

Abstract
Somatic mosaicism has been implicated as a causative mechanism in a number of genetic and genomic disorders. X-linked acrogigantism (XLAG) syndrome is a recently characterized genomic form of pediatric gigantism due to aggressive pituitary tumors that is caused by submicroscopic chromosome Xq26.3 duplications that include GPR101. We studied XLAG syndrome patients (N=18) to determine if somatic mosaicism contributed to the genomic pathophysiology.
Eighteen subjects with XLAG syndrome were identified with Xq26.3 duplications using high definition array comparative genome hybridization (HD-aCGH). We noted males with XLAG had a decreased log2 ratio compared with expected values, suggesting potential mosaicism, while females showed no such decrease. As compared with familial male XLAG cases, sporadic males had more marked evidence for mosaicism, with levels of Xq26.3 duplication between 16.1-53.8%. These characteristics were replicated using a novel, personalized breakpoint-junction specific quantification droplet digital PCR (ddPCR) technique. Using a separate ddPCR technique we studied the feasibility of identifying XLAG syndrome cases in a distinct patient population of 64 unrelated subjects with acromegaly/gigantism and identified one female gigantism patient that had increased copy number variation (CNV) threshold for GPR101 that was subsequently diagnosed as having XLAG syndrome on HD-aCGH.
Employing a combination of HD-aCGH and novel ddPCR approaches, we have demonstrated, for the first time, that XLAG syndrome can be caused by variable degrees of somatic mosaicism for duplications at chromosome Xq26.3. Somatic mosaicism was shown to occur in sporadic males but not in females with XLAG syndrome, although the clinical characteristics of the disease were similarly severe in both sexes.
Introduction
Somatic mosaicism describes a phenomenon in which two or more populations of cells compose one multicellular organism, within which each cell population is represented by its own unique genome (Lupski 2013). Somatic mosaic variants may arise from errors that occur during mitotic DNA replication. Such variants, including single nucleotides variants (SNVs), small insertions/deletions (indels), absence-of-heterozygosity (AOH) and structural variants (SVs), arise from erroneous, uncorrected mutational events and continually accumulate as cells divide during the development of a mature human individual that consists of 1016 cells amassing from a single fertilized cell (Campbell, et al. 2015). Somatic mosaic variants accompanying clonal expansion are closely related to aging and cancer in humans (Genovese, et al. 2014; Jacobs, et al. 2012; Liu, et al. 2014b). Identified as low-level mosaicism in blood-derived DNA or in specific tissues, such as those that comprise the nervous system, somatically mosaic variants may also contribute to human genetic or genomic disorders (Campbell et al. 2015; Jamuar, et al. 2014; Lindhurst, et al. 2011; Pham, et al. 2014; Poduri, et al. 2013). Alternatively, somatic mosaic variants may not have immediate clinical consequences for the carrier, but contribute to the recurrent risk of genomic disorders in offspring (Campbell, et al. 2014a; Campbell, et al. 2014b).
X-linked acrogigantism syndrome (XLAG, MIM #300942) is a recently characterized genomic disorder of early-onset gigantism caused by a submicroscopic duplication at chromosome Xq26.3 (Trivellin, et al. 2014). Affected patients are generally born normally sized following unremarkable pregnancies and develop mixed growth hormone and prolactin secreting pituitary hyperplasia and/or adenomas within the first 12-36 months of life (Beckers, et al. 2015; Trivellin et al. 2014). The XLAG syndrome phenotype of early childhood onset gigantism is aggressive and difficult to treat; it can be differentiated clinically from other forms of pituitary gigantism due to younger age and more severe hormonal hypersecretion (Rostomyan, et al. 2015). In the absence of multimodal neurosurgical and medical therapy XLAG syndrome is associated with relentless overgrowth due to GH hypersecretion (Naves, et al. 2015). The etiology of the pituitary tumor/hyperplasia in XLAG appears to be linked to a central disorder of hypersecretion of growth hormone releasing hormone (GHRH), which is a unique causative feature for pituitary gigantism in humans (Daly, et al. 2016). XLAG syndrome is caused by genomic duplications encompassing GPR101 (MIM *300393), that encodes an orphan G protein-coupled receptor; rare, potentially activating point mutations of GPR101 (e.g. p.E308D) have been identified in some patients with acromegaly, mostly in tumors (Trivellin et al. 2014).
Mosaic variants in genes encoding protein subunits involved in G-protein signaling have a recognized place in the etiology of syndromic conditions in endocrinology. McCune–Albright syndrome (MIM #174800) is caused by mosaic mutations in GNAS1 and is itself associated with pituitary gigantism and a wide spectrum of disease features (Lumbroso, et al. 2004; Vasilev, et al. 2014). Moreover, postzygotic, somatic mutational events have also been observed in other classical overgrowth syndromes, such as the AKT paralogs (AKT1, AKT2 and AKT3) that cause Proteus syndrome (MIM #176920), hypoglycemia and asymmetrical somatic growth (MIM #240900), and hemimegalencephaly (MIM #615937), respectively (Hussain, et al. 2011; Lindhurst et al. 2011; Poduri, et al. 2012).
Based on these observations of somatic mosaic mutations in several overgrowth syndromes, we hypothesized that potential somatic mosaicism might underlie XLAG syndrome in a proportion of cases. To investigate this possibility we studied an expanded series of patients with XLAG syndrome, pituitary gigantism or acromegaly to screen for, detect and quantify mosaicism for submicroscopic duplications at chromosome Xq26.3 that include GPR101.
Methods
Droplet digital polymerase chain reaction (ddPCR)
Overview of ddPCR
Two forms of droplet digital PCR (ddPCR) experiments were designed, one to assess large numbers of DNA samples for copy number variations at the GPR101 gene compared to a reference gene not included in duplications causing XLAG syndrome (“screening ddPCR”) and a second breakpoint-junction specific ddPCR (“quantification ddPCR”) to quantify somatic mosaicism at the borders of the Xq26.3 duplication in each affected XLAG syndrome case (described below). In ddPCR, target DNA molecules are distributed in droplets across multiple replicate reactions. The number of positive and negative droplets that contain a target template is used to calculate the concentration of the target and reference DNA sequences and their Poisson-based 95% confidence interval (Hindson, et al. 2011). The ddPCR methodology can readily distinguish duplication, triplication and even quadruplication of a locus (Gu, et al. 2016).
Screening ddPCR
The screening ddPCR assay was designed to quantify DNA copy number of the GPR101 gene (ENSG00000165370; X:137030148-137031674) as compared to DNA copy number of ZIC3 exon 1-intron 1/2 (ENSG00000156925; X:137566142-137577691). In previous studies of XLAG syndrome cases we have found that ZIC3 is the nearest protein-coding gene that is not included in the microduplications at Xq26.3 (Beckers et al. 2015; Trivellin et al. 2014). These screening analyses were performed on DNA derived from whole blood samples.
Screening ddPCR experiments were performed as follows. Each 21-μl reaction mixture contained 5μL of DNA template, 2X ddPCR supermix for probes (no dUTP) and GRP101 and ZIC3 exon 1 primers and probes assays. The assays were purchased as a 20X premix of primers and probes (Bio-Rad Laboratories, Marnes-la-Coquette, France) and used at 1X concentration. The 1X concentration of this assay comprised 900nM forward primer, 900nM reverse primer, and 250nM probe. Primers, hydrolysis probes sequences and ddPCR conditions are reported in Supplemental Table S1. After homogenization, the PCR reaction mixture and droplet generation oil for probes were loaded into an eight-channel droplet generator cartridge (Bio-Rad Laboratories, Marnes-la-Coquette, France). The PCR reaction mixtures were partitioned into an emulsion of approximately 15,000 droplets (∼ 1nL per droplet) that were manually transferred to a 96-well PCR plate. The PCR plate was heat-sealed and placed in a conventional thermal cycler (PRoFlex PCR systems, Life technologies) and PCR proceeded according to the manufacturer's protocol. Following the PCR, the 96-well plate was loaded on a QX100 droplet reader (Bio-Rad Laboratories, Marnes-la-Coquette, France). Analysis of the ddPCR data was performed with QuantaSoft software (version 1.7.4.0917), which analyzes each droplet individually using a two-color detection system (set to detect FAM or HEX dyes).
The absolute quantification of DNA is directly dependent on the number of accepted droplets (positive plus negative) and the DNA quantity analyzed. The calculation of the 95% confidence interval given by the Poisson law and the distribution of the CNV values according to our cohort of 91 samples and controls led us to consider a sample as duplicated if the CNV value was CNV> 2.5 and Poisson CNVmin (95% confidence) >2.0. The calculations and reporting of each CNV value ratio between GPR101 and ZIC3 and for each patient and overall groups accounts for differences in X chromosome number between males and females.
The population assessed using this screening ddPCR methodology included 36 patients with acromegaly (M/F: 24/12; age range at diagnosis: 22-50 years), six index cases from familial isolated pituitary adenoma kindreds (FIPA) with homogeneous acromegaly, and 22 patients with pituitary gigantism. None of these patients had been reported previously. All patients had pituitary adenomas diagnosed by magnetic resonance imaging and excess GH/IGF-1 secretion established before inclusion. In addition, none of the patients had mutations or deletions in genes known to cause acromegaly-gigantism, such as AIP, CDKN1B and MEN1, and none had syndromic conditions such as Carney complex or McCune Albright syndrome (Daly and Beckers 2015). As a positive control, 20 blood, tissue and tumor samples from eight previously diagnosed XLAG syndrome cases with established Xq26.3 duplication CNV on HD-CGH were included, while seven non-acrogigantism controls without GPR101 duplication CNV were also studied. All individuals and/or guardians provided informed consent and the genetic study was approved by the Ethics Committee of the Centre Hospitalier Universitaire de Liège, Belgium.
High-density array comparative genomic hybridization (HD-aCGH)
We used a custom-designed HD-aCGH to delineate high-resolution copy number variants (CNVs) in the genomic DNA samples derived from blood of subjects with XLAG. The array design and experimental procedures were reported previously (Trivellin et al. 2014; Yuan, et al. 2015).
Breakpoint junction (JCT) sequencing
JCT amplification and sequencing were performed following the protocol described in (Yuan et al. 2015).
Quantification JCT-specific ddPCR
For the quantification ddPCR we developed personalized ddPCR assays for each breakpoint junction (JCT). JCT-specific primer pairs were designed to amplify duplication JCT in each subject (Table S2). A pair of universal control primers (CTRL-F: 5′-CTCTGCCGCCTTCAACTCAACG-3′; CTRL-R: 5′-AAGGTCCGGTCGCAGCTCTTCT-3′) targeting exon 1 of ZIC3 on chromosome X, a nearby region that is apparently copy number neutral in all XLAG syndrome cases identified to date, was designed to amplify a control region in comparison to the JCT amplification. Both JCT-specific and control primers were designed with amplicons sizes of ∼ 500 bp. We performed the JCT-specific ddPCR experiments according to the manufacturer's protocol (http://www.bio-rad.com/webroot/web/pdf/lsr/literature/Bulletin_6407.pdf): 25μL master-mix containing 25ng of genomic DNA, forward/reverse primers with final concentration of 1μM, and 2x QX200 Evagreen supermix® was loaded onto QX200 AutoDG ddPCR System and followed the procedures of (1) droplet generation; (2) PCR amplification (95°C 5 minutes, [95°C 30 seconds, 60°C 1 minute, 72°C 1 minute] x40, 4°C 5 minutes, 90°C 5 minutes, and 4°C hold.); and (3) droplet reading. Data were analyzed with QuantaSoft analysis software (version 1.7.4). ddPCR can readily distinguish duplication, triplication, and even quadruplication of a locus (Gu, et al. 2015).
Workflow for mosaicism quantification by HD-aCGH and ddPCR
We utilized a workflow combining HD-aCGH, JCT sequencing and quantification ddPCR to characterize XLAG duplications and quantify their level of mosaicism (Figure S1).
HD-aCGH
The mosaicism level (αf for female, αm for male) was calculated based on HD-aCGHlog2 ratio (LR), which is the mean LR of all probes involved in the genomic segments that are duplicated:
JCT specific ddPCR
JCT-specific (JCT) or control (CTRL) ddPCR, as described above, were performed with equal DNA input (25ng) in separate reactions for each sample, and the number of positive droplets was compared between JCT and CTRL to quantify mosaicism. Theoretically, the number of positive droplets indicates the number of chromosomes with positive PCR amplification. CTRL ddPCR provides a positive signal in every droplet sequestering at least one copy of the X chromosome. JCT ddPCR specifically uses the breakpoint as a template, and only provides positive signals for the droplets sequestering at least one copy of the X chromosome with the specific XLAG syndrome duplication. Due to the random nature of partitioning in droplet generation, templates are randomly distributed in droplets. As a result, different droplets may contain different numbers of templates. Poisson distribution analysis was subsequently utilized to determined template concentration (M: concentration of JCT; N: concentration of CTRL) (http://www.bio-rad.com/webroot/web/pdf/lsr/literature/Bulletin_6407.pdf). Thus, theoretically for males (one copy of X chromosome), a JCT/CTRL ratio of M/N indicates a constitutional XLAG duplication if M=N; whereas a JCT/CTRL ratio of M/N suggests mosaicism if M<N, as M out of N cells harbor hemizygous XLAG syndrome duplication (the level of which should be calculated as M/N) (Figure S2). On the other hand, for females (copy number of X chromosome = 2), a JCT/CTRL ratio of M/N indicates a constitutional XLAG syndrome duplication if M=N/2, whereas a JCT/CTRL ratio of M/N is in keeping with mosaicism if M<N/2, as M out of N/2 cells harbor heterozygous XLAG duplication (the level of which should be calculated as 2M/N). Three technical replicates were performed for each ddPCR reaction to determine the mosaicism level.
Results
Eighteen subjects, including six males (three sporadic, three familial) and 12 females (11 sporadic, one familial), were identified with duplications encompassing GPR101 (Table 1). Among these, 15 were reported previously (Beckers et al. 2015; Naves et al. 2015; Trivellin et al. 2014) although none was studied previously for somatic mosaicism. The three new XLAG syndrome patients (2 females, 1 male) were adult sporadic pituitary gigantism cases whose disease began as very young children. We now report studies to investigate for potential mosaic duplication in these 18 subjects by orthogonal methods combining HD-aCGH, CNV breakpoint junction (JCT) sequencing and ddPCR (Campbell et al. 2014b; Gu et al. 2015) to achieve mosaic duplication detection and quantification (Figure S1).
Table 1. Mosaicism level quantification and breakpoint characterization in 18 subjects with XLAG syndrome.
| ID | Gender | Inheritance | ddPCR on parental DNA | DLRS | mosaicism level by aCGH | mosaicism level by ddPCR | Breakpoint features | Potential mechanism |
|---|---|---|---|---|---|---|---|---|
| S1 | M | Sporadic | both NA | 0.11 | 0.538 | 0.585 ± 0.016 | 12 bp microhomology | FoSTeS/MMBIR, MMEJ |
| S11 | M | Sporadic | mother negative, father NA | 0.18 | 0.256 | 0.294 ± 0.014 | 2 bp microhomology | FoSTeS/MMBIR, MMEJ |
| S15 | M | Sporadic | both NA | 0.15 | 0.161 | 0.182 ± 0.022 | 1 bp microhomology | FoSTeS/MMBIR, MMEJ |
| F2A | M | Familial | both NA | 0.27 | 0.691 | NA | 2 bp microhomology | FoSTeS/MMBIR, MMEJ |
| F1B | M | Familial | both NA | 0.14 | 0.769 | 0.854 ± 0.052 | 4 bp insertion | FoSTeS/MMBIR, NHEJ |
| F1C | M | Familial | both NA | 0.15 | 0.785 | 0.845 ± 0.059 | 4 bp insertion | FoSTeS/MMBIR, NHEJ |
| F1A | F | Familial | both NA | 0.14 | 1.030 | 0.839 ± 0.074 | 4 bp insertion | FoSTeS/MMBIR, NHEJ |
| S2 | F | Sporadic | Both Negative | 0.24 | 1.377 | 1.312 ± 0.117 | 5 bp microhomology | FoSTeS/MMBIR, MMEJ |
| S4 | F | Sporadic | Both Negative | 0.12 | 1.066 | 0.957 ± 0.010 | CGR with small insertions and microhomology at breakpoint junction | FoSTeS/MMBIR |
| S5 | F | Sporadic | both NA | 0.13 | 1.099 | 1.032 ± 0.026 | 5 bp insertion | FoSTeS/MMBIR, NHEJ |
| S6 | F | Sporadic | Both Negative | 0.13 | 0.999 | 0.944 ± 0.033 | 2 bp microhomology | FoSTeS/MMBIR, MMEJ |
| S7 | F | Sporadic | both NA | 0.14 | 1.041 | 0.944 ± 0.051 | 3 bp microhomology | FoSTeS/MMBIR, MMEJ |
| S8 | F | Sporadic | both NA | 0.14 | 0.911 | 0.968 ± 0.057 | 3 bp microhomology | FoSTeS/MMBIR, MMEJ |
| S9 | F | Sporadic | both NA | 0.24 | 0.963 | 1.017 ± 0.034 | 4 bp microhomology / 7 bp microhomeology | FoSTeS/MMBIR, MMEJ |
| S10 | F | Sporadic | both NA | 0.16 | 0.758 | 0.986 ± 0.048 | 4 bp microhomology | FoSTeS/MMBIR, MMEJ |
| S14 | F | Sporadic | both NA | 0.16 | 1.150 | 0.994 ± 0.007 | 3 bp microhomology with an 8 bp deletion nearby | FoSTeS/MMBIR, MMEJ |
| S13 | F | Sporadic | Both Negative | 0.11 | 0.806 | 0.974 ± 0.040 | 52 bp insertion | FoSTeS/MMBIR, MMEJ |
| S16 | F | Sporadic | both NA | 0.14 | 0.845 | 0.915 ± 0.033 | 291 bp insertion | FoSTeS/MMBIR, MMEJ |
On HD-aCGH all the identified XLAG duplications are unique and have apparently variable boundaries, documenting nonrecurrent duplications (Figure 1). The new XLAG duplications in the current study did not alter the smallest regions of overlap (SROs) reported previously (Beckers et al. 2015; Trivellin et al. 2014). These duplications range in size from 554 Kb to 674 Kb and all include the GPR101 gene that has been functionally demonstrated to be disease contributing (Trivellin et al. 2014). Microhomology, small insertions and one complex genomic rearrangement were identified at the JCT (Table 1, Figure S3) and are in keeping with fork stalling and template switching/microhomology-mediated break-induced replication (FoSTeS/MMBIR) as the potential mechanism for the duplication (Hastings, et al. 2009; Lee, et al. 2007; Sakofsky, et al. 2015; Trivellin et al. 2014; Zhang, et al. 2009).
Figure 1.
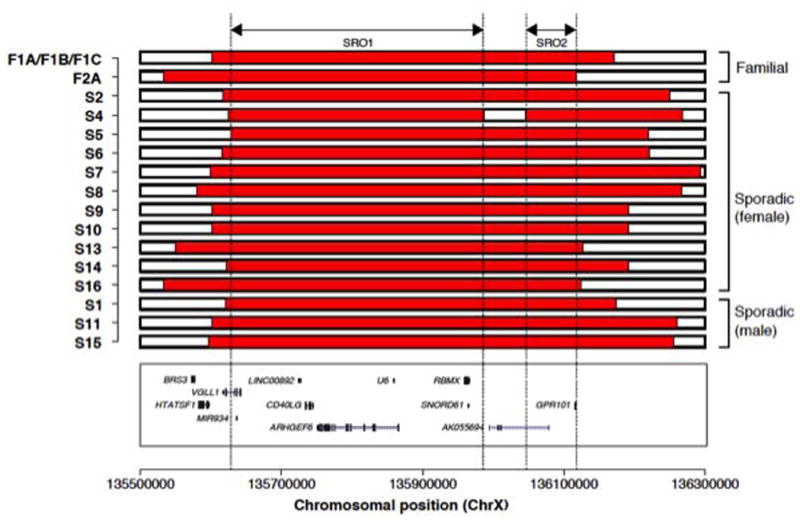
Using HD-aCGH, we observed that male subjects had a decreased log2 ratio (LR) level in comparison to the theoretical LR value of constitutional duplication CNV on the X chromosome in males (LR = 1). All the male subjects show decreased LR, in keeping with mosaicism (three sporadic males in Figure 2, and three familial males in Figure S4). Taken together, male XLAG syndrome patients, as a group, demonstrated significantly lower LR values as compared with female patients as a group (Welch Two Sample t-test, p-value = 0.003). Moreover, sporadic male XLAG patients had the clearest evidence of mosaicism on HD-aCGH. The three sporadic males had lower levels of duplication (S1 (53.8%), S11 (32.8%) and S15 (16.1%)) than the three familial XLAG males F2A (69.1%), F1B (76.9%) and F1C (78.5%). On HD-aCGH there was no evidence of mosaicism in female XLAG subjects (Table 1, Figure 3).
Figure 2.
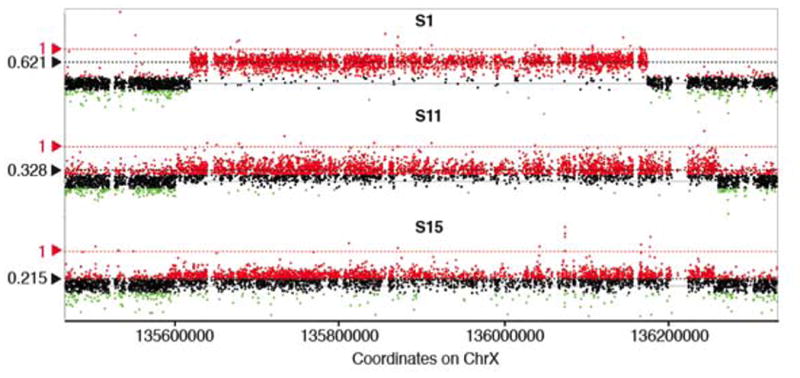
Figure 3.
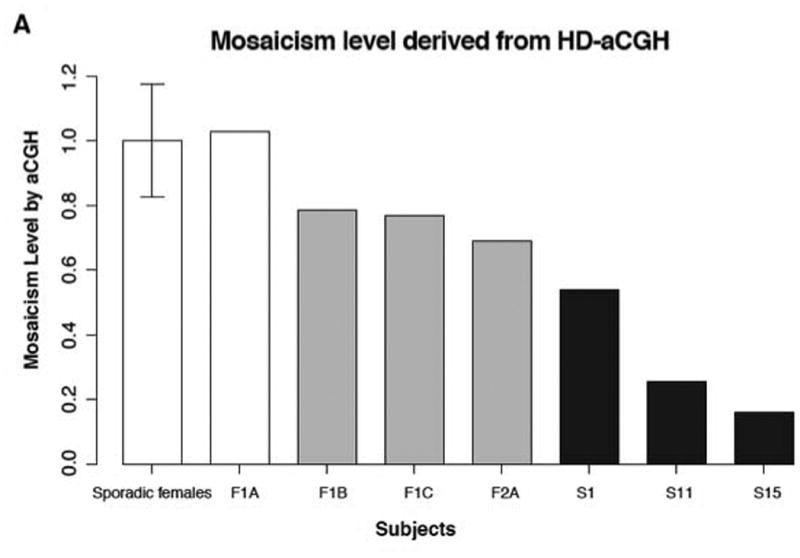
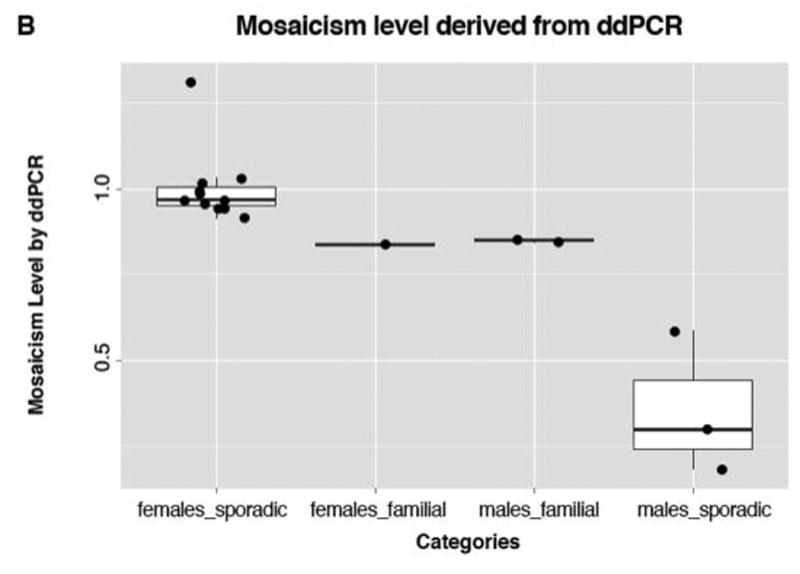
To further examine and precisely quantify the mosaicism level in the cohort we implemented personalized ddPCR assay targeting the JCT in each subject (Figure S1) and thus measuring the copy number specifically for the novel breakpoint junction (Campbell et al. 2014b; Gu et al. 2015). Using this approach, we confirmed the HD-aCGH finding that female XLAG patients had no evidence of mosaicism (Figure 3A). On ddPCR we also confirmed the HD-aCGH finding of low-level mosaicism in blood DNA of three sporadic male XLAG subjects S1, S11 and S15. The mosaicism levels in these subjects were 58.5% ± 1.6%, 29.4% ± 1.4% and 18.2% ± 2.2%, respectively, which is similar in magnitude to that obtained on HD-aCGH. In familial XLAG males, the potential mosaicism level on ddPCR was intermediate between that seen in the sporadic males and the lack of mosaicism in the female XLAG patient group (Figure 3B, Figure S5). Taken together as a group, males with XLAG had significantly lower ddPCR values than females with XLAG (Welch Two Sample t-test, p-value = 0.02677, Figure 3), which is again consistent with the HD-aCGH results.
We also developed a ddPCR assay in order to assess the feasibility of screening existing populations with acromegaly and gigantism for abnormalities in copy number at GPR101 (causative and duplicated in XLAG syndrome) versus ZIC3 (not duplicated in XLAG syndrome). We studied a total of 91 samples with a median value of 8.77 ng (95.3% CI: 7.13 to 10.13) for the ZIC3 gene (Exon 1- Intron 1/2). The median value of ΔCNV (CNVmax-CNV min) was 0.315 (95.3% CI: 0.270 to 0.350); this difference was calculated according to the Poisson law (95% confidence: CNV value ± 0.175). The CNV distribution showed very few intermediate values, reflecting the capacity of the screening ddPCR to discriminate non-duplicated samples (1.75-2.25) from duplication without intermediate values (Figure 4). Concordance results between HD-aCGH and screening ddPCR were very good (Pearson's χ2 statistic 85.78; DF 4, P<0.0001). The eight XLAG patients were positive on this screening ddPCR (CNV median: 3.050, 97.3% CI: 2.98 to 3.26; min 2.73; Figure S6A) and the seven normal patients had a normal CNV status (CNV median: 1.93, 98.4% CI: 1.710 to 2.17; Figure S6B).
Figure 4.
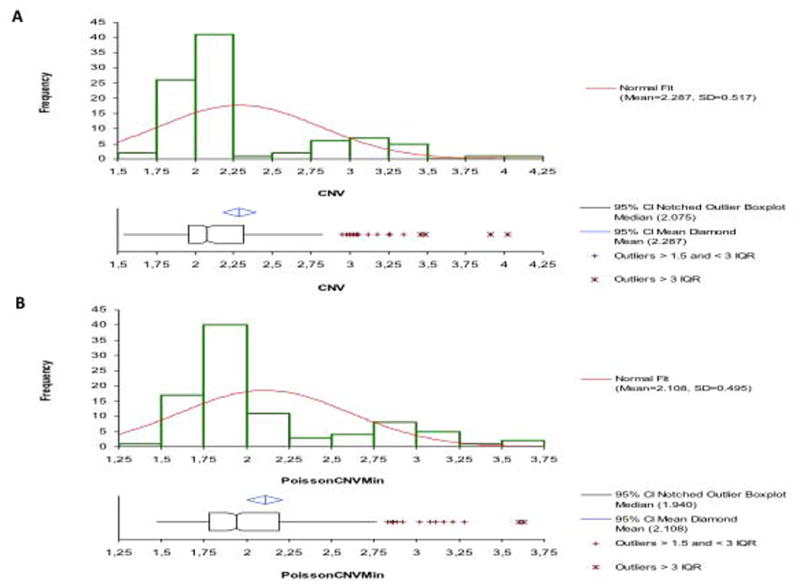
In the screened population 60/64 patients had a non-duplicated ddPCR CNV status (ddPCR CNV median: 2.05, 96.0% CI: 2.000 to 2.070). Of the four patients with CNV values outside of the thresholds empirically established for this screening assay, one female (S16) had a distant history of pediatric onset acrogigantism (diagnosed >40 years previously) and lifelong active acromegaly; her CNVmax and CNVmin values were 3.69 and 3.28, respectively. On HD-aCGH she was found to have a chromosome Xq26.3 duplication. Molecular mechanism studies revealed a 291 base pair insertion at the breakpoint, suggesting FoSTeS/MMBIR for formation of the duplications. A diagnosis of XLAG syndrome was made. A male with adolescent-onset acrogigantism was positive for potential duplication on screening ddPCR although his values were lower than those seen in XLAG cases and (CNVmax: 2.89; CNVmin: 2.33); on HD-aCGH no duplication was discerned. A female pediatric-onset acromegaly patient had inconclusive ddPCR values as her CNVmax (2.36) was below the duplication threshold of 2.5, while her CNVmin (2.36) was above the threshold of 2.0. HD-aCGH showed no duplication. A female pediatric onset gigantism patient had a screening ddPCR result with CNV levels below normal; the HD-aCGH result was normal and no abnormality was seen at the Xq26.3 locus.
Discussion
In the study we have demonstrated that somatic mosaicism plays an important role in the pathogenesis of XLAG syndrome, a newly described, severe form of pediatric onset gigantism caused by pituitary tumors. In contrast to females that have constitutional submicroscopic duplications at chromosome Xq26.3, we have shown that sporadic male patients with XLAG are somatic mosaics that display a variable degree of mosaicism. Sporadic males can demonstrate quite low levels of mosaicism for the XLAG duplication in DNA isolated from blood (e.g. 16.1%). Familial males with XLAG are intermediate between sporadic males and the female patients, which indicates that there may be variable mosaicism in males although too few familial cases are available to make that determination with certainty at this time. This finding was demonstrated first using a HD-aCGH method specifically focused around chromosome Xq (Trivellin et al. 2014). In addition we developed a new quantitative method of ddPCR that was specific to each patient and their particular unique duplication characteristics. This ddPCR technique also confirmed the existence of somatic mosaicism in male XLAG patients, with findings that were almost identical to those achieved using HD-aCGH.
Somatic mosaicism arises post-zygotically. Mutations that occur at different developmental timings may have diverse tissue distributions and impact distinctly on human genetic or genomic disorders. The number of mitoses (or cell divisions) between generations for males is estimated to be 400, whereas the number is 30 for females (Campbell et al. 2014b; Drost and Lee 1995). If a mutation occurs in the parental generation and becomes a confined gonadal mosaicism, it may be transmitted to the offspring and appear as a constitutional and apparently de novo mutation. Such mosaic mutations are confined to the germline; thus they usually do not manifest clinical phenotypes, and may evade genetic testing. However, these mutations contribute considerably to the recurrence risk of genetic disorders, a situation in which more than one child from the same family can be born with the same apparently sporadic autosomal dominant condition. On the other hand, mutations may occur during early embryonic development of an individual; particularly during the many mitoses that occur with the rapid proliferation accompanying early embryogenesis. As a result, the mutation may be segregated into a limited number of cell lineages. These somatic mosaic mutations may be associated with known genetic or genomic disorders and consequently lead to differential phenotypic consequences. Such mutations are mosaic, because they are not uniformly represented by the entire cell population throughout the human organism. If the mutation segregated in hematopoietic stem cells (HSCs) that further develop into blood cells, these may be detected by genetic testing using blood as the specimen. We present here a systematic study including the detection, molecular investigation, quantification and clinical correlation of somatic mosaicism underlying XLAG syndrome, a recently defined early-childhood onset form of pituitary gigantism.
XLAG syndrome is a form of gigantism that is likely caused by GPR101 duplication, while potentially activating and inactivating mutations of GPR101 have been identified in patients with pituitary adenomas and growth hormone deficiency, respectively (Castinetti, et al. 2016; Trivellin et al. 2014). We have identified mosaic XLAG syndrome locus duplications that are likely to be the cause of the disease phenotype. The mosaic XLAG syndrome locus duplication may arise postzygotically as a mitotic event during the early embryonic developmental stage of the sporadic males, affect growth hormone secretion thereafter, and eventually contribute to the XLAG syndrome phenotype. These XLAG locus duplications may also occur before the segregation of blood and pituitary cell lineages, and therefore affect the growth hormone secretion from the pituitary tissue and are detectable by HD-aCGH using blood DNA. Our data reveal that males had significant evidence of mosaicism while females did not (Figure 3). Moreover, familial males with XLAG syndrome had duplication levels that were intermediate between the constitutional levels seen in females and clear mosaicism seen in sporadic males. However, based on a priori hypothesis, an X-linked mutation is anticipated to be constitutional in these familial males who inherit the same mutation from the mother. The lower level of XLAG locus duplication observed in familial males in comparison to females may result from the uncertainty of measurements. In spite of this, if mosaicism is eventually confirmed in familial males, it might perhaps be explained by a somatic reversion mechanism mediated by mitotic intrachromosomal NAHR (Liehr, et al. 1996; Liu, et al. 2014a; 396 Steinmann, et al. 2007), which we do not have experimental evidence to support at this stage.
A comparison of the clinical phenotype and disease characteristics in the mosaic sporadic males as compared with sporadic females with XLAG syndrome reveals some important findings. All three mosaic males had severe early onset overgrowth due to pituitary adenomas and the disease presented and was diagnosed at a similarly young age as sporadic female XLAG syndrome cases. Furthermore, the severe hormonal hypersecretion and the subsequent overgrowth pattern required complex multimodal surgical and medical therapy in the sporadic mosaic males, again not differing from non-mosaic cases. Final height in pituitary gigantism, irrespective of genetic cause, is determined by a variety of factors, not least early control of hormonal hypersecretion (Rostomyan et al. 2015). Two of the three sporadic mosaic males with XLAG syndrome did not undergo neurosurgery or effective medical therapy during childhood and hence have extreme gigantism (209 cm at 12 years in one case, Z-score >8.7, and >230 cm final height in the other) (Naves et al. 2015). The other patient was controlled with surgery and medical therapy (growth hormone receptor antagonist, pegvisomant) during childhood and can be expected to have a normal final height (Beckers et al. 2015). Therefore the clinical profile does not seem to differ between mosaics and non-mosaic XLAG syndrome patients. Relatively low levels of duplication at the XLAG syndrome locus (16.8-32.8% as detected in blood) can lead to some of the most dramatic pediatric and adult cases of pituitary gigantism in recorded medical history. This suggests that the pathological process is highly sensitive to even minor levels of increased copy number at the XLAG syndrome locus. The pituitary findings in cases of XLAG syndrome are quite uniform (mixed GH-prolactin secreting pituitary adenomas and/or hyperplasia) irrespective of the level of mosaicism of the patient (Beckers et al. 2015; Naves et al. 2015; Trivellin et al. 2014). Moreover, as males and females are clinically similar, the impact of X-chromosome inactivation in females with XLAG syndrome should be considered, as this could hypothetically alter the level of duplication occurring in specific tissues, such as the hypothalamus.
Daly et al recently reported that GHRH hypersecretion is implicated in XLAG syndrome and that GHRH antagonism can inhibit GH and prolactin secretion from primary tumor cell culture in XLAG syndrome (Daly et al. 2016). Other rare clinical and experimental instances of chronic GHRH hypersecretion lead to similar pathological effects on the pituitary gland (Asa, et al. 1992; Borson-Chazot, et al. 2012; Mayo, et al. 1988). GHRH is a very potent physiological stimulator of GH, and GHRH secretion by a discreet population of hypothalamic neurons is tightly regulated by integrated central and peripheral signals (Gahete, et al. 2009; Veldhuis, et al. 2012). GPR101 is specifically expressed in regions of the hypothalamus and brain that are involved in integrating such signals, the dysregulation of GHRH secretion and pituitary pathology in XLAG syndrome (Bates, et al. 2006; Trivellin, et al. 2016; Trivellin et al. 2014). Taken together these findings suggest a mechanism by which even modestly increased copy number of GPR101 could lead to the severe pituitary gigantism observed in XLAG syndrome patients with an Xq26.3 duplication; some of these mosaicism levels may be beyond that which can currently be detected by our techniques. Moreover, mosaicism might occur only in nervous system tissues or only in cells from which the pituitary derives and not be present in the blood.
Somatic mosaicism, which may introduce false negative results in genetic testing, is always challenging to detect. A large number of techniques have been described for mosaicism detection (Campbell et al. 2015). While next generation sequencing has been successful in detecting somatically mosaic SNVs in patients with a specific disorder (Ansari, et al. 2014; Huisman, et al. 2013), difficulty remains for mosaic CNVs (Rahbari, et al. 2016). Conventional cytogenetic techniques, such as karyotyping and fluorescence in situ hybridization (FISH), provide direct visualization and quantification of mosaic structural variants by scoring a sufficiently large number of cells. However, the result may be biased given potential cell culture artifacts (Cheung, et al. 2007). Moreover, submicroscopic CNVs with sizes smaller than 50 kb make karyotyping and FISH unrevealing for prospective mosaicism (Pham et al. 2014). In the current study, we used aCGH with high-density probes to retrospectively interrogate the known region for XLAG syndrome and provide molecular details of the rearrangement, which allow further quantification by ddPCR. It is suggested that mosaicism may be detected at a level as low as 10-20% under ideal conditions by aCGH (Ballif, et al. 2006; Boone, et al. 2010; Pham et al. 2014), and potentially to the 5% level utilizing B-allele frequency information from SNP arrays (Conlin, et al. 2010). Driven by phenotype, a personalized assay (e.g. targeted deep sequencing or HD-aCGH) may be designed to investigate the known disease-associated loci in detail. Sampling various tissues may also benefit mosaicism detection, as demonstrated in subjects with Cornelia de Lange syndrome (Huisman et al. 2013). In females with XLAG syndrome we observe roughly equal dosage changes in pituitary tumor derived DNA as compared with DNA from blood. At this time we do not have multiple tissues sampled for the mosaic male subjects, so further analyses of the distribution of mosaicism status in different tissues of new mosaic males will be required in the future.
We used ddPCR to measure GRP101 duplication because it allows the measurement of low level mosaicism for CNV (Weaver, et al. 2010) and the accurate counting of alleles from DNA isolated from a mixture of heterogeneous cell populations. Previous studies have shown a very high level of concordance between ddPCR and exome sequencing to measure CNV (Handsaker, et al. 2015). The HD-aCGH and junction-specific ddPCR techniques provide specific information regarding duplications and mosaicism in individual cases of XLAG syndrome. Neither of these methods is, however, well suited to genetic screening of larger populations of patients with acromegaly and gigantism. To this end we developed a separate ddPCR assay and validated its use in a population of proven XLAG syndrome cases with known Xq26.3 duplications, normal individuals without Xq26.3 duplications and a large de novo patient population of acromegaly, FIPA kindreds with homogeneous acromegaly, and pituitary gigantism. Based on the recognition that none of the previously identified cases of XLAG syndrome had duplications that extended telometrically to the ZIC3 gene, we used this as a reference to compare with GPR101, which is a causative gene and is invariably duplicated. this approach we were able to rapidly “screen” a sizeable series of target patients with pituitary gigantism and acromegaly (sporadic and familial). Using this screening method we identified four acromegaly/gigantism cases with results that were abnormal compared to reference controls, of which two were above the threshold, one intermediate and one that was below the CNVmin threshold; three were normal on subsequent HD-aCGH. The other case was an adult patient with a distant history suggestive of XLAG syndrome, and the ddPCR results were confirmed by HD-aCGH as consisting of a novel duplication at the XLAG syndrome locus. This ddPCR methodology suggest that it could be used as a first step and screening assay for in studying cohorts of patients with acrogigantism for XLAG syndrome, but would require verification by HD-aCGH in indeterminate and abnormal cases (4.7% of our series).
Using a combination of standard and novel techniques we have shown that XLAG syndrome, a newly recognized form of severe acrogigantism, has a more diverse genetic pathophysiology than we originally described. Sporadic males in this study all demonstrated evidence of somatic mosaicism for the submicroscopic duplications on Xq26.3 that cause XLAG syndrome. This differs from female XLAG patients that all have apparently constitutional duplications. Results obtained using HD-aCGH were validated on CNV assay using a personalized junction-specific ddPCR technique for the unique breakpoints causing Xq26.3 duplication. Moreover, ddPCR screening based on CNV at GPR101 holds promise for identifying potential XLAG syndrome cases among larger cohorts of acrogigantism patients. Somatic mosaicism is an important pathological mechanism for genetic and genomic diseases and its contribution to the causation of both new and established disorders should be actively investigated.
Supplementary Material
Acknowledgments
We would like to thank referring physicians including particularly Catherine S Choong, Tim D Cheetham, Jacques Young, Philippe A Lysy, Andrew Cotterill, Natalia Strebkova, Nadia Mazerkina and Michael T Collins. In addition, we would like to acknowledge the patients and families for their patience and forbearance.
Footnotes
Disclosure Statement: J.R.L. has stock ownership in 23andMe, is a paid consultant for Regeneron Pharmaceuticals, has stock options in Lasergen, Inc. is a member of the Scientific Advisory Board of Baylor Miraca Genetics Laboratories, and is a co-inventor on multiple United States and European patents related to molecular diagnostics for inherited neuropathies, eye diseases and bacterial genomic fingerprinting. The Department of Molecular and Human Genetics at Baylor College of Medicine derives revenue from the chromosomal microarray analysis (CMA) and clinical exome sequencing offered in the Baylor Miraca Genetics Laboratory (BMGL: http://www.bmgl.com/BMGL/Default.aspx). Other authors have nothing to disclose.
References
- Ansari M, Poke G, Ferry Q, Williamson K, Aldridge R, Meynert AM, Bengani H, Chan CY, Kayserili H, Avci S, et al. Genetic heterogeneity in Cornelia de Lange syndrome (CdLS) and CdLS-like phenotypes with observed and predicted levels of mosaicism. J Med Genet. 2014;51:659–668. doi: 10.1136/jmedgenet-2014-102573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asa SL, Kovacs K, Stefaneanu L, Horvath E, Billestrup N, Gonzalez-Manchon C, Vale W. Pituitary adenomas in mice transgenic for growth hormone-releasing hormone. Endocrinology. 1992;131:2083–2089. doi: 10.1210/endo.131.5.1425411. [DOI] [PubMed] [Google Scholar]
- Ballif BC, Rorem EA, Sundin K, Lincicum M, Gaskin S, Coppinger J, Kashork CD, Shaffer LG, Bejjani BA. Detection of low-level mosaicism by array CGH in routine diagnostic specimens. Am J Med Genet A. 2006;140:2757–2767. doi: 10.1002/ajmg.a.31539. [DOI] [PubMed] [Google Scholar]
- Bates B, Zhang L, Nawoschik S, Kodangattil S, Tseng E, Kopsco D, Kramer A, Shan Q, Taylor N, Johnson J, et al. Characterization of Gpr101 expression and G-protein coupling selectivity. Brain Res. 2006;1087:1–14. doi: 10.1016/j.brainres.2006.02.123. [DOI] [PubMed] [Google Scholar]
- Beckers A, Lodish MB, Trivellin G, Rostomyan L, Lee M, Faucz FR, Yuan B, Choong CS, Caberg JH, Verrua E, et al. X-linked acrogigantism syndrome: clinical profile and therapeutic responses. Endocr Relat Cancer. 2015;22:353–367. doi: 10.1530/ERC-15-0038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boone PM, Bacino CA, Shaw CA, Eng PA, Hixson PM, Pursley AN, Kang SH, Yang Y, Wiszniewska J, Nowakowska BA, et al. Detection of clinically relevant exonic copy-number changes by array CGH. Hum Mutat. 2010;31:1326–1342. doi: 10.1002/humu.21360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borson-Chazot F, Garby L, Raverot G, Claustrat F, Raverot V, Sassolas G, group GTE Acromegaly induced by ectopic secretion of GHRH: a review 30 years after GHRH discovery. Ann Endocrinol (Paris) 2012;73:497–502. doi: 10.1016/j.ando.2012.09.004. [DOI] [PubMed] [Google Scholar]
- Campbell IM, Shaw CA, Stankiewicz P, Lupski JR. Somatic mosaicism: implications for disease and transmission genetics. Trends Genet. 2015;31:382–392. doi: 10.1016/j.tig.2015.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell IM, Stewart JR, James RA, Lupski JR, Stankiewicz P, Olofsson P, Shaw CA. Parent of origin, mosaicism, and recurrence risk: probabilistic modeling explains the broken symmetry of transmission genetics. Am J Hum Genet. 2014a;95:345–359. doi: 10.1016/j.ajhg.2014.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell IM, Yuan B, Robberecht C, Pfundt R, Szafranski P, McEntagart ME, Nagamani SC, Erez A, Bartnik M, Wisniowiecka-Kowalnik B, et al. Parental somatic mosaicism is underrecognized and influences recurrence risk of genomic disorders. Am J Hum Genet. 2014b;95:173–182. doi: 10.1016/j.ajhg.2014.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castinetti F, Daly AF, Stratakis CA, Caberg JH, Castermans E, Trivellin G, Rostomyan L, Saveanu A, Jullien N, Reynaud R, et al. GPR101 Mutations are not a Frequent Cause of Congenital Isolated Growth Hormone Deficiency. Horm Metab Res. 2016 doi: 10.1055/s-0042-100733. (Epub ahead of print) 10.1055/s-0042-100733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung SW, Shaw CA, Scott DA, Patel A, Sahoo T, Bacino CA, Pursley A, Li J, Erickson R, Gropman AL, et al. Microarray-based CGH detects chromosomal mosaicism not revealed by conventional cytogenetics. Am J Med Genet A. 2007;143A:1679–1686. doi: 10.1002/ajmg.a.31740. [DOI] [PubMed] [Google Scholar]
- Conlin LK, Thiel BD, Bonnemann CG, Medne L, Ernst LM, Zackai EH, Deardorff MA, Krantz ID, Hakonarson H, Spinner NB. Mechanisms of mosaicism, chimerism and uniparental disomy identified by single nucleotide polymorphism array analysis. Hum Mol Genet. 2010;19:1263–1275. doi: 10.1093/hmg/ddq003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daly AF, Beckers A. Familial isolated pituitary adenomas (FIPA) and mutations in the aryl hydrocarbon receptor interacting protein (AIP) gene. Endocrinol Metab Clin North Am. 2015;44:19–25. doi: 10.1016/j.ecl.2014.10.002. [DOI] [PubMed] [Google Scholar]
- Daly AF, Lysy PA, Desfilles C, Rostomyan L, Mohamed A, Caberg JH, Raverot V, Castermans E, Marbaix E, Maiter D, et al. GHRH excess and blockade in X-LAG syndrome. Endocr Relat Cancer. 2016;23:161–170. doi: 10.1530/ERC-15-0478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drost JB, Lee WR. Biological basis of germline mutation: comparisons of spontaneous germline mutation rates among drosophila, mouse, and human. Environ Mol Mutagen. 1995;25 Suppl 26:48–64. doi: 10.1002/em.2850250609. [DOI] [PubMed] [Google Scholar]
- Gahete MD, Duran-Prado M, Luque RM, Martinez-Fuentes AJ, Quintero A, Gutierrez-Pascual E, Cordoba-Chacon J, Malagon MM, Gracia-Navarro F, Castano JP. Understanding the multifactorial control of growth hormone release by somatotropes: lessons from comparative endocrinology. Ann N Y Acad Sci. 2009;1163:137–153. doi: 10.1111/j.1749-6632.2008.03660.x. [DOI] [PubMed] [Google Scholar]
- Genovese G, Kahler AK, Handsaker RE, Lindberg J, Rose SA, Bakhoum SF, Chambert K, Mick E, Neale BM, Fromer M, et al. Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. N Engl J Med. 2014;371:2477–2487. doi: 10.1056/NEJMoa1409405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu S, Posey JE, Yuan B, Carvalho CM, Luk HM, Erikson K, Lo IF, Leung GK, Pickering CR, Chung BH, et al. Mechanisms for the Generation of Two Quadruplications Associated with Split-Hand Malformation. Hum Mutat. 2016;37:160–164. doi: 10.1002/humu.22929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Handsaker RE, Van Doren V, Berman JR, Genovese G, Kashin S, Boettger LM, McCarroll SA. Large multiallelic copy number variations in humans. Nat Genet. 2015;47:296–303. doi: 10.1038/ng.3200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hastings PJ, Ira G, Lupski JR. A microhomology-mediated break-induced replication model for the origin of human copy number variation. PLoS Genet. 2009;5:e1000327. doi: 10.1371/journal.pgen.1000327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hindson BJ, Ness KD, Masquelier DA, Belgrader P, Heredia NJ, Makarewicz AJ, Bright IJ, Lucero MY, Hiddessen AL, Legler TC, et al. High-throughput droplet digital PCR system for absolute quantitation of DNA copy number. Anal Chem. 2011;83:8604–8610. doi: 10.1021/ac202028g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huisman SA, Redeker EJ, Maas SM, Mannens MM, Hennekam RC. High rate of mosaicism in individuals with Cornelia de Lange syndrome. J Med Genet. 2013;50:339–344. doi: 10.1136/jmedgenet-2012-101477. [DOI] [PubMed] [Google Scholar]
- Hussain K, Challis B, Rocha N, Payne F, Minic M, Thompson A, Daly A, Scott C, Harris J, Smillie BJ, et al. An activating mutation of AKT2 and human hypoglycemia. Science. 2011;334:474. doi: 10.1126/science.1210878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobs KB, Yeager M, Zhou W, Wacholder S, Wang Z, Rodriguez-Santiago B, Hutchinson A, Deng X, Liu C, Horner MJ, et al. Detectable clonal mosaicism and its relationship to aging and cancer. Nat Genet. 2012;44:651–658. doi: 10.1038/ng.2270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jamuar SS, Lam AT, Kircher M, D'Gama AM, Wang J, Barry BJ, Zhang X, Hill RS, Partlow JN, Rozzo A, et al. Somatic mutations in cerebral cortical malformations. N Engl J Med. 2014;371:733–743. doi: 10.1056/NEJMoa1314432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JA, Carvalho CM, Lupski JR. A DNA replication mechanism for generating nonrecurrent rearrangements associated with genomic disorders. Cell. 2007;131:1235–1247. doi: 10.1016/j.cell.2007.11.037. [DOI] [PubMed] [Google Scholar]
- Liehr T, Rautenstrauss B, Grehl H, Bathke KD, Ekici A, Rauch A, Rott HD. Mosaicism for the Charcot-Marie-Tooth disease type 1A duplication suggests somatic reversion. Hum Genet. 1996;98:22–28. doi: 10.1007/s004390050154. [DOI] [PubMed] [Google Scholar]
- Lindhurst MJ, Sapp JC, Teer JK, Johnston JJ, Finn EM, Peters K, Turner J, Cannons JL, Bick D, Blakemore L, et al. A mosaic activating mutation in AKT1 associated with the Proteus syndrome. N Engl J Med. 2011;365:611–619. doi: 10.1056/NEJMoa1104017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu P, Gelowani V, Zhang F, Drory VE, Ben-Shachar S, Roney E, Medeiros AC, Moore RJ, DiVincenzo C, Burnette WB, et al. Mechanism, prevalence, and more severe neuropathy phenotype of the Charcot-Marie-Tooth type 1A triplication. Am J Hum Genet. 2014a;94:462–469. doi: 10.1016/j.ajhg.2014.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu P, Kaplan A, Yuan B, Hanna JH, Lupski JR, Reiner O. Passage number is a major contributor to genomic structural variations in mouse iPSCs. Stem Cells. 2014b;32:2657–2667. doi: 10.1002/stem.1779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lumbroso S, Paris F, Sultan C, European Collaborative S Activating Gsalpha mutations: analysis of 113 patients with signs of McCune-Albright syndrome--a European Collaborative Study. J Clin Endocrinol Metab. 2004;89:2107–2113. doi: 10.1210/jc.2003-031225. [DOI] [PubMed] [Google Scholar]
- Lupski JR. Genetics. Genome mosaicism--one human, multiple genomes. Science. 2013;341:358–359. doi: 10.1126/science.1239503. [DOI] [PubMed] [Google Scholar]
- Mayo KE, Hammer RE, Swanson LW, Brinster RL, Rosenfeld MG, Evans RM. Dramatic pituitary hyperplasia in transgenic mice expressing a human growth hormone-releasing factor gene. Mol Endocrinol. 1988;2:606–612. doi: 10.1210/mend-2-7-606. [DOI] [PubMed] [Google Scholar]
- Naves LA, Daly AF, Dias LA, Yuan B, Zakir JC, Barra GB, Palmeira L, Villa C, Trivellin G, Junior AJ, et al. Aggressive tumor growth and clinical evolution in a patient with X-linked acro-gigantism syndrome. Endocrine. 2015;51(2):236–44. doi: 10.1007/s12020-015-0804-6. 2016 Feb. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pham J, Shaw C, Pursley A, Hixson P, Sampath S, Roney E, Gambin T, Kang SH, Bi W, Lalani S, et al. Somatic mosaicism detected by exon-targeted, high-resolution aCGH in 10,362 consecutive cases. Eur J Hum Genet. 2014;22:969–978. doi: 10.1038/ejhg.2013.285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poduri A, Evrony GD, Cai X, Elhosary PC, Beroukhim R, Lehtinen MK, Hills LB, Heinzen EL, Hill A, Hill RS, et al. Somatic activation of AKT3 causes hemispheric developmental brain malformations. Neuron. 2012;74:41–48. doi: 10.1016/j.neuron.2012.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poduri A, Evrony GD, Cai X, Walsh CA. Somatic mutation, genomic variation, and neurological disease. Science. 2013;341:1237758. doi: 10.1126/science.1237758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahbari R, Wuster A, Lindsay SJ, Hardwick RJ, Alexandrov LB, Al Turki S, Dominiczak A, Morris A, Porteous D, Smith B, et al. Timing, rates and spectra of human germline mutation. Nat Genet. 2016;48:126–133. doi: 10.1038/ng.3469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rostomyan L, Daly AF, Petrossians P, Nachev E, Lila AR, Lecoq AL, Lecumberri B, Trivellin G, Salvatori R, Moraitis AG, et al. Clinical and genetic characterization of pituitary gigantism: an international collaborative study in 208 patients. Endocr Relat Cancer. 2015;22:745–757. doi: 10.1530/ERC-15-0320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakofsky CJ, Ayyar S, Deem AK, Chung WH, Ira G, Malkova A. Translesion Polymerases Drive Microhomology-Mediated Break-Induced Replication Leading to Complex Chromosomal Rearrangements. Mol Cell. 2015;60:860–872. doi: 10.1016/j.molcel.2015.10.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinmann K, Cooper DN, Kluwe L, Chuzhanova NA, Senger C, Serra E, Lazaro C, Gilaberte M, Wimmer K, Mautner VF, et al. Type 2 NF1 deletions are highly unusual by virtue of the absence of nonallelic homologous recombination hotspots and an apparent preference for female mitotic recombination. Am J Hum Genet. 2007;81:1201–1220. doi: 10.1086/522089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trivellin G, Bjelobaba I, Daly A, Larco D, Faucz F, THIRY A, Wu T, Stojilkovic S, Feldman B, Costanzi S. Characterization of GPR101 transcripts structure, expression and signaling. Abstract book-Keystone Symposia on GPCRs 2016 [Google Scholar]
- Trivellin G, Daly AF, Faucz FR, Yuan B, Rostomyan L, Larco DO, Schernthaner-Reiter MH, Szarek E, Leal LF, Caberg JH, et al. Gigantism and acromegaly due to Xq26 microduplications and GPR101 mutation. N Engl J Med. 2014;371:2363–2374. doi: 10.1056/NEJMoa1408028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasilev V, Daly AF, Thiry A, Petrossians P, Fina F, Rostomyan L, Silvy M, Enjalbert A, Barlier A, Beckers A. McCune-Albright syndrome: a detailed pathological and genetic analysis of disease effects in an adult patient. J Clin Endocrinol Metab. 2014;99:E2029–2038. doi: 10.1210/jc.2014-1291. [DOI] [PubMed] [Google Scholar]
- Veldhuis JD, Iranmanesh A, Erickson D, Roelfsema F, Bowers CY. Handbook of Neuroendocrinology. San Diego: Academic Press; 2012. Chapter 10 - Lifetime Regulation of Growth Hormone (GH) Secretion A2 - Levine, George FinkDonald W. PfaffJon E; pp. 237–257. [Google Scholar]
- Weaver S, Dube S, Mir A, Qin J, Sun G, Ramakrishnan R, Jones RC, Livak KJ. Taking qPCR to a higher level: Analysis of CNV reveals the power of high throughput qPCR to enhance quantitative resolution. Methods. 2010;50:271–276. doi: 10.1016/j.ymeth.2010.01.003. [DOI] [PubMed] [Google Scholar]
- Yuan B, Harel T, Gu S, Liu P, Burglen L, Chantot-Bastaraud S, Gelowani V, Beck CR, Carvalho CM, Cheung SW, et al. Nonrecurrent 17p11.2p12 Rearrangement Events that Result in Two Concomitant Genomic Disorders: The PMP22-RAI1 Contiguous Gene Duplication Syndrome. Am J Hum Genet. 2015;97:691–707. doi: 10.1016/j.ajhg.2015.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang F, Khajavi M, Connolly AM, Towne CF, Batish SD, Lupski JR. The DNA replication FoSTeS/MMBIR mechanism can generate genomic, genic and exonic complex rearrangements in humans. Nat Genet. 2009;41:849–853. doi: 10.1038/ng.399. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.