

Abstract
The discovery and characterization of a novel chemical series of phosphorothioyl-containing imidazopyridines as potent Neuropeptide S Receptor antagonists is presented. The synthesis of analogs and their SAR with respect to the Gq, Gs, and ERK pathways is detailed. Pharmacokinetics and in vivo efficacy of a potent analog in a food intake rodent model are also included underscoring its potential therapeutic value for the treatment of sleep, anxiety, and addiction disorders.
Keywords: Neuropeptide S Receptor antagonist, sleep disorders, addiction disorders, imidazo-pyridinium
Graphical Abstract

INTRODUCTION
Neuropeptides are short chains of amino acids or small proteins that play a crucial role in controlling intracellular neuronal signaling. They bind to cell surface receptors eliciting a variety of physiological responses.1 The 21-amino acid neuropeptide S (NPS), named “S” due to a conserved N-terminus serine residue found across vertebrate species, has received considerable attention in the recent past. The cognate receptor for NPS, Neuropeptide S receptor (NPSR), was first described by Sato in 2002 but the endogenous peptide was left uncharacterized.2 The laboratories of Rainer Reinscheid deorphanized the receptor and also identified regions of the brain where the peptide and its receptor were expressed.3 They also detailed the pharmacology of the NPS/NPSR system using Chinese hamster ovary (CHO) and Human Embryonic Kidney 293 (HEK 293) cells stably expressing human and murine variants of NPSR. NPS acted as an agonist in these cell lines causing a dose-dependent increase in the levels of intracellular calcium, cyclic adenosine monophosphate (cAMP), and p42/p44 Mitogen-activated protein kinase (MAPK) phosphorylation. This suggested that NPSR was coupled to the Gq, Gs proteins, and the MAPK pathway respectively. Furthermore, they synthesized the human [125I] Y10-hNPS radiolabeled peptide and showed that it was an agonist that was equipotent to the natural ligand NPS and could be used in radiolabeled ligand displacement assays.4 The pharmacological characterization was extended to an Asn107Ile mutant and a C-terminal splice variant that were linked to asthma.5,6
Leading in vivo studies published by Xu et al. showed an increase in locomotor activity and reduction of all stages of sleep on supraspinal administration of NPS in mice.3 Paradoxically they also demonstrated a reduction in anxiety in four different validated mouse models of anxiety earning NPS the unique distinction of a novel activating anxiolytic.7 Experiments with Long–Evans rats have also shown administration of NPS in the lateral brain ventricle leads to anorexia-like symptoms with suppression of food intake.8 Ciccocioppo et al. have reported that administration of NPS in the lateral hypothalamus of the rat brain resulted in a significant increase in cue-induced alcohol seeking behavior in an alcohol relapse rat model.9 This effect was reduced by the selective oxerin receptor antagonist SB-33486710 indicating the presence of a strong link between the Hypocretin1/OrexinA and the NPS signaling pathways. Similar in vivo work has also implicated this circuitry’s relevance towards addiction to cocaine and morphine.11 The types of physiological changes that are observed in various rodent models after central administration on NPS have been summarized in several reviews.12
Systematic exploration of amino acids along the SFRNGVGTGMKKTSFQRAKS peptide sequence of NPS has not only identified key residues essential for agonist activity but also led to the discovery of novel peptide antagonists.13 Recently several small molecule antagonists of the NPS/NPSR pathway have also been reported in the literature. This has been a promising development as these molecules serve not only as vital tools to understand pharmacology but also hold potential for drug development towards anxiety, food, and addiction disorders. In this regard two molecules, SHA66 (1a) and SHA68 (1b), disclosed by Takeda Pharmaceuticals,14 have been characterized extensively as potent inhibitors of the NPS/NPSR signaling pathway (Figure 1). A 50 milligrams per kilogram (mpk) intraperitoneal (IP) dose of 1b was also able to reduce NPS-induced hyperlocomotion in a 90 minute mouse model by about 50%.15 Reported pharmacokinetic (PK) studies showed brain levels (∼6 µM at 15 min, ∼2 µM @ 2 h) well beyond its in vitro half maximal inhibitory concentration (IC50). Moreover, the bioactive enantiomer of 1b has recently been identified.16 Merck has disclosed potent inhibitors from two separate chemical series (2 and 3).17, 18 The best compound from a series of quinolinones (2) demonstrated high ex vivo occupancy of NPSR, after a 30 mpk IP dose, in discrete regions of the rat brain. GSK and Actelion Pharmaceutical Ltd. have also reported novel NPSR antagonists around the scaffolds represented by pyrroloimidazolone (4) and indanone (5) respectively.19,20 Our efforts within the NIH have resulted in the disclosure of a naphtha pyrano pyrimidine probe (6) where structural changes in the same chemical series led to the discovery of selective modulators of either the Gq or Gs pathway.21 Dal Ben has summarized the SAR around these small NPSR antagonists in a recent review.22 Herein we disclose the discovery of another structurally uncommon chemotype, which after medicinal chemistry optimization produced a compound with potent antagonism of the NPS/NPSR signaling pathway.
Figure 1.

Potent Small Molecule Antagonists of the NPSR pathways.
aActivity described in literature; bActivity determined in house.
RESULTS AND DISCUSSION
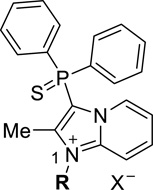
Our efforts to discover novel antagonists for the NPS pathway started with the development of a high-throughput HTRF (Homogeneous Time Resolved Fluorescence) assay that could measure the formation of cAMP on binding of NPS to NPSR using CHO cells stably expressing NPSR. This cAMP assay was used to screen 222,256 compounds in the quantitative high throughput screening (qHTS) format.23 This effort led to the discovery of the inhibitor 3-(diphenylphosphorothioyl)-1,2-dimethylimidazo[1,2-a]pyridin-1-ium 7 (Figure 1).24 The antagonistic activity of this compound was further confirmed in orthogonal assays measuring the reduction of calcium mobilization in the same cell line and in a NPS-radiolabeled displacement assay. The unusual functionality within the molecule prompted us to evaluate its stability in various aqueous buffers. We found that 7 did not decompose to any other species after exposure to Hank’s balanced buffer solution (HBBS) or Phosphate buffered saline (PBS) over a period of 48 hours. This confirmed its robust chemical stability and spurred a medicinal chemistry effort aimed at examining the key structural elements that were necessary for the antagonistic activity. In the previous chemotype reported by us (6),21 we observed a structure-related differential in antagonist activity toward Gs or Gq pathways within the same NPS antagonistic series.21 At the present moment, it is not clear which signaling pathway is linked to different biological responses observed by NPSR activation. Therefore, we decided to examine and report here inhibition of both Gs and Gq pathways with the cAMP and Ca2+ assays. In addition, we also analyzed the inhibition of ERK activation as the NPSR is also coupled to the MAPK pathway. To that end a synthetic route (Scheme 1) was developed for our SAR exploration. While the desmethyl compound 10a was commercially available, 2-methylimidazo[1,2-a]pyridine 10b could be accessed via a known palladium catalyzed internal cyclization of the amino group on the pendant alkyne in the 2-amino-1-(propargyl)pyridinium salt 9.25 The Tolmachev protocol allowed for the electrophilic aromatic substitution at the 3-position for the introduction of the diphenyl phosphino group.26 While 10a could be reacted with chlorodiphenylphosphine the 2-methyl counterpart 10b required use of the more reactive iododiphenylphosphine which was generated from chlorodiphenyl phosphine and trimethylsilyliodide.27 Treatment of the crude phosphine (11) with either elemental sulfur or hydrogen peroxide provided the diphenylphosphorothioyl (12b,d) or the diphenylphosphoryl species (12a,c) respectively. A final alkyation with dimethyl sulfate led to imidazopyridiniums 13a,b.28 These imidazopyridinium salts could be purified by normal or reverse phase chromatographic techniques and the nature of the counter anion proved to be inconsequential towards the bioactivity. The generation of the indole variants 15a,b were carried out in similar fashion from commercially available 14a,b.29
Scheme 1.

aSynthetic Scheme towards screening hit and analogs.
aReagents and conditions. a) EtOH, CHCCH2Br, reflux, 47% yield; b) Pd(PPh3)4, CuI, DMF, rt, 72% yield; c) TMSI, toluene; d) py, Et3N, PPh2I; e) 30% H2O2, 30–50% yield over 2 steps; f) sulfur, 30–50% yield over 2 steps; g) Me2SO4, 1,4-dioxane, sealed tube, 100–120 °C, 30–50% yield.
We were also interested in evaluating a tertiary diphenyl alcohol group as a possible bioisosteric replacement for the phosphorothioyl group. To that end the 3-bromo imidazopyrides 17a,b were generated from 16a,b with N-bromosuccinimide. Subsequent halogen-metal exchange using the chemistry described by Knochel was followed by addition of benzophenone to generate the tertiary alcohols 18a,b. These alcohols could be chemoselectively alkylated at the N-1 nitrogen in dioxane to yield analogs 19a,b.30
As initial confirmation of our HTS data, we obtained similar IC50’s to our previous values in both assays for our resynthesized original hit compound 7. In addition, the activity data in Table 1 indicates the sensitivity of the 2-methyl imidazopyridine core and the phosphorothioyl moieties towards structural change. Thus, elimination of the 2-methyl substituent reduced the activity between 3 to 25 times (for example 12b vs. 12d and 13a vs. 13b) in the cAMP assay. Replacement of the sulfur atom in the thiophosphoryl functional group by an oxygen (12c vs. 12d) or its elimination (11a) abolished the antagonist activity. Similarly, the diphenyl alcohols 18a,b and their corresponding N1 methylated analogs 19a,b were inactive. The indole replacement for the imidazopyridium core in 15a,b also proved deleterious towards activity. In general the active compounds appeared to have enhanced potencies in the Ca2+ and ERK assays.31
Table 1.
SAR of initial analogs.
| # | Structure | R c | AMP IC50 µM | Ca2+ IC50 µM | ERK IC50 µM |
|---|---|---|---|---|---|
| 11a | 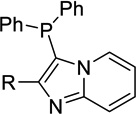 |
H | Inact | 35a | <50%b |
| 12a | 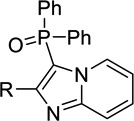 |
H | Inact | Inact | <50%b |
| 12c | Me | Inact | 27 ± 13a | <50%b | |
| 12b | 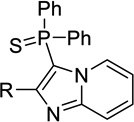 |
H | 63 ± 0a | 4.03 ± 0.07 | <50%b |
| 12d | Me | 18 ± 3 | 1.0 ± 0.16 | 0.0091, 0.030c | |
| 13a | 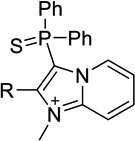 |
H | 39 ± 0a | 2.5 ± 0.4 | <50%b |
| 7 | Me | 1.7 ± 0.1 | 0.051 ± 0.008 | 0.030 ± 0.006 | |
| 15a | 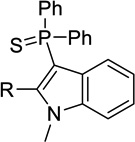 |
H | Inact | 7.3± 2.3 | <50%b |
| 15b | Me | Inact | 2.8 ± 0.0 | <50%b | |
| 18a | 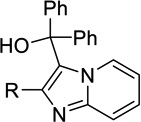 |
H | Inact | Inact | <50%b |
| 18b | Me | Inact | Inact | <50%b | |
| 19a | 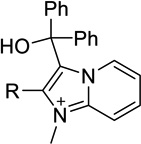 |
H | Inact | Inact | <50%b |
| 19b | Me | Inact | Inact | <50%b |
The cAMP and Ca2+ assay activity is the mean IC50 with SD from two separate experiments, each ran in duplicate. The ERK assay activity is the mean IC50 with SD from one experiment run in duplicate.
These compounds showed partial efficacy in their antagonistic behavior.
These compounds showed <50% inhibition at the highest concentration (50 µM) tested.
Both IC50 values are reported here as we feel that the standard deviation between the replicates is significant. (Inact=Inactive)
The activity results of the first set of synthetic compounds (Table 1) convinced us to preserve the 2-methylimidazopyridium core and the thiophosphorus moiety in our subsequent analog design. The striking structural similarity between the diphenylphospho group in our screening hit and the geminal diphenyl group in 1a and 1b prompted us to consider simple overlap models between these compounds (Figure 2). Moreover, in house [125I]Tyr 10-NPS displacement experiments had disclosed that both chemical series are able to compete with the natural ligand and therefore we hypothesized that both orthosteric antagonists would bind to the same region, probably with similar three dimensional geometry. We overlapped the geminal diphenyl substituents to realize that the oxazolidinone dipole aligned well with the phosphorothioyl moiety. The piperidine in 1b also overlapped reasonably with the imidazole of the imidazopyridine. However, it seemed that we were probably missing a hydrophobic phenyl ring at the lower portion of our compound which may the critical element in 1a and 1b’s higher potency. Fortunately we could test this hypothesis out by utilizing the chemistry that had been developed to resynthesize screening hit 7. To that end 12d would be an ideal intermediate for alkylation at N1 with various commercially available bromides which would place a phenyl ring at different spacer units away from the core of the molecule (Scheme 3).
Figure 2.

An overlap model of 1b and screening hit 7 (green).
Scheme 3.

Synthetic Route to analogs with chemical diversity in position 1.
Table 2 summarizes the activity for the analogs generated to test our hypothesis. Compounds with a phenyl ring one methylene unit away from N1 (20a) had similar potency as the original hit (7). This also held true for 20b and 20c which had an ethylene and ethanone spacer units between a phenyl ring and N1. Remarkably, the compound with the phenyl propyl chain 20d recorded substantial improvements in potency with 110, 1 and 2 nM IC50s in the cAMP, calcium and ERK assays respectively. The same trend was observed for the more constrained propenylbenzene appendage 20e which recorded a benchmark potency of 45 nM in the cAMP assay. Extending the phenyl ring by four (20f) or five (20g) methylene units led to erosion of the potent activity which was achieved by three spacer units. Each analog in Table 2 showed at least a 25-fold drop in its potency in the cAMP assay, compared to its individual potency in the calcium or ERK assay. At this juncture we also evaluated additional substitution in the phenyl ring separated optimally with a propenyl group from the N1 of the imidazopyridine core. Table 3 displays the activity of such compounds in the context of the Gs and Gq pathways. Although many types of substitutions were tolerated, unsubstituted 20e remained the most potent compound. It appeared that meta substitution was slightly less tolerated compared to para substitution as exemplified by compounds 21g and 21c when compared to 21f and 21d respectively. The weakest inhibitor on this series was the napthyl compound 21i indicating a size restriction that might be associated with the hydrophobic region that is accommodating the phenyl group in 20e.
Table 2.
SAR of analogs in position one.
 | ||||
|---|---|---|---|---|
| R | # | cAMP IC50 µM | Ca2+ IC50 µM | ERK IC50 µM |
| 7 | 1.7 ± 0.1 | 0.051 ± 0.008 | 0.030 ± 0.006 | |
| 20a | 1.3 ± 0.0 | 0.032 ± 0.005 | 0.016 ± 0.001 | |
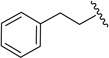 |
20b | 2.1 ± 0.2 | 0.073 ± 0.023 | 0.016, 0.063b |
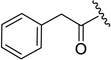 |
20c | 2.1 ± 0.0 | 0.080 ± 0.013 | 0.013, 0.028b |
| 20d | 0.11 ± 0.01 | 0.0016, <0.0005a | 0.0035, <0.0005a | |
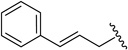 |
20e | 0.045 ± 0.000 | 0.00096 ± 0.00023 | 0.0013 ± 0.0007 |
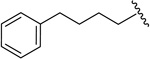 |
20f | 0.94 ± 0.08 | 0.0011, < 0.0005a | 0.015 ± 0.000 |
| 20g | 0.71 ± 0.00 | 0.013 ± 0.002 | 0.0062 ± 0.0007 | |
The cAMP and Ca2+ assay activity is the mean IC50 with SD from two separate experiments, each ran in duplicate. The ERK assay is the mean IC50 with SD from one experiment run in duplicate.
The two replicate IC50s are reported here, one (<0.5 nM) being less than that could be detemined accurately by the assay.
Both IC50 values are reported here as we feel that the standard deviation between the replicates is significant.
Table 3.
SAR of substituted phenyl ring.
 | |||
|---|---|---|---|
| # | R | cAMP IC50 (µM) | Ca2+ IC50 (µM) |
| 20e | 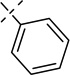 |
0.044 ± 0.000 | 0.00096 ± 0.00023 |
| 21a | 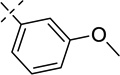 |
0.11 ± 0.00 | 0.0032 ± 0.0005 |
| 21b | 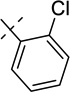 |
0.094 ± 0.008 | 0.0034 ± 0.0016 |
| 21c | 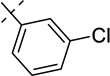 |
0.22 ± 0.00 | 0.0092 ± 0.0029 |
| 21d | 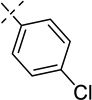 |
0.12 ± 0.01 | 0.0022, 0.0071a |
| 21e | 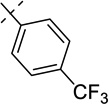 |
0.40 ± 0.00 | 0.013, 0.0045a |
| 21f | 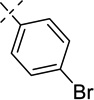 |
0.17 ± 0.01 | 0.0057 ± 0.0000 |
| 21g | 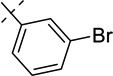 |
0.67 ± 0.05 | 0.023 ± 0.000 |
| 21h | 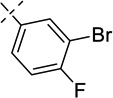 |
0.84 ± 0.07 | 0.045± 0.000 |
| 21i | 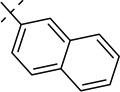 |
1.3 ± 0.0 | 0.064 ± 0.010 |
| 21j | 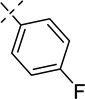 |
0.071 ± 0.000 | 0.0034 ± 0.0003 |
| 21k | 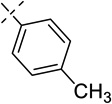 |
0.33 ± 0.03 | 0.0090 ± 0.0000 |
The cAMP and Ca2+ assay activity is the mean IC50 with SD from two separate experiments, each ran in duplicate.
Both IC50 values are reported here as we feel that the standard deviation between the replicates is significant.
While we had discovered potent antagonists of the NPS/NPSR signaling pathway, it was crucial to compare the activity of this structural class with the best-in-class small molecule inhibitors in literature. We did this in the context of our functional assays as well the radiolabeled binding assay. We observed that 20e was 5-fold more potent than 1b in the calcium and displacement assay and 9-fold more potent in the cAMP assay. On the other hand the potencies in these assays were comparable to the active enantiomer of 2 reported by Merck. 20e appeared to be more potent inhibitor of ERK phosphorylation. We also decided to compare the intrinsic stabilities of these key NPSR antagonists and to that end we analyzed the percentage of parent compound remaining at 15, 30 and 60 min after incubation in mouse liver microsomes. We were pleased to learn that 20e had the slowest rate of intrinsic clearance, which boded well for prospective in vivo studies and was a clear advantage over 1b and 2.
The discovery of the potent NPSR antagonist 20e warranted an in vivo evaluation of its activity. As a preliminary study we chose to examine the efficacy of this compound in a rat food intake model where the NPSR peptide antagonist [D-Cys(tBU)5]NPS was able to reverse the anorectic effect of NPS.32 While we predicted a reasonable exposure in plasma we were unsure of the blood brain barrier penetration of 20e. Hence, at the outset, we decided to dose 20e by the intracerebroventricular (icv) route as well. To that end Wistar rats were habituated to palatable food consumption and then treated with icv injections of NPS which lead to a marked reduction in food intake measured at 15, 30 and 60 min. Gratifying a single 10µg dose of 20e was able to reverse the suppression of food intake, induced by NPS, at all three time points (Figure 3).
Figure 3.

Reversal of NPS-induced suppression of palatable food intake.
Three groups (each N=7) of rats were treated with two successive, 10 min apart, 10 uL i.c.v. injections, of a) Vehicle (10µl 2% DMSO in distilled water) followed by vehicle (veh / veh); b) Vehicle followed by 10 µg NPS in vehicle (veh / NPS); and c) 10 µg 20e in vehicle followed by 10 µg NPS in vehicle (antagonist / NPS). Following the second injection, animals were placed back in their cages. Palatable food (17% H2O, 33% sweetened condensed milk, 51% rat chow) was introduced 20 min thereafter, and intake was measured at the timepoints indicated. #p<0.5 veh / NPS group vs. veh / veh group; *p<0.05 antagonist / NPS group vs. veh / NPS group.
The success in the food intake in vivo model experiment led to further characterization of 20e. While intracerebroventicluar (icv) administration of the drug was a robust way to check for proof of principle, the evaluation of pharmacokinetics after intraperitoneal administration of drug was a more suitable path forward towards more long term in vivo studies. Table 5 enumerates the exposure levels that were achieved after a 10 mpk dose in mice. In plasma, a Cmax of 1.5 µM was reached 15 min post dose and the concentrations steadily declined with a half life of 8.8 h to about 54 nM at 24 h. More importantly, the drug crossed the blood brain barrier and drug levels (52 nM) that were above the in vitro IC50s in all three functional assays were observed even at 24 h. No adverse reactions were observed in this single dose study. This bodes well for further in vivo characterization of this chemical series in other disease models. While we are aware that the total drug concentration in the brain may not be a good indicator of a pharmacodynamic effect,33 detailed in vivo studies in rat alcohol models with 20e show efficacy with an IP dose of 1.0 mpk.34
Table 5.
Mouse Pharmacokinetics of 20e35 The concentration at each time point presented here is the mean derived from N=3.
| 10 mpk IP | ||||||
|---|---|---|---|---|---|---|
| Plasma | Brain | |||||
| Sampling | Meana | SD | Mean | Meana | SD | Mean |
| time(hr) | ng/mL | ng/mL | nM | ng/g | ng/g | nmol/g |
| 0 | BQL | N/A | BQL | BQL | N/A | BQL |
| 0.083 | 700 | 306 | 1503 | 14 | 2.41 | 30 |
| 0.25 | 617 | 111 | 1325 | 24 | 8.16 | 52 |
| 0.5 | 536 | 235 | 1150 | 25 | 3.57 | 53 |
| 1 | 259 | 5.03 | 556 | 21 | 5.14 | 44 |
| 2 | 180 | 7.64 | 386 | 27 | 6.95 | 57 |
| 4 | 114 | 18.3 | 245 | 24 | 3.31 | 53 |
| 8 | 99 | 34.6 | 214 | 28 | 8.06 | 60 |
| 12 | 58 | 9.66 | 126 | 43 | 17.8 | 93 |
| 24 | 25 | 4.33 | 54 | 24 | 2.45 | 52 |
Mean from N=3
| PK parameters | Plasma | Brain |
|---|---|---|
| Tmax (hr) | 0.083 | 12 |
| Cmax (ng/mL) | 700 | 43 |
| T1/2 (hr) | 8.8 | N/A |
| AUClast (hr*ng/mL) | 2240 | 746 |
| AUCinf (hr*ng/mL) | 2560 | N/A |
| AUCbrain/AUCplasma (%) | 33 |

20e was also profiled at 10 µM against 55 targets. We observed >90% inhibition of control in seven targets which were followed with IC50 determination studies (Table 1, Supplementary Information). We were particularly concerned with the activity in the µ-agonist displacement assay. We decided to compare this activity with compounds having potent affinity towards the µ-receptor (Figure 4). Thus in an in-house assay which monitored the displacement of radiolabeled DAMGO, a peptide with potent affinity for the µ-opioid receptor, we observed that 20e was <200-fold active than naloxone or morphine. With an IC50 of 1 nM in the 125I-NPS displacement assay, it appears that a reasonable therapeutic window exists between the probe’s affinity towards NPSR and the µ-opioid receptor.
Figure 4.

µ-opiod receptor affinity of 20e compared to naloxone and morphine in Human Recombinant µ-Opioid Receptor OPRM1 Stable Cell Line.a.
aMorphine IC50 2.5±0.05 nM ( ), Naloxone IC50 1.87±0.07 nM (
), Naloxone IC50 1.87±0.07 nM ( ), 20e IC50 580±0.11 nM (
), 20e IC50 580±0.11 nM ( )
)
CONCLUSIONS
In summary the qHTS paradigm at NCGC was used to identify a structurally novel small molecule as an antagonist to the NPS-NPSR neurocircuitry. Further medicinal chemistry revealed the uniqueness of this chemotype towards receptor binding and that even slight modifications to the structure would result in dramatic loss of activity. Simple overlap models with a previously disclosed inhibitor prompted a SAR study which led to the synthesis of the potent analogs 20d and 20e. The latter was characterized as a potent antagonist in functional and binding assays. Administration of this compound by the icv route completely reversed NPS-induced suppression of palatable food intake. In vitro stability in mouse liver microsomes showed that this compound had a slower rate of metabolism compared to the most characterized compounds in literature. This translated to a 10 mpk IP dose in mice which proved to be safe and maintained high levels of drug in the plasma with slow elimination. This experiment also demonstrated that the drug was able to cross the blood brain barrier and maintain concentrations higher than the in vitro functional assay IC50 for 24h in the brain. Thus, in spite of its unusual structure we have described the progression of these phosphorothioyl-containing imidazopyridium compounds to physiologically relevant antagonists of the NPS/NPSR pathway. Recently, we completed the evaluation of 20e in animal models of alcoholism where antagonism of NPSR is predicted to have an effect.34 Results of these studies show a supression of alcohol self administartion in rats with a dose as low as 1 mpk. We also show that, at the same dose, 20e is able to reduce alcohol induced ERK phosphorylation in the central amygdala of these rats.
EXPERIMENTAL SECTION
cAMP assay
A Chinese hamster ovary cell line stably expressing the NPSR was generated in Dr. Heilig’s lab. The cells were maintained in F12 medium containing 10% FBS, 100units/ml Penicillin, 100µg/ml Streptomycin, and 200µg/ml Geneticin at 37 °C, 5% CO2. Suspended CHO-NPSR cells were seeded into 1536-well tissue culture-treated white plates at a density of 1800cells/well in 4µl media without Geneticin and incubated at 37°C, 5% CO2 for overnight. After 1µl of stimulation buffer (1X PBS buffer, 0.1% BSA, 0.05% Tween-20, 500µM Ro 20-1724, EC80 of NPS) was added to each well, cells were incubated at 37°C, 5% CO2 for 30 min. 1.25µl of d2 conjugated cAMP and 1µf cryptate conjugated anti-cAMP antibody were then added. D2 conjugated cAMP and cryptate conjugated anti-cAMP antibody were both prepared in cell lysis buffer according to the manufacturer’s instruction. After 30 minutes, plates were then read in Viewlux plate reader (Perkin Elmer) using the TRF detection mode optimized for HTRF. This assay was done routinely as compounds were generated. The data presented in the manuscript is the mean IC50 with SD from two independent experiments that were each run in duplicate.
ERK assay
CHO-NPS cells were cultured using F12K media (ATCC) supplemented with 10% FBS and 250µg/ml hygromycin b (Life Technologies) selection antibiotic. Cells were harvested in Opti-MEM (ATCC) supplemented with 1% FBS and plated in Greiner 96 well white tissue culture treated clear bottom plates at a seeding density of 25,000 cells per well and grown overnight at 37°C, 5% CO2. The media in wells was replaced with 100µl Opti-MEM only and the cells incubated at 37°C , 5% CO2 for additional 4 hours. 1µl of compound (at the appropriate dilution in DMSO) was added to the wells and incubated for 10 minutes. 100µl of 1nM NPS final concentration (EC80) was added to every well prepared in Opti-MEM supplemented with 0.2% BSA and 0.005% Tweeen-20 (Sigma). After incubation for 20min. all media was removed and the plate palced on ice for 5min. Cellul’erk kit (Cisbio) instructions were then followed. Briefly, 50µl of 1X lysis solution was added per well and incubated at room temperature while gently rocking for 15 minutes. 16µl of lysed cells were transferred to Greiner white medium binding half well plates. 2µl per well of a 1X solution of deuterium conjugated anti-ERK1/2 antibody was added and incubated for 2 hours at room temperature in the dark. 2µl per well of a 1X solution of europium cryptate conjugated anti–ERK1/2 antibody was added and incubated over night at ambient temperature in the dark. The plate was read using HTRF settings on EnVision (PerkinElmer). The ERK assay activity presented in the manuscript is the mean IC50 with SD from one experiment that was run in duplicate.
Calcium Assay
The CHO-NPSR cell line used in the cAMP assay was also used in this calcium mobilization assay. The suspended cells were plated at 3 µl/well with 2000 cells in the black, tissue culture treated, clear bottom 1536-well plates. After overnight incubation at 37 °C, 5% CO2, 3 µl of the calcium dye (no wash High Performance PBX Calcium Assay Kit, BD Biosciences) was loaded to each well and the plates were incubated at 37 °C, 5 % CO2 for 1 hour followed by 10-min incubation with 23 nl compound prepared in DMSO solution. The assay plates were then placed onto the FDSS-7000 kinetic fluorescence plate reader for measuring the changes of intracellular free calcium. The basal fluorescence signal was recorded for 6 sec at 1 Hz followed by an addition of 1 µl of NPS stimulation buffer (1X PBS buffer, 0.1% BSA, 0.05% Tween-20, 500 µM Ro 20-1724, EC80 of NPS) and 4-minute continuous recording at 1 Hz. This assay was done routinely as compounds were generated. The data presented in the manuscript is the mean IC50 with SD from two independent experiments that were each run in duplicate.
[125I] Y10-hNPS Binding Assay
The assay was carried out as described earlier with minor modification.3 Y10-NPS labeled with 125I was bought from NEN Perkin Elmer (Boston, MA). CHO cells stably expressing human NPSR were seeded into 24-well plates and cultured until reaching 90–95% confluency. Cells were washed with 1ml PBS once, and then incubated with radioligand with or without compounds or in DMEM medium containing 0.1% bovine serum albumin at 20°C for 1.5 hr. Increasing concentrations of compounds or unlabeled human NPS were used to compete with 0.15 nM [125I] Y10-NPS. Nonspecific binding was determined in the presence of 1 µM unlabeled human NPS. Cells were washed twice with cold PBS and lysed with 1 N NaOH. Bound radioactivity was counted in a liquid scintillation counter. The activity presented in the manuscript is the mean IC50 with SD from one experiment that was run in duplicate.
General Methods
Unless otherwise stated, all reactions were carried out under an atmosphere of dry argon or nitrogen in dried glassware. Indicated reaction temperatures refer to those of the reaction bath, while room temperature (rt) is noted as ∼25 °C. All anhydrous solvents, commercially available starting materials, and reagents were purchased from Aldrich Chemical Co. and used as received. Analytical thin layer chromatography (TLC) was performed with Sigma Aldrich TLC plates (5 × 20 cm, 60 Å, 250 µm). Visualization was accomplished by irradiation under a 254 nm UV lamp. Chromatography on silica gel was performed using forced flow (liquid) of the indicated solvent system on Biotage KP-Sil pre-packed cartridges and using the Biotage SP-1 automated chromatography system. Reverse phase preparative purification was performed on a Waters semi-preparative HPLC. The column used was a Phenomenex Luna C18 (5 micron, 30 × 75 mm) at a flow rate of 45 mL/min. The mobile phase consisted of acetonitrile and water (each containing 0.1% trifluoroacetic acid). A gradient of 10% to 50% acetonitrile over 8 minutes was used during the purification. Fraction collection was triggered by UV detection (220 nM). 1H spectra were recorded on a Varian Inova 400 MHz spectrometer. Chemical shifts are reported in ppm with the solvent resonance as the internal standard (CDCl3 7.27 ppm, DMSO-d6 2.50 ppm, for 1H). Data are reported as follows: chemical shift, multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, br s = broad singlet, m = multiplet), coupling constants, and number of protons.
Analytical purity analysis and retention times reported here were performed by the following two methods on an Agilent LC/MS (Agilent Technologies, Santa Clara, CA): Method 1: A 7 minute gradient of 4 to 100% acetonitrile (containing 0.025% trifluoroacetic acid) in water (containing 0.05% trifluoroacetic acid) was used with an 8 minute run time at a flow rate of 1 mL/min. A Phenomenex Luna C18 column (3 micron, 3 × 75 mm) was used at a temperature of 50°C. Method 2: A 3 minute gradient of 4 to 100% acetonitrile (containing 0.025% trifluoroacetic acid) in water (containing 0.05% trifluoroacetic acid) was used with a 4.5 minute run time at a flow rate of 1 mL/min. A Phenomenex Luna C18 column (3 micron, 3 × 100 mm) was used at a temperature of 50°C.
Unless otherwise stated, all analogs were determined to have >95% purity based on the above methods. Mass determination was performed using an Agilent 6130 mass spectrometer with electrospray ionization in the positive mode.
Molecular weight confirmation was confirmed using an Agilent Time-Of-Flight Mass Spectrometer (TOF, Agilent Technologies, Santa Clara, CA). A 3 minute gradient from 4 to 100% acetonitrile (0.1% formic acid) in water (0.1% formic acid) was used with a 4 minute run time at a flow rate of 1 mL/min. A Zorbax SB-C18 column (3.5 micron, 2.1 × 30 mm) was used at a temperature of 50°C. Confirmation of molecular formula was confirmed using electrospray ionization in the positive mode with the Agilent Masshunter software (version B.02).
2-Amino-1-(prop-2-yn-1-yl)pyridin-1-ium, Br- (9)
Pyridin-2-amine (9.5 g, 0.10 mol) and 3-bromoprop-1-yne (10 ml, 0.10 mol) were taken in ethanol (50 ml) and refluxed for 2 h. The reaction was left to cool overnight. A light yellow solid precipitated out of the reaction. This was filtered via a Buchner funnel, washed with cold ethanol, and air dried to obtain 2-amino-1-(prop-2-ynyl)pyridinium, Br− (10 g, 47 mmol, 47 % yield). 1H NMR (400 MHz, DMSO-d6) δ ppm 3.82 (t, J=2.5 Hz, 1 H), 5.08 (d, J=2.5 Hz, 2 H), 6.95 (td, J=6.9, 1.4 Hz, 1 H), 7.11 (m, 1 H), 7.91 (ddd, J=8.9, 7.1, 1.7 Hz, 1 H), 8.17 (m, 1 H), 8.63 (br. s., 2 H).
2-Methylimidazo[1,2-a] pyridine (10b)
2-Amino-1-(prop-2-ynyl)pyridinium bromide 9 (6.0 g, 28 mmol) was mixed with copper(I) iodide (0.537 g, 2.82 mmol), PdCl2(PPh3)2 (0.495 g, 0.705 mmol) in DMF (50 mL), treated with triethylamine (12 mL, 87 mmol) and stirred overnight by which the light orange solution had turned brown. The reaction was concentrated, adsorbed on to silica gel and then purified by flash silica gel chromatography (0 to 100% DCM/EtOAc) to provide 2-methylimidazo[1,2-a]pyridine (2.69 g, 20.4 mmol, 70.5 % yield). 1H NMR (400 MHz, CHLOROFORM-d) δ ppm 2.46 (s, 3 H), 6.72 (m, 1 H), 7.11 (ddd, J=9.0, 6.8, 1.4 Hz, 1 H), 7.33 (s, 1 H), 7.54 (m, 1 H), 8.03 (dt, J=6.8, 1.2 Hz, 1 H).
3-(Diphenylphosphino)imidazo[1,2-a]pyridine (11a)
Chlorodiphenylphosphine (1.40 g, 6.35 mmol) and iodotrimethylsilane (1.27 g, 6.35 mmol) were mixed in toluene (4 ml) and stirred for about 2 h. This mixture was transferred to a premixed solution of imidazo[1,2-a]pyridine (500 mg, 4.23 mmol) and triethylamine (2.35 ml, 16.9 mmol) in pyridine (10 ml). The reaction was allowed to stir overnight, concentrated in vacuo, and concentrated with toluene (X2) to remove pyridine. The crude mixture was dissolved in DCM and purified by silica gel chromatography (0 to 100% EtOAc/DCM) to provide 3-(diphenylphosphino)imidazo [1,2-a]pyridine (550 mg, 1.82 mmol, 43.0 % yield). 1H NMR (400 MHz, DMSO-d6) δ ppm 6.94 (td, J=6.8, 1.4 Hz, 1 H), 7.36 (m, 12 H), 7.71 (m, 1 H), 8.16 (m, J=6.8, 2.1, 1.1, 1.1 Hz, 1 H); LC/MS: Method 1, retention time 4.500 min; HRMS (m/z) calcd for C19H16N2P+ (M+H)+ 303.1046, found 303.1049.
3-(Diphenylphosphoryl) imidazo[1,2-a]pyridine, HCl (12a)
3-(Diphenylphosphino)imidazo[1,2-a]pyridine 11a (125 mg, 0.413 mmol) was dissolved in THF and treated with excess 30% hydrogen peroxide (300 mg, 2.65 mmol). The reaction was stirred for 16 h, then diluted with EtOAc and washed with water. The organic layer was dried (MgSO4), filtered, conc, and purified by reverse phase HPLC. The TFA salt obtained was dissolved in CH2Cl2, treatment with excess HCl (4M in Et2O) and then concentration in vacuo (twice) to obtain the HCl salt 3-(diphenylphosphoryl) imidazo[1,2-a]pyridine, HCl (45 mg, 0.13 mmol, 31 % yield). 1H NMR (400 MHz, DMSO-d6) δ ppm 7.39 (m, 1 H), 7.63 (m, 4 H), 7.75 (m, 6 H), 7.87 (m, 1 H), 7.94 (s, 1 H), 8.00 (m, 1 H), 8.78 (d, J=6.8 Hz, 1 H); LC/MS: Method 1, retention time 3.693 min; HRMS (m/z) calcd for C19H16N2OP (M+H)+ 319.0995, found 319.0995.
3-(Diphenylphosphorothioyl)imidazo[1,2-a]pyridine, HCl (12b)
3-(Diphenylphosphino)imidazo [1,2-a]pyridine 11a (125 mg, 0.413 mmol) was dissolved in THF and treated with excess sulfur (50 mg, 1.6 mmol).The reaction was stirred for 16 h, then diluted with EtOAc and washed with water. The organic layer was dried (MgSO4), filtered, concentrated, and purified by reverse phase HPLC. The TFA salt obtained was dissolved in CH2Cl2, treatment with excess HCl (4M in Et2O) and then concentration in vacuo (twice) to obtain the HCl salt 3-(diphenylphosphorothioyl)imidazo[1,2-a]pyridine, HCl (50 mg, 0.135 mmol, 33 % yield). 1H NMR (400 MHz, DMSO-d6) δ ppm 7.19 (t, J=6.8 Hz, 1 H), 7.38 (br. s., 1 H), 7.66 (m, 7 H), 7.80 (m, 4 H), 7.89 (d, J=9.2 Hz, 1 H), 8.52 (d, J=6.7 Hz, 1 H); LC/MS: Method 1, retention time 4.845 min; HRMS (m/z) calcd for C19H16N2PS (M+H)+ 335.0766, found 335.0771.
(2-Methylimidazo[1,2-a]pyridin-3-yl)diphenylphosphine oxide, HCl (12c)
This could be prepared from 2-methylimidazo[1,2-a]pyridine using the procedure to make 12a or using the following procedure: 3-Bromo-2-methylimidazo[1,2-a]pyridine (290 mg, 1.37 mmol, not completely pure) was dissolved in THF (5 ml), cooled to −15 °C under nitrogen. Isopropylmagnesiumchloride.lithium chloride (2 ml, 2 mmol) was added and the reaction allowed to warm to 10 °C at which point it was determined by tlc (1:1 DCM:EtOAc) that the halogen metal exchange was complete. A solution of chlorodiphenylphosphine (300 mg, 1.36 mmol) in THF (1 mL) was added via syringe and the reaction was allowed to gradually warm to rt. The reaction was quenched by addition of sat aq. NH4Cl, extracted with EtOAc. The organic layer was separated, purified by silica gel chromatography (0 to 100% EtOAc/DCM). The product, which was not completely pure by 1H NMR, was taken in THF and treated with excess 30% hydrogen peroxide, stirred overnight. Water was added, extracted with EtOAc, concentrated, and the residue purified by reverse phase HPLC. The fractions were treated with sat. aq. NaHCO3, extracted with EtOAc, dried (MgSO4), filtered, concentrated, redissolved in DCM, converted to the purported HCl salt by treatment with excess 1M HCl in Et2O and then concentration. The residue was redissolved in DCM and treated with Et2O to cause precipitation. The solid obtained was filtered and washed with Et2O to obtaine 3-(diphenylphosphoryl)-2-methylimidazo[1,2-a]pyridine, HCl (30 mg, 0.08 mmol, 5.9 % yield). 1H NMR (400 MHz, DMSO-d6) δ ppm 1.82 (d, J=1.2 Hz, 3 H), 7.24 (m, 1 H), 7.68 (m, 11 H), 7.85 (d, J=8.6 Hz, 1 H), 8.84 (d, J=6.7 Hz, 1 H); LC/MS: Method 1, retention time 3.734 min; HRMS (m/z) calcd for C20H18N2OP+ (M+H)+ 333.1151, found 333.1157.
3-(Diphenylphosphorothioyl)-2-methylimidazo[1,2-a]pyridine (12d)
General Procedure A
Chlorodiphenylphosphine (3.89 ml, 26.4 mmol) and iodotrimethylsilane (5.29 g, 26.4 mmol) were stirred in toluene (10 ml) for 2 h. This was added to a premixed solution of 2-methylimidazo[1,2-a]pyridine 10b (2.33 g, 17.6 mmol) and triethylamine (9.78 ml, 70.5 mmol) in pyridine (10.0 ml) and stirred 12 h. Sulfur (0.565 g, 17.6 mmol) was added and stirred for another 6 h. The reaction was concentrated, concentrated again with toluene (to remove pyridine), and diluted with benzene. The solids (presumed to be Et3N and py salts) were filtered, and the filtrate, adsorbed over silica and subjected to purification by flash silica gel chromatography (0 to 75% EtOAc in DCM) to provide 3-(diphenylphosphorothioyl)-2-methylimidazo[1,2-a]pyridine (3.02 g, 8.67 mmol, 49.2 % yield). 1H NMR (400 MHz, DMSO-d6) δ ppm 1.57 (m, 3 H), 6.96 (m, J=6.7, 6.7, 1.0, 0.7 Hz, 1 H), 7.44 (m, 1 H), 7.60 (m, 4 H), 7.68 (m, 3 H), 7.78 (m, 4 H), 8.35 (dt, J=6.8, 1.2 Hz, 1 H).
3-(Diphenylphosphorothioyl)-1,2-dimethylimidazo[1,2-a]pyridin-1-ium, MeSO4− (13b)
General procedure B
3-(Diphenylphosphorothioyl)-2-methylimidazo[1,2-a]pyridine 12d (90 mg, 0.26 mmol) and dimethyl sulfate (0.05 ml, 0.52 mmol) were taken in dioxane (2 ml). The contents were heated in a sealed tube at 100 °C for 16 h. The reaction was cooled and the solid obtained was filtered, washed with diethyl ether, and air dried to obtain 3-(diphenylphosphorothioyl)-1,2-dimethyl-1H-imidazo[1,2-a]pyridin-4-ium, MeSO4− (50 mg, 0.11 mmol, 41 % yield). 1H NMR (400 MHz, DMSO-d6) δ ppm 1.67 (m, 3 H), 3.91 (s, 3 H), 7.54 (t, J=7.0 Hz, 1 H), 7.68 (td, J=7.6, 3.5 Hz, 4 H), 7.77 (m, 2 H), 7.87 (m, 4 H), 8.15 (t, J=8.3 Hz, 1 H), 8.39 (m, 1 H), 8.66 (d, J=6.8 Hz, 1 H); LC/MS: Method 1, retention time 4.545 min; HRMS (m/z) calcd for C21H20N2PS+ (M)+ 363.1079, found 363.1080.
3-(Diphenylphosphorothioyl)-1-methylimidazo[1,2-a]pyridin-1-ium, MeSO4− (13a)
3-(Diphenylphosphorothioyl)imidazo[1,2-a]pyridine 12b (150 mg, 0.449 mmol) was reacted with dimethyl sulfate (0.086 ml, 0.897 mmol), according to general procedure B, to obtain 3-(diphenylphosphorothioyl)-1-methyl-1H-imidazo[1,2-a]pyridin-4-ium, MeSO4− (70 mg, 0.15 mmol, 34 % yield). 1H NMR (400 MHz, DMSO-d6) δ ppm 4.02 (s, 3 H), 7.65 (m, 5 H), 7.77 (m, 2 H), 7.86 (m, 4 H), 8.08 (dd, J=2.0, 0.4 Hz, 1 H), 8.21 (m, 1 H), 8.3 (m, 1 H), 8.72 (m, 1 H); LC/MS: Method 1, retention time 4.468 min; HRMS (m/z) calcd for C20H18N2PS+ (M)+ 349.0923, found 349.0925.
Imidazo[1,2-a]pyridin-3-yldiphenylmethanol (18a)
3-Bromoimidazo[1,2-a]pyridine (172 mg, 0.873 mmol) 17a was dissolved in THF (5 ml), cooled to −15 °C under nitrogen.
Isopropylmagnesiumchloride.lithium chloride (1.5 ml, 1.5 mmol) was added and the reaction gradually warmed to 10 °C. A solution of benzophenone (175 mg, 0.960 mmol) in THF (1 ml) was added via syringe and the reaction warmed to rt. The reaction was quenched by addition of sat aq. NH4Cl and then extracted with EtOAc. The organic layer was separated, concentrated, and purified by reverse phase HPLC which provided the TFA salt. This was converted to its free base by dissolution in EtOAc and then treatment with sat aq. NaHCO3. The organic phase was dried (MgSO4), filtered, and concentrated. The oily residue was dissolved in minimal CH2Cl2, treated with Et2O, and sonicated to cause precipitation of a solid which was filtered and air dried to provide imidazo[1,2-a]pyridin-3-yldiphenylmethanol (80 mg, 0.27 mmol, 18% yield). 1H NMR (400 MHz, CHLOROFORM-d) δ ppm 3.05 (s, 1 H), 6.63 (m, 1 H), 7.01 (s, 1 H), 7.18 (m, 1 H), 7.35 (m, 8 H), 7.61 (m, 1 H), 8.06 (m, 1H); LC/MS: Method 1, retention time 4.143 min; HRMS (m/z) calcd for C20H17N2O+ (M+H)+ 301.1335, found 301.1337.
(2-Methylimidazo[1,2-a]pyridin-3-yl)diphenylmethanol (18b)
3-Bromo-2-methylimidazo[1,2-a]pyridine (220 mg, 1.04 mmol, not completely pure) was dissolved in THF (5 ml), cooled to −15 °C under nitrogen. Isopropylmagnesiumchloride.lithium chloride (2.7 ml, 2.70 mmol) was added and the reaction was allowed to warm to 10 °C. A solution of benzophenone (208 mg, 1.141 mmol) in THF (1 ml) was added via syringe and the reaction warmed to rt. The reaction was quenched by addition of sat. aq. NH4Cl and then extracted with EtOAc. The organic layer was separated and then purified by silica gel chromatography (0 to 100% EtOAc in DCM) to provide (2-methylimidazo[1,2-a]pyridin-3-yl)diphenylmethanol (100 mg, 0.318 mmol, 30.5 % yield). 1H NMR (400 MHz, DMSO-d6) δ ppm 1.38 (s, 3 H), 6.65 (m, 1 H), 6.93 (s, 1 H), 7.14 (ddd, .J=9.0, 6.7, 1.4 Hz, 1 H), 7.23 (m, 4 H), 7.33 (m, 6 H), 7.43 (dt, .J=9.0, 1.2 Hz, 1 H), 8.11 (dt, .J=7.0, 1.3 Hz, 1H); LC/MS: Method 1, retention time 4.290 min; HRMS (m/z) calcd for C21H19N2O+ (M+H)+ 315.1492, found 315.1495.
3-(Hydroxydiphenylmethyl)-1,2-dimethylimidazo[1,2-a]pyridin-1-ium, TFA (19b)
General Procedure C
(2-Methylimidazo[1,2-a]pyridin-3-yl)diphenylmethanol (32 mg, 0.102 mmol) and dimethyl acetate (15 µl, 0.157 mmol) in dioxane (3.0 ml) were heated in a sealed tube overnight. An oily residue was observed suspended in dioxane. The reaction was concentrated, purified with reverse phase HPLC (25 to 70% MeCN in Water, 0.1% TFA) to provide 3-(hydroxydiphenylmethyl)-1,2-dimethylimidazo[1,2-a]pyridin-1-ium, TFA- (25 mg, 0.057 mmol, 56 % yield) . The counter anion (MeSO4-) was assumed to be exchanged out with TFA-. 1H NMR (400 MHz, DMSO-d6) δ ppm 1.49 (s, 3 H), 3.88 (s, 3 H), 7.37 (m, 11 H), 7.60 (m, 1 H), 7.99 (m, 1 H), 8.25 (d, J=9.2 Hz, 1 H), 8.58 (d, .J=7.0 Hz, 1 H); LC/MS: Method 1, retention time 4.321 min; HRMS (m/z) calcd for C22H21N2O+ (M)+ 329.1648, found 329.1656
3-(Hydroxydiphenylmethyl)-1-methylimidazo[1,2-a]pyridin-1-ium, TFA (19a)
Imidazo[1,2-a]pyridin-3-yldiphenylmethanol 18a (48 mg, 0.16 mmol) and dimethyl sulfate (25 µl, 0.26 mmol) were reacted according to general procedure C to obtain 3-(hydroxydiphenylmethyl)-1-methylimidazo[1,2-a]pyridin-1-ium, TFA− (35 mg, 0.082 mmol, 51 % yield). 1H NMR (400 MHz, DMSO-d6) δ ppm 7.41 (m, 11 H), 7.56 (s, 1 H), 8.05 (ddd, J=9.1, 7.1, 1.2 Hz, 1 H), 8.24 (dt, J=9.2, 1.1 Hz, 1 H), 8.39 (m, 1H); LC/MS: Method 1, retention time 4.074 min; HRMS (m/z) calcd for C21H19N2O+ (M)+ 315.1492, found 315.1497.
(1,2-Dimethyl-1H-indol-3-yl)diphenylphosphine (15b)
Chlorodiphenylphosphine, iodotrimethylsilane, commercially available 1,2-dimethyl-1H-indole, and triethylamine were reacted according to general procedure A to obtain 3-(diphenylphosphorothioyl)-1,2-dimethyl-1H-indole in 40 % yield. 1H NMR (400 MHz, DMSO-d6) δ ppm 2.17 (d, J=1.4 Hz, 3 H), 3.72 (s, 3 H), 6.36 (dt, J=8.0, 1.0 Hz, 1 H), 6.78 (ddd, J=8.2, 7.1, 1.2 Hz, 1 H), 7.10 (ddd, J=8.2, 7.0, 1.2 Hz, 1 H), 7.53 (m, 5 H), 7.59 (m, 2 H), 7.82 (m, 4 H); LC/MS: Method 1, retention time 6.915 min; HRMS (m/z) calcd for C22H21NPS+ (M+H)+ 362.1127, found 362.1134.
(1-Methyl-1H-indol-3-yl)diphenylphosphine sulfide (15a)
Chlorodiphenylphosphine, iodotrimethylsilane, 1-methyl-1H-indole, triethylamine and sulfur were reacted according to general procedure A to produce 3-(diphenylphosphoro thioyl)-1-methyl-1H-indole in 39 % yield. 1H NMR (400 MHz, DMSO-d6) δ ppm 3.85 (s, 3 H), 7.05 (ddd, J=8.1, 7.0, 1.1 Hz, 1 H), 7.25 (ddd, J=8.3, 7.1, 1.3 Hz, 1 H), 7.30 (dt, J=8.0, 1.0 Hz, 1 H), 7.44 (d, J=4.5 Hz, 1 H), 7.56 (m, 7 H), 7.74 (m, 4 H); LC/MS: Method 1, retention time 6.793 min; HRMS (m/z) calcd for C21H19NPS+ (M+H)+ 348.0970, found 348.0974.
The following compounds were prepared by General procedure B or C:
1-Benzyl-3-(diphenylphosphorothioyl)-2-methylimidazo[1,2-a]pyridin-1-ium, TFA− (20a)
1H NMR (400 MHz, DMSO-d6) δ ppm 1.64 (m, 3 H), 5.76 (m, 2 H), 7.30 (m, 2 H), 7.39 (m, 3 H), 7.60 (m, 1 H), 7.68 (m, 4 H), 7.77 (m, 2 H), 7.91 (m, 4 H), 8.19 (m, 1 H), 8.44 (m, 1 H), 8.70 (d, J=6.8 Hz, 1 H); LC/MS: Method 1, retention time: 5.160 min; HRMS (m/z) calcd for C27H24N2PS+ (M)+ 439.1392, found 439.1398.
3-(Diphenylphosphorothioyl)-2-methyl-1-phenethylimidazo[1,2-a]pyridin-1-ium, Br−(20b)
1H NMR (400 MHz, DMSO-d6) δ ppm 1.41 (d, J=1.4 Hz, 3 H), 3.07 (t, J=6.6 Hz, 2 H), 4.66 (t, J=6.8 Hz, 2 H), 7.11 (m, 2 H), 7.23 (m, 3 H), 7.49 (td, J=7.1, 1.3 Hz, 1 H), 7.66 (m, 4 H), 7.78 (m, 6 H), 8.04 (m, 1 H), 8.22 (m, 1 H), 8.65 (m, 1 H); LC/MS: Method 1, retention time: 5.180 min; HRMS (m/z) calcd for C28H26N2PS+ (M)+ 453.1549, found 453.1555.
3-(Diphenylphosphorothioyl)-2-methyl-1-(2-oxo-2-phenylethyl)imidazo[1,2-I]pyridin-1-ium, Br−(20c)
1H NMR (400 MHz, DMSO-d6) δ ppm 1.62 (s, 3 H), 6.37 (s, 2 H), 7.66 (m, 7 H), 7.79 (m, 3 H), 7.91 (m, 4 H), 8.11 (dd, J=8.4, 1.4 Hz, 2 H), 8.18 (m, 1 H), 8.44 (m, 1 H), 8.76 (d, J=6.8 Hz, 1 H); LC/MS: Method 1, retention time 5.391 min; HRMS (m/z) calcd for C28H24N2OPS+ (M)+ 467.1341, found 467.1346.
3-(Diphenylphosphorothioyl)-2-methyl-1-(3-phenylpropyl)imidazo[1,2-a]pyridin-1-ium, TFA−(20d)
1H NMR (400 MHz, DMSO-d6) δ ppm 1.65 (m, 3 H), 2.05 (qd, J=8.0, 7.7 Hz, 2 H), 2.71 (m, 2 H), 4.42 (t, J=7.7 Hz, 2 H), 7.17 (m, 5 H), 7.52 (tt, J=7.0, 0.6 Hz, 1 H), 7.64 (m, 4 H), 7.74 (m, 2 H), 7.83 (m, 4 H), 8.13 (m, 1 H), 8.41 (m, 1 H), 8.62 (d, J=7.0 Hz, 1 H); LC/MS: Method 1, retention time: 5.407 min; HRMS (m/z) calcd for C29H28N2PS+ (M)+ 467.1705, found 467.1709.
1-Cinnamyl-3-(diphenylphosphorothioyl)-2-methylimidazo[1,2-a]pyridin-1-ium, Br−(20e)
1H NMR (400 MHz, DMSO-d6) δ ppm 1.75 (d, J=1.4 Hz, 3 H), 5.28 (m, 2 H), 6.43 (m, 1 H), 6.86 (d, J=16.0 Hz, 1 H), 7.32 (m, 3 H), 7.44 (m, 2H), 7.59 (td, J=7.1, 1.3 Hz, 1 H), 7.68 (m, 4 H), 7.78 (m, 2 H), 7.91 (m, 4 H), 8.20 (ddd, J=9.2, 7.2, 1.2 Hz, 1 H), 8.48 (m, 1 H), 8.70 (m, 1 H) ); LC/MS: Method 1, retention time 5.744 min; HRMS (m/z) calcd for C29H26N2PS+ (M)+ 465.1549, found 465.1553.
3-(Diphenylphosphorothioyl)-2-methyl-1-(4-phenylbutyl)imidazo[1,2-a]pyridin-1-ium, Br− (20f)
1H NMR (400 MHz, DMSO-d6) δ ppm 1.73 (m, 7 H), 2.62 (m, 2 H), 4.43 (m, 2 H), 7.18 (m, 3 H), 7.28 (m, 2 H), 7.55 (td, J=7.0, 1.4 Hz, 1 H), 7.68 (m, 4 H), 7.78 (m, 2 H), 7.89 (m, 4 H), 8.15 (ddd, J=9.1, 7.1, 1.2 Hz, 1 H), 8.42 (dd, J=9.1, 1.1 Hz, 1 H), 8.64 (m, 1 H); LC/MS: Method 1, retention time 5.638 min; HRMS (m/z) calcd for C30H30N2PS+ (M)+ 481.1868, found 481.1871.
3-(Diphenylphosphorothioyl)-2-methyl-1-(5-phenylpentyl)imidazo[1,2-a]pyridin-1-ium, TFA−(20g)
1H NMR (400 MHz, DMSO-d6) δ ppm 1.39 (m, 2 H), 1.61 (m, 2 H), 1.70 (s, 3 H), 1.76 (m, 2 H), 2.57 (t, J=7.4 Hz, 2 H), 4.39 (t, J=7.7 Hz, 2 H), 7.16 (m, 3 H), 7.26 (m, 2 H), 7.55 (m, 1 H), 7.68 (td, J=7.6, 3.5 Hz, 4 H), 7.78 (m, 2 H), 7.89 (ddd, J=14.7, 8.3, 1.3 Hz, 4 H), 8.15 (m, 1 H), 8.43 (m, 1 H), 8.65 (d, J=7.0 Hz, 1 H); LC/MS: Method 1, retention time 5.738 min; HRMS (m/z) calcd for C31H32N2PS+ (M)+ 495.2030, found 495.2033.
(E)-3-(Diphenylphosphorothioyl)-1-(3-(4-fluorophenyl)allyl)-2-methylimidazo[1,2-a]pyridin-1-ium, TFA− (21a)
1H NMR (400 MHz, DMSO-d6) δ ppm 1.75 (d, J=1.4 Hz, 3 H), 3.75 (s, 3 H), 5.27 (m, 2 H), 5.75 (s, 1 H), 6.86 (m, 2 H), 7.01 (m, 2 H), 7.26 (m, 1 H), 7.59 (m, 1 H), 7.69 (m, 4 H), 7.78 (td, J=7.6, 1.6 Hz, 2 H), 7.91 (ddd, J=14.7, 8.4, 1.4 Hz, 4 H), 8.20 (m, 1 H), 8.48 (d, J=9.2 Hz, 1 H), 8.70 (m, 1 H); LC/MS: Method 1, retention time 5.305min; HRMS (m/z) calcd for C30H28N2OPS+ (M)+ 495.1661, found 495.1661.
(E)-1-(3-(2-Chlorophenyl)allyl)-3-(diphenylphosphorothioyl)-2-methylimidazo[1,2-a]pyridin-1-ium, TFA− (21b)
1H NMR (400 MHz, DMSO-d6) δ ppm 1.76 (d, J=1.4 Hz, 3 H), 5.39 (m, 2 H), 6.46 (dt, J=15.8, 6.2 Hz, 1 H), 7.10 (d, J=16.0 Hz, 1 H), 7.33 (m, 2 H), 7.46 (m, 1 H), 7.65 (m, 6 H), 7.78 (m, 2 H), 7.91 (m, 4 H), 8.21 (ddd, J=8.9, 7.3, 1.2 Hz, 1 H), 8.50 (dd, J=9.2, 1.2 Hz, 1 H), 8.70 (m, 1 H); LC/MS: Method 1, retention time 5.440 min; HRMS (m/z) calcd for C29H25ClN2PS+ (M)+ 499.1168, found 499.1170.
(E)-1-(3-(3-Chlorophenyl)allyl)-3-(diphenylphosphorothioyl)-2-methylimidazo[1,2-a]pyridin-1-ium, TFA− (21c)
1H NMR (400 MHz, DMSO-d6) δ ppm 1.75 (m, 3 H), 5.29 (m, 2 H), 6.53 (dt, J=15.9, 6.1 Hz, 1 H), 6.82 (d, J=15.8 Hz, 1 H), 7.36 (m, 3 H), 7.54 (m, 1 H), 7.60 (m, 1 H), 7.69 (m, 4 H), 7.78 (m, 2 H), 7.90 (m, 4 H), 8.20 (m, 1 H), 8.46 (m, 1 H), 8.71 (d, J=7.0 Hz, 1 H); LC/MS: Method 1, retention time 5.520 min; HRMS (m/z) calcd for C29H25ClN2PS+ (M)+ 499.1168, found 499.1163.
(E)-1-(3-(4-Chlorophenyl)allyl)-3-(diphenylphosphorothioyl)-2-methylimidazo[1,2-a]pyridin-1-ium, TFA− (21d)
1H NMR (400 MHz, DMSO-d6) δ ppm 1.75 (d, 3 H), 5.28 (m, 2 H), 6.47 (m, 1 H), 6.84 (d, J=16.0 Hz, 1 H), 7.41 (m, 2 H), 7.47 (m, 2 H), 7.59 (td, J=7.1, 1.2 Hz, 1 H), 7.69 (m, 4 H), 7.78 (m, 2 H), 7.91 (m, 4 H), 8.20 (ddd, J=9.2, 7.2, 1.2 Hz, 1 H), 8.47 (m, 1 H), 8.70 (m, 1 H); LC/MS: Method 1, retention time 5.503 min; HRMS (m/z) calcd for C29H25ClN2PS+ (M)+ 499.1168, found 499.1166.
(E)-3-(Diphenylphosphorothioyl)-2-methyl-1-(3-(4-(trifluoromethyl)phenyl)allyl) imidazo[1,2-a]pyridin-1-ium, TFA− (21e)
1H NMR (400 MHz, DMSO-d6) δ ppm 1.75 (d, J=1.4 Hz, 3 H), 5.32 (dd, J=6.7, 0.9 Hz, 2 H), 6.62 (dt, J=16.1, 6.0 Hz, 1 H), 6.92 (d, J=16.0 Hz, 1 H), 7.59 (td, J=7.0, 1.2 Hz, 1 H), 7.68 (m, 8 H), 7.78 (m, 2 H), 7.91 (m, 4 H), 8.20 (ddd, J=8.9, 7.3, 1.2 Hz, 1 H), 8.47 (dd, J=9.2, 1.0 Hz, 1 H), 8.71 (m, 1 H); LC/MS: Method 1, retention time 5.592 min; HRMS (m/z) calcd for C30H25F3N2PS+ (M)+ 533.1427, found 533.1426.
(E)-1-(3-(4-Bromophenyl)allyl)-3-(diphenylphosphorothioyl)-2-methylimidazo[1,2-a]pyridin-1-ium, TFA− (21f)
1H NMR (400 MHz, DMSO-d6) δ ppm 1.74 (d, J=1.4 Hz, 3 H), 5.26 (dd, J=6.1, 1.2 Hz, 2 H), 6.47 (m, 1 H), 6.81 (d, J=16.2 Hz, 1 H), 7.40 (d, J=8.6 Hz, 2 H), 7.57 (m, 3 H), 7.68 (m, 4 H), 7.78 (m, 2 H), 7.90 (m, 4 H), 8.19 (m, 1 H), 8.46 (dd, J=9.2, 1.2 Hz, 1 H), 8.70 (dd, J=6.9, 1.1 Hz, 1 H); LC/MS: Method 1, retention time 5.555 min; HRMS (m/z) calcd for C29H25BrN2PS+ (M)+ 543.0660, found 543.0657.
(E)-1-(3-(3-Bromophenyl)allyl)-3-(diphenylphosphorothioyl)-2-methylimidazo[1,2-a]pyridin-1-ium, TFA− (21g)
1H NMR (400 MHz, CHLOROFORM-d) δ ppm 1.79 (d, J=0.4 Hz, 3 H), 5.35 (m, 2 H), 6.20 (m, 1 H), 6.57 (d, J=15.1 Hz, 1 H), 7.18 (t, J=7.7 Hz, 1 H), 7.26 (m, 2 H), 7.40 (m, 1 H), 7.47 (t, J=1.7 Hz, 1 H), 7.63 (m, 6 H), 7.96 (m, 5 H), 8.13 (m, 1 H), 8.81 (d, J=6.8 Hz, 1 H); LC/MS: Method 1, retention time 5.552 min; HRMS (m/z) calcd for C29H25BrN2PS+ (M)+ 543.0660, found 543.0662.
(E)-1-(3-(3-bromo-4-fluorophenyl)allyl)-3-(diphenylphosphorothioyl)-2-methylimidazo[1,2-a]pyridin-1-ium, TFA− (21h)
1H NMR (400 MHz, CHLOROFORM-d) δ ppm 1.79 (d, J=1.4 Hz, 3 H), 5.35 (m, 2 H), 6.14 (m, 1 H), 6.58 (d, J=15.8 Hz, 1 H), 7.07 (t, J=8.3 Hz, 1 H), 7.27 (m, 2 H), 7.53 (dd, J=6.6, 2.2 Hz, 1 H), 7.59 (m, 4 H), 7.67 (m, 2 H), 7.96 (m, 5 H), 8.13 (d, J=9.2 Hz, 1 H), 8.80 (d, J=6.7 Hz, 1 H); LC/MS: Method 1, retention time 5.566 min; HRMS (m/z) calcd for C29H24BrFN2PS+ (M)+ 561.0564, found 561.0565.
(E)-3-(Diphenylphosphorothioyl)-2-methyl-1-(3-(naphthalen-2-yl)allyl)imidazo[1,2-a]pyridin-1-ium, Br− (21i)
LC/MS: Method 1, retention time 5.633 min; HRMS (m/z) calcd for C33H28N2PS+ (M)+, 515.1712 found 515.1706.
(E)-3-(Diphenylphosphorothioyl)-1-(3-(4-fluorophenyl)allyl)-2-methylimidazo[1,2-a]pyridin-1-ium, TFA− (21j)
LC/MS: Method 1, retention time 5.313 min; HRMS (m/z) calcd for C29H25FN2PS+ (M)+ 483.1466, found 483.1465.
(E)-3-(diphenylphosphorothioyl)-2-methyl-1-(3-p-tolylallyl)imidazo[1,2-a]pyridin-1-ium, Br−(21k)
HRMS (m/z) calcd for C30H28N2PS+ (M)+ 479.1712, found 479.1716; LC/MS: Method 1, retention time 5.491 min.
Supplementary Material
Scheme 2.

aSynthetic Scheme towards tertiary alcohols 19a,b.
aReagents and conditions. a) NBS; b) iPrMgCl.LiCl, THF, Ph2CO, 18% yield for 18a, 31% for 18b; c) MeI, 1,4-dioxane, sealed tube, 100–120 °C, 50–60% yield.
Table 4.
A comparison of key potent NPSR antagonists.
| % parent compd remaining after incubation in mouse liver microsomes |
|||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| [125I] | with NADPH | Without NADPH | |||||||||||
| Ca2+ | cAMP | ERK | Y10- | ||||||||||
| # | IC50 | IC50 | IC50 | hNPS | |||||||||
| nMa | nMa | nMa | IC50 | ||||||||||
| nMa | |||||||||||||
| T min |
0 | 15 | 30 | 60 | 0 | 15 | 30 | 60 | |||||
| 1b | 5.3 | 420 | 14 | 25 | 100 | 29 | 9 | 2 | 98 | 105 | 96 | 101 | |
| 2 | 1.2 | 55 | 11 | 6.7 | 100 | 0 | 0 | 0 | 96 | 103 | 97 | 94 | |
| 20e | 0.96 | 45 | 1.3 | 3.5 | 100 | 72 | 46 | 26 | 94 | 102 | 102 | 102 | |
The cAMP and Ca2+ assay activity reported here is the mean IC50 from two separate experiments, each ran in duplicate. The ERK, [125I] Y10-hNPS displacement, and microsome assay results reported here are the mean values from respective single experiments that were run in duplicate.
Acknowledgments
This research was supported by the Molecular Libraries Initiative of the National Institutes of Health Roadmap for Medical Research and the Intramural Research Programs of the National Institute on Alcohol Abuse and Alcoholism, NIH. We thank Dr. Reinscheid for generously providing the NPSR clone originally used to generate NPSR-CHO cell line; Sam Michael for assistance in robotic screen; William Leister, Paul Shinn, Jeremy Smith, Danielle van Leer, and James Bougie for compound management.
ABBREVIATIONS USED
- NPS
neuropeptide S
- NPSR
neuropeptide S receptor
- cAMP
cyclic adenosine monophosphate
- MAPK
Mitogen-activated protein kinase
- SAR
Structure Activity Relationship
- qHTS
quantitative high throughput screening
- HTRF
Homogeneous Time Resolved Fluorescence
- ERK
extracellular signal-regulated kinase
- Tyr
Tyrosine
- G-protein
Guanosine nucleotide-binding protein
- icv
intracerebroventricular
- IP
intraperitoneal
- mpk
milligrams per kilogram
- DAMGO
[D-Ala2, N-MePhe4, Gly-ol]-enkephalin
Footnotes
ASSOCIATED CONTENT
Supporting Information. This contains the profile of 20e against 55 other targets and 2D NMR spectra for 13b. This material is available free of charge via the Internet at http://pubs.acs.org.”
References
- 1.Holmes A, Heilig M, Rupniak NMJ, Steckler T, Griebel G. Neuroropeptide systems as novel therapeutic targets for depression and anxiety disorders. Trends Pharmacol. Sci. 2003;24:580–588. doi: 10.1016/j.tips.2003.09.011. [DOI] [PubMed] [Google Scholar]
- 2.Sato S, Shintani Y, Miyajima N, Yoshimura K. Novel G protein-coupled receptor protein and dna thereof. PCT Int. Appl. 2002 WO 2002031145 A1 20020418. [Google Scholar]
- 3.Xu YL, Reinscheid RK, Huitron-Resendiz S, Clark SD, Wang Z, Lin SH, Brucher FA, Zeng J, Ly NK, Henriksen SJ, de Lecea L, Civelli O. Neuropeptide S: a neuropeptide promoting arousal and anxiolytic-like effects. Neuron. 2004;43:487–497. doi: 10.1016/j.neuron.2004.08.005. [DOI] [PubMed] [Google Scholar]
- 4.Xu YL, Gall CM, Jackson VR, Civelli O, Reinscheid RK. Distribution of neuropeptide S receptor mRNA and neurochemical characteristics of neuropeptide S-expressing neurons in the rat brain. J. Comp. Neurol. 2007;500:84–102. doi: 10.1002/cne.21159. [DOI] [PubMed] [Google Scholar]
- 5.Reinscheid RK, Xu YL, Okamura N, Zeng J, Chung S, Pai R, Wang Z, Civelli O. Pharmacological characterization of human and murine neuropeptide s receptor variants. J. Pharmacol. Exp. Ther. 2005;315:1338–1345. doi: 10.1124/jpet.105.093427. [DOI] [PubMed] [Google Scholar]
- 6.Laitinen T, Polvi A, Rydman P, Vendelin J, Pulkkinen V, Salmikangas P, Makela S, Rehn M, Pirskanen A, Rautanen A, Zucchelli M, Gullstén H, Leino M, Alenius H, Petäys T, Haahtela T, Laitinen A, Laprise C, Hudson TJ, Laitinen LA, Kere J. Characterization of a common susceptibility locus for asthma-related traits. Science. 2004;304:300–304. doi: 10.1126/science.1090010. [DOI] [PubMed] [Google Scholar]
- 7.Koob GF, Greenwell TN. Neuropeptide S: a novel activating anxiolytic? Neuron. 2004;43:441–442. doi: 10.1016/j.neuron.2004.08.007. [DOI] [PubMed] [Google Scholar]
- 8.Beck B, Fernette B, Stricker-Krongrad A. Peptide S is a novel potent inhibitor of voluntary and fast-induced food intake in rats. Biochem. Biophys. Res. Commun. 2005;332:859–865. doi: 10.1016/j.bbrc.2005.05.029. [DOI] [PubMed] [Google Scholar]
- 9.Cannella N, Economidou D, Kallupi M, Stopponi S, Heilig M, Massi M, Ciccocioppo R. Persistent increase of alcohol-seeking evoked by neuropeptide S: an effect mediated by the hypothalamic hypocretin system. Neuropsychopharmacology. 2009;34:2125–2134. doi: 10.1038/npp.2009.37. [DOI] [PubMed] [Google Scholar]
- 10.Smart D, Sabido-David C, Brough SJ, Jewitt F, Johns A, Porter RA, Jerman JC. SB-334867-A: the first selective orexin-1 receptor antagonist. Br. J. Pharmacol. 2001;132:1179–1182. doi: 10.1038/sj.bjp.0703953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.(a) Pañeda C, Huitron-Resendiz S, Frago LM, Chowen JA, Picetti R, de Lecea L, Roberts AJ. Neuropeptide S reinstates cocaine-seeking behavior and increases locomotor activity through corticotropin-releasing factor receptor 1 in mice. J. Neurosci. 2009;29:4155–4161. doi: 10.1523/JNEUROSCI.5256-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Li W, Gao Y, Chang M, Peng Y, Yao J, Han R, Wang R. Neuropeptide S inhibits the acquisition and the expression of conditioned place preference to morphine in mice. Peptides. 2009;30:234–240. doi: 10.1016/j.peptides.2008.10.004. [DOI] [PubMed] [Google Scholar]
- 12.(a) Guerrini R, Salvadori S, Rizzi A, Regoli D, Calo G. Neurobiology, pharmacology, and medicinal chemistry of neuropeptide S and its receptor. Med. Res. Rev. 2010;30:751–777. doi: 10.1002/med.20180. [DOI] [PubMed] [Google Scholar]; (b) Pape H-C, Juengling K, Seidenbecher T, Lesting J, Reinscheid RK. Neuropeptide S: a transmitter system in the brain regulating fear and anxiety. Neuropharmacology. 2009;58:29–34. doi: 10.1016/j.neuropharm.2009.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Lei Z, Yao Y, Li Y, Yang G. Xumu Yu Shouyi. 2008;40:97–100. [Google Scholar]; (d) Okamura N, Reinscheid RK. Neuropeptide S: a novel modulator of stress and arousal. Stress. 2007;10:221–226. doi: 10.1080/10253890701248673. [DOI] [PubMed] [Google Scholar]
- 13.(a) Guerrini R, Camarda V, Trapella C, Calo G, Rizzi A, Ruzza C, Fiorini S, Marzola E, Reinscheid RK, Regoli D, Salvadori S. Further studies at neuropeptide s position 5: discovery of novel neuropeptide S receptor antagonists. J. Med. Chem. 2009;52:4068–4071. doi: 10.1021/jm900604g. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Guerrini R, Camarda V, Trapella C, Calo G, Rizzi A, Ruzza C, Fiorini S, Marzola E, Reinscheid RK, Regoli D, Salvadori S. Synthesis and biological activity of human neuropeptide S analogues modified in position 5: identification of potent and pure neuropeptide S receptor antagonists. J. Med. Chem. 2009;52:524–529. doi: 10.1021/jm8012294. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Camarda V, Trapella C, Calo G, Guerrini R, Rizzi A, Ruzza C, Fiorini S, Marzola E, Reinscheid RK, Regoli D, Salvadori S. Synthesis and biological activity of human neuropeptide S analogues modified in position 2. J. Med. Chem. 2008;51:655–658. doi: 10.1021/jm701204n. [DOI] [PubMed] [Google Scholar]
- 14.Fukatsu K, Nakayama Y, Tarui N, Mori M, Matsumoto H, Kurasawa O, Banno H. Bicyclic piperazine compound and use thereof. PCT Int. Appl. 2005 WO 2005021555 A1 20050310. [Google Scholar]
- 15.Okamura N, Habay SA, Zeng J, Chamberlin AR, Reinscheid RK. Synthesis and pharmacological in vitro and in vivo profile of 3-oxo-1,1-diphenyl-tetrahydro-oxazolo[3,4-a]pyrazine-7-carboxylic acid 4-fluoro-benzylamide (SHA 68), a selective antagonist of the neuropeptide S receptor. J. Pharmacol. Exp. Ther. 2008;325:893–901. doi: 10.1124/jpet.107.135103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Trapella C, Pela M, Del Zoppo L, Calo G, Camarda V, Ruzza C, Cavazzini A, Costa V, Bertolasi V, Reinscheid RK, Salvadori S, Guerrini R. Synthesis and separation of the enantiomers of the neuropeptide S receptor antagonist (9R/S)-3-oxo-1,1-diphenyl-tetrahydro-oxazolo[3,4-a]pyrazine-7-carboxylic acid 4-fluoro-benzylamide (SHA 68) J. Med. Chem. 2011;54:2738–2744. doi: 10.1021/jm200138r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Melamed JY, Zartman AE, Kett NR, Gotter AL, Uebele VN, Reiss DR, Condra CL, Fandozzi C, Lubbers LS, Rowe BA, McGaughey GB, Henault M, Stocco R, Renger JJ, Hartman GD, Bilodeau MT, Trotter BW. Synthesis and evaluation of a new series of Neuropeptide S receptor antagonists. Bioorg. Med. Chem. Lett. 2010;20:4700–4703. doi: 10.1016/j.bmcl.2010.04.143. [DOI] [PubMed] [Google Scholar]
- 18.Trotter BW, Nanda KK, Manley PJ, Uebele VN, Condra CL, Gotter AL, Menzel K, Henault M, Stocco R, Renger JJ, Hartman GD, Bilodeau MT. Tricyclic imidazole antagonists of the Neuropeptide S Receptor. Bioorg. Med. Chem. Lett. 2010;20:4704–4708. doi: 10.1016/j.bmcl.2010.04.016. [DOI] [PubMed] [Google Scholar]
- 19.Micheli F, Di Fabio R, Giacometti A, Roth A, Moro E, Merlo G, Paio A, Merlo-Pich E, Tomelleri S, Tonelli F, Zarantonello P, Zonzini L, Capelli AM. Synthesis and pharmacological characterization of 5-phenyl-2-[2-(1-piperidinylcarbonyl)phenyl]-2,3-dihydro-1H–pyrrolo[1,2-c]imidazol-1-ones: a new class of Neuropeptide S antagonists. Bioorg. Med. Chem. Lett. 2010;20:7308–7311. doi: 10.1016/j.bmcl.2010.10.067. [DOI] [PubMed] [Google Scholar]
- 20.Fretz H, Gatfield J, Isler M, Kimmerlin T, Koberstein R, Lyothier I, Monnier L, Pothier J, Valdenaire A. Indanone and indandione derivatives and heterocyclic analogs. PCT Int. Appl. 2013 WO 2013068785 A1 20130516. [Google Scholar]
- 21.McCoy JG, Marugan JJ, Liu K, Zheng W, Southall N, Heilig M, Austin CA. Selective Modulation of Gq/Gs pathways by Naphtho Pyrano Pyrimidines as antagonists of the Neuropeptide S Receptor. ACS Chem. Neurosci. 2010;1:559–574. doi: 10.1021/cn100040h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dal Ben D, Antonini I, Buccioni M, Lambertucci C, Marucci G, Thomas A, Volpini R, Cristalli G. Neuropeptide S receptor: recent updates on nonpeptide antagonist discovery. Neuropeptide S receptor: recent updates on nonpeptide antagonist discovery. ChemMedChem. 2011;6:1163–1171. doi: 10.1002/cmdc.201100038. [DOI] [PubMed] [Google Scholar]
- 23.Pub Chem AID: 1461 (http://pubchem.ncbi.nlm.nih.gov/assay/assay.cgi?aid=1461&loc=ea_ras)
- 24.Available commercially at Enamine, Cat# T0504-5950.
- 25.Bakherad M, Nasr-Isfahani H, Keivanloo A, Bakherad M, Doostmohammadi N. Pd-Cu catalyzed heterocyclization during Sonogashira coupling: synthesis of 2-benzylimidazo[1,2-a]pyridine. Tetrahedron Lett. 2008;49:3819–3822. [Google Scholar]
- 26.(a) Tolmachev AA, Yurchenko AA, Kozlov ES, Merkulov AS, Semenova MG, Pinchuk AM. Phosphorylated Imidazo[1,2-a] pyridines. Heteroatom Chem. 1995;5:419–432. [Google Scholar]; (b) Tolmachev AA, Yurchenko AA, Kozlov ES. Phosphorylation of 2-methylimidazo[1,2-a]pyridine. Rare cases of nitrogen-atom alkylation of a phosphine and phosphonite. J. Gen. Chem. USSR (Engl. Transl.) 1992;62:1369–1371. [Google Scholar]
- 27.Romanenko VD, Tovstenko VI, Markovski LN. Applications of iodotrimethylsilane for the Synthesis of iodophosphines, iodophosphoranes, and iodomethylphosphine oxides. Synthesis. 1980;10:823–825. [Google Scholar]
- 28.The alkylation proceeds at the N1 position as expected from similar compounds described earlier in the literature (ref. 25). The regiochemistry of alkylation was also proved by the interpretation of COSY, HMQC, HMBC, and NOESY spectra of 13b (see Supplementary information). The masses of fragments acquired via mass spectroscopy of 13a, 13b, and 20e also support alkylation at N1 of the imidazopyridine.
- 29.Benincori T, Piccolo O, Rizzo S, Sannicolò F. 3,3’-Bis(diphenylphosphino)-1,1’-disubstituted-2,2’-biindoles: easily accessible, electron-rich, chiral diphosphine ligands for homogeneous enantioselective hydrogenation of oxoesters. J. Org. Chem. 2000;65:8340–8347. doi: 10.1021/jo001207d. [DOI] [PubMed] [Google Scholar]
- 30.Krasovskiy A, Knochel P. A LiCl-mediated Br/Mg exchange reaction for the preparation of functionalized aryl- and heteroarylmagnesium compounds from organic bromides. Angew. Chem. Int. Ed. 2003;43:3333–3336. doi: 10.1002/anie.200454084. [DOI] [PubMed] [Google Scholar]
- 31.The IC50s reported in Tables 1–3 are calculated from assays that were performed in 1536-well format. We observed slightly weaker potencies while testing in a 386-well format. Also, it appeared that the chemical series discussed in this communication, as exemplified by 7 and 20e, had enhanced potencies towards inhibition of ERK phosphorylation in the 386-well format. This observation has been emphasized in a separate communication.34
- 32.Fedeli A, Braconi S, Economidou D, Cannela N, Kallupi M, Guerrini R, Calò G, Cifani C, Massi M, Ciccocioppo R. The paraventricular nucleus of the hypothalamus is a neuroanatomical substrate for the inhibition of palatable food intake by neuropeptide S. Eur. J. Neurosci. 2009;30:1594–1602. doi: 10.1111/j.1460-9568.2009.06948.x. [DOI] [PubMed] [Google Scholar]
- 33.Li Di L, Rong H, Feng B. Demystifying brain penetration in central nervous system drug discovery. Miniperspective. J. Med. Chem. 2013;56:2–12. doi: 10.1021/jm301297f. [DOI] [PubMed] [Google Scholar]
- 34.Thorsell A, Tapocik JD, Liu K, Zook M, Bell L, Flanigan M, Patnaik S, Marugan J, Damadzic R, Dehdashti S, Schwandt ML, Southall N, Austin CA, Eskay R, Ciccocioppo R, Zheng W, Heilig M. A novel brain penetrant NPS receptor antagonist, NCGC00185684, blocks alcohol-induced ERK-phosphorylation in the central amygdala and decreases operant alcohol self-administration in rats. J. Neurosci. 2013;33:10132–10142. doi: 10.1523/JNEUROSCI.4742-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Calculated properties for 20e as per ChemBioDraw Ultra 12.0: ClogP 3.33, tPSA 6.25. Measured solubility of 20e in PBS: kinetic solubility >150 µM, equilibrium solubility ∼45 µM.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.