

Abstract
Perfringolysin O (PFO) is a member of the cholesterol-dependent cytolysin (CDC) family of bacterial pore-forming proteins, which are highly efficient in delivering exogenous proteins to the cytoplasm. However, the indiscriminate and potent cytotoxicity of PFO limits its practical use as an intracellular delivery system. In this study, we describe the design and engineering of a bispecific, neutralizing antibody against PFO, which targets reversibly attenuated PFO to endocytic compartments via receptor-mediated internalization. This PFO-based system efficiently mediated the endosomal release of a co-targeted gelonin construct with high specificity and minimal toxicity in vitro. Consequently, the therapeutic window of PFO was improved by more than 5 orders of magnitude. Our results demonstrating that the activity of pore-forming proteins can be controlled by antibody-mediated neutralization present a novel strategy for utilizing these potent membrane-lytic agents as a safe and effective intracellular delivery vehicle.
Keywords: perfringolysin O, cholesterol-dependent cytolysin, intracellular delivery, protein delivery, endosomal release, neutralizing antibody
Graphical abstract
INTRODUCTION
The ability to safely and efficiently deliver exogenous proteins to the cytoplasm of target cells is highly desired for allowing potential therapeutic interventions. While much progress has been made in the development of delivery systems that utilize mechanical disruption1,2 or various materials,3–8 their practical implementation remains a significant challenge.9
Members of the cholesterol-dependent cytolysin (CDC) family of bacterial pore-forming toxins have previously been demonstrated to deliver a wide range of payloads to both established and primary cell types,10 but their widespread use as a delivery system has been limited by their cytotoxicity. Generally, soluble CDC monomers are thought to bind to the cell membrane via cholesterol or other cell surface receptors, oligomerize into a prepore ring structure composed of 35–50 monomers, and undergo a conformational change where amphiphilic β hairpins insert into the membrane to create a pore 25–30 nm in diameter.11 Early studies showed that CDCs such as streptolysin O (SLO), perfringolysin O (PFO), and listeriolysin O (LLO) can be used as versatile transfection regents to introduce diverse membrane-impermeable payloads into cells, including plasmid DNA,12 antisense oligonucleotides,13 siRNA,14 glycopeptides (bleomycin),15 and various proteins.16 However, the cytotoxicity of the CDCs often required them to be removed after a brief incubation to avoid cell killing.17,18 Because such manipulations are not possible in an in vivo setting, alternative delivery methods are needed.
Among such methods proposed are encapsulating or conjugating LLO into or onto liposomes, which are modified with targeting antibodies in some cases, to shield or inactivate the protein until they are internalized into target cells.19,20 Although the specificity of delivery was greatly increased when using these approaches in in vitro models, such nanoparticulate formulations often suffer from poor pharmacokinetics and biodistribution, accumulating in the reticuloendothelial system21 to cause dose-limiting toxicity. Indeed, in vivo demonstrations of LLO-encapsulating liposomes have been limited to vaccination applications targeting phagocytic cells.22,23 Alternatively, to allow specific targeting of CDCs with favorable biodistribution properties, we previously generated targeted LLO and PFO constructs fused to binding moieties against cancer antigens. While the targeted constructs delivered macromolecular payloads such as the ribosome-inactivating toxin gelonin24 and siRNA25 to antigen-positive cells more efficiently than their untargeted counterparts, they remained equally toxic.
In this study, we report a novel, nonparticulate engineering strategy that widens the therapeutic window of PFO by more than 5 orders of magnitude, substantially improving its potential translatability. The guiding principle of this engineering strategy, first attempted by Lee et al. with liposomal delivery,10 is to direct pore formation to preferentially occur in endosomal compartments rather than on the plasma membrane, to eliminate the deleterious toxicities associated with breaching the latter while efficiently releasing co-endocytosed payloads to the cytoplasm. To such ends, we created a bispecific neutralizing antibody capable of binding to PFO, inhibiting its pore-forming activity in the extracellular space, and the cancer-associated antigen EGFR, promoting receptor-mediated internalization into target cells. In vitro, complexed with an attenuated PFO mutant, this antibody/PFO system delivered the payload gelonin with an efficacy comparable to that of the previously reported targeted PFO construct, while achieving unprecedented low levels of cytotoxicity. Antibody-mediated internalization of PFO was necessary for efficient delivery, supporting the model of endosomal release.
Our findings support the exploration of CDCs as a versatile, safe, and effective delivery vehicle that can enhance the intracellular access of exogenous proteins. Furthermore, we demonstrate the concept of antibody-mediated neutralization as a novel strategy for controlling the activity of potent membrane-disrupting agents. This approach can potentially be extended to other pore-forming proteins, including human perforin, to further advance the practical implementation of highly efficient, pore-forming protein-based intracellular delivery systems.
MATERIALS AND METHODS
Cell Lines
The A431 and CHO-K1 cell lines (ATCC, Manassas, VA) were cultured in DMEM and F-12K medium (ATCC), respectively, supplemented with 10% heat-inactivated FBS (Life Technologies, Grand Island, NY). HEK 293F cells were cultured in suspension in FreeStyle 293 expression medium (Life Technologies). All cell lines were maintained at 37°C and 5% CO2 in a humidified incubator.
Protein Expression and Purification
Fn3, E6rGel, and PFO variants were expressed using the pE-SUMO vector (LifeSensors, Malvern, PA) in Rosetta 2 (DE3) Escherichia coli (Novagen, San Diego, CA). Point mutations in PFO and E6rGel (C459A/T490A/L491V and Y74A/Y133A/E166K/R169Q, respectively) were introduced by QuikChange site-directed mutagenesis (Agilent, Santa Clara, CA). Briefly, bacterial cultures were grown to an OD600 of 2 in Terrific Broth (TB) and induced with 1 mM isopropyl β-D-1-thiogalactopyranoside at 20 °C overnight. The proteins of interest were purified from sonicated pellets using Talon metal affinity chromatography (Clontech, Mountain View, CA) per the manufacturer’s protocol. Following an overnight digestion with SUMO protease at 4 °C, cleaved SUMO and SUMO protease were removed by Talon metal affinity chromatography. C225.2 was expressed and purified from HEK 293F cells as previously described.26 All proteins were subjected to endotoxin removal as described below and stored in PBS.
Endotoxin Removal
The protein of interest was exchanged into a 20 mM buffer suitable for anion exchange chromatography at a pH one unit below its isoelectric point, with 150 mM NaCl in addition. The isoelectric points were calculated using Geneious software (Biomatters, Auckland, New Zealand). The flow-through was collected after repeated passages through a 5 mL HiTrap Q HP anion exchange column (GE Healthcare, Pittsburgh, PA), and endotoxin levels were measured using the QCL-1000 LAL assay following the manufacturer’s instructions (Lonza, Basel, Switzerland).
Purification of C225.2/PFO Complexes
PFO mutants were precomplexed with C225.2 at a molar ratio of 1:1.25 (PFO:C225.2) with C225.2 (and corresponding PFO binding sites) in excess to ensure complete capture of PFO. The mixture was incubated at 4 °C for 30 min, diluted 5-fold into running buffer [50 mM sodium phosphate and 100 mM ammonium sulfate (pH 7.4)] and purified using a 1 mL HiTrap Butyl HP column (GE Healthcare) to separate unbound C225.2 captured on the column from the C225.2/PFO complexes present in the flow-through. Purified complexes were analyzed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) stained with Sypro Orange (Life Technologies) and imaged on the Typhoon Trio imager (GE Healthcare). The concentrations of C225.2 and PFO were determined by densitometry using a standard curve generated with known amounts of PFO.
Sedimentation Velocity Analytical Ultracentrifugation
Velocity analytical ultracentrifugation (AUC) of the C225.2/PFO complex was performed in a Beckman XL-I analytical ultracentrifuge (Beckman Coulter, Fullerton, CA) using an An-60 Ti rotor at 42000 rpm and 4 °C. The proteins were at an AU of 0.2 in PBS. Data were collected at radial steps of 0.003 cm and analyzed with Sedfit using the c(s) method.27
Isolation of Inhibitory Binders against PFO
Binders to PFO based on the fibronectin scaffold (Fn3) were engineered using standard yeast surface display techniques as previously described,28 with modified selection schemes to identify inhibitory clones. A pooled combination of the YS, G2, and G4 Fn3 libraries previously developed29 were first screened with biotinylated PFO captured on magnetic beads,30 followed by fluorescence-activated cell sorting (FACS). Random mutagenesis was performed after every two or three selections to maintain a high library diversity. For FACS, one round of positive selection at pH 7.4 was followed by one round of negative selection at pH 5.5, to enrich for binders with favorable pH sensitivity. Individual clones from the resulting library were expressed solubly and screened for their ability to inhibit the hemolytic activity of PFO at pH 7.4 as described in Hemolysis Assays. The most effective clone was subjected to additional rounds of mutagenesis and FACS-based selections.
The binding affinities of all individual clones were analyzed by yeast surface titration using biotinylated PFO as previously described.29 Sorting was performed on MoFlo (Beckman Coulter, Brea, CA) or Aria (BD Biosciences, San Jose, CA) instruments.
Binding Assays
To compare the binding affinity of Fn3 clone 1.2 for different PFO mutants, EBY100 yeast were transformed with 1.2 using the EZ Yeast transformation kit (Zymo Research, Irvine, CA) and induced to display the Fn3 as previously described.28 Soluble PFO clones were labeled with Alexa Fluor 647 following the manufacturer’s instructions (Life Technologies). 0.3 million yeast displaying 1.2 were incubated with 500 nM PFO for 30 min at 4 °C and analyzed on an Accuri C6 cytometer (BD Accuri Cytometers, Ann Arbor, MI). The measured fluorescence intensities were normalized to that of PFO.
To analyze the binding specificity of the C225.2/PFO complex for cell lines of interests, fluorescently labeled PFO clones were preincubated with C225.2 or the control sm3e.2 at an equimolar ratio of PFO to its binding site, for 30 min at 4 °C. The PFO mixtures were then diluted in PBSA to the desired concentrations, incubated with A431 or CHO-K1 cells for 1.5 h at 4 °C in suspension, and analyzed on the Accuri C6 cytometer (BD Accuri Cytometers). Fluorescence intensities were normalized to the maximal value obtained for each PFO clone.
Hemolysis Assays
CPDA-1-stabilized human red blood cells (Research Blood Components, Boston, MA) were washed with and resuspended as a 50% suspension in PBSA (to a density of approximately 1010 cells/mL). 50 μL of red blood cells was incubated with 50 μL of PFO or the C225.2/PFO complex at varying concentrations for 30 min at 37 °C. Following centrifugation at 2500g for 10 min, the absorbance of the supernatant was measured at 541 nm using the Infinite 200 Pro plate reader (Tecan, Mannedorf, Switzerland). The background absorbance from the negative control (PBSA) was subtracted from all measurements, after which all values were normalized to that of the positive control (1% Triton X-100).
To identify PFO binders that inhibit the pore forming activity of PFO, hemolysis assays were performed as described, but varying concentrations of PFO were first complexed with 3 μM (1.1) or 300 nM (1.2) Fn3s in PBSA for 20 min at 4 °C before incubation with red blood cells. The Fn3s were present at these concentrations also during the incubation to drive binding to PFO.
Viability Assays
Cells were plated at a density of 12000 (A431) or 10000 (CHO-K1) cells/well in 96-well plates 16–20 h prior to the experiment. PFO or C225.2/PFO complexes were diluted to the desired concentrations in complete medium with or without 10 nM E6rGel and incubated with cells for 16 h overnight. To measure viability, cells were incubated with 100 μL of WST-1 cell proliferation reagent (Roche, Indianapolis, IN) diluted 1:10 in complete medium, for 30 min at 37 °C. The absorbance of the supernatant was measured at 450 nm using the Infinite 200 Pro plate reader (Tecan). The background absorbance of the reagent in medium was subtracted from all measurements, after which all values were normalized to that of untreated cells.
Translation Inhibition Assays
A431 cells were plated at a density of 20000 cells/well in 96-well plates 16–20 h prior to the experiment. The appropriate protein samples were prepared in complete medium and incubated with cells for 2 h. Cells were then incubated with DMEM containing 1 μCi/mL of [1-14C]leucine for 30 min at 37 °C. The medium used was free of unlabeled leucine and supplemented with 10% dialyzed serum. [1-14C]Leucine was used to track protein synthesis exclusively. Following washes with PBS, cells were lysed with RIPA buffer and transferred to a LumaPlate-96 (PerkinElmer, Waltham, MA) for counting on a TopCount NXT Microplate Scintillation and Luminescence Counter (PerkinElmer). The background counts from untreated cells incubated with medium lacking [1-14C]leucine was subtracted from all measurements, after which all values were normalized to that of untreated cells.
To determine the importance of PFO binding to and internalizing with EGFR, translation inhibition assays were performed as described above with the following modifications. First, to block the C225.2/PFO complex from associating with EGFR, the assay was performed in the presence of C225 maintained at a 10-fold molar excess relative to C225.2. To reduce the rate of clathrin-mediated endocytosis, cells were pretreated with 2.5 μM Pitstop 2 (Abcam, Cambridge, MA) or an equivalent concentration of DMSO in serum-free medium for 15 min, incubated with protein samples at 37 °C for 45 min, and labeled with [1-14C]leucine (1 μCi/mL) at 37 °C for 45 min. Pitstop 2 or DMSO was present throughout the incubation and radiolabeling steps. Following background subtraction as described above, all measurements were normalized to that of cells treated with Pitstop 2 or DMSO only.
RESULTS
Engineering of a PFO Binder for Reversible Neutralization
PFO is capable of forming pores very efficiently, but its activity cannot be readily tuned in a rational manner because its mechanism of action requires multiple intermolecular interactions and conformational change.11 Yeast surface display, a well-established technique for directed evolution, could not be applied to engineer PFO directly because the protein was too toxic to express in yeast (data not shown). In contrast, yeast surface display techniques can be applied in a straightforward manner to obtain proteins that bind to PFO. We reasoned that binders that interact with functionally important epitopes on PFO would affect its activity, similar to neutralizing antibodies,31,32 and thus, the binding properties could be tuned to control function (Figure 1a).
Figure 1.

Engineered PFO binder reversibly inhibits the hemolytic activity of PFO. (a) Schematic of PFO neutralization. Binders to PFO were engineered on the Fn3 scaffold using yeast surface display techniques and selected for those that inhibit PFO function. The binder prevents pore formation when it is associated with PFO but allows normal activity following dissociation, allowing reversible inhibition. (b) Reduced hemolytic activity of PFO in the presence of PFO binders. Fn3s 1.1 and 1.2 were precomplexed with PFO at saturating concentrations (3 μM and 300 nM, respectively) and maintained as such after red blood cells were added. Hemoglobin release was measured after a 30 min incubation at 37 °C. (c) Clone 1.2 was displayed on the surface of yeast and tested for its ability to bind fluorescently labeled, soluble PFO mutants at 10 nM. The relative fluorescence units were normalized to that measured with PFO.
To isolate neutralizing binders against PFO, we first sorted through yeast libraries previously developed on the fibronectin scaffold29 to obtain a collection of distinct binders. The 10th type III domain of fibronectin (Fn3) is a stable, cysteine-free scaffold that has been shown to effectively mediate molecular recognition33 and to be readily expressed as a genetic fusion with other proteins of interest. The Fn3 libraries were subjected to multiple rounds of mutagenesis via error-prone PCR and enriched for binders via magnetic bead selection and fluorescence-activated cell sorting (FACS). Selections were performed using PFO of which the cysteine at position 459 was mutated to an alanine34 to prevent cross-linking. This PFOC459A mutant is herein termed “PFO”. Eighteen individual clones from the resulting library could be grouped into six distinct families on the basis of sequence similarity, suggesting that binders were isolated against multiple epitopes on PFO.
To investigate whether any of these PFO binders interacted with functionally important epitopes, representative members from each group were expressed as soluble proteins and analyzed for their ability to inhibit the hemolytic activity of PFO. The Fn3 clones were preincubated with PFO at saturating concentrations and maintained as such after red blood cells were added. Among the binders tested, clone 1.1 (Figure S1) had an equilibrium dissociation constant (KD) of 13.5 ± 2.9 nM as measured by yeast surface titrations (Figure S2), and displayed the greatest degree of functional inhibition by reducing the EC50 of hemolysis by approximately 3-fold (from 0.24 ± 0.02 nM in the absence of 1.1 to 0.74 ± 0.23 nM in the presence of 1.1) (Figure 1b).
We further matured the affinity of clone 1.1 via additional rounds of mutagenesis and FACS to obtain clone 1.2 (Figure S1) with a KD of 0.53 ± 0.26 nM (Figure S2). 1.2 inhibited the hemolytic activity of PFO by approximately 11-fold to an EC50 of 2.7 ± 0.21 nM (Figure 1b). Interestingly, increasing the affinity of the PFO binder increased the degree of inhibition even at saturating concentrations. This observation suggested that the binding interaction between the Fn3 and PFO, the duration of which is prolonged by a reduced off-rate in the higher-affinity binder, prevented hemolysis. Furthermore, it suggested that the reversible binding interaction between the Fn3 and PFO can allow pores to form over time as PFO dissociates from the binder and becomes sequestered on the cell membrane before rebinding to 1.2. Both 1.1 and 1.2 were engineered to bind tighter to PFO at pH 7.4 than at pH 5.5 (Figure S2 and Figure 2b), reducing the degree of inhibition at endosomal pH (Figure S3).
Figure 2.

C225.2 forms monodisperse complexes with PFO and potently inhibits its activity. (a) Schematic of the bispecific, neutralizing antibody C225.2 against PFO and EGFR. PFO binder 1.2 was genetically fused to the heavy chain N-terminus of C225 via a flexible (Gly4Ser)2 linker. (b) Distribution of sedimentation coefficients in the purified C225.2/PFO complex obtained from sedimentation velocity AUC. (c) Reduced hemolytic activity of PFO following complexation with C225.2. PFO or C225.2/PFO complexes were incubated with red blood cells at varying concentrations for 30 min at 37 °C. The molar concentrations denote that of PFO, in free form or complexed to C225.2. The corresponding concentrations of C225.2 can be calculated as half of that of PFO.
To better understand the molecular basis of how clone 1.2 is inhibiting the activity of PFO, we measured how well this Fn3 bound to known PFO mutants. Previously, it has been proposed that residues T490 and L491 of PFO mediate binding to cholesterol,35 the natural cell surface receptor of PFO, whereas residues Y181 and F318 provide stacking interactions between neighboring monomers to stabilize the oligomeric complex.36 Thus, 1.2 was displayed on the surface of yeast and incubated with fluorescently labeled PFO clones harboring point mutations in the aforementioned residues.
1.2 bound significantly weaker to PFOY181A than to PFO (Figure 1c), suggesting it may prevent the formation or subsequent stabilization of the oligomeric complex by sterically occluding the PFO stacking surface. PFOY181A was previously shown to bind to and oligomerize on liposomal membranes,37 indicating that it was unlikely that the observed loss of binding was due to gross misfolding of the mutant. Mutants of F318 were not tested because this residue is not surface-exposed in the soluble monomer. There was no difference in the binding of 1.2 to PFO and PFOT490G,L491G, suggesting that the epitope of 1.2 is physically distinct and separated in distance from the membrane binding region of PFO.
Engineering of a Bispecific Neutralizing Antibody for Specific Targeting of PFO
Next, to target the reversibly attenuated PFOs to endosomal compartments, we genetically fused 1.2 to Cetuximab (C225), a monoclonal antibody against EGFR (Figure 2a). C225 was chosen as the antibody backbone because EGFR has been reported to be constitutively internalized with a half-time of approximately 30 min and undergo multiple rounds of recycling and internalization.38 Thus, C225 was well suited for implementing our engineering strategy promoting pore formation in endocytic vesicles. Furthermore, EGFR is therapeutically relevant because of its overexpression in various cancer types. The N-terminus of C225’s heavy chain was chosen as the site of fusion because the resulting bispecific antibody had superior yields compared to those of other fusion topologies (data not shown). 1.2 was linked to C225 by a flexible (Gly4Ser)2 linker to allow modular binding to PFO without interfering with C225’s interaction with EGFR.
To remove uncomplexed proteins that can obscure downstream analysis, we developed a hydrophobic interaction chromatography (HIC)-based purification strategy for obtaining C225.2/PFO complexes with high monodispersity. In the purified complex, the molar ratio of PFO to C225.2 was consistently close to 2 (2.2 ± 0.2:1 PFO:C225.2, from 20 separate preparations), as analyzed by fluorescence densitometry following SDS-PAGE (Figure S4). In addition, 89% of the complex fit to a single peak as analyzed by sedimentation velocity (SV) analytical ultracentrifugation (AUC) (Figure 2b and Figure S5), illustrating that the complex was highly monodisperse.
Next, we confirmed that the 1.2 portion of C225.2 maintained its ability to bind to and inhibit the hemolytic activity of PFO. Indeed, PFO was 31-fold less active when complexed to C225.2, with an EC50 of hemolysis of 7.57 ± 0.37nM (Figure 2c). To confirm that C225.2 maintained its ability to engage EGFR and improve the targeting specificity of PFO, fluorescently labeled PFO was complexed with C225.2 or a control bispecific antibody (sm3e.2) where 1.2 was fused to an antibody against the carcinoembryonic antigen (CEA) (sm3e) in the same topology as C225.2. A431 cells express undetectable levels of surface CEA, making sm3e.2 a suitable negative control. Binding was measured on the A431 cell line, which expresses high levels of EGFR and is thus widely used as a model system to study EGFR-based therapeutics.39 The CHO-K1 cell line, which does not express human EGFR (C225 is not cross-reactive with murine EGFR), served as an antigen-negative control. All binding measurements were performed at 4 °C to prevent cell lysis, as the activity of PFO is significantly attenuated at lower temperatures (data not shown).
Free PFO bound with moderate affinity to both cell lines as expected because of its interaction with membrane cholesterol, which is ubiquitous (Figure 3a). Interestingly, binding was detectable only at concentrations much higher than those required for cell lysis (Figure 1b), highlighting that even a few pores can potently influence membrane barrier properties. sm3e.2 prevented PFO from binding to both cell lines, likely because of steric hindrance provided by the bulky antibody framework. In contrast, the C225.2/PFO complex bound only to A431s in a specific manner, as the signal was abrogated with excess C225.
Figure 3.
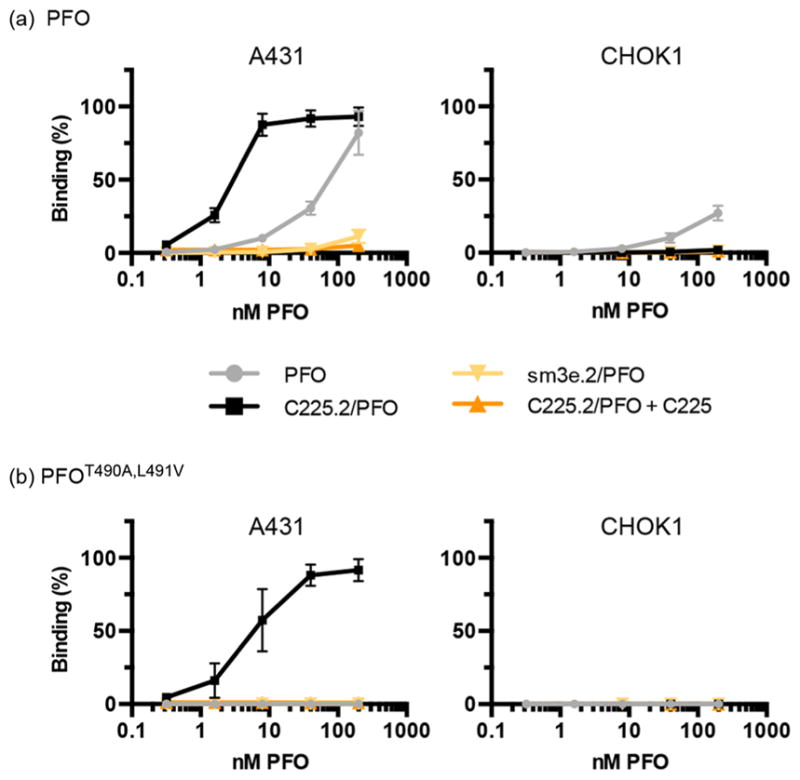
C225.2 improves the targeting specificity of PFO. (a) Fluorescently labeled PFO was incubated with the indicated cell lines at 4 °C for 1.5 h, in free form or as a complex with neutralizing antibody C225.2 or sm3e.2. To determine the specificity of the binding interaction, C225 was included at a 10-fold molar excess over C225.2 to compete for EGFR. All fluorescence intensities were normalized to the maximal value obtained on A431 cells. (b) Equivalent analysis with fluorescently labeled PFOT490A,L491V. The figure legends are identical to those of panel a.
Next, on the basis of our previous observations that suggested that (a) PFO, after dissociating from C225.2, may associate with either C225.2 or the cell membrane with competing kinetics and (b) free PFO is capable of binding to membrane cholesterol and lysing cells with high efficiency, we reasoned that decreasing the affinity of PFO for the membrane will favor its rebinding to C225.2 and promote endocytosis. On the basis of previous reports that demonstrated that this affinity can be fine-tuned via mutagenesis of residues T490 and L491,35 we employed the mutant PFOT490A,L491V, which showed no detectable binding to either cell line by itself in the concentration range tested (Figure 3b). PFOT490A,L491V was less hemolytically active than PFO as expected (Figure S6), based on its attenuated ability to anchor onto the cell membrane. However, we anticipated that this attenuation would not significantly affect the performance of the C225.2/ PFO complex as a delivery system as even a few pores may potently permeabilize the membrane, and the complex may reach high local concentrations in endocytic compartments following clathrin-mediated concentration and endocytosis of the receptors. As 1.2 does not interact with residues T490 and L491 (Figure 1c), PFOT490A,L491V could be complexed with C225.2 and purified to monodispersity in a manner identical to that of PFO. As expected, the resulting C225.2/PFOT490A,L491V complex bound specifically to A431s, but not CHO-K1s (Figure 3b).
Specific and Efficient Intracellular Delivery Enabled by the C225.2/PFO System
To determine whether the neutralized and endosomally targeted C225.2/PFO system retained PFO’s ability to deliver exogenous payloads, we analyzed its dose-dependent cytotoxicity and efficacy in vitro. The efficacy of delivery was assessed with the payload E6rGel, an EGFR-targeted gelonin construct developed previously.40 As a type I ribosome-inactivating protein (RIP) that lacks its own membrane translocation domain, gelonin is modestly cytotoxic as a single agent but can potently kill cells when artificially introduced into the cytoplasm, as demonstrated by prior studies.41,42 In addition, the targeting moiety, E6, a binder against EGFR engineered on the Fn3 scaffold, does not to compete with C225 for binding,43 allowing the C225.2/PFO complex and E6rGel to engage the same receptor molecule. This setup allowed maximal overlap of the C225.2/PFO complex and E6rGel in endosomal compartments, the intended site of release, following EGFR-mediated internalization.
The concentration of E6rGel was fixed at a value (10 nM) above the KD of E6 (0.26 nM43) to saturate the available receptors. E6rGel by itself caused no cell death under these conditions (Figure S7a). Consequently, any synergistic cell killing between PFO and E6rGel could be attributed to PFO enhancing gelonin’s access to the cytoplasm (in the absence of cell killing caused by PFO itself). Minimal synergy was observed when PFO was combined with E6rGel in either cell line, highlighting how the intrinsic cytotoxicity of PFO can cause cell death before sufficient quantities of E6rGel can be delivered. Similarly, the targeted PFO construct reported previously,25 in which PFO is fused to an Fn3 that binds to EGFR, increased the level of E6rGel-mediated cell killing in A431 cells but remained severely cytotoxic to both cell lines (Figure S7b). In contrast, C225.2/PFO and C225.2/ PFOT490A,L491V complexes markedly synergized with E6rGel only in A431s, but not CHO-K1s, consistent with our model in which PFO mediates the release of gelonin from endocytic compartments following EGFR-mediated internalization. Notably, in A431s, the C225.2/PFOT490A,L491V complex efficiently delivered E6rGel at low (picomolar) concentrations while being completely inert by itself in the concentration range tested (=3 μM), creating a therapeutic window spanning more than 5 orders of magnitude. Furthermore, equivalent delivery was observed in CHO-K1 cells only at very high (micromolar) concentrations. Overall, the C225.2/PFOT490A,L491V system was a significant improvement over the starting PFO clone, in terms of the safety, efficacy, and specificity of delivery.
To confirm that the observed cell killing was indeed caused by E6rGel in the cytoplasm, we measured protein synthesis levels in A431s following treatment with the C225.2/ PFOT490A,L491V complex and E6rGel (Figure 4b). A significant reduction was observed only when wild-type gelonin (E6rGelwt), but not its enzymatically inactive counterpart (E6rGelmut44), was used, demonstrating that gelonin in the cytoplasm cleaved the corresponding ribosomes via its catalytic residues. Cell viability judged by gross cell morphology was not significantly affected during the incubation period (Figure S8a), and thus, the observed reduction in the level of protein synthesis was not a result of fewer cells being analyzed. Protein synthesis was unaffected when cells were treated with C225.2/ PFOT490A,L491V or E6rGel alone (Figure S8b).
Figure 4.

The C225.2/PFO system efficiently mediates the cytosolic delivery of targeted gelonin with low toxicity and high specificity. (a) PFO or PFOT490A,L491V, in free form or in complex with C225.2, was incubated with the indicated cell line at 37 °C overnight in the presence or absence of 10 nM E6rGel in complete medium. Cytotoxicity was measured using the WST-1 reagent. (b) C225.2/PFOT490A,L491V complexes were incubated with A431 cells for 2 h at 37 °C, in combination with 10 nM E6rGel (E6rGelwt) or an inactive mutant (E6rGelmut; contains Y74A, Y133A, E166K, and R169Q mutations). Cells were then incubated with 1 μCi/mL of [1-14C]leucine for 20 min, and the incorporated radioactivity was measured by solid scintillation.
To gain a deeper understanding of the delivery mechanism, we further probed the role of EGFR-mediated endocytosis in the C225.2/PFOT490A,L491V system. First, we repeated the translation inhibition assay in the presence of excess C225 to block the specific binding interaction between C225.2 and EGFR. C225 completely abolished the reduction in protein synthesis levels (Figure 5a), illustrating that this binding interaction is necessary to allow intracellular delivery. Furthermore, decreasing the rate of EGFR-mediated internalization using the clathrin inhibitor Pitstop 2 significantly increased the level of protein synthesis, indicating that clathrin-mediated endocytosis is required for efficient delivery (Figure 5b). Protein synthesis did not return to basal levels, however, which may be due to incomplete inhibition of endocytosis by Pitstop 2, or residual delivery of gelonin across the cell membrane in addition to its endosomal release. Pitstop 2 did not affect the translational inhibition produced by the analogous but endocytosis-independent system composed of untargeted PFOT490A,L491V and untargeted gelonin (Figure S8c), confirming that it does not influence the gelonin-mediated inactivation of ribosomes. Eliminating the EGFR-targeting Fn3 moiety (E6) of E6rGel also caused a significant increase in protein synthesis levels (Figure 5b). Collectively, these results support the model in which C225.2/PFOT490A,L491V and E6rGel bind to and internalize with EGFR via clathrin-mediated endocytosis, followed by PFOT490A,L491V mediating gelonin’s release from endocytic compartments.
Figure 5.

EGFR-mediated binding and internalization are critical for efficient delivery. (a) The C225.2/PFOT490A,L491V complex and E6rGel (10 nM) were incubated with A431s at 37 °C for 2 h, with C225 in 10-fold molar excess over C225.2 at all points. The concentrations denote those of PFOT490A,L491V in the C225.2/PFOT490A,L491V complex. Protein synthesis levels were measured using [1-14C]leucine as described above. (b) A431 cells were pretreated with 2.5 μM Pitstop 2 or DMSO, followed by C225.2/PFOT490A,L491V in combination with targeted or untargeted gelonin. Pitstop 2 or DMSO was present throughout the incubation and radiolabeling steps. The asterisks denote P = 0.05 between E6rGel with vehicle and E6rGel with Pitstop 2, and between E6rGel with vehicle and rGel with vehicle, as analyzed by two-way analysis of variance. The concentrations denote those of PFOT490A,L491V in the C225.2/PFOT490A,L491V complex.
DISCUSSION
In this study, we have described the development of a novel PFO-based intracellular delivery system, which successfully expanded the therapeutic window of PFO from being negligible to spanning more than 5 orders of magnitude in vitro. Specifically, we engineered a bispecific, neutralizing antibody composed of an inhibitory binder against PFO and a monoclonal antibody against an internalizing antigen to deliver reversibly inhibited PFO to endosomal compartments. This system allows pore formation in endocytic vesicles and blocks such on the cell membrane, greatly reducing the cytotoxicity associated with PFO while allowing the efficient release of co-targeted macromolecular payloads into the cytoplasm.
The delivery system described herein is highly modular and can be expanded to alternative antigens, pore-forming proteins, and payloads. First, as the PFO binder can be fused with other antibody frameworks using standard molecular biology techniques, bispecific antibodies analogous to C225.2 can be created for other antibody-antigen pairs. Of note, while we have investigated a system in which PFO and the payload were targeted to the same internalizing receptor, previous work has demonstrated that the two agents can also be targeted to distinct receptors that colocalize in endosomal compartments.24 Second, the antibody-based neutralization strategy is compatible with other pore-forming proteins, particularly with those for which neutralizing antibodies have already been obtained.45–47 The neutralizing antibody, converted to a single-chain variable fragment (scFv), is functionally analogous to the inhibitory PFO binder reported in our study. Perforin, which is remarkably similar in structure to CDCs,48 is of particular interest as it can potentially circumvent immunogenicity concerns that arise when considering bacterial proteins for therapeutic applications. Lastly, we anticipate this delivery system to be seamlessly compatible with nonproteinaceous payloads in addition to proteins, as demonstrated with unmodified CDCs.
The exact mechanism of how PFO was preferentially activated in endosomal compartments in this system remains to be elucidated, although several explanations can be proposed. First, 1.2 bound weaker to PFO at pH 5.5 than at pH 7.4 (Figure S2), producing a modest but consistent increase in PFO’s hemolytic activity (by 4-fold) at the lower pH (Figure S3). Given the potent membrane-permeabilizing ability of PFO pores, moderate differences in hemolytic activity may result in amplified differences in biological activity. Second, the superior stability of PFO over C225.2 may have led to preferential unfolding and degradation of C225.2 in maturing endosomes and lysosomes, releasing PFO from inhibition. Third, following the internalization of the C225.2/PFO complex into endocytic compartments, the reversible binding interaction between the two components may have been simply competed off over time by the irreversible process of pore formation. Finally, any combination of the aforementioned possibilities could have collectively activated PFO in endocytic vesicles. Incorporating additional responsive elements for finer control of pore-forming activity before and after endocytosis will be a subject of future studies.
The C225.2/PFO system described in this study involves multiple kinetic processes, including the internalization and recycling of EGFR, the association and dissociation between multiple components (C225.2 and EGFR, C225.2 and PFO, and PFO and the lipid membrane), the gradual maturation of endosomes into lysosomes and subsequent degradation of proteins, and the uptake and release of payloads into and out of such intracellular vesicles. Thus, quantitative modeling of the system can be beneficial for better understanding the relative importance of each kinetic process and refining the system for in vivo delivery. For example, in an in vivo setting, prolonged circulation of the C225.2/PFO complex in the blood compartment prior to reaching the target site is expected to greatly reduce the probability of dissociated PFO rebinding to C225.2, particularly in the presence of active clearance mechanisms such as renal filtration and reticuloendothelial uptake. Thus, we anticipate the need for a tighter PFO binder that can complex and neutralize PFO for a longer period of time.
In addition, as a two-component system in which the payload (E6rGel) is physically separate from the agent mediating endosomal release (C225.2/PFO), this system is expected to have unique advantages and disadvantages as a delivery strategy in vivo. Notably, as the two agents are expected to have differing clearance and tumor accumulation kinetics, their dosing scheme will require empirical optimization to achieve maximal colocalization in target cells. Prior work demonstrating proof of concept that non-neutralized PFO, in trans, can potentiate the cytotoxicity of gelonin in a xenograft tumor model24 supports the feasibility of colocalizing two agents in vivo. In addition, the minimal toxicity of the C225.2/PFO complex and E6rGel toward bystander cells is expected to allow large dosages to be administered, driving the accumulation of both agents to working concentrations in the tumor interstitium. Of note, separating the payload from PFO may provide additional benefits. First, such a trans administration strategy preserves the modularity of the system, as it allows E6rGel to be substituted with other payloads without further adjusting the C225.2/PFO component. Second, physically separating the two agents may further improve the specificity of targeting, as the likelihood of bystander cells receiving both agents simultaneously decreases. Individually, the C225.2/PFO complex and E6rGel are inert within the concentration ranges we have tested.
Overall, this study provides strong evidence that the activity of pore-forming proteins can be successfully controlled via neutralizing binders, antibody-mediated targeting, and mutagenesis approaches, to allow the safe and specific delivery of macromolecular payloads to the cytosol. As EGFR is already a validated cancer antigen and C225 a validated therapeutic antibody, considerable therapeutic opportunities exist in combining C225-mediated antibody therapy with C225.2/ PFO-mediated delivery of various macromolecular drugs for synergistic treatment.
Supplementary Material
Acknowledgments
This work was funded by the National Institutes of Health Grant CA101830. N.J.Y. was supported by the MIT/NIGMS Biotechnology Training Program. The Koch Institute Flow Cytometry core and the MIT Biophysical Instrumentation facilities provided equipment and technical assistance with fluorescence-activated cell sorting and analytical ultracentrifugation, respectively. All schematics were prepared using Servier Medical Art.
References
- 1.Shalek AK, Robinson JT, Karp ES, Lee JS, Ahn DR, Yoon MH, et al. Vertical silicon nanowires as a universal platform for delivering biomolecules into living cells. Proc Natl Acad Sci US A. 2010;107:1870–1875. doi: 10.1073/pnas.0909350107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sharei A, Zoldan J, Adamo A, Sim WY, Cho N, Jackson E, et al. A vector-free microfluidic platform for intracellular delivery. Proc Natl Acad Sci US A. 2013;110:2082–2087. doi: 10.1073/pnas.1218705110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lo S, Wang S. Organelle-Specific Pharmaceutical Nanotechnology. John Wiley & Sons; New York: 2010. Peptide-Based Nanocarriers for Intracellular Delivery of Biologically Active Proteins. [Google Scholar]
- 4.Cronican JJ, Thompson DB, Beier KT, McNaughton BR, Cepko CL, Liu DR. Potent Delivery of Functional Proteins into Mammalian Cells in Vitro and in Vivo Using a Supercharged Protein. ACS Chem Biol. 2010;5:747–752. doi: 10.1021/cb1001153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yan M, Du J, Gu Z, Liang M, Hu Y, Zhang W, et al. A novel intracellular protein delivery platform based on single-protein nanocapsules. Nat Nanotechnol. 2010;5:48–53. doi: 10.1038/nnano.2009.341. [DOI] [PubMed] [Google Scholar]
- 6.Zelphati O, Wang Y, Kitada S, Reed JC, Felgner PL, Corbeil J. Intracellular Delivery of Proteins with a New Lipid-mediated Delivery System. J Biol Chem. 2001;276:35103–35110. doi: 10.1074/jbc.M104920200. [DOI] [PubMed] [Google Scholar]
- 7.Wang M, Alberti K, Sun S, Arellano CL, Xu Q. Combinatorially Designed Lipid-like Nanoparticles for Intracellular Delivery of Cytotoxic Protein for Cancer Therapy. Angew Chem, Int Ed. 2014;53:2893–2898. doi: 10.1002/anie.201311245. [DOI] [PubMed] [Google Scholar]
- 8.Malmsten M. Inorganic nanomaterials as delivery systems for proteins, peptides, DNA, and siRNA. Curr Opin Colloid Interface Sci. 2013;18:468–480. doi: 10.1016/j.cocis.2013.06.002. [DOI] [Google Scholar]
- 9.Gu Z, Biswas A, Zhao M, Tang Y. Tailoring nanocarriers for intracellular protein delivery. Chem Soc Rev. 2011;40:3638–3655. doi: 10.1039/C0CS00227E. [DOI] [PubMed] [Google Scholar]
- 10.Provoda CJ, Lee KD. Bacterial pore-forming hemolysins and their use in the cytosolic delivery of macromolecules. Adv Drug Delivery Rev. 2000;41:209–221. doi: 10.1016/S0169-409X(99)00067-8. [DOI] [PubMed] [Google Scholar]
- 11.Hotze EM, Tweten RK. Membrane assembly of the cholesterol-dependent cytolysin pore complex. Biochim Biophys Acta. 2012;1818:1028–1038. doi: 10.1016/j.bbamem.2011.07.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gottschalk S, Tweten RK, Smith LC, Woo SL. Efficient gene delivery and expression in mammalian cells using DNA coupled with perfringolysin O. Gene Ther. 1995;2:498–503. [PubMed] [Google Scholar]
- 13.Barry EL, Gesek FA, Friedman PA. Introduction of antisense oligonucleotides into cells by permeabilization with streptolysin O. BioTechniques. 1993;15:1016–1018. 1020. [PubMed] [Google Scholar]
- 14.Brito JLR, Davies FE, Gonzalez D, Morgan GJ. Streptolysin-O reversible permeabilisation is an effective method to transfect siRNAs into myeloma cells. J Immunol Methods. 2008;333:147–155. doi: 10.1016/j.jim.2008.01.009. [DOI] [PubMed] [Google Scholar]
- 15.Kerr DE, Wu GY, Wu CH, Senter PD. Listeriolysin O Potentiates Immunotoxin and Bleomycin Cytotoxicity. Bioconjugate Chem. 1997;8:781–784. doi: 10.1021/bc970124+. [DOI] [PubMed] [Google Scholar]
- 16.Walev I, Bhakdi SC, Hofmann F, Djonder N, Valeva A, Aktories K, et al. Delivery of proteins into living cells by reversible membrane permeabilization with streptolysin-O. Proc Natl Acad Sci US A. 2001;98:3185–3190. doi: 10.1073/pnas.051429498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Walev I. Cell permeabilization with Streptolysin O. In: Harris JR, Graham J, Rickwood D, editors. Cell Biology Protocols. John Wiley & Sons, Ltd; Chichester, West Sussex, England: 2006. [accessed July 17, 2014]. pp. 248–249. ( http://onlinelibrary.wiley.com/doi/10.1002/0470033487.fmatter/summary) [Google Scholar]
- 18.Brito JLR, Brown N, Morgan GJ. Transfection of siRNAs in Multiple Myeloma Cell Lines. In: Min W-P, Ichim T, editors. RNA Interference. Humana Press; Totowa, NJ: 2010. [accessed July 17, 2014]. pp. 299–309. ( http://link.springer.com/protocol/10.1007/978-1-60761-588-0_19) [DOI] [PubMed] [Google Scholar]
- 19.Lee KD, Oh YK, Portnoy DA, Swanson JA. Delivery of Macromolecules into Cytosol Using Liposomes Containing Hemolysin from Listeria monocytogenes. J Biol Chem. 1996;271:7249–7252. doi: 10.1074/jbc.271.13.7249. [DOI] [PubMed] [Google Scholar]
- 20.Kullberg M, Mann K, Anchordoquy TJ. Targeting Her-2+ Breast Cancer Cells with Bleomycin Immunoliposomes Linked to LLO. Mol Pharmaceutics. 2012;9:2000–2008. doi: 10.1021/mp300049n. [DOI] [PubMed] [Google Scholar]
- 21.Li SD, Huang L. Pharmacokinetics and Biodistribution of Nanoparticles. Mol Pharmaceutics. 2008;5:496–504. doi: 10.1021/mp800049w. [DOI] [PubMed] [Google Scholar]
- 22.Mandal M, Lee KD. Listeriolysin O-liposome-mediated cytosolic delivery of macromolecule antigen in vivo: Enhancement of antigen-specific cytotoxic T lymphocyte frequency, activity, and tumor protection. Biochim Biophys Acta. 2002;1563:7–17. doi: 10.1016/S0005-2736(02)00368-1. [DOI] [PubMed] [Google Scholar]
- 23.Sun X, Provoda C, Lee KD. Enhanced in vivo gene expression mediated by listeriolysin O incorporated anionic LPDII: Its utility in cytotoxic T lymphocyte-inducing DNA vaccine. J Controlled Release. 2010;148:219–225. doi: 10.1016/j.jconrel.2010.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pirie CM, Liu DV, Wittrup KD. Targeted Cytolysins Synergistically Potentiate Cytoplasmic Delivery of Gelonin Immunotoxin. Mol Cancer Ther. 2013;12:1774–1782. doi: 10.1158/1535-7163.MCT-12-1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liu DV, Yang NJ, Wittrup KD. A Nonpolycationic Fully Proteinaceous Multiagent System for Potent Targeted Delivery of siRNA. Mol Ther—Nucleic Acids. 2014;3:e162. doi: 10.1038/mtna.2014.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Spangler JB, Manzari MT, Rosalia EK, Chen TF, Wittrup KD. Triepitopic Antibody Fusions Inhibit Cetuximab-Resistant BRAF and KRAS Mutant Tumors via EGFR Signal Repression. J Mol Biol. 2012;422:532–544. doi: 10.1016/j.jmb.2012.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schuck P. Size-Distribution Analysis of Macromolecules by Sedimentation Velocity Ultracentrifugation and Lamm Equation Modeling. Biophys J. 2000;78:1606–1619. doi: 10.1016/S0006-3495(00)76713-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chen TF, de Picciotto S, Hackel BJ, Wittrup KD. Methods in Enzymology. Elsevier; Amsterdam: 2013. [accessed September 30, 2013]. Engineering Fibronectin-Based Binding Proteins by Yeast Surface Display; pp. 303–326. ( http://europepmc.org/abstract/MED/23422436/reload=0,jsessionid=xhkszUwKAERUOnMvcOR6.16) [DOI] [PubMed] [Google Scholar]
- 29.Hackel BJ, Kapila A, Wittrup KD. Picomolar Affinity Fibronectin Domains Engineered Utilizing Loop Length Diversity, Recursive Mutagenesis, and Loop Shuffling. J Mol Biol. 2008;381:1238–1252. doi: 10.1016/j.jmb.2008.06.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ackerman M, Levary D, Tobon G, Hackel B, Orcutt KD, Wittrup KD. Highly avid magnetic bead capture: An efficient selection method for de novo protein engineering utilizing yeast surface display. Biotechnol Prog. 2009;25:774–783. doi: 10.1002/btpr.174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Maynard JA, Maassen CBM, Leppla SH, Brasky K, Patterson JL, Iverson BL, et al. Protection against anthrax toxin by recombinant antibody fragments correlates with antigen affinity. Nat Biotechnol. 2002;20:597–601. doi: 10.1038/nbt0602-597. [DOI] [PubMed] [Google Scholar]
- 32.Orth P, Xiao L, Hernandez LD, Reichert P, Sheth PR, Beaumont M, et al. Mechanism of Action and Epitopes of Clostridium difficile Toxin B-neutralizing Antibody Bezlotoxumab Revealed by X-ray Crystallography. J Biol Chem. 2014;289:18008–18021. doi: 10.1074/jbc.M114.560748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bloom L, Calabro V. FN3: A new protein scaffold reaches the clinic. Drug Discovery Today. 2009;14:949–955. doi: 10.1016/j.drudis.2009.06.007. [DOI] [PubMed] [Google Scholar]
- 34.Shepard LA, Heuck AP, Hamman BD, Rossjohn J, Parker MW, Ryan KR, et al. Identification of a Membrane-Spanning Domain of the Thiol-Activated Pore-Forming Toxin Clostridium perfringens Perfringolysin O: An α,-Helical to β,-Sheet Transition Identified by Fluorescence Spectroscopy. Biochemistry. 1998;37:14563–14574. doi: 10.1021/bi981452f. [DOI] [PubMed] [Google Scholar]
- 35.Farrand AJ, LaChapelle S, Hotze EM, Johnson AE, Tweten RK. Only two amino acids are essential for cytolytic toxin recognition of cholesterol at the membrane surface. Proc Natl Acad Sci US A. 2010;107:4341–4346. doi: 10.1073/pnas.0911581107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ramachandran R, Heuck AP, Tweten RK, Johnson AE. Structural insights into the membrane-anchoring mechanism of a cholesterol-dependent cytolysin. Nat Struct Mol Biol. 2002;9:823–827. doi: 10.1038/nsb855. [DOI] [PubMed] [Google Scholar]
- 37.Hotze EM, Heuck AP, Czajkowsky DM, Shao Z, Johnson AE, Tweten RK. Monomer-Monomer Interactions Drive the Prepore to Pore Conversion of a β,-Barrel-forming Cholesterol-dependent Cytolysin. J Biol Chem. 2002;277:11597–11605. doi: 10.1074/jbc.M111039200. [DOI] [PubMed] [Google Scholar]
- 38.Wiley HS. Trafficking of the ErbB receptors and its influence on signaling. Exp Cell Res. 2003;284:78–88. doi: 10.1016/S0014-4827(03)00002-8. [DOI] [PubMed] [Google Scholar]
- 39.Goldstein NI, Prewett M, Zuklys K, Rockwell P, Mendelsohn J. Biological efficacy of a chimeric antibody to the epidermal growth factor receptor in a human tumor xenograft model. Clin Cancer Res. 1995;1:1311–1318. [PubMed] [Google Scholar]
- 40.Pirie CM, Hackel BJ, Rosenblum MG, Wittrup KD. Convergent Potency of Internalized Gelonin Immunotoxins across Varied Cell Lines, Antigens, and Targeting Moieties. J Biol Chem. 2011;286:4165–4172. doi: 10.1074/jbc.M110.186973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Provoda CJ, Stier EM, Lee KD. Tumor Cell Killing Enabled by Listeriolysin O-liposome-mediated Delivery of the Protein Toxin Gelonin. J Biol Chem. 2003;278:35102–35108. doi: 10.1074/jbc.M305411200. [DOI] [PubMed] [Google Scholar]
- 42.Selbo PK, Weyergang A, Høgset A, Norum OJ, Berstad MB, Vikdal M, et al. Photochemical internalization provides time-and space-controlled endolysosomal escape of therapeutic molecules. J Controlled Release. 2010;148:2–12. doi: 10.1016/j.jcon-rel.2010.06.008. [DOI] [PubMed] [Google Scholar]
- 43.Hackel BJ, Neil JR, White FM, Wittrup KD. Epidermal growth factor receptor downregulation by small heterodimeric binding proteins. Protein Eng, Des Sel. 2012;25:47–57. doi: 10.1093/protein/gzr056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.de Virgilio M, Lombardi A, Caliandro R, Fabbrini MS. Ribosome-Inactivating Proteins: From Plant Defense to Tumor Attack. Toxins. 2010;2:2699–2737. doi: 10.3390/toxins2112699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nato F, Reich K, Lhopital S, Rouyre S, Geoffroy C, Mazie JC, et al. Production and characterization of neutralizing and nonneutralizing monoclonal antibodies against listeriolysin O. Infect Immun. 1991;59:4641–4646. doi: 10.1128/iai.59.12.4641-4646.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Schlesinger BC, Cheng L. Characterization of a novel monoclonal antibody against human perforin using transfected cell lines. Immunology. 1994;81:291–295. [PMC free article] [PubMed] [Google Scholar]
- 47.Sawada-Hirai R, Jiang I, Wang F, Sun SM, Nedellec R, Ruther P, et al. Human anti-anthrax protective antigen neutralizing monoclonal antibodies derived from donors vaccinated with anthrax vaccine adsorbed. J Immune Based Ther Vaccines. 2004;2:5. doi: 10.1186/1476-8518-2-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Law RHP, Lukoyanova N, Voskoboinik I, Caradoc-Davies TT, Baran K, Dunstone MA, et al. The structural basis for membrane binding and pore formation by lymphocyte perforin. Nature. 2010;468:447–451. doi: 10.1038/nature09518. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.