed above, tHcy still demonstrated significant mediation on TPA (P = 0.03), but not on stenosis (P = 0.16).
Conclusions
tHcy accounts for a significant part, but not all of the effect of renal impairment on atherosclerosis. Other uremic toxins including metabolic products of the intestinal microbiome may explain residual effects of renal failure on atherosclerosis. Therapeutic approaches arising from that hypothesis are discussed.
Keywords: ADMA, cardiovascular risk, indoles, intestinal microbiome, TMAO
INTRODUCTION
Cardiovascular risk is markedly increased in patients with renal failure. Wheeler indicated in 1996 [1] that their risk of cardiovascular events was increased ∼17-fold. A more recent analysis [2] showed that life expectancy from age 55 declines from 19.9 years with normal or only slightly impaired renal function to 5.6 years with severe renal impairment [estimated glomerular filtration rate (eGFR) <15 mL/min/1.73 m² or renal replacement therapy].
A strong candidate for a role in the relationship between renal impairment and cardiovascular risk is elevation of plasma total homocysteine (tHcy) [3]. In patients on hemodialysis, levels of tHcy are markedly elevated (to ∼30 µmol/L) and do not respond to therapy with folic acid [4]. Increased tHcy is a risk factor for cardiovascular disease, particularly stroke [3]. Although it is widely believed that B vitamin therapy to lower tHcy does not reduce the risk of cardiovascular disease [5], it is now apparent that key issues of adequate B12 dosing and impaired renal function obscured the effect of vitamin therapy [6], and lowering levels of tHcy does indeed appear to reduce the risk of stroke, particularly among patients with normal renal function [7]. This has been substantiated in recent meta-analyses [8, 9]. A recent large trial in China showed a significant reduction of stroke in primary prevention with folic acid [10].
Previous reports from this study population have shown that carotid stenosis and carotid plaque burden [measured as total plaque area (TPA)] are biologically distinct phenotypes [11, 12], affected differentially by some cardiovascular risk factors. Plaque progression may be related to factors such as elevated low-density lipoprotein (LDL) cholesterol, oxidative stress and impaired reverse cholesterol transport, whereas stenosis may be related more to factors predisposing to plaque rupture such as inflammation and matrix metalloproteinase activity, and to thrombosis at the site of plaque rupture. As an example, lipoprotein (a) is associated with stenosis, but not plaque burden [13]. It has been suggested that this may be due to increased thrombosis after plaque rupture [13, 14].
In this study we explored the relationship of eGFR calculated by CKD/EPI equations and plasma tHcy as two key independent/explanatory variables, to carotid plaque burden and carotid stenosis as two dependent variables. The purpose was to assess to what extent elevated levels of tHcy may contribute to an effect of renal impairment on atherosclerosis burden and stenosis.
MATERIALS AND METHODS
Study population
The study was conducted from the clinical database of the Stroke Prevention & Atherosclerosis Research Centre, Robarts Research Institute, London, Ontario. Patients in the database were referred to one of several clinics conducted at University Hospital, London, Ontario: a Stroke Prevention Clinic, an Urgent TIA Clinic and a Premature Atherosclerosis Clinic. The protocol was approved by the Human Subjects Research Ethics Board of Western University. All the biochemical analytes included in the analyses were measured in the Biochemistry Department of the London Health Sciences Centre, using standard methods. We excluded patients who had creatinine >300 or <15 mmol/L, because patients with very high serum creatinine do not respond to vitamin therapy for homocysteine, and because serum creatinine of <15 mmol/L seemed unlikely to be valid. We also excluded patients with tHcy of >40 μmol/L, because such high levels are likely to be due to hereditary causes rather than to renal function. Serum creatinine was measured using Roche Modular Chemistry analyzer, using the CREA plus diagnostic kit. This method is based on enzymatic determination of creatinine and is standardized against ID-MS. eGFR was calculated from CKD/EPI equations because of superior performance in older white patients (most of our study population) [15].
Ultrasound methods
Total plaque area was measured by two registered vascular ultrasound technologists using a high-resolution ultrasound scanner as previously described [16]. An Advanced Technology Laboratories (ATL) Mark 9 was used before 2000 and an ATL HDI 5000 thereafter (Phillips, Bothell, WA). The technologists scanned along the length of the right and left common, internal and external carotid arteries between the angle of the jaw and the clavicle. They then determined the largest extent of each plaque present and traced the outline of each plaque in a longitudinal view with a cursor. For branches that were occluded, the entire cross-sectional area of the branch was regarded as occupied by plaque. A microprocessor in the machine computed the TPA for each plaque; summing the individual plaque areas yielded the TPA. Intra- and inter-observer reliability measured by intraclass correlation coefficient was 0.94 and 0.85, respectively [16]. We and others have shown that TPA is a strong predictor of cardiovascular events [16], and a stronger predictor of risk than carotid intima-media thickness [17, 18].
Total carotid stenosis was defined as the sum of the percent stenosis in the right and left internal carotid arteries; the upper limit of total stenosis (for a patient with bilateral carotid occlusion) was thus 200%. Stenosis was measured by Doppler peak frequency shift before 2003 and Doppler peak velocity after 2003 and was calibrated angiographically from 100 angiograms (200 arteries) measured in the North American Symptomatic Carotid Endarterectomy Trial [19]. Carotid occlusion was defined by absence of flow on Doppler ultrasound with color flow.
Statistical methods
We summarized continuous variables by mean and standard deviation, and categorical variables by frequency and percent.
To study the relationship among variables, we used linear regression using all available (i.e. non-missing) data. The two study outcomes (TPA and stenosis) were modeled separately with the same set of covariates including the exposure of interest (eGFR) and a potential mediator (tHcy). We fitted three regression models: Model 1 has eGFR in the model, Model 2 has tHcy in the model and Model 3 has both eGFR and tHcy in the model, whereas all models are adjusted for age, sex, systolic blood pressure (SBP), smoking (in pack years), LDL, high-density lipoprotein (HDL) and weight. We also fitted and present unadjusted models. We examined the regression coefficient, standard error, P-value and R-square from each regression model. Here, we log-transformed TPA and homocysteine due to severe skewness, but not stenosis and eGFR.
We visualized the association of key independent and dependent variables by age group and sex. Associations of some key variables were also visualized via scatter plot with LOcal regrESSion (LOESS) fit and assessed by correlation coefficient. Since Pearson (linear) and Spearman (rank-based) correlation coefficients were very similar, we decided to report only the latter.
In addition, we conducted a mediation analysis. The relationship between the independent variable and the dependent variable is hypothesized to be an indirect effect that exists due to the influence of a mediator. We used the Sobel test, basically a specialized t-test that provides a method to determine whether the reduction in the effect of the independent variable, after including the mediator in the model, is statistically significant [20, 21].
SAS 9.3 was used for data analysis (SAS institute, Cary, NC). All P-values are two-sided.
RESULTS
There were 3967 patients in the database with data on eGFR, TPA and stenosis; of those, 1967 patients had complete data for the adjusted models; characteristics are shown in Table 1.
Table 1.
Characteristics of the study population (n = 1967a)
| Continuous variables | N | Mean | Standard deviation |
|---|---|---|---|
| Age (years) | 1967 | 64.8 | 14.1 |
| Weight (kg) | 1886 | 79.5 | 19.0 |
| Systolic blood pressure (mmHg) | 1962 | 143.8 | 22.0 |
| Diastolic blood pressure (mmHg) | 1962 | 81.8 | 13.0 |
| Total cholesterol (mmol/L) | 1828 | 4.72 | 1.21 |
| Triglycerides (mmol/L) | 1828 | 1.80 | 1.21 |
| HDL cholesterol (mmol/L) | 1822 | 1.34 | 0.43 |
| LDL cholesterol (mmol/L) | 1775 | 2.59 | 1.02 |
| Serum creatinine (mmol/L) | 1967 | 83.9 | 25.7 |
| eGFR (mL/min per 1.73 m2) | 1967 | 77.4 | 20.8 |
| Serum B12 (pmol/L) | 1671 | 332.1 | 180.1 |
| Plasma total homocysteine (µmol/L) | 1967 | 11.1 | 4.49 |
| Smoking (pack years) | 1950 | 15.2 | 20.5 |
| Stenosis % total (right and left ICA) | 1965 | 75.9 | 28.5 |
| TPA (mm2) | 1967 | 123 | 133 |
| Categorical variables | N | % | |
| Male | 1967 | 995 | 50.6 |
| Diabetic | 1957 | 368 | 18.8 |
| Smoking status | 1967 | ||
| Never | 766 | 38.9 | |
| Quit | 830 | 42.2 | |
| Still smoking | 371 | 18.9 |
We excluded patients with creatinine of >300 or of <15 mmol/L and patients with homocysteine of >40 μmol/L.
If B12 was >1000 pmol/L, we replaced the value by 1000. eGFR was derived from CKD/EPI formulae; ICA, internal carotid artery.
aNumber of patients with variables used in the regression models.
As shown in Figure 1, eGFR declined, and TPA and total carotid stenosis (the sum of % stenosis in the right and left internal carotid artery) increased approximately linearly with age in both sexes. In the LOESS, fit between log tHcy and eGFR was stronger (Spearman's R = −0.43, P < 0.001) than for log tHcy and serum B12 (R = −0.19, P < 0.001) (Figure 2). Strong nonlinear fit by LOESS is notable with eGFR and log TPA (R = −0.41, P < 0.001), and with log tHcy and log TPA (R = 0.28, P < 0.001) (Figure 3).
FIGURE 1:
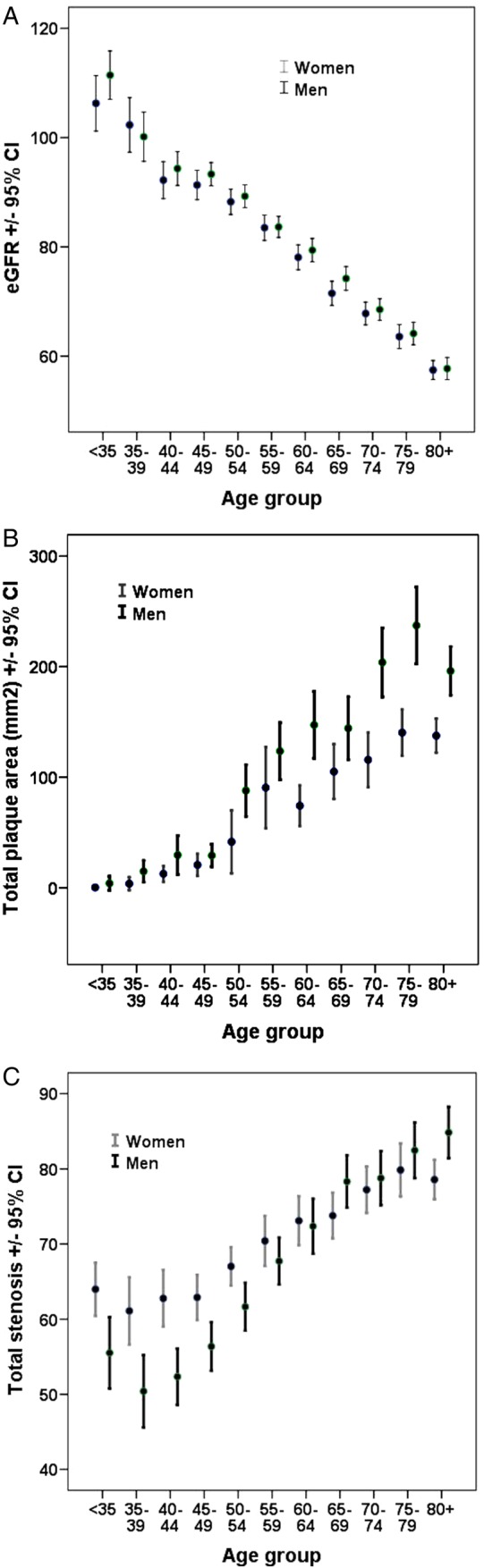
eGFR, carotid TPA and total stenosis by age and sex. With age, eGFR (CKD/EPI) (A) declines, and carotid TPA (mm2) (B) and total stenosis (the sum of % stenosis in the right and left internal carotid arteries, C) increase, in approximately linear fashion (n = 3967).
FIGURE 2:

LOESS of eGFR and tHcy serum B12. A stronger fit by LOESS is noted with tHcy and eGFR (Spearman R = −0.43, P < 0.001) (A) than with tHcy and serum B12 (Spearman R = −0.19, P < 0.001) (B). Although in the setting of folate fortification serum B12 is often considered the predominant determinant of plasma tHcy, it is more strongly correlated with eGFR. Impaired renal function should be taken into consideration in studies of tHcy and vascular disease. tHcy is in log scale.
FIGURE 3:

LOESS of eGFR and tHcy with TPA. Strong nonlinear fit by LOESS is notable in eGFR and TPA (Spearman R = −0.41, P < 0.001) (A), and a less strong fit of tHcy and TPA (Spearman R = 0.28, P < 0.001) (B). TPA and tHcy are in log scale.
When modeled separately, adjusted for age, sex, SBP, smoking (in pack years), LDL cholesterol, HDL cholesterol and weight, both eGFR and tHcy were significant predictors of baseline TPA, with tHcy being a somewhat stronger predictor (P = 0.006 versus 0.003). When both were added to the model, the magnitude of effect of each was reduced, i.e. tHcy was still significant (P = 0.03) but creatinine was only borderline significant (P = 0.06) (Table 2). After adjustment for the same set of covariates, eGFR and tHcy were weakly associated with total stenosis when they were modeled separately. When both were added to the model both became non-significant (Table 2). Notably, the variability in the outcome explained by the set of covariates included in the regression model, measured by R-square, was 44% for TPA and 6% for stenosis; thus, substantially higher model determination or predictability is demonstrated for TPA.
Table 2.
Association of eGFR versus homocysteine with vascular outcomes
| Beta (SE) | P-value | |
|---|---|---|
| A. Outcome = TPA | ||
| Unadjusted regression (N = 1967) | ||
| Model 1 (R2 = 0.17) | ||
| eGFR | −0.03 (0.002) | <0.0001 |
| Model 2 (R2 = 0.08) | ||
| tHcy | 1.24 (0.10) | <0.0001 |
| Model 3 (R2 = 0.18) | ||
| eGFR | −0.03 (0.002) | <0.0001 |
| tHcy | 0.51 (0.10) | <0.0001 |
| Adjusted regression (N = 1690) | ||
| Model 1 (R2 = 0.44) | ||
| eGFR | −0.005 (0.002) | 0.006 |
| Model 2 (R2 = 0.44) | ||
| tHcy | 0.26 (0.09) | 0.003 |
| Model 3 (R2 = 0.44) | ||
| eGFR | −0.004 (0.002) | 0.06 |
| tHcy | 0.20 (0.09) | 0.03 |
| B. Outcome = stenosis | ||
| Unadjusted regression (N = 1965) | ||
| Model 1 (R2 = 0.02) | ||
| eGFR | −0.17 (0.03) | <0.0001 |
| Model 2 (R2 = 0.01) | ||
| tHcy | 5.99 (1.67) | 0.0003 |
| Model 3 (R2 = 0.02) | ||
| eGFR | −0.15 (0.03) | <0.0001 |
| tHcy | 2.38 (1.86) | 0.20 |
| Adjusted regression (N = 1688) | ||
| Model 1 (R2 = 0.06) | ||
| eGFR | −0.08 (0.04) | 0.08 |
| Model 2 (R2 = 0.06) | ||
| tHcy | 3.61 (1.88) | 0.06 |
| Model 3 (R2 = 0.06) | ||
| eGFR | −0.05 (0.04) | 0.23 |
| tHcy | 2.80 (2.00) | 0.16 |
Carotid plaque burden is measured by TPA and total stenosis is measured by the sum of % stenosis in the right and left internal carotid arteries. TPA and tHcy, not eGFR and stenosis, are in log scale due to skewness. Model 1 has eGFR in the model, Model 2 has tHcy in the model and Model 3 has both eGFR and tHcy in the model. Adjusted models are adjusted for age, sex, SBP, smoking (in pack years), LDL, HDL and weight.
The Spearman's rank correlation coefficient of eGFR and TPA was −0.41 and that of tHcy and TPA was 0.28 (P < 0.0001 for both) but partial correlation coefficient controlling for the other covariates was −0.09 and 0.07 (P = 0.0002 and 0.003, respectively; results not in tables). Numerically different but qualitatively similar results were obtained in the regression analyses; see above and Table 2.
A mediation analysis showed that tHcy determined a significant proportion of the effect of eGFR (Table 3) on TPA but not total stenosis (P < 0.0001 and 0.20, respectively, by the Sobel test). When we adjusted for the same panel of covariates listed earlier, tHcy determined a significant proportion of the effect of eGFR on TPA (0.03), but not stenosis (P = 0.16). The proportion of the total effect mediated by tHcy was estimated to be 12 and 31% in these analyses.
Table 3.
Detecting and quantifying mediating effect of homocysteine in the association between eGFR as independent variable and vascular outcomes
| P-value | % of total effect mediated | Ratio of the indirect to the direct effect | |
|---|---|---|---|
| Unadjusted regression | |||
| Outcome = TPA | <0.0001 | 0.12 | 0.14 |
| Outcome = stenosis | 0.20 | 0.12 | 0.13 |
| Adjusted regression | |||
| Outcome = TPA | 0.03 | 0.31 | 0.44 |
| Outcome = stenosis | 0.16 | 0.31 | 0.45 |
TPA is in log scale.
Unadjusted regression included three covariates (exposure, mediator and outcome) in the model.
Adjusted regression further included age, sex, SBP, smoking (in pack years), LDL, HDL and weight in the model.
DISCUSSION
We found that eGFR was associated with both TPA and stenosis and that this association was partly and significantly mediated by tHcy. Addition of tHcy to the multiple regression model substantially reduced the association between eGFR and plaque area, and both became non-significant when modeled together for stenosis.
Strengths of the study are the relatively large number of patients and measurement of carotid plaque burden in addition to stenosis. The principal weakness is that the study is cross-sectional.
Vitamin B12 is thought to be the main determinant of tHcy since folate fortification of the grain supply in North America [22]; however, it is seldom recognized that renal function could be a more important determinant of tHcy, as indicated in Figure 2. Thus, by increasing the levels of tHcy and other metabolites discussed below, the decline in renal function with age, as shown in Figure 1A, is an important aspect of accelerating cardiovascular risk with age, and of plaque progression with age, as shown in Figure 1B. Spence reported [23] that among patients attending a stroke prevention clinic, the proportion with tHcy of ≥14 µmol/L rose from 20% below age 60, to >40% above age 80. This has importance particularly for the risk of atrial fibrillation with aging: in the Framingham study [24], strokes attributable to atrial fibrillation increased from 1.5 to 23.5% between age 50 and age 80–89 and elevated tHcy quadruples the risk of stroke in atrial fibrillation [25].
When modeled separately or jointly, without or with accounting for other risk factors, we could not find consistent evidence for which of tHcy versus eGFR is a stronger predictor of the outcomes. This suggests the relative importance of eGFR versus tHcy is difficult to judge but it is clear that their roles should be understood together and with other risk factors. This raises interesting questions about what other metabolites might be of importance in the effect of renal failure on atherosclerosis. For example, there may be other metabolites that are elevated in renal failure that predispose more to stenosis than to plaque burden, by increasing plaque rupture and/or thrombosis. What might these be? Gansevoort et al. [2] discussed many of the mechanisms by which chronic renal failure increases cardiovascular risk, including a high prevalence of traditional cardiovascular risk factors, left ventricular hypertrophy, dyslipidemia, chronic inflammation, activation of the renin-angiotensin system, hyperphosphatemia and increased levels of asymmetric dimethylarginine (ADMA). Here we focus on possible candidates that may lead to approaches to therapy in the context of end-stage renal disease.
ADMA
As mentioned by Gansevoort et al. [2], one candidate culprit is ADMA, an antagonist of nitric oxide that is often elevated in parallel with tHcy [26], but has effects independent of tHcy [27]. Besides increasing thrombosis, tHcy has additional adverse effects on endothelial function [28], but it has been suggested that this may be mediated by ADMA [29]. Wilcox [30] reviewed the relationship of renal impairment, reactive oxygen species (ROS) and ADMA, pointing out that ADMA is generated by ROS, to which hyperhomocysteinemia contributes, and ADMA is cleared from plasma primarily by renal elimination. Potter et al. [31] found that adjusting for renal function eliminated the effect of tHcy on flow-mediated vasodilation and carotid intima-media thickness.
Thiocyanate
Another candidate culprit is thiocyanate. Vitamin therapy with cyanocobalamin to lower tHcy reduced the risk of stroke in a subgroup of the Vitamin Intervention for Stroke Prevention (VISP) trial from which patients with a eGFR in the lowest decile (<47 mL/min/1.73 m2) were excluded but was harmful among patients with diabetic nephropathy, in the Diabetic Intervention with Vitamins in Nephropathy (DIVINe) trial. In patients with diabetic nephropathy, high doses of B vitamins including cyanocobalamin 1000 μg daily accelerated the decline of eGFR and doubled the risk of vascular events [32]. All the events occurred among patients with a eGFR of <50 mL/min/1.73 m2 [33]. A possible mechanism for toxicity of high-dose B vitamins in patients with renal failure, accumulation of thiocyanate from cyanocobalamin, was also suggested [6].
Koyama et al. [34] showed that dialysis patients accumulate cyanide in the form of thiocyanate from cyanocobalamin; they also showed in dialysis patients [35] that methylcobalamin lowered levels of both tHcy and ADMA. In contrast, Løland et al. [36] found that cyanocobalamin did not lower levels of ADMA in the Western Norway B Vitamin Intervention Trial. Similarly, hydroxycobalamin, but not cyanocobalamin, is effective in alcohol/tobacco amblyopia, a condition in which cyanide plays a key role [37]. Therefore, in patients with renal failure, methylcobalamin may be superior to cyanocobalamin. A possible connection between cyanide and vascular disease is that cyanide consumes hydrogen sulfide in its elimination as thiocyanate, and hydrogen sulfide is a recently recognized endothelium-derived relaxing factor, analogous to nitric oxide [38]. Related issues were reviewed in 2012 by Perna and Ingrosso [39]. Thiocyanate also catalyzes oxidation of LDL cholesterol [40] and is thought to be important in atherosclerosis [41].
Trimethylamine N-oxide (TMAO)
Recent understanding of the role of the intestinal microbiome in nutrition and cardiovascular disease [42] points to trimethylamine and its oxidative product, TMAO as likely culprits. Uremic patients have high plasma levels of trimethylamine, thought to account for their fishy breath odor [43]. Both lecithin [44] (from egg yolk and other sources) and l-carnitine [45] (from animal flesh, particularly red meat) are converted by the intestinal microbiome to trimethylamine, which undergoes hepatic oxidation to TMAO. TMAO promotes atherosclerosis in animal models [44, 45]. In patients referred for coronary angiography, after a test dose of two hard-boiled eggs, levels of TMAO in the top quartile increased the 3-year risk of stroke, myocardial infarction or death 2.5-fold [46].
Levels of TMAO are markedly elevated among patients with renal failure [47], and among patients with renal failure, levels of TMAO in the top quartile increase cardiovascular mortality 1.93-fold after adjustment for other risk factors [47]. In a murine model, TMAO led to progressive renal tubulointerstitial fibrosis and dysfunction [47].
Because of intestinal metabolism of lecithin and carnitine to trimethylamine, patients with renal failure should probably limit their intake of egg yolk and red meat [48]. However, l-carnitine has some beneficial effects on glucose and lipid metabolism [49], so further research in this area is needed.
Other uremic toxins produced by the intestinal microbiome
Indoxyl sulfate is a gut-derived uremic toxin derived from the metabolism of dietary tryptophan. As kidney function declines, there is a gradual increase in the concentration of plasma indoxyl sulfate [50, 51]. Similar to tHcy, indoxyl sulfate is a highly protein bound uremic toxin so clearance by dialysis is minimal. Indoxyl sulfate promotes the generation of ROS in endothelial cells [52] and likely plays a role in the endothelial dysfunction observed in uremic patients [53]. Clinical studies have confirmed the contribution of indoxyl sulfate to cardiovascular disease in patients with decreased renal function. Indoxyl sulfate concentration is associated with aortic calcification, vascular stiffness and overall and cardiovascular mortality [54]. Indole 3-acetic acid is another protein-bound uremic toxin derived from the metabolism of dietary tryptophan. Similar to indoxyl sulfate, indole 3-acetic acid causes an increase in ROS and is a predictor of major cardiovascular events and mortality in patients with compromised renal function [55]. Serum p-cresyl sulfate is another gut-derived, protein-bound uremic toxin that is derived from metabolism of dietary tyrosine. Similar to indoles, p-cresyl sulfate has also been implicated in mediating the cardiovascular complications consistently observed in patients with decreased renal function. Studies have demonstrated that p-cresyl sulfate induces oxidative stress in endothelial cells [56]. Multiple clinical investigations have linked elevated levels of p-cresyl sulfate with cardiovascular and all-cause mortality in patients with chronic kidney disease [57].
Possible clinical implications
Possible clinical implications of these hypotheses are that the very high cardiovascular risk of dialysis patients might be reduced by several maneuvers: treatment with thiols such as mesna to lower tHcy [58], dietary restriction of l-carnitine from red meat and phosphatidylcholine from egg yolks to reduce levels of TMAO [42] and overnight daily dialysis, which normalizes levels of tHcy [59]. It seems likely that daily dialysis may also reduce levels of ADMA, as well as TMAO and other metabolic products of the intestinal microbiome. Besides dialysis, approaches based on absorption of bacterial metabolic products are another possibility.
AST-120 is a non-absorbable oral adsorbent made of high purity porous carbon. It functions by adsorbing precursors of uremic toxins (e.g. indole) in the intestine, decreasing their absorption and promoting excretion into the feces. AST-120 has been shown to decrease serum concentration of indoxyl sulfate, p-cresyl sulfate and ROS in patients with chronic kidney disease. AST-120 has been used in Japan for years to slow the progression of kidney disease. Although a recent study showed no beneficial effect on progression of kidney disease in North American patients [60], randomized trials to evaluate the effect of AST-120 on cardiovascular complications in patients with kidney disease are warranted.
Because of the very high cardiovascular risk in patients with renal failure, all of these potential approaches may be worthy of further study. Of particular interest would be clinical trials in patients with Grade 4 and 5 renal failure.
CONCLUSION
The effect of renal dysfunction on carotid plaque burden and stenosis is only partly explained by levels of tHcy. There are a number of other likely candidates to explain the high risk of cardiovascular disease in renal failure, and therapeutic approaches to these toxins are being developed. Clinical trials should be carried out to test the therapeutic hypotheses arising from these findings.
CONFLICTS OF INTEREST STATEMENT
None declared.
(See related article by Perna and Ingrosso. Atherosclerosis determinants in renal disease: how much is homocysteine involved? Nephrol Dial Transplant 2016; 31: 860–863)
ACKNOWLEDGEMENTS
H.B. was partly supported by the National Center for Advancing Translational Sciences, National Institutes of Health, through grant number UL1 TR000002. This paper was presented at the February 2015 meeting of the American Society for Arteriosclerosis, Thrombosis and Vascular Biology, and published as an abstract of that meeting.
REFERENCES
- 1.Wheeler DC. Cardiovascular disease in patients with chronic renal failure. Lancet 1996; 348: 1673–1674 [DOI] [PubMed] [Google Scholar]
- 2.Gansevoort RT, Correa-Rotter R, Hemmelgarn BR et al. Chronic kidney disease and cardiovascular risk: epidemiology, mechanisms, and prevention. Lancet 2013; 382: 339–352 [DOI] [PubMed] [Google Scholar]
- 3.Spence JD. Homocysteine-lowering therapy: a role in stroke prevention? Lancet Neurol 2007; 7: 830–838 [DOI] [PubMed] [Google Scholar]
- 4.Spence JD, Cordy P, Kortas C et al. Effect of usual doses of folate supplementation on elevated plasma homocyst(e)ine in hemodialysis patients: No difference between 1 and 5 mg daily. Am J Nephrol 1999; 18: 405–410 [DOI] [PubMed] [Google Scholar]
- 5.Rodionov RN, Lentz SR. The homocysteine paradox. Arterioscler Thromb Vasc Biol 2008; 28: 1031–1033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Spence JD, Stampfer MJ. Understanding the complexity of homocysteine lowering with vitamins: the potential role of subgroup analyses. JAMA 2011; 306: 2610–2611 [DOI] [PubMed] [Google Scholar]
- 7.Spence JD. B vitamin therapy for homocysteine: renal function and vitamin B12 determine cardiovascular outcomes. Clin Chem Lab Med 2013; 51: 633–637 [DOI] [PubMed] [Google Scholar]
- 8.Huang T, Chen Y, Yang B et al. Meta-analysis of B vitamin supplementation on plasma homocysteine, cardiovascular and all-cause mortality. Clin Nutr 2012; 31: 448–454 [DOI] [PubMed] [Google Scholar]
- 9.Ji Y, Tan S, Xu Y et al. Vitamin B supplementation, homocysteine levels, and the risk of cerebrovascular disease: a meta-analysis. Neurology 2013; 81: 1298–1307 [DOI] [PubMed] [Google Scholar]
- 10.Huo Y, Li J, Qin X et al. Efficacy of folic acid therapy in primary prevention of stroke among adults with hypertension in China: the CSPPT randomized clinical trial. JAMA 2015; 313: 1325–1335 [DOI] [PubMed] [Google Scholar]
- 11.Spence JD, Hegele RA. Noninvasive phenotypes of atherosclerosis. Arterioscler Thromb Vasc Biol 2004; 24: e188–e189 [DOI] [PubMed] [Google Scholar]
- 12.Al Shali K, House AA, Hanley AJ et al. Differences between carotid wall morphological phenotypes measured by ultrasound in one, two and three dimensions. Atherosclerosis 2005; 178: 319–325 [DOI] [PubMed] [Google Scholar]
- 13.Klein JH, Hegele RA, Hackam DG et al. Lipoprotein(a) is associated differentially with carotid stenosis, occlusion, and total plaque area. Arterioscler Thromb Vasc Biol 2008; 28: 1851–1856 [DOI] [PubMed] [Google Scholar]
- 14.Spence JD, Koschinsky M. Mechanisms of lipoprotein(a) pathogenicity: prothrombotic, proatherosclerotic, or both? Arterioscler Thromb Vasc Biol 2012; 32: 1550–1551 [DOI] [PubMed] [Google Scholar]
- 15.Koppe L, Klich A, Dubourg L et al. Performance of creatinine-based equations compared in older patients. J Nephrol 2013; 26: 716–723 [DOI] [PubMed] [Google Scholar]
- 16.Spence JD, Eliasziw M, DiCicco M et al. Carotid plaque area: a tool for targeting and evaluating vascular preventive therapy. Stroke 2002; 33: 2916–2922 [DOI] [PubMed] [Google Scholar]
- 17.Johnsen SH, Mathiesen EB. Carotid plaque compared with intima-media thickness as a predictor of coronary and cerebrovascular disease. Curr Cardiol Rep 2009; 11: 21–27 [DOI] [PubMed] [Google Scholar]
- 18.Mathiesen EB, Johnsen SH, Wilsgaard T et al. Carotid plaque area and intima-media thickness in prediction of first-ever ischemic stroke: a 10-year follow-up of 6584 men and women: the Tromso Study. Stroke 2011; 42: 972–978 [DOI] [PubMed] [Google Scholar]
- 19.Barnett HJM, Taylor DW, Eliasziw M et al. Benefit of carotid endarterectomy in patients with symptomatic moderate or severe carotid stenosis. N Engl J Med 1998; 339: 1415–1425 [DOI] [PubMed] [Google Scholar]
- 20.Sobel ME. Asymptotic confidence intervals for indirect effects in structural equation models. Sociol Methodol 1982; 13: 290–312 [Google Scholar]
- 21.MacKinnon DP, Dwyer JH. Estimating mediated effects in prevention studies. Eval Rev 1993; 17: 144–158 [Google Scholar]
- 22.Robertson J, Iemolo F, Stabler SP et al. Vitamin B12, homocysteine and carotid plaque in the era of folic acid fortification of enriched cereal grain products. CMAJ 2005; 172: 1569–1573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Spence JD. Mechanisms of thrombogenesis in atrial fibrillation. Lancet 2009; 373: 1006. [DOI] [PubMed] [Google Scholar]
- 24.Wolf PA, Abbott RD, Kannel WB. Atrial fibrillation as an independent risk factor for stroke: the Framingham Study. Stroke 1991; 22: 983–988 [DOI] [PubMed] [Google Scholar]
- 25.Poli D, Antonucci E, Cecchi E et al. Culprit factors for the failure of well-conducted warfarin therapy to prevent ischemic events in patients with atrial fibrillation: the role of homocysteine. Stroke 2005; 36: 2159–2163 [DOI] [PubMed] [Google Scholar]
- 26.Boger RH, Bode-Boger SM, Sydow K et al. Plasma concentration of asymmetric dimethylarginine, an endogenous inhibitor of nitric oxide synthase, is elevated in monkeys with hyperhomocyst(e)inemia or hypercholesterolemia. Arterioscler Thromb Vasc Biol 2000; 20: 1557–1564 [DOI] [PubMed] [Google Scholar]
- 27.Rodionov RN, Dayoub H, Lynch CM et al. Overexpression of dimethylarginine dimethylaminohydrolase protects against cerebral vascular effects of hyperhomocysteinemia. Circ Res 2010; 106: 551–558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lentz SR. Mechanisms of homocysteine-induced atherothrombosis. J Thromb Haemost 2005; 3: 1646–1654 [DOI] [PubMed] [Google Scholar]
- 29.Boger RH, Lentz SR, Bode-Boger SM et al. Elevation of asymmetrical dimethylarginine may mediate endothelial dysfunction during experimental hyperhomocyst(e)inaemia in humans. Clin Sci (Lond) 2001; 100: 161–167 [PubMed] [Google Scholar]
- 30.Wilcox CS. Asymmetric dimethylarginine and reactive oxygen species: unwelcome twin visitors to the cardiovascular and kidney disease tables. Hypertension 2012; 59: 375–381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Potter K, Hankey GJ, Green DJ et al. Homocysteine or renal impairment: which is the real cardiovascular risk factor? Arterioscler Thromb Vasc Biol 2008; 28: 1158–1164 [DOI] [PubMed] [Google Scholar]
- 32.House AA, Eliasziw M, Cattran DC et al. Effect of B-vitamin therapy on progression of diabetic nephropathy: a randomized controlled trial. JAMA 2010; 303: 1603–1609 [DOI] [PubMed] [Google Scholar]
- 33.Spence JD, Eliasziw M, House AA. B-Vitamin therapy for diabetic nephropathy: reply. JAMA 2010; 304: 636–637 [DOI] [PubMed] [Google Scholar]
- 34.Koyama K, Yoshida A, Takeda A et al. Abnormal cyanide metabolism in uraemic patients. Nephrol Dial Transplant 1997; 12: 1622–1628 [DOI] [PubMed] [Google Scholar]
- 35.Koyama K, Ito A, Yamamoto J et al. Randomized controlled trial of the effect of short-term coadministration of methylcobalamin and folate on serum ADMA concentration in patients receiving long-term hemodialysis. Am J Kidney Dis 2010; 55: 1069–1078 [DOI] [PubMed] [Google Scholar]
- 36.Løland KH, Bleie O, Borgeraas H et al. The association between progression of atherosclerosis and the methylated amino acids asymmetric dimethylarginine and trimethyllysine. PLoS One 2013; 8: e64774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Freeman AG. Hydroxocobalamin versus cyanocobalamin. J R Soc Med 1996; 89: 659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang R. Is H2S a stinky remedy for atherosclerosis? Arterioscler Thromb Vasc Biol 2009; 29: 156–157 [DOI] [PubMed] [Google Scholar]
- 39.Perna AF, Ingrosso D. Low hydrogen sulphide and chronic kidney disease: a dangerous liaison. Nephrol Dial Transplant 2012; 27: 486–493 [DOI] [PubMed] [Google Scholar]
- 40.Exner M, Hermann M, Hofbauer R et al. Thiocyanate catalyzes myeloperoxidase-initiated lipid oxidation in LDL. Free Radic Biol Med 2004; 37: 146–155 [DOI] [PubMed] [Google Scholar]
- 41.Rader DJ, Ischiropoulos H. ‘Multipurpose oxidase’ in atherogenesis. Nat Med 2007; 13: 1146–1147 [DOI] [PubMed] [Google Scholar]
- 42.Spence JD. Effects of the intestinal microbiome on constituents of red meat and egg yolks: a new window opens on nutrition and cardiovascular disease. Can J Cardiol 2014; 30: 150–151 [DOI] [PubMed] [Google Scholar]
- 43.Simenhoff ML, Burke JF, Saukkonen JJ et al. Biochemical profile or uremic breath. N Engl J Med 1977; 297: 132–135 [DOI] [PubMed] [Google Scholar]
- 44.Wang Z, Klipfell E, Bennett BJ et al. Gut flora metabolism of phosphatidylcholine promotes cardiovascular disease. Nature 2011; 472: 57–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Koeth RA, Wang Z, Levison BS et al. Intestinal microbiota metabolism of l-carnitine, a nutrient in red meat, promotes atherosclerosis. Nat Med 2013; 19: 576–585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tang WHW, Wang Z, Levison BS et al. Intestinal microbial metabolism of phosphatidylcholine and cardiovascular risk. N Engl J Med 2013; 368: 1575–1584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tang WH, Wang Z, Kennedy DJ et al. Gut microbiota-dependent trimethylamine N-oxide (TMAO) pathway contributes to both development of renal insufficiency and mortality risk in chronic kidney disease. Circ Res 2015; 116: 448–455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Moraes C, Fouque D, Amaral AC et al. Trimethylamine N-oxide from gut microbiota in chronic kidney disease patients: focus on diet. J Ren Nutr 2015; 25: 459–465 [DOI] [PubMed] [Google Scholar]
- 49.Johri AM, Heyland DK, Hetu MF et al. Carnitine therapy for the treatment of metabolic syndrome and cardiovascular disease: evidence and controversies. Nutr Metab Cardiovasc Dis 2014; 24: 808–814 [DOI] [PubMed] [Google Scholar]
- 50.Poesen R, Viaene L, Verbeke K et al. Renal clearance and intestinal generation of p-cresyl sulfate and indoxyl sulfate in CKD. Clin J Am Soc Nephrol 2013; 8: 1508–1514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Barreto FC, Barreto DV, Liabeuf S et al. Serum indoxyl sulfate is associated with vascular disease and mortality in chronic kidney disease patients. Clin J Am Soc Nephrol 2009; 4: 1551–1558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Dou L, Jourde-Chiche N, Faure V et al. The uremic solute indoxyl sulfate induces oxidative stress in endothelial cells. J Thromb Haemost 2007; 5: 1302–1308 [DOI] [PubMed] [Google Scholar]
- 53.Dou L, Sallee M, Cerini C et al. The cardiovascular effect of the uremic solute indole-3 acetic Acid. J Am Soc Nephrol 2015; 26: 876–887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bammens B, Evenepoel P, Keuleers H et al. Free serum concentrations of the protein-bound retention solute p-cresol predict mortality in hemodialysis patients. Kidney Int 2006; 69: 1081–1087 [DOI] [PubMed] [Google Scholar]
- 55.Liabeuf S, Barreto DV, Barreto FC et al. Free p-cresylsulphate is a predictor of mortality in patients at different stages of chronic kidney disease. Nephrol Dial Transplant 2010; 25: 1183–1191 [DOI] [PubMed] [Google Scholar]
- 56.Wu IW, Hsu KH, Hsu HJ et al. Serum free p-cresyl sulfate levels predict cardiovascular and all-cause mortality in elderly hemodialysis patients–a prospective cohort study. Nephrol Dial Transplant 2012; 27: 1169–1175 [DOI] [PubMed] [Google Scholar]
- 57.Schulman G, Agarwal R, Acharya M et al. A multicenter, randomized, double-blind, placebo-controlled, dose-ranging study of AST-120 (Kremezin) in patients with moderate to severe CKD. Am J Kidney Dis 2006; 47: 565–577 [DOI] [PubMed] [Google Scholar]
- 58.Urquhart BL, Freeman DJ, Spence JD et al. The effect of mesna on plasma total homocysteine concentration in hemodialysis patients. Am J Kidney Dis 2007; 49: 109–117 [DOI] [PubMed] [Google Scholar]
- 59.Nesrallah G, Suri R, Moist L et al. Volume control and blood pressure management in patients undergoing quotidian hemodialysis. Am J Kidney Dis 2003; 42 (1 Suppl): 13–17 [DOI] [PubMed] [Google Scholar]
- 60.Schulman G, Berl T, Beck GJ et al. Randomized Placebo-Controlled EPPIC Trials of AST-120 in CKD. J Am Soc Nephrol 2015; 26: 1732–1746 [DOI] [PMC free article] [PubMed] [Google Scholar]