

SUMMARY
Sudden unexplained death in epilepsy (SUDEP) is the most common cause of premature mortality in epilepsy and was linked to mutations in ion channels; however, genes within the channel protein interactome might also represent pathogenic candidates. Here we show that mice with partial deficiency of Sentrin/SUMO-specific protease 2 (SENP2) develop spontaneous seizures and sudden death. SENP2 is highly enriched in the hippocampus, often the focus of epileptic seizures. SENP2 deficiency results in hyper-SUMOylation of multiple potassium channels known to regulate neuronal excitability. We demonstrate that the depolarizing M-current conducted by Kv7 channel is significantly diminished in SENP2-deficient hippocampal CA3 neurons, primarily responsible for neuronal hyperexcitability. Following seizures, SENP2-deficient mice develop atrioventricular conduction blocks and cardiac asystole. Both seizures and cardiac conduction blocks can be prevented by retigabine, a Kv7 channel opener. Thus, we uncover a disease-causing role for hyper-SUMOylation in the nervous system and establish an animal model for SUDEP.
INTRODUCTION
Epilepsy is a major medical condition with inherited and acquired forms. Among those epileptic conditions linked to channelopathies, mutations in potassium channel subunits represent the largest category (Brenner and Wilcox, 2012; Cooper, 2012; Noebels, 2003). It has been estimated that the rate of sudden death is 20-fold higher in epilepsy patients than in the general population, and sudden unexplained death in epilepsy (SUDEP) represents the most common epilepsy-related cause of death (Sillanpaa and Shinnar, 2010). The cause of SUDEP in human has not been determined. In animal models, inactivation of potassium channels genes has been linked to SUDEP (Goldman et al., 2009; Glasscock et al., 2010). These animal models demonstrated an important connection between the brain and the heart. However, it remains unclear whether seizure and sudden death are two separate manifestations of potassium channel deficiency in the brain and the heart or seizure predisposes the heart to lethal cardiac arrhythmia and death.
Inherited disorders of ion channels are a major source of human disease in excitable tissues. Mutation of individual subunits of these heteromeric complexes gives rise to a wide variety of neural and cardiac excitability disorders, yet genes involved in their posttranslational modification within the membrane may also be disease producing (Herren et al., 2013). For example, Kv2.1, a voltage-gated potassium channel, contains 16 serine phosphorylation sites in the cytoplasmic domain. Mutational analysis revealed that phosphorylation at multiple series provided graded activity-dependent regulation of channel activity (Park et al., 2006). Another posttranslational modification that has been shown to affect Kv2.1 channel activity is SUMOylation (Dai et al., 2009; Plant et al., 2011). Small ubiquitin-like modifier (SUMO) covalently modifies a large number of cellular proteins; SUMOylation is implicated in the regulation of multiple cellular processes through its ability to alter protein localization, function, or protein-protein interaction (Yeh, 2009).
SUMOylation is catalyzed by SUMO-specific E1, E2, and E3s and can be reversed by a family of Sentrin/SUMO-specific proteases (SENPs) (Yeh et al., 2000). There are three different SUMOs: SUMO-1, SUMO-2, and SUMO-3. SUMO-2 and SUMO-3 are closely related and are usually called SUMO-2/3. SUMO-1 modifies its substrate as a monomer, whereas SUMO-2/3 forms polymeric chains. The SUMOylation status of a particular substrate is dictated by the balance among SUMO E1, E2, E3, and SENPs. There are six SENPs with different substrate specificities (Yeh, 2009). Although the biochemical properties of SENPs have been well documented, their specific targets and physiological roles are known only in a limited number of cases. SENP1 or SENP2 knockout mouse embryos do not survive to birth (Cheng et al., 2007; Kang et al., 2010), suggesting that the SENPs are not redundant and have distinct substrate specificity during development. SENP1 plays a key role in the hypoxic response by regulating HIF1a stability (Cheng et al., 2007), whereas SENP2 is involved in the binding of polycomb complex to H3K27me3 (Kang et al., 2010) and in regulating myostatin expression and myogenesis (Qi et al., 2014).
SUMOylation has also been shown to regulate ion channel activity. For example, SUMOylation of K2P1 or Kv1.5 can inactivate these potassium channels in Xenopus oocytes and myocytes, and SUMOylation of Kv2.1 increases the excitability of hippocampal neurons (Benson et al., 2007; Plant et al., 2010; Plant et al., 2011; Rajan et al., 2005). However, it is not known whether ion channel regulation by SUMO in vitro may be implicated in common disease phenotypes, and whether it is translatable to animal models. In this report, we show that SENP2 deficiency produces an animal model of seizures and sudden death closely resembling SUDEP. We provide evidence that SENP2 deficiency leads to hyper-SUMOylation of both Kv1.1 and Kv7.2/Kv7.3 in hippocampal neurons. However, SUMOylation of Kv1.1 does not significantly affect its channel activity. Remarkably, hyper-SUMOylation of Kv7.2/Kv7.3 significantly diminishes the M-current, which is primarily responsible for neuronal hyperexcitability. The identification of the M-current as a main culprit in SENP2 deficiency is supported by the observations that both seizures and AV blocks can be prevented by the Kv7 channel opener retigabine. Our findings unravel an important physiological role of SENP2 in regulation of the M-current and that reversing SUMOylation may present a new approach for treating subsets of individuals with SUDEP.
RESULTS
Seizures and Sudden Death in SENP2-Deficient Mice
Because SENP2 is required for expression of key developmental genes, SENP2 deletion is embryonic lethal (Kang et al., 2010). To explore the potential role of SENP2 in the regulation of biological processes in adult mice, we generated mice homozygous for a floxed SENP2 allele that contained a neomycin insert (SENP2fxN/fxN), which displayed a postnatal phenotype (Figures S1A and S1B available online). The SENP2fxN/fxN mice appeared normal at birth; however, by the sixth postnatal week, these mice developed spontaneous convulsive seizures, followed by sudden death at 6–8 weeks postpartum with 100% penetrance (Figure 1A; Movie S1). To determine whether retention of the neomycin cassette in the floxed allele may have contributed to early lethality in SENP2fxN/fxN mice due to neighborhood effects (Ren et al., 2002), we removed the cassette by FLP-mediated recombination. The homozygous mice without neomycin cassette (SENP2fx/fx) had a normal lifespan.
Figure 1. SENP2 Deficiency Causes Seizures and Sudden Death.
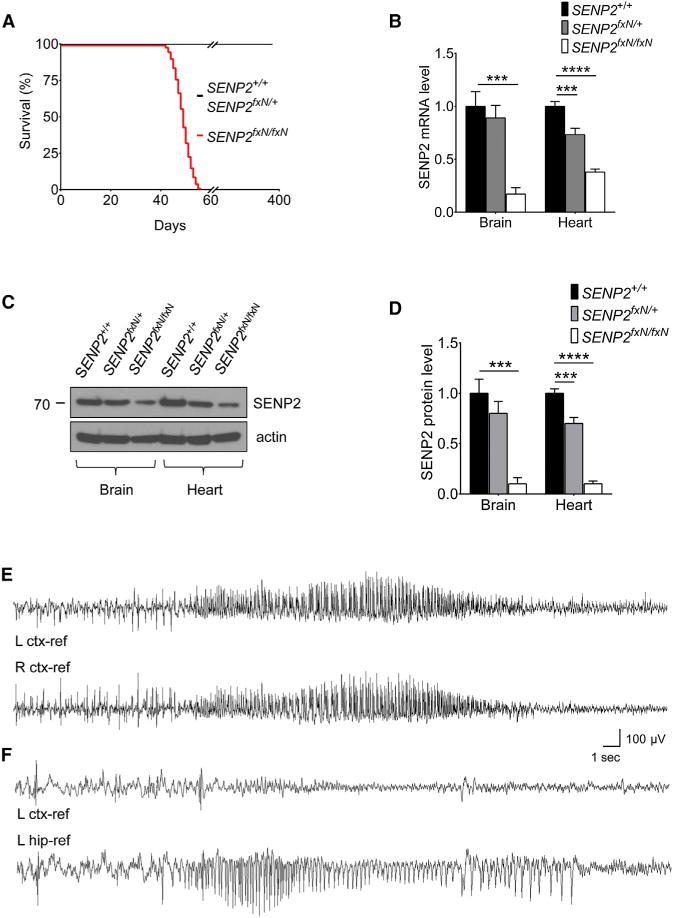
(A) Lifespan of SENP2+/+ (n = 120), SENP2fxN/+ (n = 108), and SENP2fxN/fxN (n = 110) mice.
(B) Expression level of SENP2 transcripts in brain and heart of SENP2+/+, SENP2fxN/+ and SENP2fxN/fxN mice. SENP2 transcript level was measured by real-time PCR, normalized to SENP2+/+ control (n = 3 mice/group). The ANOVA revealed a significant differences between the groups from brain (F[2,6] = 48.94, p < 0.001) and heart (F[2,6] = 137.1, p < 0.0001).
(C) Western blot analysis of brain and heart extracts of SENP2+/+, SENP2fxN/+, and SENP2fxN/fxN mice with anit-SENP2 and anti-actin antibodies.
(D) The ratios of SENP2 protein to actin of SENP2+/+, SENP2fxN/+, and SENP2fxN/fxN mice were normalized to SENP2+/+ mice (n = 3 mice/group). The ANOVA revealed a significant differences between the groups from brain (F[2,6] = 53.9, p < 0.001) and heart (F[2,6] = 296.8, p < 0.0001).
(E) Representative EEG seizures recorded from awake SENP2fxN/fxN mice at 7 weeks of age (n = 3). EEG traces revealed baseline low-amplitude desynchronized activity with frequent sharp interictal spikes followed by onset of spontaneous non-convulsive bilateral seizure.
(F) Representative traces recorded simultaneously from the left cortex (upper) and the left hippocampus (lower) of SENP2fxN/fxN mice (n = 3). EEG traces showed isolated electrographic hippocampal seizure; overlying cortex displays only a decremental activity EEG pattern.
Values are shown as mean ± SEM. ***p < 0.001; ****p < 0.0001 by one-way ANOVA followed by Dunnett’s test. See also Figures S1–S3 and Movies S1, S2, and S3.
Inclusion of the neomycin cassette caused a reduction in SENP2 transcripts (Figure 1B), and SENP2 protein level was markedly reduced in SENP2fxN/fxN mice when compared with SENP2+/+ or SENP2fxN/+ mice (Figures 1C and 1D), while the SENP2 mRNA and protein levels were restored to normal in SENP2fx/fx mice after removal of the neomycin cassette (Figures S2A–S2C). The neomycin cassette insertion primarily affected the SENP2 transcription because transcription of the Gm4943 gene, which overlapped with SENP2 gene, was not affected by insertion of neomycin cassette (Figure S2D). The reduction of SENP2 protein led to accumulation of SUMO-1- or SUMO-2/3-conjugated proteins in both the brain and cardiac tissues (Figures S3A–S3C). This was not due to an increase in the expression of either SUMO-1 or SUMO-2 message in the SENP2fxN/fxN mice (Figures S3D and S3E). Thus, SENP2 deficiency resulted in a hyper-SUMOylation state.
To determine the origin of the seizure phenotype, we monitored freely moving adult mice implanted with chronic cortical and hippocampal electroencephalogram (EEG) electrodes. Video EEG recordings revealed low-amplitude desynchronized cortical EEG activity and frequent bilateral interictal spike discharges (10–120/hr) in SENP2fxN/fxN, but not in SENP2+/+, mice (Figure 1E). Two types of spontaneous seizures, typically 10–30 s in duration, were observed. The first type was bilaterally generalized electrographic episodes with high voltage, rhythmic spiking, and slower poly-spike/waves, terminating with minimal postictal depression. During these purely electrographic seizures, the mice continued to engage in exploratory behavior, grooming, or remained motionless or asleep. Depth electrodes revealed the presence of isolated hippocampal seizures that failed to propagate to overlying neocortex (Figure 1F; Movie S2), where only a decremental EEG pattern was evident without interruption of behavior, lasting about 30 s. The second seizure type consisted of wild running fits, involving convulsive tonic and clonic movements of all limbs and trunk, with absent minimal cortical discharge, likely of brainstem origin without high-amplitude cortical discharges, followed by postictal rigidity (Movie S3), lasting about 15 s, and ultimately death.
Hyperexcitability in SENP2-Deficient Hippocampal Neurons
Since SENP2 is highly expressed in the hippocampal formation (Allen Brain Atlas; http://www.brain-map.org), we first examined the excitability of these neuronal networks in brain slices. SENP2+/+ or SENP2fxN/fxN brain slices did not demonstrate spontaneous field discharges in the CA3 pyramidal cell region when bathed in normal saline solution. However, elevating the extracellular K+ concentration from 2.5 mM to 7.5 mM induced a significantly higher discharge rate in SENP2fxN/fxN slices as compared to SENP2+/+, indicating an intrinsic network hyperexcitability in SENP2fxN/fxN mice (Figures 2A and 2B).
Figure 2. Hyperexcitability of Neuronal Network in SENP2-Deficient Neurons.
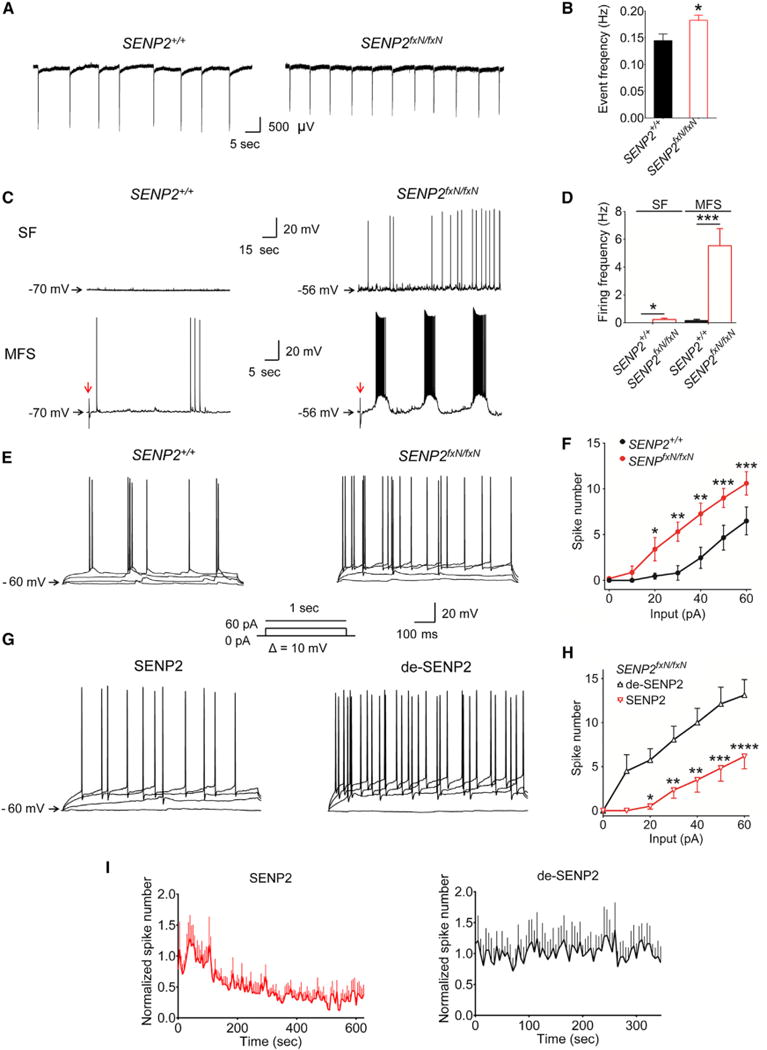
(A) Representative traces of firing frequency in the CA3 neurons from the field recordings in SENP2+/+ and SENP2fxN/fxN brain slices superfused with artificial CSF containing 7.5 mM potassium.
(B) Mean value of firing frequency in the CA3 neurons in the SENP2fxN/fxN brain slices (n = 33) was markedly increased compared with that in the SENP2+/+ slices (n = 34).
(C) Representative traces of spontaneous firing (SF) and evoked firing of hippocampal CA3 neurons by single mossy fiber stimulation (MFS) recorded from SENP2+/+ and SENP2fxN/fxN brain slices. The red arrows indicate the peaks elicited by MFS. The repetitive bursting behavior can only be evoked by MFS in the SENP2fxN/fxN brain slices.
(D) Mean values of the SF and MFS-evoked firing frequencies in SENP2fxN/fxN neurons (n = 12 in SF and 9 in MFS) were markedly increased when compared with the SENP2+/+ neurons (n = 15 in SF and 10 in MFS).
(E) Representative traces of CA3 neuronal firing evoked by current injection (input = 0, 20, 40, and 60 pA) in SENP2+/+ and SENP2fxN/fxN mice (current injection protocol shown in the insert).
(F) Mean values of the current injection evoked firing of CA3 neurons were significantly increased in SENP2fxN/fxN brain slices (n = 16) compared to SENP2+/+ controls (n = 17). The ANOVA revealed a significant differences between the groups (F[1,217] = 34.89, p < 0.0001).
(G) Representative traces of CA3 neuronal firing evoked by current injection (input = 0, 20, 40, and 60 pA) with SENP2 (250 pM) or heat-denatured, inactive SENP2 (de-SENP2, 250 pM) peptide in pipette solution (current injection protocol in the insert).
(H) Inclusion of SENP2 (n = 10) but not heat-denatured SENP2 (n = 8) peptide in the pipette solution reduced hyperexcitability of CA3 neurons in SENP2fxN/fxN mice. The ANOVA revealed a significant differences between the groups (F[1,77] = 65.22, p < 0.0001).
(I) Time course of SENP2 or de-SENP2 on CA3 neuronal firing frequency. Application of SENP2 (n = 9) but not de-SENP2 (n = 5) peptide significantly reduced the firing frequency in CA3 neurons of SENP2fxN/fxN mice. Peptides were dissolved in the pipette solution, and the firings were evoked by injecting currents (from 50 to 100 pA). The mean values of spike number (Bin is 5 s) were normalized with the average of first min as 1. Values are shown as mean ± SEM. *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001 by unpaired t test in (B) and (D) or two-way ANOVA followed by Bonferroni’s test in (F) and (H). See also Figure S4 and Table S1.
We next examined firing properties of hippocampal CA3 pyramidal neurons with whole-cell patch-clamp recordings. The spontaneous firing and the firing rate evoked by current injection in CA3 neurons were much higher in SENP2fxN/fxN neurons than in SENP2+/+ controls. Moreover, a repetitive burst behavior was triggered by mossy fiber stimulation in CA3 neurons in SENP2fxN/fxN mice but not in the control mice (Figures 2C–2F and S4; Table S1). In light of both intrinsic and synaptically driven neuronal hyperexcitability in SENP2fxN/fxN mice, we asked whether addition of SENP2 could rescue the hyperexcitability phenotype in a cell-autonomous way. When SENP2 peptide was included in the pipette solution, we observed a marked reduction in evoked firings of CA3 neurons in SENP2fxN/fxN mice, while heat-denatured SENP2 (de-SENP2) did not change excitability (Figures 2G and 2H). The SENP2 peptide markedly reduced neuronal hyperexcitability around 2 min after administration, while de-SENP2 had no effect (Figure 2I). Taken together, these results indicate that aberrant burst firing and network hyperexcitability in hippocampal neurons are attributable to SENP2 deficiency.
Hyper-SUMOylation of Kv1.1 and Kv7.2/Kv7.3 in SENP2-Deficient Neurons
Interestingly, SENP2 is highly expressed in the hippocampus, a critical brain region for genesis of seizures. Moreover, the brain expression pattern of SENP2 precisely overlaps those of a number of voltage-gated potassium channels linked to the development of temporal lobe epilepsy and early lethality (Allen Brain Atlas). We hypothesized that SENP2 deficiency resulted in hyper-SUMOylation of multiple potassium channels leading to increased neuronal excitability in the hippocampal circuitry.
SENP2 proteins are present at the nuclear envelope or in a subnuclear domain in HeLa cells and endothelial cells (Kang et al., 2010; Zhang et al., 2002a). However, the localization of SENP2 in hippocampal neurons has not yet been determined. We cultured hippocampal neurons from SENP2+/+ and SENP2fxN/fxN mice and analyzed the expression of Kv1.1 and Kv7.2, two voltage-gated potassium channels implicated in epileptic seizures (Glasscock et al., 2010; Smart et al., 1998; Singh et al., 1998, 2003). Immunofluorescence analyses showed that SENP2, Kv1.1, and Kv7.2 colocalized in hippocampal neurons (Figure 3A). The intensity of SENP2 was markedly reduced in hippocampal neurons of SENP2fxN/fxN mice, while SENP2 deficiency did not alter the apparent localization of Kv1.1 and Kv7.2 (Figure 3A) or Kv7.2 distribution between membrane and cytosolic areas (Figures S5A and S5B).
Figure 3. Hyper-SUMOylation of Kv1.1 and Kv7.2 in SENP2fxN/fxN Neurons.
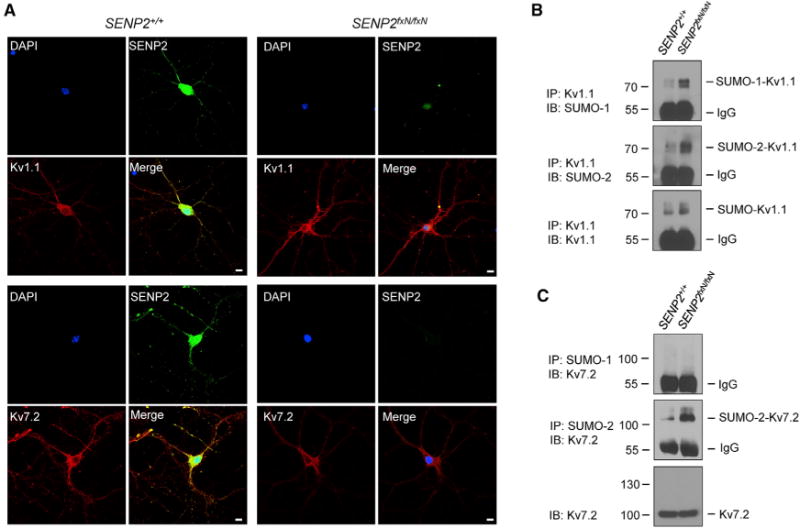
(A) SENP2 colocalizes with Kv1.1 and Kv7.2 in hippocampal neurons. Neurons from the brains of SENP2+/+ and SENP2fxN/fxN mice were cultured for immunocytochemistry with SENP2 (green) and Kv1.1- (red) or Kv7.2 (red)-specific antibodies. DAPI (blue) was used to show nuclei. Bars in all panels are 5 μm.
(B) Kv1.1 channels were modified by SUMO-1 and SUMO-2. The immunoprecipitates (IPs) with anti-Kv1.1 from brain lysates were detected by immunoblotting (IB) with anti-SUMO-1, anti-SUMO-2, and anti-Kv1.1 antibodies.
(C) Kv7.2 channels were modified by SUMO-2, but not SUMO-1. IPs with anti-SUMO-1 or anti-SUMO-2/3 from brain lysates were detected by IB with anti-Kv7.2 antibody.
See also Figure S5.
We next examined SUMOylation of Kv1.1 and Kv7 channels in mouse brain tissue. Immunoprecipitation-western blot analysis showed that SUMO-1- and SUMO-2-modified Kv1.1 bands were highly enriched in the SENP2fxN/fxN brain (Figure 3B). Only the SUMO-2-, but not the SUMO-1-, modified Kv7.2 band showed enrichment in the SENP2fxN/fxN brain (Figure 3C). Kv7.1 and Kv7.3 were also modified by SUMO-2, and their SUMOylations were enhanced in the SENP2fxN/fxN brain (Figures S5C and S5D). Thus, SENP2 deficiency leads to hyper-SUMOylation of both Kv1.1 and Kv7.2/Kv7.3 in hippocampal neurons.
Hyper-SUMOylation of Kv7.2/Kv7.3 Diminishes M-Currents and Contributes to Neuronal Hyperexcitability
The SENP2fxN/fxN neurons had a less negative resting membrane potential than wild-type controls (Figure S6A; Table S1). This could be due to reduction of potassium currents from hyper-SUMOylation of potassium channels. Indeed, we found that the amplitude of whole-cell potassium currents of CA3 pyramidal neurons was significantly reduced in hippocampal neurons from SENP2fxN/fxN mice compared to SENP2+/+ littermates (Figures S6B and S6C). We asked whether reduction in the whole-cell potassium current was due to reduced Kv1 and/or Kv7 currents in the SENP2fxN/fxN mice. Unexpectedly, using the Kv1.1-specific blocker dendrotoxin-K (DTX-K), we found that the amplitude of Kv1.1 currents and firings of CA3 neurons did not differ significantly between the SENP2fxN/fxN and SENP2+/+ mice (Figures S6D–S6H). In the hippocampus, Kv7.2 and Kv7.3 form a heteromeric channel, which conducts the hyperpolarizing M-current to control membrane potential and neuronal activity (Brown and Passmore, 2009; Hansen et al., 2008). Using a standard deactivation voltage protocol (Passmore et al., 2003) with 10,10-Bis(4-pyridinylmethyl)-9(10H)-anthracenone dihydrochloride (XE-991), a specific Kv7 blocker, we observed prominent basal M-currents in the CA3 neurons in SENP2+/+ mice (Figures 4A and 4C). In contrast, the basal M-currents in the CA3 neurons in the SENP2fxN/fxN mice were significantly diminished (Figures 4B and 4C). Using retigabine, a specific Kv7 opener, we observed that the M-currents were potentiated in the CA3 neurons in both SENP2+/+ and SENP2fxN/fxN mice (Figures 4A–4C). These results were also consistent with our data collected by using an activation voltage protocol (Figures S6I–S6L).
Figure 4. Reduction in M-Current Contributes to Neuronal Hyperexcitability in SENP2fxN/fxN Mice.
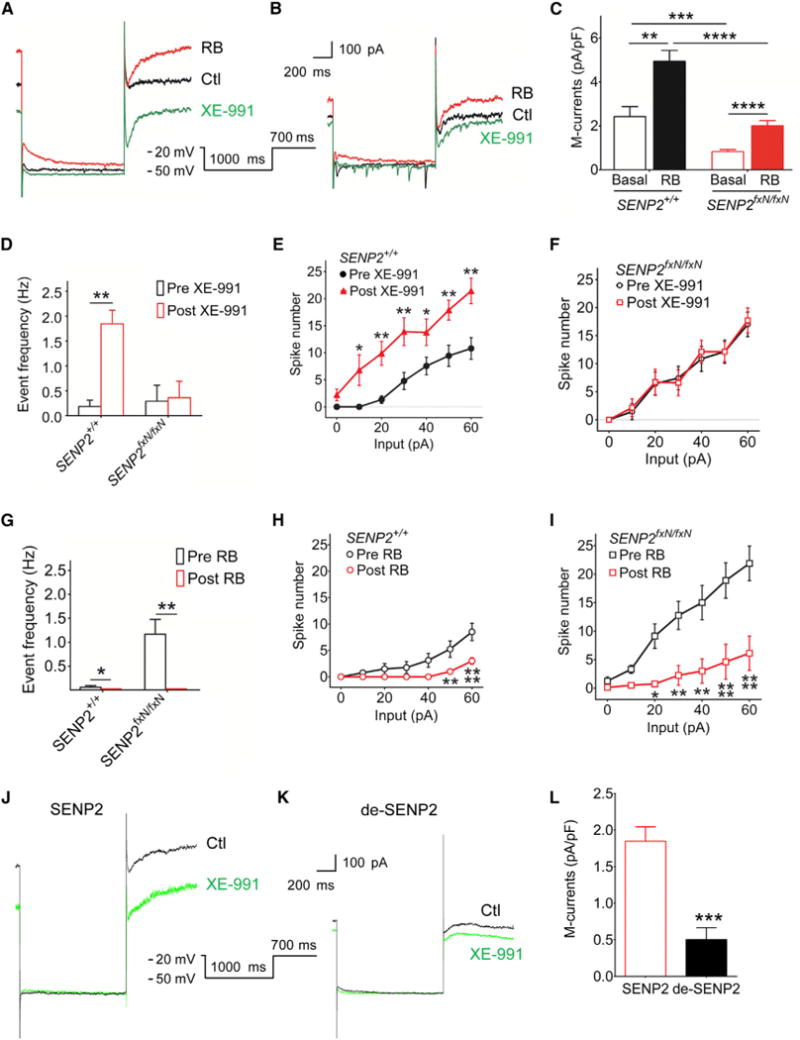
(A and B) Representative traces of deactivation tail currents in the absence (Ctl) or presence of Kv7 opener retigabine (RB) (10 μM) and blocker XE-991 (3 μM) in the CA3 neurons of the SENP2+/+ mice (A) and in their SENP2fxN/fxN littermates (B). The deactivation tail currents were elicited with the voltage protocol shown in the insert.
(C) Mean values of the M-current densities show that the basal M-currents, which are defined as XE-991-sensitive tail currents, were significantly diminished in the SENP2fxN/fxN neurons. Application of RB significantly potentiated the M-currents in neurons from both SENP2+/+ (n = 8) and SENP2fxN/fxN (n = 12) mice. The ANOVA revealed a significant differences between the groups (F[3,36] = 19.65, p < 0.001).
(D) The mean spontaneous firing frequency of CA3 neurons was significantly increased only in SENP2+/+ mice (n = 8/group) following XE-991 application. There is no significant difference in SENP2fxN/fxN mice (n = 7/group).
(E and F) XE-991 application resulted in a significant increase in the current injection evoked firing only in SENP2+/+ mice (E) (n = 12/group) but not in SENP2fxN/fxN mice (F) (n = 16/group). The ANOVA revealed a significant differences between the groups in SENP2+/+ (F[1,112] = 54.78, p < 0.0001), but not in SENP2fxN/fxN, mice (F[1,84] = 0.09, p > 0.05).
(G) The mean spontaneous firing frequency of CA3 neurons was significantly decreased by retigabine (RB) in both SENP2+/+ (n = 7) and SENP2fxN/fxN (n = 8) mice. (H and I) RB application resulted in a significant decrease in the mean spontaneous firing frequency of CA3 neurons from both SENP2+/+ (I) (n = 8/group) and SENP2fxN/fxN (J) (n = 8/group) mice. The ANOVA revealed a significant differences between the groups in SENP2+/+ (F[1,98] = 28.85, p < 0.0001) and SENP2fxN/fxN mice (F[1,98] = 63.57, p < 0.0001).
(J and K) Representative traces of deactivation tail currents in the absence (Ctl) or presence of Kv7 opener RB (10 μM) and blocker XE-991 (3 μM) in the CA3 neurons of the SENP2fxN/fxN mice with SENP2 (250 pM) or heat-denatured inactive SENP2 (de-SENP2, 250 pM) peptide in pipette solution. The deactivation tail currents were elicited with the voltage protocol shown in the insert. To minimize the contamination of other currents, all recordings were carried out in the presence of tetrodotoxin (0.5 μM) to block sodium currents, CdCl2 (100 μM) to block calcium currents, DTX-K (100 nM) to block Kv1.1 currents, and Cs+ (1 mM) to block hyperpolarization-activated currents.
(L) Mean values of the M-current densities show that the M-currents, which are defined as XE-991-sensitive tail currents, were significantly potentiated by SENP2 (n = 8) but not de-SENP2 (n = 6) in the SENP2fxN/fxN neurons.
Values are shown as mean ± SEM. *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001 by paired t test in (D), (G), and (L) or two-way ANOVA followed by Tukey’s test in (C) or followed by Bonferroni’s test in (E), (F), (H), and (I). See also Figure S6 and Table S1.
To determine the role of Kv7 in the hyperexcitability of CA3 neurons in SENP2fxN/fxN mice, we examined the effects of XE-991 on spontaneous and evoked firings by current injection. When Kv7 channels were blocked by XE-991, the spontaneous and evoked firings were markedly increased in SENP2+/+ neurons (Figures 4D and 4E). CA3 neurons in SENP2fxN/fxN mice displayed a high firing rate at baseline, but their firings did not further increase during XE-991 application (Figures 4D and 4F). Likewise, potentiation of the M-currents by retigabine in the SENP2fxN/fxN neurons led to substantial suppression of spontaneous and evoked firings by current injection (Figures 4G–4I). Since SENP2 peptide reduced neuronal hyperexcitability in the SENP2fxN/fxN neurons, we asked whether SENP2 could also restore the diminished M-currents. As shown, there was a marked increase in M-currents in CA3 neurons of SENP2fxN/fxN mice after infusion of SENP2 peptide, whereas de-SENP2 had no effect (Figures 4J–4L). These results suggest that Kv7 channel function in the hippocampus is profoundly impaired in SENP2fxN/fxN mice, and the impaired Kv7 channel function can be restored by retigabine or SENP2 peptide. Taken together, SENP2 deficiency in hippocampal neurons of SENP2fxN/fxN mice diminishes the M-currents conducted by Kv7 channels, contributing to the more positive resting membrane potential and hyperexcitability of hippocampal neurons.
Recapitulation of the Sudden Death Phenotype with Brain-Specific Deletion of SENP2
Since SENP2fxN/fxN mice develop epilepsy and die between 6 and 8 weeks of age, we hypothesized that SENP2fxN/fxN mice may possess potentially lethal cardiac defects predisposing them to early death. In order to identify the proximal cause of death, we monitored the electrocardiogram (ECG) from freely moving SENP2fxN/fxN mice and SENP2+/+ controls over a continuous 1 week period. The ECG recordings showed that the SENP2fxN/fxN mice displayed episodes of severe prolonged bradycardia (Figures 5A, 5B, S7B, and S7D) and higher activity (Figures S7A and S7C). In addition to sinus bradycardia, the SENP2fxN/fxN mice also developed high-degree AV conduction blocks and AV dissociation prior to lethal asystole (Figures 5A and 5B). During the interictal periods between seizures, SENP2fxN/fxN mice displayed longer AV block durations (Figure 5C) and higher frequency of blocks (Figure 5D) compared to SENP2+/+ littermates.
Figure 5. Severe Bradycardia and AV Conduction Blocks in SENP2fxN/fxN Mice.
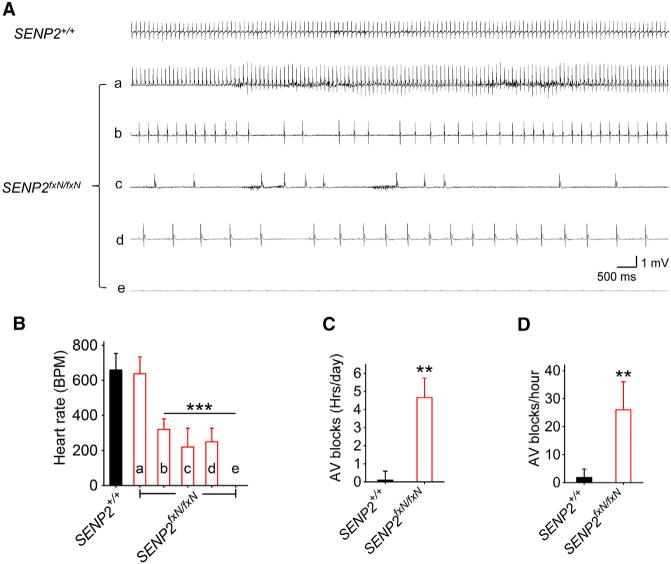
(A) Representative telemetric ECG traces recorded from awake SENP2+/+ and SENP2fxN/fxN mice. The ECG traces corresponded to time points as indicated by arrows in Figures S7B and S7D: The upper trace was recorded from a SENP2+/+ mouse; the bottom traces ([Aa]–[Ae]) were recorded at different time points from a representative SENP2fxN/fxN mouse; trace (Ae) indicates death.
(B) Mean heart rates of SENP2+/+ (n = 3) and SENP2fxN/fxN (n = 5) mice calculated from a 2-hr periods at time intervals defined in Figures S7B and S7D. The ANOVA revealed a significant differences between the groups (F[5,22] = 65.91, p < 0.0001).
(C) Mean AV blocks were counted from ECGs recorded continuously for 7 days in SENP2+/+ (n = 3) and SENP2fxN/fxN (n = 5) mice.
(D) Mean AV blocks were counted from the ECGs recorded within the last 24 hr prior to the death in SENP2+/+ (n = 3) and SENP2fxN/fxN (n = 5) mice.
Values are shown as mean ± SEM. *p < 0.05; ***p < 0.001 by one-way ANOVA followed by Dunnett’s test in (B) or unpaired t test in (C) and (D). See also Figure S7.
Since SENP2 deficiency resulted in hyper-SUMOylation of both brain and cardiac proteins (Figures S3A–S3C), it is unclear whether sudden death is primarily neurogenic or cardiogenic. Thus, we generated SENP2fx/fx mice and crossed with either tamoxifen-inducible Thy1 or myosin heavy chain (MHC) promoter-specific Cre mice. SENP2 mRNA was reduced to 15% of baseline in the brain following five daily injections of tamoxifen in the SENP2fx/fx-Thy1 mice (Figure 6A). On the other hand, SENP2 mRNA dropped to 28% of baseline level in the heart in SENP2fx/fx-MHC mice after identical tamoxifen treatment (Figure 6B). Depletion of SENP2 in cardiomyocytes had no apparent effect on the development of behavioral seizures or lifespan of the mice; however, depletion of SENP2 in the projection neuron populations in central and peripheral nervous system resulted in 100% mortality within 11 days (Figure 6C). To determine cause of death, we monitored freely moving adult mice implanted with chronic cortical EEG electrodes. Video EEG recordings revealed development of cortical seizures in the SENP2fx/fx-Thy1 mice 5 days after tamoxifen induction (Figure 6D). These mice also developed severe bradycardia, high-degree AV block, and eventually asystole (Figures 6E–6H, S7F, and S7H) with lower activity (Figures S7E and S7G). Thus, brain-specific, but not cardiomyocyte-specific, deletion of SENP2 recapitulated the phenotype of global SENP2 deficiency.
Figure 6. Neuronal-Specific Deletion of SENP2 Causes Seizure and Sudden Death.
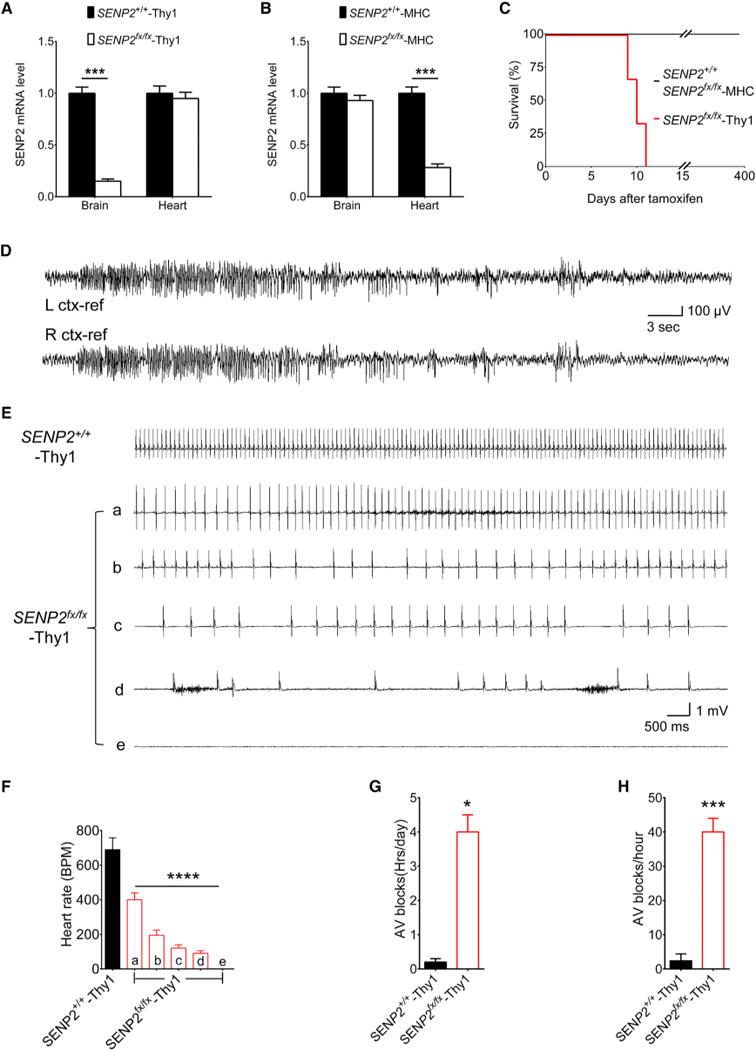
(A) Expression level of SENP2 transcripts in brain and heart of SENP2+/+-Thy1 and SENP2fx/fx-Thy1 mice. SENP2 transcript levels were measured by real-time PCR, normalized to SENP2+/+-Thy1 mice (n = 3 mice/group).
(B) Expression level of SENP2 transcripts in brain and heart of SENP2+/+-MHC and SENP2fx/fx- MHC mice. SENP2 transcript levels were measured by real-time PCR, normalized to SENP2+/+- MHC mice (n = 3mice/group).
(C) Lifespan of SENP2+/+ (n = 125), SENP2fx/fx-MHC (n = 130), and SENP2fx/fx-Thy1 (n = 120) mice.
(D) Representative EEG seizures recorded from awake SENP2fx/fx-Thy1 mouse after SENP2 neuronal-specific deletion. EEG traces revealed baseline low-amplitude desynchronized activity with frequent sharp interictal spikes followed by onset of spontaneous nonconvulsive bilateral seizure.
(E) Representative telemetric ECG traces recorded from awake SENP2+/+-Thy1 and SENP2fx/fx-Thy1 mice. The ECG traces corresponded to time points as indicated by arrows in Figures S7F and S7H. The upper trace was recorded from a SENP2+/+-Thy1 mouse; the bottom traces ([Ea]–[Ee]) were recorded at different times from a representative SENP2fx/fx-Thy1 mouse; trace (Ee) indicates death.
(F) Mean heart rates in SENP2+/+-Thy1 (n = 3) and SENP2fx/fx-Thy1 (n = 5) mice calculated from a 2-hr period at indicated time intervals in Figures S7F and S7H. The ANOVA revealed a significant differences between the groups; F(5,22) = 130, p < 0.0001.
(G) Mean AV blocks per day were counted from ECGs recorded continuously for 5 days in SENP2+/+-Thy1 (n = 3) and SENP2fx/fx-Thy1 (n = 5) mice.
(H) Mean AV blocks were counted from the ECGs recorded within the last 24 hr before death in SENP2+/+-Thy1 (n = 3) and SENP2fx/fx-Thy1 (n = 5) mice. The SENP2fx/fx-Thy1 mice have longer AV block duration (F) and higher AV block frequency (G) when compared to their SENP2+/+-Thy1 littermates.
Values are shown as mean ± SEM. *p < 0.05: ***p < 0.001; ****p < 0.0001 by unpaired t test in (A), (B), (G), and (H) or one-way ANOVA followed by Dunnett’s test in (F). See also Figure S7.
Prevention of Seizures and Cardiac Conduction Blocks with Retigabine
We discovered that auditory stimulation (22.5 kHz for 10 s) can reliably induce seizures in the SENP2fxN/fxN mice when they reached 6 weeks of age (Movie S4). Seizures could be induced 1–5 s following activation of the acoustic signals and lasted for 10–20 s (indicated by noisy ECG baseline in bracket). This was followed by a period of sinus pauses or AV conduction blocks (Figure 7A). The sinus pauses and AV blocks were due to seizure-induced increase in the parasympathetic tone because atropine (1 mg/kg) was able to prevent postseizure sinus pauses and AV blocks (Figure 7B). On the other hand, pretreatment with propranolol (4mg/kg) caused more postseizure AV blocks (Figure 7B). As expected, neither atropine nor propranolol prevented seizure induction (Figure 7B).
Figure 7. Prevention of Acoustics-Induced Seizures and AV Block in SENP2fxN/fxN Mice with Retigabine.
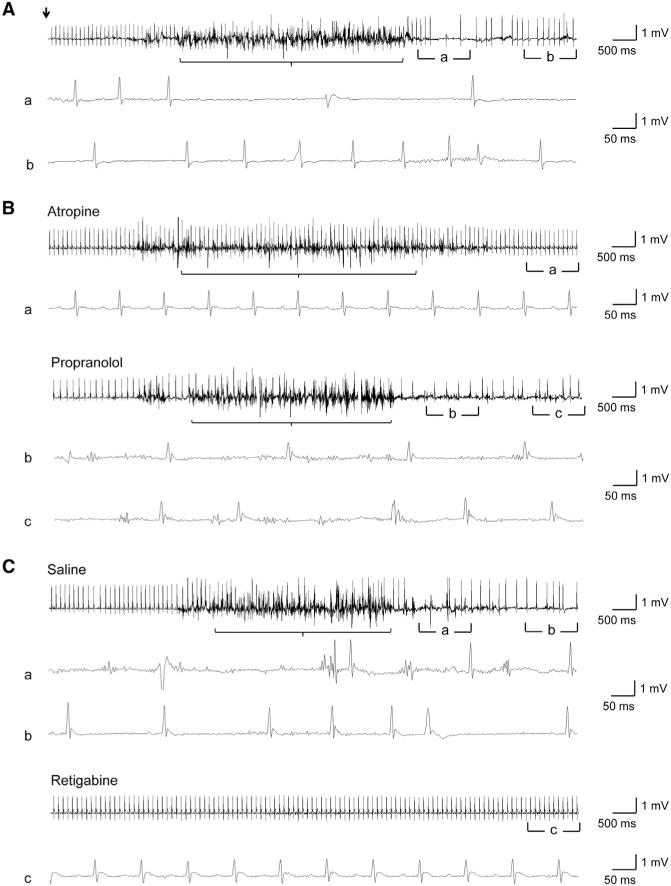
(A) Representative telemetric ECG traces recorded from SENP2fxN/fxN mice following acoustics-induced seizures. The onset of acoustic induction (22.5 kHz for 10 s) is indicated by an arrow. The noisy baseline indicated seizure activity (bracket). The acoustic induction caused seizure followed by AV blocks in saline administrated mice (n = 4). (Aa and Ab) Magnified ECG traces showing AV conduction blocks and sinus pauses.
(B) Representative telemetric ECG traces recorded from SENP2fxN/fxN mice after acoustic stimulation following 30 min pretreatment of atropine (1 mg/kg, n = 6) or propranolol (4 mg/kg, n = 7). The administration of atropine eliminated interictal AV blocks, while pretreatment with propranolol worsened AV blocks. Neither of them prevents the acoustic-induced seizures. (Ba) Magnified normal ECG traces; (Bb and Bc) Magnified traces showing AV conduction blocks and sinus pauses.
(C) Representative telemetric ECG traces recorded from SENP2fxN/fxN mice with pretreatment of retigabine (10 mg/kg, n = 8) or saline (n = 5) followed 30 min later with acoustic stimulation (22.5 kHz for 10 s). The 30 min pretreatment of retigabine prevented both acoustic-induced seizures and AV blocks. (Ca and Cb) Magnified traces showing AV conduction blocks and sinus pauses; (Cc) Magnified normal ECG traces. See also Movie S4.
Since retigabine is able to open the Kv7 channels in the SENP2fxN/fxN neurons, we asked whether retigabine can prevent seizure induced by acoustic stimulation in the SENP2fxN/fxN mice. To test this hypothesis, we first determined that seizures can be induced in the 6-week-old SENP2fxN/fxN mice with acoustic stimulation. These mice were rested for 6 hr and injected intraperitoneally with either saline or retigabine (10 mg/kg) in a blinded fashion. Thirty minutes following saline or retigabine injection, mice were stimulated acoustically (22.5 kHz for 10 s). We found that seizures can only be induced in saline-treated mice but not in the retigabine-treated mice (Figure 7C). Since the retigabine-treated mice never developed seizure, there were no postseizure AV blocks. This is consistent with the ability of retigabine to open the Kv7 channel and to suppress hyperexcitability of the SENP2-deficient neurons (Figures 4G–4I).
DISCUSSION
SUMO conjugation has been reported to modulate the function of several neuronal K+ channels, including K2P1 (Rajan et al., 2005), Kv1.5 (Benson et al., 2007), and Kv2.1 (Dai et al., 2009; Plant et al., 2011). Kainate receptor-mediated excitatory post-synaptic currents are decreased by SUMOylation and enhanced by de-SUMOylation (Martin et al., 2007), suggesting that SUMOylation is a general mechanism for regulating ion channel physiology (Martin et al., 2007). However, these studies were all based on in vitro findings, and the functional role of SUMOylation on ion channel activity in living animals has not been previously determined. The epileptic and sudden death phenotype of the SENP2-deficient mice closely resembles that of Kv1.1-null mice (Glasscock et al., 2010). Despite their coexpression in CA3 neurons, hyper-SUMOylation of Kv1.1 channels due to SENP2 deficiency did not cause a detectable reduction in Kv1.1 current amplitude. On the other hand, the M-current, which plays a critical role in controlling membrane potentials and firing activity (Brown and Passmore, 2009; Hansen et al., 2008), was significantly diminished in the hippocampal neurons of SENP2-deficient mice (Figures 4B and 4C). Although we cannot exclude the involvement of other ion channels or receptors, defective M-current can account for the neuronal hyperexcitability observed in the SENP2-deficient mice.
There are five Kv7 channels, encoded by the KCNQ genes (Brown and Passmore, 2009). Kv7.2 and Kv7.3 are the major components of the M-current, which regulates neuronal excitability (Scott and Holmes, 2012; Surti and Jan, 2005). Mutation in the KCNQ1 gene at the LQT1 locus (11p15.5) is the most common cause of long QT syndrome (LQTS), which predisposes patients to lethal cardiac arrhythmias (Goldenberg et al., 2008; Wang et al., 1996). In the current study, SENP2-deficient mice developed profound bradycardia and high-degree AV conduction blocks, but not ventricular arrhythmia (Figure 5A). The cardiac conduction blocks usually followed by seizure activity and can be prevented by atropine, but not propranolol (Figure 7B). Thus, the high-degree AV blocks observed in SENP2-deficient mice are likely due to an increase in parasympathetic tone following seizures. Interestingly, the Kv7.1 channel encoded by the KCNQ1 gene is also expressed in forebrain and brainstem nuclei, and mice carrying human LQTS mutations in the KCNQ1 gene also develop seizures and sudden death due to an increase in parasympathetic tone (Goldman et al., 2009). Furthermore, we found that mice with cardiomyocyte-specific knockout of SENP2 had a normal lifespan (Figure 6C). In contrast, brain-specific deletion of SENP2 resulted in 100% mortality 11 days after tamoxifen induction. These mice also developed severe seizures, high-degree AV blocks, and asystole. Thus, the sudden death phenotype due to SENP2 deficiency is most likely neurogenic. Taken together, defective Kv7 channel function can explain both seizures and sudden death exhibited by the SENP2-deficient mice.
Retigabine is a third-generation antiepileptic drug that received approval from the Food and Drug Administration (FDA) for the treatment of partial-onset seizures. It is unique among the antiepilepsy drugs approved by FDA because of its specific mechanism of action. Retigabine activates Kv7.2–Kv7.5, but not the Kv7.1, channel, through a hyperpolarization shift of the voltage dependence of channel activation and an increase of the channel maximal opening probability (Tatulian et al., 2001). We showed that retigabine restored the M-currents (Figures 4B and 4C), prevented spontaneous firings (Figure 4G), and abolished current injection evoked firings (Figure 4I) in the hippocampal neurons of the SENP2-deficient mice. Furthermore, retigabine can prevent stimulator-induced seizure and AV block in the SENP2-deficient mice (Figure 7C). Notably, we did not observe the repetitive bursting behavior in the CA3 neurons of the SENP2-deficient mice in the presence of retigabine. The burst firing behavior was triggered only by mossy fiber stimulation, but not current injection at the soma of the CA3 neurons, in the SENP2-deficient mice. The burst firing may originate from increased dendritic excitability following synaptic stimulation. Consistent with this interpretation, direct paired dendrite-soma recordings have shown that current injection at the soma elicits single action potentials, whereas current injection into the apical trunk initiates burst firing in the dendrites of hippocampal neurons (Wong and Stewart, 1992). Taken together, these results provide strong support that the molecular mechanism accounting for seizures and sudden death in the SENP2-deficient mice is due to hyper-SUMOylation of the Kv7 channels.
In summary, we have identified an animal model of SUDEP, which is caused by alteration of a posttranslational protein modification pathway (SUMOylation) on a unique voltage-gated potassium channel (Kv7.2/Kv7.3). This finding expands the number of biological pathways leading to critical ion channel disorders in the central nervous system and validates SENP2 as a candidate gene for SUDEP. Patients with epilepsy could be screened for genetic variations of SENP2 that either reduces its expression or activity. Retigabine or other Kv7 channel-specific openers should be explored for their utility in prevention of seizures and sudden death in these individuals.
EXPERIMENTAL PROCEDURES
Generation of Mouse Strains and Animal Care
SENP2fxN/fxN mice were generated as shown in Figure S1. In brief, a genomic clone containing exons 11–16 of the mouse SENP2 gene was isolated from a BAC library and cloned into a vector using homologous recombination (Zhang et al., 2002b). A cassette containing a loxP-flanked NeoR gene expressed from the phosphoglycerate kinase promoter (PGK-NeoR) was cloned into intron 14 to obtain the final targeting vector. Following germline transmission, SENP2fxN/+ mice were mated to obtain SENP2fxN/fxN knocking in mice and wild-type littermates. After SENP2fxN/fxN crossing with FLP mice (Jackson lab), the final allele without Neo cassette (SENP2fx/fx) was obtained. We established mouse models with neuron-specific (SENP2fx/fx-Thy1) and cardiomyocyte-specific (SENP2fx/fx-MHC) deletion of SENP2. SENP2fx/fx mice were crossed with mice carrying Thy1 promoter (Thy1-cre/ERT2, -EYFP, Jackson lab) or MHC promoter (α-MHC-MerCreMer, Jackson lab) and a fusion protein containing Cre and mutated estrogen receptor.
Hippocampal Neuronal Culture
Hippocampal neuronal cultures were prepared from 1-day-old SENP2+/+ and SENP2fxN/fxN pups. The hippocampal neurons were cultured in Neurobasal supplemented with 2% B27 and 1% Glutamax-I (Invitrogen) in a humidified atmosphere containing 5% CO2 at 37°C. Subsequent feeding occurred twice weekly, each time replacing half the volume with medium including FGF2 and AraC. The neurons were used for immunocytochemistry at DIV 14.
Immunocytochemistry
For immunolabeling, the fixed cells were permeabilized with 0.1% Triton X-100 in PBS, incubated with antibodies, and then examined by confocal laser scanning microscopy (Leica).
RNA Isolation and Real-Time PCR
For mice genotyping, we isolated genomic DNA from tail clips using DiretPCR lysis reagent (Viagen) and determined the genotypes of SENP2fxN/fxN, SENP2fx/fx -Thy1, and SENP2fx/fx -MHC using PCR amplification of specific alleles with indicated primers (Table S2). Quantitative PCR was then performed using reaction mixtures of 20 ng total RNA, 100 nM primers (Table S3), and SYBR Green reagent (Applied Biosystems) with the ABI PRISM 7300 system (Perkin-Elmer). PCR was done in triplicate, and SDs representing experimental errors were calculated. All data were analyzed using ABI PRISM SDS 2.0 software (Perkin-Elmer).
Western Blotting and Immunoprecipitation
Whole mouse brain and heart were extracted, frozen in liquid nitrogen, and subsequently homogenized on ice in lysis buffer with protease inhibitors. Western blotting was done with primary anti-SENP2 (Santa Cruz), anti-SUMO-1 (Cell Signaling), anti-SUMO-2 (Abacm), and anti-Kv7.2 (Alomone labs) antibodies. For immunoprecipitation experiments, brain lysate was incubated overnight with anti-Kv1.1 (UC Davis/NIH NeuroMab Facility), anti-SUMO-1, and anti-SUMO-2 antibodies and followed by western immunoblotting with anti-Kv7.2 and antibodies described above.
In Vivo EEG/ECG or Telemetric ECG Recordings
For EEG/ECG recordings, silver wire electrodes soldered to a microminiature connector were implanted bilaterally into the subdural space over frontal and parietal cortex of mice under avertin anesthesia several days prior to recording. Behavioral video EEG/ECG monitoring and analysis were performed using a digital electroencephalograph (Stellate Systems). For telemetric ECG recording, a telemetry transmitter (ETA-F10, DSI) was implanted in the abdominal cavity of each mouse with subcutaneous electrodes tunneled to the right upper and left lower thorax (Kannankeril et al., 2006). ECG, heart rate, and activity monitoring were performed using Dataquest software (ART 4.0, DSI).
In Vitro Hippocampal Electrophysiological Recordings
Field recordings were made from the visually identified CA3 pyramidal cell layer using a Getting Instruments Model 5A amplifier, digitized by a Digidata 1322A, and collected using Clampex. To calculate the burst frequency, we let the slices equilibrate to 7.5 mM KCl for 5–15 min after beginning to burst and then calculated the burst frequency over a 5 min interval using Clampfit and Origin software.
For single-neuron current and firing recording, whole-cell configuration (in current- or voltage-clamp mode) in hippocampal CA3 pyramidal neurons was established under visual control using an upright microscope. The total Kv currents were recorded in voltage-clamp mode in the presence of TTX to block sodium channels and cadmium to block calcium channels. To dissect Kv1.1 currents from the total Kv currents, the neurons were perfused with a Kv1.1 channel blocker DTX-K after the total Kv currents were recorded, such that the DTX-K-sensitive currents represent the Kv1.1 currents. To evaluate the Kv7/M-currents, the deactivation tail currents (Passmore et al., 2003) were recorded sequentially in the presence of retigabine and XE-991. To assess whether SENP2 can reverse the hyperexcitability in the SENP2fxN/fxN neurons, SENP2 peptide (250 pM) in the pipette solution was applied when recording the current injection evoked neuronal firings. To evaluate the contributions of Kv1.1 or Kv7 channels to the hyperexcitability manifesting in the SENP2fxN/fxN neurons, the same recordings in the current-clamp mode were performed in the absence and presence of DTX-K (100 nM), XE-991 (3 μM), or retigabine (10 μM) in bath solutions, respectively.
Pharmacology and Acoustic Seizure Induction
SENP2fxN/fxN mice (6 weeks old) were administrated atropine (1 mg/kg; Sigma) or propranolol (4 mg/kg; Sigma) for selective parasympathetic or sympathetic blockade and retigabine (10 mg/kg; Alomone labs) for seizure blockade. For each setting, resting telemetry ECG were continuously recorded before drug administration to establish the baseline rate of seizure and AV blocks, injected with drug, and induced acoustic seizure via auditory stimulation (Microson, 22.5 kHz) for 10 s half hour after injection, and then recorded to determine the drug’s effect on the rate of seizure and AV blocks. The seizures were induced with acoustic stimulation, and then the SENP2fxN/fxN mice were rested for 6 hr and were injected with either saline or each drug in a blinded fashion, and 30 min following injection, all mice were stimulated with stimulator.
Statistical Analysis
All data were presented as mean ± SEM or SD of at least three separate experiments. For electrophysiological experiments, both Clampex 9.0 software (Molecular Devices) and MiniAnalysis 6.0 software (Synaptosoft Inc.) were used to analyze firing and current waveform parameters. Statistical significance was defined as p < 0.05 (*p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001).
Supplementary Material
Acknowledgments
This work was supported by the McNair Medical Foundation (E.T.H.Y.), NS29709 and NS0769 (J.L.N.), HL077400 (H.-L.P.), and National Natural Science Foundation of China (91019021 to J.C.).
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information includes seven figures, three tables, four movies, and Supplemental Experimental Procedures and can be found with this article online at http://dx.doi.org/10.1016/j.neuron.2014.07.042.
AUTHOR CONTRIBUTIONS
Y.Q. generated the mice and conducted the molecular experiments; J.W. conducted the electrophysiology studies. V.C.B., D.-P.L., S.-R.C., H.S., Y.X., and J.G.R. contributed to parts of the electrophysiology studies. J.C. advised on the genetic studies. J.L.N. planed the EEG studies, and H.-L.P. designed the electrophysiology experiments. E.T.H.Y. conceived the project. Y.Q. and E.T.H.Y. wrote the paper with editorial input from J.W., H.-L.P., and J.L.N.
References
- Benson MD, Li QJ, Kieckhafer K, Dudek D, Whorton MR, Sunahara RK, Iniguez-Lluhi JA, Martens JR. SUMO modification regulates inactivation of the voltage-gated potassium channel Kv1.5. Proc Natl Acad Sci USA. 2007;104:1805–1810. doi: 10.1073/pnas.0606702104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenner R, Wilcox KS. Potassium Channelopathies of Epilepsy. In: Noebels JL, Avoli M, Rogawski MA, et al., editors. Jasper’s Basic Mechanisms of the Epilepsies. Bethesda: National Center for Biotechnology Information; 2012. [PubMed] [Google Scholar]
- Brown DA, Passmore GM. Neural KCNQ (Kv7) channels. Br J Pharmacol. 2009;156:1185–1195. doi: 10.1111/j.1476-5381.2009.00111.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng J, Kang X, Zhang S, Yeh ET. SUMO-specific protease 1 is essential for stabilization of HIF1alpha during hypoxia. Cell. 2007;131:584–595. doi: 10.1016/j.cell.2007.08.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper EC. Potassium channels (including KCNQ) and epilepsy. In: Noebels JL, Avoli M, Rogawski MA, Olsen RW, Delgado-Escueta AV, editors. Jasper’s Basic Mechanisms of the Epilepsies. Bethesda: National Center for Biotechnology Information; 2012. http://www.ncbi.nlm.nih.gov/books/NBK98164/ [PubMed] [Google Scholar]
- Dai XQ, Kolic J, Marchi P, Sipione S, Macdonald PE. SUMOylation regulates Kv2.1 and modulates pancreatic beta-cell excitability. J Cell Sci. 2009;122:775–779. doi: 10.1242/jcs.036632. [DOI] [PubMed] [Google Scholar]
- Glasscock E, Yoo JW, Chen TT, Klassen TL, Noebels JL. Kv1.1 Potassium Channel Deficiency Reveals Brain-Driven Cardiac Dysfunction as a Candidate Mechanism for Sudden Unexplained Death in Epilepsy. J Neurosci. 2010;30:5167–5175. doi: 10.1523/JNEUROSCI.5591-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldenberg I, Zareba W, Moss AJ. Long QT Syndrome. Curr Probl Cardiol. 2008;33:629–694. doi: 10.1016/j.cpcardiol.2008.07.002. [DOI] [PubMed] [Google Scholar]
- Goldman AM, Glasscock E, Yoo J, Chen TT, Klassen TL, Noebels JL. Arrhythmia in heart and brain: KCNQ1 mutations link epilepsy and sudden unexplained death. Sci Transl Med. 2009;1:2ra6. doi: 10.1126/scitranslmed.3000289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen HH, Waroux O, Seutin V, Jentsch TJ, Aznar S, Mikkelsen JD. Kv7 channels: interaction with dopaminergic and serotonergic neurotransmission in the CNS. J Physiol. 2008;586:1823–1832. doi: 10.1113/jphysiol.2007.149450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herren AW, Bers DM, Grandi E. Post-translational modifications of the cardiac Na channel: contribution of CaMKII-dependent phosphorylation to acquired arrhythmias. Am J Physiol Heart Circ Physiol. 2013;305:H431–H445. doi: 10.1152/ajpheart.00306.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang X, Qi Y, Zuo Y, Wang Q, Zou Y, Schwartz RJ, Cheng J, Yeh ET. SUMO-specific protease 2 is essential for suppression of polycomb group protein-mediated gene silencing during embryonic development. Mol Cell. 2010;38:191–201. doi: 10.1016/j.molcel.2010.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kannankeril PJ, Mitchell BM, Goonasekera SA, Chelu MG, Zhang W, Sood S, Kearney DL, Danila CI, De Biasi M, Wehrens XH, et al. Mice with the R176Q cardiac ryanodine receptor mutation exhibit catecholamine-induced ventricular tachycardia and cardiomyopathy. Proc Natl Acad Sci USA. 2006;103:12179–12184. doi: 10.1073/pnas.0600268103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin S, Nishimune A, Mellor JR, Henley JM. SUMOylation regulates kainate-receptor-mediated synaptic transmission. Nature. 2007;447:321–325. doi: 10.1038/nature05736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noebels JL. The biology of epilepsy genes. Annu Rev Neurosci. 2003;26:599–625. doi: 10.1146/annurev.neuro.26.010302.081210. [DOI] [PubMed] [Google Scholar]
- Park KS, Mohapatra DP, Misonou H, Trimmer JS. Graded regulation of the Kv2.1 potassium channel by variable phosphorylation. Science. 2006;313:976–979. doi: 10.1126/science.1124254. [DOI] [PubMed] [Google Scholar]
- Passmore GM, Selyanko AA, Mistry M, Al-Qatari M, Marsh SJ, Matthews EA, Dickenson AH, Brown TA, Burbidge SA, Main M, Brown DA. KCNQ/M currents in sensory neurons: significance for pain therapy. J Neurosci. 2003;23:7227–7236. doi: 10.1523/JNEUROSCI.23-18-07227.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plant LD, Dementieva IS, Kollewe A, Olikara S, Marks JD, Goldstein SAN. One SUMO is sufficient to silence the dimeric potassium channel K2P1. Proc Natl Acad Sci USA. 2010;107:10743–10748. doi: 10.1073/pnas.1004712107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plant LD, Dowdell EJ, Dementieva IS, Marks JD, Goldstein SAN. SUMO modification of cell surface Kv2.1 potassium channels regulates the activity of rat hippocampal neurons. J Gen Physiol. 2011;137:441–454. doi: 10.1085/jgp.201110604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi Y, Zuo Y, Yeh ET, Cheng J. An essential role of small ubiquitin-like modifier (SUMO)-specific Protease 2 in myostatin expression and myogenesis. J Biol Chem. 2014;289:3288–3293. doi: 10.1074/jbc.M113.518282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajan S, Plant LD, Rabin ML, Butler MH, Goldstein SA. Sumoylation silences the plasma membrane leak K+ channel K2P1. Cell. 2005;121:37–47. doi: 10.1016/j.cell.2005.01.019. [DOI] [PubMed] [Google Scholar]
- Ren SY, Angrand PO, Rijli FM. Targeted insertion results in a rhombomere 2-specific Hoxa2 knockdown and ectopic activation of Hoxa1 expression. Dev Dyn. 2002;225:305–315. doi: 10.1002/dvdy.10171. [DOI] [PubMed] [Google Scholar]
- Scott RC, Holmes GL. Before epilepsy unfolds: opening up the potassium door in neonatal seizures. Nat Med. 2012;18:1624–1625. doi: 10.1038/nm.2987. [DOI] [PubMed] [Google Scholar]
- Sillanpaa M, Shinnar S. Long-term mortality in childhood-onset epilepsy. N Engl J Med. 2010;363:2522–2529. doi: 10.1056/NEJMoa0911610. [DOI] [PubMed] [Google Scholar]
- Singh NA, Charlier C, Stauffer D, DuPont BR, Leach RJ, Melis R, Ronen GM, Bjerre I, Quattlebaum T, Murphy JV, et al. A novel potassium channel gene, KCNQ2, is mutated in an inherited epilepsy of newborns. Nat Genet. 1998;18:25–29. doi: 10.1038/ng0198-25. [DOI] [PubMed] [Google Scholar]
- Singh NA, Westenskow P, Charlier C, Pappas C, Leslie J, Dillon J, Anderson VE, Sanguinetti MC, Leppert MF, Consortium BP. KCNQ2 and KCNQ3 potassium channel genes in benign familial neonatal convulsions: expansion of the functional and mutation spectrum. Brain. 2003;126:2726–2737. doi: 10.1093/brain/awg286. [DOI] [PubMed] [Google Scholar]
- Smart SL, Lopantsev V, Zhang CL, Robbins CA, Wang H, Chiu SY, Schwartzkroin PA, Messing A, Tempel BL. Deletion of the K(V)1.1 potassium channel causes epilepsy in mice. Neuron. 1998;20:809–819. doi: 10.1016/s0896-6273(00)81018-1. [DOI] [PubMed] [Google Scholar]
- Surti TS, Jan LY. A potassium channel, the M-channel, as a therapeutic target. Curr Opin Investig Drugs. 2005;6:704–711. [PubMed] [Google Scholar]
- Tatulian L, Delmas P, Abogadie FC, Brown DA. Activation of expressed KCNQ potassium currents and native neuronal M-type potassium currents by the anti-convulsant drug retigabine. J Neurosci. 2001;21:5535–5545. doi: 10.1523/JNEUROSCI.21-15-05535.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, Curran ME, Splawski I, Burn TC, Millholland JM, VanRaay TJ, Shen J, Timothy KW, Vincent GM, de Jager T, et al. Positional cloning of a novel potassium channel gene: KVLQT1 mutations cause cardiac arrhythmias. Nat Genet. 1996;12:17–23. doi: 10.1038/ng0196-17. [DOI] [PubMed] [Google Scholar]
- Wong RK, Stewart M. Different firing patterns generated in dendrites and somata of CA1 pyramidal neurones in guinea-pig hippocampus. J Physiol. 1992;457:675–687. doi: 10.1113/jphysiol.1992.sp019401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeh ET. SUMOylation and De-SUMOylation: wrestling with life’s processes. J Biol Chem. 2009;284:8223–8227. doi: 10.1074/jbc.R800050200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeh ET, Gong L, Kamitani T. Ubiquitin-like proteins: new wines in new bottles. Gene. 2000;248:1–14. doi: 10.1016/s0378-1119(00)00139-6. [DOI] [PubMed] [Google Scholar]
- Zhang H, Saitoh H, Matunis MJ. Enzymes of the SUMO modification pathway localize to filaments of the nuclear pore complex. Mol Cell Biol. 2002a;22:6498–6508. doi: 10.1128/MCB.22.18.6498-6508.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang P, Li MZ, Elledge SJ. Towards genetic genome projects: genomic library screening and gene-targeting vector construction in a single step. Nat Genet. 2002b;30:31–39. doi: 10.1038/ng797. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.