

Abstract
Discovered a little over two decades ago, small interfering RNAs (siRNAs) and microRNAs (miRNAs) are noncoding RNAs with important roles in gene regulation. They have recently been investigated as novel classes of therapeutic agents for the treatment of a wide range of disorders including cancers and infections. Clinical trials of siRNA- and miRNA-based drugs have already been initiated. siRNAs and miRNAs share many similarities, both are short duplex RNA molecules that exert gene silencing effects at the post-transcriptional level by targeting messenger RNA (mRNA), yet their mechanisms of action and clinical applications are distinct. The major difference between siRNAs and miRNAs is that the former are highly specific with only one mRNA target, whereas the latter have multiple targets. The therapeutic approaches of siRNAs and miRNAs are therefore very different. Hence, this review provides a comparison between therapeutic siRNAs and miRNAs in terms of their mechanisms of action, physicochemical properties, delivery, and clinical applications. Moreover, the challenges in developing both classes of RNA as therapeutics are also discussed.
Keywords: cancer therapy, gene silencing, microRNA, RNA interference, small interfering RNA
The term “non-coding RNA” is commonly employed for RNA that does not encode a protein.1 Although the current understanding of these RNA molecules represents perhaps only the tip of the iceberg, with the rapid development of molecular biotechnology, noncoding RNAs are increasingly found to have far more important functions than previously recognized and many new classes of noncoding RNA have been identified. Among them, small interfering RNAs (siRNAs) and microRNAs (miRNAs) have attracted considerable attention because their role in gene regulation makes them likely targets for drug discovery and development. Indeed, the therapeutic potential of siRNAs and miRNAs has been demonstrated in the treatment of many different diseases including cancers2,3,4 and infections.5,6,7 Compared with conventional small therapeutic molecules, siRNAs and miRNAs offer the advantages of being highly potent and able to act on “non-druggable” targets (for example, proteins which lack an enzymatic function or have a conformation that is inaccessible to traditional drug molecules),8 as they can be designed to affect virtually any gene of interest.
Therapeutic approaches based on siRNA involve the introduction of a synthetic siRNA into the target cells to elicit RNA interference (RNAi), thereby inhibiting the expression of a specific messenger RNA (mRNA) to produce a gene silencing effect.9 By contrast, miRNA-based therapeutics comprise two approaches: miRNA inhibition and miRNA replacement. The former approach resembles antisense therapy,10 with synthetic single stranded RNAs acting as miRNA antagonists (also known as antagomirs or anti-miRs) to inhibit the action of the endogenous miRNAs. In the replacement approach, synthetic miRNAs (also known as miRNA mimics) are used to mimic the function of the endogenous miRNAs.11 It thus leads to mRNA degradation/inhibition, and produces a gene silencing effect. This review focuses on the therapeutic approach achieved by gene silencing, and so only the miRNA replacement approach is discussed and compared with siRNA. The therapeutic potentials and applications of the miRNA inhibition approach have been reviewed previously.12,13,14
siRNAs and miRNAs have similar physicochemical properties but distinct functions (Table 1). Both are short RNA duplexes that target mRNA(s) to produce a gene silencing effect, yet their mechanisms of action are distinct. As a result, the requirements for sequence design and therapeutic applications of siRNAs and miRNAs are different. On the other hand, for clinical development, the two types of small RNA molecules face a similar set of barriers: poor stability in vivo, delivery challenges and off-target effects15; and so, the same strategies can be employed to improve their in vivo efficacy.
Table 1. Comparison of general properties between siRNA and miRNA.

Gene silencing mechanism of siRNA and miRNA
RNA interference and siRNA
RNAi is a natural cellular process that silences gene expression by promoting the degradation of mRNA. It plays an important role in gene regulation and innate defense against invading viruses.16 RNAi was first described by Fire and Mello based on their Nobel prize winning study investigating the mechanisms for effective gene inhibition by exogenous RNA in C. elegans.17 According to their observations, long double-stranded RNA (dsRNA) mediates potent and specific silencing of homologous genes. It appeared later that a similar process also occurs in mammals.18 After years of investigation, the mechanism underlying RNAi is better understood (Figure 1). In general, the dsRNA (either transcribed from cellular genes or infecting pathogens, or artificially introduced into the cells) is processed by a specialized ribonuclease (RNase) III-like enzyme named Dicer in the cytoplasm into a smaller dsRNA molecule. This short dsRNA molecule is known as the siRNA, which has 21–23 nucleotides with 3′ two-nucleotide overhangs. The siRNA interacts with and activates the RNA-induced silencing complex (RISC). The endonuclease argonaute 2 (AGO2) component of the RISC cleaves the passenger strand (sense strand) of the siRNA while the guide strand (antisense strand) remains associated with the RISC. Subsequently, the guide strand guides the active RISC to its target mRNA for cleavage by AGO2. As the guide strand only binds to mRNA that is fully complementary to it, siRNA causes specific gene silencing.16,19
Figure 1.

Gene silencing mechanisms of siRNA and miRNA. siRNA: dsRNA (either transcribed or artificially introduced) is processed by Dicer into siRNA which is loaded into the RISC. AGO2, which is a component of RISC, cleaves the passenger strand of siRNA. The guide strand then guides the active RISC to the target mRNA. The full complementary binding between the guide strand of siRNA and the target mRNA leads to the cleavage of mRNA. miRNA: Transcription of miRNA gene is carried out by RNA polymerase II in the nucleus to give pri-miRNA, which is then cleaved by Drosha to form pre-miRNA. The pre-miRNA is transported by Exportin 5 to the cytoplasm where it is processed by Dicer into miRNA. The miRNA is loaded into the RISC where the passenger strand is discarded, and the miRISC is guided by the remaining guide strand to the target mRNA through partially complementary binding. The target mRNA is inhibited via translational repression, degradation or cleavage.
Since the discovery of RNAi, dsRNAs have been used as research tools to study the gene functions of different cell types. However, in mammalian cells, the delivery of exogenous, long dsRNAs (over 30 nucleotides) is associated with the activation of the interferon (IFN) pathway,20 which is part of the defense mechanism against viral infection. The long dsRNAs bind and activate protein kinase R (PKR), which in turn stimulate a plethora of genes belonging to the IFN pathway, resulting in nonspecific mRNA degradation and apoptosis.21 An in vitro study in mammalian cells, including human cell cultures, showed that the direct introduction of synthetic siRNAs, instead of the long dsRNAs (thus skipping the step of Dicer processing), leads to effective RNAi without the complication of activating the IFN response.18 In view of this finding, siRNAs have become useful tools to inactivate target gene expression. However, later studies suggest that synthetic siRNAs may also induce partial IFN response and innate immune responses.22,23 As this effect can be either sequence-dependent or -independent, special care must be taken when designing siRNA therapeutics. Alternatively, short hairpin RNAs (shRNAs) can be used to achieve a specific gene silencing effect via the RNAi mechanism.24 shRNAs are stem-loop RNAs, which are expressed in the nucleus, typically through the delivery of viral vectors. Once expressed, they are transported to the cytoplasm for further processing, and subsequently loaded into the RISC for specific gene silencing activity in the same manner as synthetic siRNAs. However, the requirement of viral vectors for shRNA expression poses safety concerns in therapeutic applications, which are discussed in section “Viral vectors”.
Gene silencing mediated by miRNA
Similarly to siRNAs, miRNAs also inhibit gene expression in a post-transcriptional manner. Although the gene silencing effects of siRNAs and miRNAs are distinct, the distinction has been obscured because they are associated with common enzymes (e.g., Dicer and RISC) and their functions overlap with each other to a certain extent. The major difference between siRNAs and miRNAs is that the former inhibit the expression of one specific target mRNA while the latter regulate the expression of multiple mRNAs. A considerable body of literature now classifies miRNAs as RNAi molecules.15,25,26,27
The first miRNA was discovered in 1993 in a study examining developmental regulatory genes in C. elegans.28 Soon after its discovery, miRNA was quickly found to be a class of small RNA molecules that negatively regulate gene expression (Figure 1). miRNA gene transcription is carried out by RNA polymerase II in the nucleus to give primary miRNA (pri-miRNA), which is 5′ capped, 3′ polyadenylated RNA with double-stranded stem-loop structure. The pri-miRNA is then cleaved by a microprocessor complex (comprising Drosha and microprocessor complex subunit DCGR8) to form precursor miRNA (pre-miRNA), which is a duplex that contains 70–100 nucleotides with interspersed mismatches and adopts a loop structure. The pre-miRNA is subsequently transported by Exportin 5 from the nucleus to the cytoplasm, where it is further processed by Dicer into a miRNA duplex of 18–25 nucleotides. The miRNA duplex then associates with the RISC forming a complex called miRISC. The miRNA duplex is unwound, releasing and discarding the passenger strand (sense strand)—unlike in the processing of siRNA, in which the AGO2 of the RISC causes the cleavage of the passenger strand of siRNA. The mature single-stranded miRNA guides the miRISC to the target mRNAs. The miRNA binds to the target mRNAs through partial complementary base pairing with the consequence that the target gene silencing occurs via translational repression, degradation, and/or cleavage.25,29
Recognition of mRNA targets by siRNA and miRNA
To elicit RNAi, the siRNA must be fully complementary to its target mRNA (Figure 2). The complementary binding activates the AGO2, which then cleaves the phosphodiester backbone of the mRNA between bases 10 and 11 relative to the 5′end of the guide strand.30 The mRNA fragments generated are subsequently degraded by different exonucleases.31 By contrast, the target recognition of miRNA is more complex, as different binding sites and different degree of complementarity between the miRNA and the target RNA exist. This is a consequence of imperfect base pairing; miRNA only needs to be partially complementary to its target mRNA. The complementary pairing between mRNA and the mature miRNA typically occurs at the 3′ untranslated region (UTR) of the former and the seed region (nucleotides 2–7 from the 5′ end) of the latter (Figure 2).32,33 Other miRNA binding sites, such as the centered sites, 3′ supplementary sites and bulged sites, are considered to be atypical.32,34,35 Since miRNA-mRNA recognition does not require perfect pairing, one miRNA strand can recognize an array of mRNAs, and hence miRNA has the characteristic of having multiple targets. For example, a microarray analysis showed that miRNA-124, which is preferentially expressed in brain tissues, can downregulate 174 annotated genes.33 Due to the partially complementary base pairing between mRNA and miRNA, AGO2 of the miRISC is not activated. Instead, the silencing of the mRNA targets of miRNA occurs through translation repression, or degradation by deadenylation, decapping or exonuclease action.36 In rare cases, high level of complementary between mRNA and miRNA leads to the endonucleolytic cleavage of mRNA by AGO protein, a mechanism that is similar to siRNA-mediated gene silencing.37
Figure 2.

Target recognition by siRNA and miRNA. (a) siRNA is usually fully complementary to the coding region of its target mRNA; (b) miRNA is partially complementary to its target miRNA. Complementary binding usually occurs at the seed region (nucleotides (nt) 2–7 of the 5' end) of miRNA and the 3' UTR of the target mRNA.
siRNA and miRNA as therapeutic agents
The specific gene silencing effect of siRNAs makes them useful tools for target identification and validation in drug discovery and development.38,39 Since miRNAs have multiple mRNA targets and the disruption of their functions contributes to the development of many diseases including cancers, neurodegenerative disorders and cardiovascular diseases, their clinical use as biomarkers and in diagnostics is rapidly developing.40 Furthermore, both siRNAs and miRNAs have huge potential as therapeutic agents. They can overcome the major limitation of traditional small drug molecules, which can only target certain classes of proteins. Even for protein-based drugs including monoclonal antibodies that are highly specific, their targets are mainly limited to cell-surface receptors or circulating proteins. By contrast, siRNAs and miRNAs can downregulate the expression of virtually all genes and their mRNA transcripts. Since many diseases result from the expression of undesired or mutated genes, or from overexpression of certain normal genes, the discovery of siRNA and miRNA opens up a whole new therapeutic approach for the treatment of diseases by targeting genes that are involved causally in the pathological process. A comparison between conventional small molecules, protein-based therapeutic agents and siRNA/miRNA-based drugs is summarized in Table 2. Although the therapeutic potential of siRNAs and miRNAs is promising, different sets of hurdles retard their development into clinical use. Some of these challenges, such as problems regarding stability and poor efficiency of delivery, are similar for both RNA molecules.
Table 2. A comparison between small molecules, protein-based drugs (including monoclonal antibodies) and siRNA/miRNA-based drugs.

Design of therapeutic siRNA
The first essential step for successful siRNA therapy is the design of a siRNA sequence that is potent and specific to the intended mRNA to minimize any off-target effect. A conventional siRNA consists of 19–21 nucleotides with two nucleotide overhangs at the 3′ end, usually TT and UU, which are important for recognition by the RNAi machinery.41 Increasing the length of the dsRNA may enhance its potency, as demonstrated by an in vitro study that dsRNAs with 27 nucleotides were up to 100 times more potent than the conventional siRNAs with 21 nucleotides.42 The long dsRNAs require processing by Dicer into the shorter siRNAs (hence they are termed as “Dicer-ready” or “Dicer-substrate” siRNAs), which are more efficiently loaded into the RISC, thus facilitating the subsequent gene silencing mechanism.21,42,43,44 On the other hand, dsRNAs longer than 30 nucleotides can activate the IFN pathway20 and should be avoided for therapeutic applications.
The gene silencing efficiency of the siRNA varies greatly, depending on the region of the mRNA to which they are complementary. An understanding of this relationship can permit the design of a siRNA sequence with optimal efficacy, and hence the rational design of effective siRNA sequences has been a focus of research. While many siRNA design algorithms have emerged in recent years to predict efficacy,45,46 it is nevertheless essential to validate the gene silencing efficiency of siRNA experimentally. Some commonly used strategies for the design of therapeutic siRNAs are summarized in Table 3.
Table 3. A summary of commonly employed strategies to enhance the efficacy and specificity of siRNAs, and to reduce the off-target effects.

Strand selection. To ensure effective gene silencing, the siRNA must be correctly orientated and loaded into the AGO of the RISC in order for the passenger strand to be cleaved and discarded, so that the guide strand that is complementary to the target mRNA remains bound to the active RISC and directs it to the target mRNA. The guide strand of the RNA duplex is determined during the AGO loading step.47 However, both strands in the RNA duplex could potentially be loaded into the AGO as the guide strand. An incorrect loading orientation results in the intended guide strand being discarded and off-target effects are produced as the remaining strand (the intended passenger strand) base-pairs to the nonintended mRNA. Since this phenomenon can occur with both siRNA and miRNA,48,49,50 the RNA duplex needs to be carefully designed to warrant correct guide strand selection by the RISC. Two major sequence parameters are known to determine the guide strand selection: (i) the asymmetry rule and (ii) 5′ nucleotide preference; both of which can be applied to siRNA as well as miRNA design.
The asymmetry rule is based on the finding that the relative thermodynamic stability of the two ends of the duplex contributes to the selection of the strand to be loaded into AGO.48,51 The strand with a relatively unstable 5′ end (i.e., higher A/U content) is selected as guide strand while the strand with a more stable 5′ end is discarded as the passenger strand. For this reason, RNA duplexes should always be designed with the intended guide strand having the less stable 5′ end. In addition to the asymmetry rule, the 5′ nucleotide preference is also important in correct AGO loading. AGO proteins appear to have a preference for the strand with a U, or less favorably, an A at position one at the 5′ end as the guide strand. Therefore, the guide strand should ideally contain a U or A at the 5′end, whereas the passenger strand should always contain C and G at the 5′ end to minimize the risk of being incorrectly selected as a guide strand.52
Efficiency affected by G/C content. The overall G/C content of the siRNA influences the siRNA activity,53 although the importance of this influence is still debated. The G/C content affects the overall duplex thermodynamic stability as well as target site accessibility; siRNAs with very high G/C content appear to be less functional.54 Some studies suggest that the optimal G/C content of siRNA is around 30–50%, while others show that siRNAs with G/C contents of about 60% are highly efficient.55,56 As a general guideline, the G/C content of siRNA is ideally between 30 and 64%.57 Furthermore, sequences with G/C stretches of nine or more nucleotides should be avoided as this may reduce the gene silencing efficiency of siRNA.58
Minimizing off-target effect. Although one of the distinctive features that differentiate siRNA from miRNA is that siRNA is designed to silence the expression of a specific target mRNA, siRNA may lead to the downregulation of unintended, unpredicted targets, resulting in off-target effects. Indeed, one of the major challenges of siRNA therapy is to reduce off-target effects, as these compromise the therapeutic effect, specificity and can even lead to cell death.59
The most common type of off-target effect of siRNA is the miRNA-like effect.60,61,62,63 This occurs when the 5′ end of the guide strand of siRNA is complementary to the 3′UTR of the mRNA (reminiscent of the target recognition by the seed region of miRNA).64 In some situations, this off-target effect occurs simply due to the poor design of the siRNA, as siRNA can tolerate several mismatches at the mRNA (imperfect complementarity) without losing gene silencing ability.65 Under these circumstances, siRNA behaves like a miRNA molecule: it enters the natural miRNA pathway leading to the inhibition or degradation of multiple mRNAs. In certain cases, this type of off-target effect is nearly as efficient as the on-target effect in reducing the protein levels.66 Another type of off-target effect is not sequence-dependent, but due to the saturation of the RNAi machinery.61 When synthetic siRNAs (or miRNAs) are introduced into the cells, they compete with the endogenous miRNAs for common proteins such as RISC and other factors. As a result, gene regulation by endogenous miRNAs is perturbed, leading to unpredictable off-target effects.67
Reduction of siRNA off-target effects is one of the research priorities in siRNA therapeutics development. Several strategies have been proposed to mitigate such off-target effects. One approach is to use the lowest possible siRNA concentrations, as the off-target effects due to the miRNA-like effect and RNAi machinery saturation are concentration-dependent.60,67 Pooling of multiple siRNAs targeting the same mRNA is another strategy, which allows the gene silencing effect to be achieved at low concentrations of each siRNA in the pool. As each siRNA has a unique off-target signature, the off-target effects can be selectively reduced.61,68 However, there is also a risk that siRNA pool may cause more off-target effects than the beneficial effects from on-target activity.52
To avoid miRNA-like off-target effect, the logical approach is to reduce complementarity between the seed region (2–7 nucleotides of 5′ end) of siRNA and the 3′UTR of mRNA. Clearly, the seed sequences of miRNA (identified with miRNA databases) should be avoided in siRNA design.69 In addition, the siRNA should have a low thermodynamic stability of the duplex between the seed region of the guide strand of siRNA and its target mRNA, since a low seed-target duplex stability reduces the capability of siRNA to induce seed-dependent off-target effects.58,70
Although the seed region of siRNA is implicated in the miRNA-like off-target effect, only some mRNAs, with this type of sequence complementarity, are silenced by siRNA. By analogy, miRNAs regulate the expression of multiple mRNAs through binding of the seed region to the target 3′UTR, but not all mRNAs with the same degree of sequence complementarity are the targets of a given miRNA.61 Therefore, it is speculated that several characteristics, other than sequence complementarity of the mRNA, are involved in defining the mRNA as a siRNA and/or miRNA target. However, these characteristics are still poorly understood and the identification of these features could contribute to the design of siRNA with minimal off-target effects. Another way to avoid the occurrence of off-target effects is by chemical modification, which is discussed in more details in section “Chemical modification”.
Avoidance of immune response. Initial studies suggested that long dsRNAs (over 30 nucleotides) could trigger an immune response by activating the IFN pathway.20 This led to the development of synthetic siRNAs (with smaller number of nucleotides), in the hope to generate therapeutic gene silencing without immunogenic adverse effect.18 However, it was soon discovered that siRNAs could also activate innate immunity71,72; this complication creates another major hurdle to the development of siRNAs as therapeutic agents.
Indeed, siRNAs can cause immune responses in a sequence-independent and sequence-dependent manner. The former involves the PKR and toll-like receptor (TLR) 3 signaling pathways, although they may play only minor roles.73 The latter is mediated by TLR 7 and TLR 8 on dendritic cells and monocytes, respectively.74 These receptors are transmembrane receptors present in the endosomes of immune cells. Several immune-stimulatory sequence motifs have been reported. They include (5′ to 3′) “GUCCUUCAA”, “UGUGU”, “UGU”, and “UGGC”.59,71 Moreover, the presence of U-rich sequences correlates with TLR 7/8 activation.75 While avoiding these immune-stimulatory sequence motifs could reduce the siRNA immunogenicity, it may be impractical to exclude U from the primary siRNA sequence. Alternatively, stimulation of the TLR 7/8 mediated-immune response could be minimized by the use of delivery agents that exclude siRNA endosomal delivery (e.g., electroporation) or by chemical modification of the immune-stimulatory sequences to render them unrecognizable by TLR.76 At present, the rules of sequence-dependent immune activation are still poorly understood. Therefore, all therapeutic siRNAs must be carefully tested for any possible immunostimulatory adverse effects.
Design of therapeutic miRNA
Compared with siRNAs, miRNAs have a broader therapeutic application. Over 2,500 human miRNAs have been recorded in the miRBase (version 20 accessed June 2015), a searchable online miRNA database. Since more than 60% of the human protein-coding genes contain at least one conserved miRNA-binding site, together with the presence of numerous nonconserved sites, the majority of protein-coding genes are under the control of miRNAs.29 The extensive involvement of miRNAs across many human diseases makes them attractive targets for therapeutic strategies, as well as prognostic and predictive biomarkers.77
The goal of miRNA replacement therapy using synthetic miRNAs (or miRNA mimics) is to achieve the same biological functions as the endogenous miRNAs. Therefore the synthetic miRNAs should possess the ability to be loaded to RISC and silence the target mRNAs through the natural miRNA signaling pathway. In theory, a single-stranded RNA molecule containing the sequence that is identical to the guide strand of the mature miRNA could be functioned as miRNA mimic. However, the double stranded miRNA containing both guide and passenger strands was found to be 100 to 1,000 times more potent than the single stranded one.4,14 The double stranded structure can facilitate the proper loading of the RNA molecule into the RISC, thereby enhancing the gene silencing effect. Therefore, designing miRNA mimics with a duplex structure has become the direction of therapeutic development. Synthetic miRNA precursors with longer sequences (from a few extra nucleotides to a full-length pri-miRNA) have also been proposed as therapeutic agents.78 Since pri-miRNAs require processing in the nucleus, whereas pre-miRNAs and miRNAs do not, different strategies are required for the delivery of different types of miRNA mimics to their cellular targets.79 Similarly to shRNAs, viral vectors can be used to express miRNAs inside the cells. This review only discusses exogenously delivered, synthetic miRNAs.
The design of therapeutic miRNA is more straightforward than that of siRNA, as the sequence of the former should be almost, if not entirely, identical to the endogenous miRNA of interest. Nevertheless, the development of miRNA therapeutics faces similar hurdles that are encountered by siRNAs. In vivo administration of miRNAs can activate the innate immune system via TLR,80 leading to significant undesirable effects. As the sequence variation for therapeutic miRNA is limited, chemical modification is the major approach to tackle this problem. Furthermore, therapeutic miRNAs also face the barriers of poor stability and inefficient delivery. The strategies to overcome these barriers are discussed in the following sections.
Chemical modification
RNAs are extremely vulnerable to serum nucleases. Although double-stranded RNA is more resistant to nuclease degradation than single-stranded RNA, naked RNAs in their unmodified forms are degraded rapidly following administration by the abundant nuclease present in the bloodstream, which contributes to a short half-life in vivo.81 Poor stability is one of the major obstacles toward the successful application of siRNAs and miRNAs as therapeutic agents. Chemical modifications of RNA were developed initially to address this issue. In addition, chemical modification of the RNA duplexes can minimize immunogenicity and reduce off-target effects.82
A wide variety of RNA modification approaches have been investigated since the development of antisense therapies in the 1980s. The techniques have matured over the years and some are also applicable to siRNAs and miRNAs. Successful chemical modification should not compromise the gene silencing efficiency of these RNA molecules. In order for the modified siRNAs or miRNAs to be compatible with the endogenous silencing pathways and be loaded into the RISC in the correct orientation, a number of factors, including the position and the type of modification, and its effect on the charge of the RNA duplex, needs to be considered.76 Since the 5′ phosphate, the 5′ proximal part, and the central positions of the guide strand mirror the important areas of the RNA duplex for the interaction with and the action of the RISC and AGO proteins,83,84 RNA duplexes are less tolerant to chemical modifications at these sites. By contrast, chemical modifications at the entire passenger strand and the 3′ proximal part and 3′ overhang of the guide strand would have the least influence on the specificity and/or function of the RNA.85
The major types of chemical modification that are commonly investigated in siRNA and miRNA design include: (i) ribose 2′- OH group modification; (ii) locked and unlocked nucleic acids; and (iii) phosphorothioate (PS) modification (Figure 3). Different RNA modification approach may be employed to serve different functions. Combinations of different modification strategies are also commonly used.
Figure 3.

Structures of chemically modified RNA. (i) In the ribose 2' –OH group modification, the 2' –OH group is modified with 2' –O-methyl (2' O-Me), 2' –fluoro (2' –F) or 2' –methoxyethyl (3' –O-MOE). (ii) In locked nucleic acid (LNA) modification, the ribose is locked in a C3' endo conformation by introducing a 2'-O and 4'-C methylene bridge; In unlocked nucleic acid (UNA), the ribose ring is cleaved between 2' -C and 3' -C. (iii) In backbone modification, the phosphodiester backbone linkage is being substituted. The nonbridging phosphate atom is replaced with a sulfur atom to give a phosphorothioate modification, or replaced with a borane (BH3) moiety to give a boranophosphate modification.
Ribose 2′-OH group modification. Modification of the ribose 2′-OH group is the most diverse and also the most popular type of modification in RNA duplex design, as the gene silencing activity of siRNAs or miRNAs does not depend on this group.86 This strategy involves the substitution of the ribose 2′-OH group with other chemical groups, including 2′-O-methyl (2′-O-Me), 2′-fluoro (2′-F) and 2′-methoxyethyl (2′-O-MOE) (Figure 3), and can effectively enhance the stability of the RNA duplex in serum. In particular, substitutions with 2′-O-Me and 2′-F are the two most extensively studied modifications in siRNA. Although these modifications are generally well-tolerated at most siRNA positions, extensive or full modification may lead to significant loss of silencing efficiency.60 By alternating 2′-O-Me and 2′-F substitutions in a fully substituted siRNA, nuclease-resistant and highly potent modified siRNA can be produced.87 Bulky substitution such as 2′-O-MOE may enhance nuclease resistance, but is poorly tolerated in terms of activity.85,88,89,90
Apart from enhancing nuclease stability, ribose 2′-OH modifications also reduce immune activation of the RNA duplex.91 The substitution of the 2′-OH at only the U sites with either 2′-O-Me or 2′-F can abrogate immune responses without affecting siRNA potency.92 Such modification is believed to render the RNA duplex unrecognizable by TLR 7/8, which is responsible for siRNA-mediated immune response, as U-rich RNA sequences are associated with TLR 7/8 activation.75 Another approach proposed to reduce TLR 7 activation is to alternate 2′-O-Me modification of the passenger strand thereby preserving the gene silencing potency of the guide strand of the siRNA.93
LNA and UNA modifications. Locked nucleic acid (LNA) is a type of chemically modified nucleic acid containing a methylene bridge between the 2′-O and the 4′-C of the sugar to create a stable “locked” ring conformation94 (Figure 3). This modification improves RNA duplex stability by increasing its resistance against nuclease degradation.95,96 However, multiple LNA modifications may lead to decreased efficacy in vitro and in vivo.95,97,98 In order to function properly, siRNA or miRNA must be designed in a way that favors the selection of the intended guide strand by the RISC to minimize off-target effects resulting from the passenger strand being wrongly selected. LNA modification can avoid this type of off-target effect as the modification of the passenger strand at the 5′ end precludes its incorporation into the RISC.95 Moreover, LNA modification in general can reduce RNA duplex-induced immunogenicity by preventing the immunogenic sequence-motifs from being recognized by TLR 7/8, without affecting its silencing activity.99
In recent years, unlocked nucleic acid (UNA) modification has been introduced to siRNA100,101 (Figure 3). UNA monomers are acyclic derivatives of RNA, lacking the C2′ and C3′-bond of the RNA ribose ring, but structurally similar to unmodified RNA upon incorporation into RNA duplexes. Single UNA modifications are well-tolerated at most tested positions in the passenger and guide strands, exhibiting efficient gene silencing and improved performance and stability both in vitro and in vivo.100 However, additional UNA modification, especially in the guide strand, results in reduced silencing efficiency, possibly by destabilizing the siRNA duplex or by interactions with the target mRNA.100 Furthermore, UNA modification in the seed region of the guide strand can prevent miRNA-like off-target effect without compromising siRNA activity.101 Overall, results from various studies suggested that UNA modification represent an important modification with potential for future therapeutic RNA design.
Backbone modification. Backbone modifications are commonly used to improve the stability of nucleic acids against nuclease resistance by substituting the phosphodiester backbone linkages with other types of linkage. Among those, phosphorothioate (PS) modification is the most widely used strategy, in which one of the nonbridging phosphate oxygen atoms is replaced with a sulfur atom (Figure 3).102 This approach was first described in the development of antisense oligonucleotides, and is very efficient in increasing exonuclease resistance following parenteral administration.103 The PS modification also promotes plasma protein binding, thereby reducing clearance by glomerular filtration and urinary excretion, and hence improving the pharmacokinetic profile of nucleotides.103 This technique was successfully employed in antisense PS drug fomivirsen, which was approved by the Food and Drug Administration in the late 90s.104
When this approach was applied to siRNAs, the stability of the modified siRNA was successfully enhanced in vivo.105,106 However, increased toxicity and reduction of gene silencing was also observed.86,89,105,106 This is probably because siRNAs, unlike antisense oligonucleotides, tolerate only limited modifications to remain RISC-compatible. It has been suggested that PS modification at the center of the duplex, especially at the scissile phosphate position, impairs the activity of the RISC.49 Therefore, partial PS modification was recommended, together with other types of modifications to enhance exonuclease resistance of siRNAs.107 This strategy has not been popular due to the limitations mentioned above. However, by carefully controlling the stereochemistry of phosphorothioate siRNA during synthesis, nuclease resistance could be improved without compromising biological activity.108 Alternatively, boranophosphate modification, which involves the replacement of nonbridging phosphate oxygen atoms with an isoelectronic borane (BH3) moiety (Figure 3),109 is more nuclease-resistant and less toxic compared to its PS counterparts. Whether or not this modification can retain the biological activity of siRNAs remains to be determined, and the application of this modification in miRNA therapeutics is yet to be studied.
Delivery of siRNA and miRNA Therapeutics
While chemical modification can improve the stability and reduce off-target effects of siRNAs and miRNAs, poor delivery is still a major challenge in translating therapeutic siRNAs and miRNAs into the clinic. Both types of RNA molecules have an intracellular site of action, but their intrinsic properties, including hydrophilic nature, negative charge and high molecular weight (~14–15 kDa), render them poorly permeable across biological membranes. The primary role of a delivery system is to facilitate the cellular uptake of siRNAs or miRNAs to their target sites.110 A delivery system can also protect the nucleic acids from premature nuclease degradation, thereby reducing the need for chemically modifications, which may affect the specificity and functionality of the RNA molecules. Since siRNAs and miRNAs have similar physicochemical properties (double-stranded RNAs with 21–23 nucleotides) and the same intracellular site of actions (both require enzymatic functions of the RISC to be active against the target mRNAs), similar delivery technologies can be applied to both types of RNA molecules.
Viral vectors
Viral vectors encoding shRNAs or miRNAs have been used to trigger RNAi and gene silencing effects.111 Viruses that are commonly employed for this purpose include lentiviruses,112,113,114 adenoviruses,115,116,117 and adeno-associated viruses (AAVs).118,119,120 They are extremely efficient in transferring the RNA-encoding vectors into the nucleus of mammalian cells to ensure high expression of RNA. Almost 70% of all gene therapy clinical trials have involved the use of viral vectors to deliver nucleic acids because of their high transduction efficiency.110 Viruses that are used to carry therapeutic RNA are genetically engineered to remove their virulence, and their tropism can be altered by genetic manipulation of the viral capsid for targeting to specific cell types.27 In addition, long-term expression can be achieved by using viruses, such as lentiviruses, that can integrate into the host genome. However, there are serious safety concerns associated with the use of viral vectors, including high immunogenicity (especially in adenoviruses)121 and the risk of insertional mutagenesis (especially in lentiviruses).122,123 In addition, low packaging capacity (especially in AAVs)118 and high production cost have also limited their clinical applications.124 Therefore, despite their inferior transfection efficiency, nonviral vectors have become attractive alternatives in delivering synthetic siRNAs and miRNAs due to their better safety profile and lower production cost.
Nonviral vectors
Most of the nonviral vectors that have been investigated for RNA delivery are also used to deliver other types of nucleic acids including plasmid DNA and antisense oligonucleotides. Since the development of nonviral vectors has been extensively reviewed, readers who are interested in the design and structure of different types of nonviral delivery systems are referred to a number of recent articles.79,110,125,126,127 Polymer-based and lipid-based systems are the two main categories of RNA delivery systems. Apart from the advantages mentioned above (i.e., relatively good safety profile and low production cost), nonviral vectors are highly versatile. They can be easily modified to improve their delivery efficiency, e.g., to achieve site-specific delivery by incorporating targeting ligands, or to improve serum stability and extend the circulation time by PEGylation (attachment of polyethylene glycol (PEG) polymer chains).110 Despite the effort to develop suitable RNA delivery systems for clinical use, a lack of correlation between in vitro and in vivo efficacies is observed. It is often reported that a delivery system worked efficiently in vitro but failed in vivo either due to toxicity problems, poor pharmacokinetic profiles, nonspecific uptake or immune responses.128 The success of therapeutic siRNAs and miRNAs is highly dependent on the availability of a safe and efficient delivery system. Selected examples of nonviral delivery systems that have been investigated to deliver therapeutic siRNAs and miRNAs in animal and preclinical studies are summarized in Table 4.
Table 4. A summary of selected examples of nonviral vectors investigated for delivery of therapeutic siRNAs and miRNAs in animal and preclinical studies in recent years.

Polymer-based delivery systems. Cationic polymers can form polyplexes with the negatively charged RNA through electrostatic interactions. The preparation of polyplexes is simple, and the nanosized polyplexes can facilitate cellular uptake through endocytosis. In addition, polymers that exhibit high proton buffering capacity can promote endosomal escape, thereby avoiding endosomal-lysosomal RNA degradation. Synthetic polyethylenimine (PEI), which has an extensive pH buffering capacity, is one of the early generation polymers studied for nucleic acid delivery.129 It is the most widely investigated polymer for siRNA and miRNA delivery in vivo.130,131 Because of its high transfection efficiency, PEI is regarded as the gold standard among the nonviral vectors. Apart from PEI, dendrimers, which are highly branched synthetic polymers with well-defined molecular architecture, are also frequently studied for nucleic acid delivery.132,133 Polyplexes that are formed between dendrimers and nucleic acids are also known as dendriplexes. Poly(amidoamine) (PAMAM)134,135,136 and polypropylenimine137 are cationic dendrimers that have been evaluated for delivering RNA in vivo. However, because of their high charge density, cationic PEI and dendrimers are often associated with high toxicity which has limited their clinical use. Therefore, modified versions of PEI or dendrimers are developed to address this issue and to further improve their delivery efficiency.138,139,140 Alternatively, natural cationic polymers such as chitosan, which is derived from chitin (commonly found in the exoskeleton of crustaceans), and atelocollagen, which is highly purified protein derived from calf dermis, are considered to be safer options for RNA delivery.141,142,143,144,145
Cyclodextrins, the cyclic oligomers of glucose, have been used in pharmaceutical formulations and their long-term effects in humans are well-established.146 Due to their low toxicity, high stability and lack of immune stimulation, cyclodextrin-based nanoparticles were investigated as a carrier of siRNA.147,148 Poly(lactic-co-glycolic acid) (PLGA) is a Food and Drug Administration-approved synthetic biodegradable polymer that is widely studied for delivering various types of therapeutic molecules including RNA due to its low toxicity and good safety profile.126 The rate of drug release can be controlled by the molecular weight and composition of PLGA. Since PLGA is a neutral polymer, it does not form polyplexes with nucleic acids. Instead, RNA can be loaded in PLGA nanoparticles or microparticles.126 Due to the hydrophilic nature of RNA and the hydrophobic nature of PLGA, it is always a challenge to obtain a high loading efficiency. Moreover, the neutral PLGA particles do not promote cellular uptake as effectively as cationic polyplexes. Incorporating small amount of cationic polymers such as PEI into the PLGA nanoparticles can enhance the encapsulation and transfection efficiency. This can also lower the toxicity when compared to the use of cationic polymers alone.149,150,151 Silica-based nanoparticles are biocompatible, biodegradable with low toxicity, and have wide biomedical applications.152 Their high internal pore volume and high capacity for functionalization make them attractive materials for drug delivery,153 and they have been investigated in recent years for RNA delivery in vivo.154,155,156
Lipid-based delivery systems. Similarly to cationic polymers, cationic lipids and liposomes can form lipoplexes with RNA through electrostatic interactions.157 In general, lipids used for nucleic acid delivery are composed of a cationic head group and a hydrophobic chain. The choice of the head group and the hydrophobic chain may dramatically affect the transfection efficiency and toxicity level of the lipoplexes. Examples of commonly used cationic lipids for nucleic acid delivery include 1,2-dioleoyloxy-3-trimethylammonium propane (DOTAP) and 1,2-di-O-octadecenyl-3-trimethylammonium propane (DOTMA), which are often used in combination with neutral lipids such as dioleoylphosphatidylethanolamine (DOPE) and cholesterol to enhance transfection efficiency.157,158,159 Many of the commercial available transfection reagents such as Lipofectamine, Oligofectamine, DharmaFECT, siPORT, and TransIT-TKO are lipid-based systems and have been frequently used for RNA delivery. Despite their high transfection efficiency in vitro, the in vivo performance of most lipid-based delivery systems is not satisfactory due to toxicity, nonspecific uptake, and unwanted inflammatory and immune responses.128,158 Incorporation of PEG is a commonly employed strategy to reduce immunogenicity as well as prolong circulation following systemic delivery, but PEGylation may lead to reduction of cellular uptake. The incorporation of D-α-tocopheryl polyethylene glycol succinate into the delivery system has been reported to overcome this problem. D-α-tocopheryl polyethylene glycol succinate is a water-soluble derivative of natural vitamin E, which is formed by esterification of vitamin E succinate with PEG. It has the advantages of PEG, but also promotes cellular uptake.160,161 To improve specificity, targeting moieties such as antibodies and small peptides can be employed.162,163
Lipid-based nanoparticles with low toxicity and high efficiency are developed to produce a more sophisticated delivery system for RNA. For example, “stabilized nucleic acid lipid particles” in which RNA is encapsulated inside the highly PEGylated liposomes prepared by ethanol dilution method have been proposed.164 In some studies, RNA is loaded into the cationic solid lipid nanoparticles for sustained release.165,166 The low toxic liposome-polycation (or protamine)-hyaluronic acid nanoparticles, in which the negatively charged RNA and hyaluronic acid are complexed with the cationic protamine to form the core of the liposomes, are also being developed for RNA delivery.167,168
Lipolyplexes. In recent years, lipolyplexes have emerged as a new generation delivery system for nucleic acids. Typically, such a system is composed of both polymers and lipids in an attempt to address the limitations of polymer-based and lipid-based delivery systems by combining the advantageous characteristics of both.126,169,170 This approach has been employed for the delivery of RNA in vivo. For example, stearic acid was incorporated into the backbone of PEI for siRNA delivery.171 Modification of PEI with stearic acid led to better transfection efficiency in vivo compared to unmodified PEI. Similarly, cholesterol and deoxycholic acid-modified PEI improved the transfection efficiency of siRNA in vivo.172,173,174
siRNA AND miRNA therapeutics in clinical studies
The first clinical trial of siRNA therapeutics was initiated in 2004,175 merely 6 years after the discovery of RNAi. The rapid progress of siRNA advancing into clinical trials is perhaps due to the experience gained during the development of antisense and other nucleic acid-based therapies. To date, around 30 siRNA candidates have reached various stages of clinical trials for the treatment of different diseases (Table 5). In comparison, the clinical development of miRNA as therapeutics is lagging behind, with only two miRNA therapeutics, both of which are indicated for the treatment of cancers, being registered in clinical trial to date (Table 6). The first miRNA therapeutic trial began in 2013 with the second one starting in early 2015. Although siRNAs share many similarities with miRNAs, the relatively slow progress of miRNA therapeutics could be due to their uncertain mechanism of action and specificity. The diverse potential applications of miRNAs (e.g., as drug target and biomarkers) may also have distracted from their development as therapeutic agents.
Table 5. A summary of siRNA therapeutics in clinical trials (registered with clinicaltrials.gov, last accessed 13 June 2015).
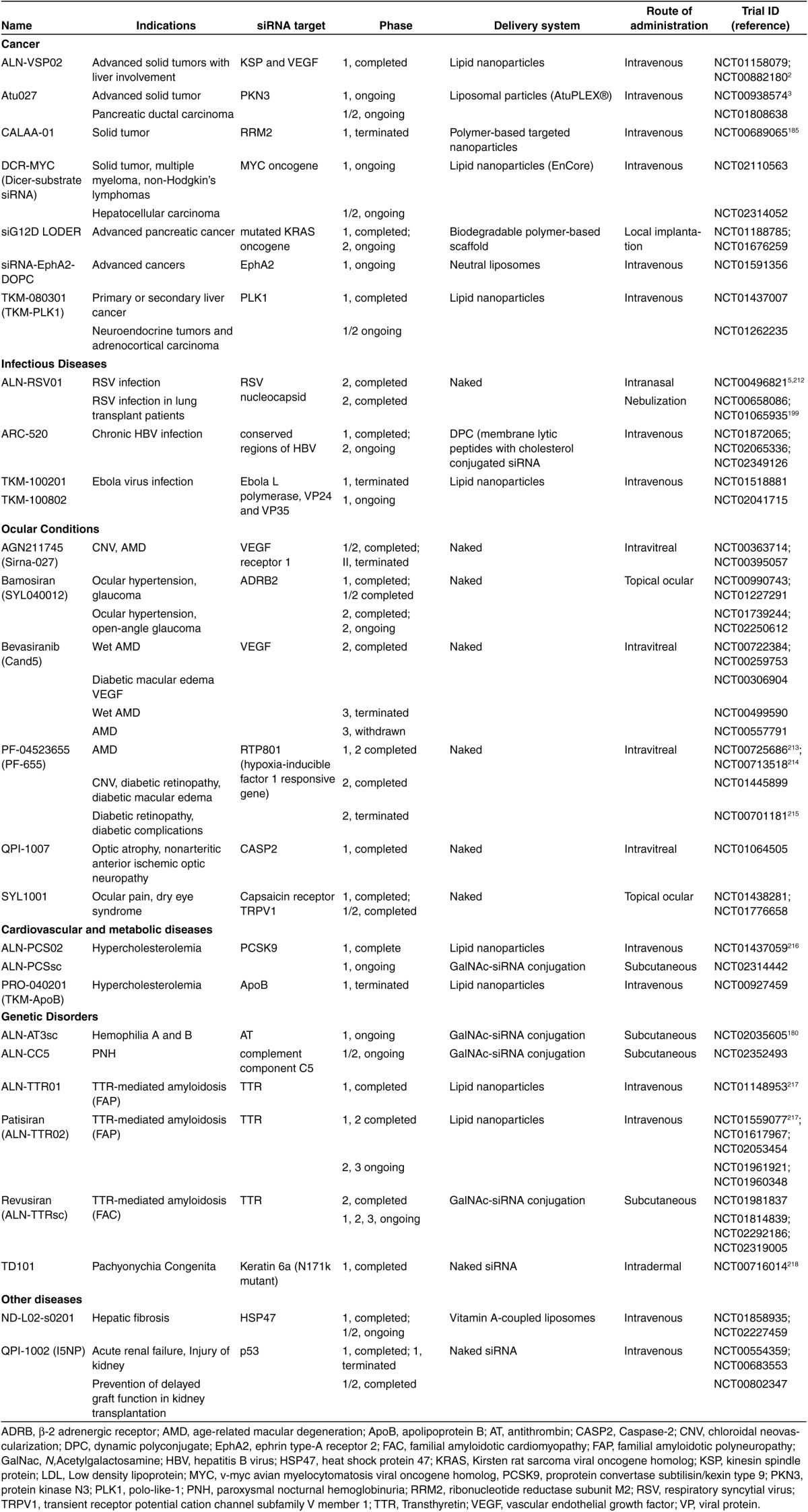
Table 6. A summary of miRNA therapeutics in clinical trials (registered with clinicaltrials.gov, last accessed 13 June 2015).

However, miRNAs have an advantage over siRNAs as the therapeutics for complex multigenic diseases such as cancers and neurodegenerative disorders, which require modulation of multiple pathways for effective treatment. With the ability to inhibit the expression of a number of target genes, which often work together as a network within the same cellular pathway, a whole disease phenotype can potentially be changed by a single miRNA sequence.176,177 By contrast, the therapeutic potential of siRNAs is limited by its ability to target only one specific gene. It will be challenging to use siRNAs to modulate complex diseases, although the strategy of employing multiple siRNA sequences in a single formulation has been reported in clinical studies for the treatment of cancers and viral infections.178 On the other hand, siRNAs are extremely useful for targeting single gene disorders such as hemophilia and hereditary amyloidosis.179,180 Clinical trials of siRNA and miRNA therapeutics that have been registered with clinicaltrials.gov are summarized in Tables 5 and 6, respectively. The proportion of conditions addressed by siRNA and miRNA therapeutics in clinical trials is illustrated in Figure 4. Since cancer is the only intensively researched condition for which both siRNA and miRNA therapeutics have reached the clinical trial stage, special attention is paid to the discussion of their use in oncology.
Figure 4.

Therapeutic indications of siRNA and miRNA therapeutics.
Cancer
Cancer is a leading cause of death worldwide. Almost one-third of the siRNA and miRNA based therapeutics in clinical trials are targeted at cancer. Both siRNAs and miRNAs aim to silence cancer-related gene(s) in order to inhibit tumor cell growth, angiogenesis, metastasis and/or drug resistance. Oncogenes, mutated tumor suppressor genes and other genes that contribute to tumor progression are potential targets for gene silencing. The specificity of siRNAs makes them possible to serve as a platform for personalized medicine in cancer therapy.8 On the other hand, since miRNA therapeutics can target multiple genes, typically in the context of a network, they are efficient in regulating distinct biological cell processes relevant to malignant cell biology. This characteristic makes them particularly attractive in cancer treatment,77 and may explain why the two miRNA clinical trials to date aim at cancer therapy.
By targeting the mRNA of cell-cycle proteins, tumor cell growth can be inhibited. An overexpression of polo-like kinase 1 (PLK1), a cell-cycle protein that is important in mitosis and cytokinese, is observed in many human tumors, and the inhibition of its activity induces apoptosis and tumor cell death.181,182 A lipid nanoparticle-based delivery system containing therapeutic siRNA targeting PLK1, TKM-080301, has been developed. It is currently in phase 1/2 clinical trial for the treatment of neuroendocrine and adrenocortical cancers. Ribonucleotide reductase is an enzyme involved in DNA replication. The M2 subunit of ribonucleotide reductase (RRM2) is an established anticancer target.183,184 Inhibition of RRM2 by siRNAs reduces the growth potential of cancer cells in vitro and in vivo.184 CALAA-01 is a siRNA therapeutic targeting RRM2 for the treatment of solid tumors. Adopting a transferrin-receptor targeting cyclodextrin nanoparticle delivery system, CALAA-01 prevents the proliferation of transferrin receptor-expressing tumor cells. A phase 1 study showed that CALAA-01, following systemic administration, silenced the cancer-associated gene by RNAi mechanism in targeted tumor cells.185 However two patients experienced dose-limiting toxic events in a later trial, possibly due to the formulation problems (mis-folded, aggregated or degraded transferrin); this outcome thus underlines the significance of quality control assays of protein-targeted nanoparticle-based therapeutics.186
Other siRNA therapeutics currently undergoing clinical trials include siG12D LODER, which targets the mutated KRAS oncogene for the treatment of locally advanced pancreatic cancer and is delivered by a biodegradable polymer matrix for sustained release; siRNA-EphA2-DOPC, which targets cancer-related EphA2 gene using liposomal delivery for the treatment of advanced solid tumors; and DCR-MYC, a Dicer-substrate siRNA that targets the MYC oncogene carried by lipid nanoparticles for the treatment of various types of cancer.
Angiogenesis is a key process that promotes tumor growth and survival. One of the major regulators of this process is vascular endothelial growth factor (VEGF), which thus is an attractive target for inhibiting tumor angiogenesis.187 ALN-VSP02 is a dual targeted siRNA therapeutics carried by lipid nanoparticles that suppresses not only the cell-cycle protein kinesin spindle protein (KSP) to promote cell-cycle arrest and eventually cell death,188 but also VEGF.2 It is indicated for advanced solid tumors with liver metastasis, and the initial data from a completed phase I trial showed that ALN-VSP02 possessed antitumor activity while being well-tolerated by patients.2 The siRNA was detected in tumor biopsies following intravenous administration of loaded lipid nanoparticles. Moreover, siRNA-mediated mRNA cleavage in the liver and complete regression of liver metastases was observed in patients with endometrial cancer. Protein kinase N3 (PKN3), a downstream effector of phosphoinositide 3-kinase (PI3K) signaling, is another validated target in cancer. PI3K is only transiently activated after growth factor stimulation in normal cells. Excessive and/or chronic activation of this pathway occurs in many cancer types, and is believed to be involved in the process of metastasis.189 Inhibition of PKN3 resulted in the significant inhibition of tumor growth and the reduction of lymph node metastases in vivo.190,191 Atu027, a liposomal siRNA targeting PKN3, is currently undergoing a phase 1/2 clinical trial for the treatment of advanced or metastatic pancreatic tumor. Early results showed that Atu027 was safe in patients with advanced solid tumors, with 41% of patients showed no further progression of tumors after 8 weeks of treatment.3
The role of miRNA in cancer has been a focus of research in recent years, evidenced by the number of registered clinical trials evaluating the use of miRNA as biomarkers for patient diagnosis, prognosis, and response to treatment.40,192 Several miRNAs are upregulated or downregulated in various tumors. They can act either as oncogenes (also known as oncomiRs) or tumor suppressors (tumor suppressor miRNAs),116 and the latter are employed in miRNA replacement therapy for the treatment of cancer.
MRX34 is a first-in-class cancer therapy and the first synthetic miRNA to enter clinical trials.193 MRX34 was designed to deliver miRNA-34 mimic by liposomal formulation. Indeed, miRNA-34 is a well-characterized, naturally occurring regulator of tumor suppression,194 and it is downregulated in many cancers. It inhibits a series of signaling molecules that contribute to cancer processes including cell proliferation, antiapoptosis, metastasis, and chemoresistance.194 Currently, in phase 1 study, MRX34 is indicated for primary liver cancer or liver metastasis from other solid tumors, and the study is expected to be completed in late 2015. The other miRNA therapeutic that has also reached the clinical trial stage is TargomiRs, which is indicated for malignant pleural mesothelioma and non–small-cell lung cancer. TargomiRs consists of three components, namely a miRNA-16 mimic, a nanoparticle drug delivery system using nonliving bacterial minicells, and an antiepidermal growth factor receptor antibody as a targeting moiety. miRNA-16 is another tumor suppressor.195 Restoration of miRNA-16 leads to inhibition of tumor-promoting gene transcription and hence tumor growth, as well as sensitization of tumor cells to certain chemotherapeutic agents. The phase 1 clinical study of TargomiRs is expected to be completed in mid-2016.
Although only two miRNA replacement therapies have reached clinical trials, many tumor suppressor miRNAs, such as miRNA-7, miRNA-126, miRNA-143/145, miRNA-200, miRNA-355, and the members of the let-7 families,40,192 have been identified with the ability to downregulate oncogenes. Some of these tumor suppressor miRNAs are currently in the preclinical stage and ready to enter phase 1 clinical trials very soon. In terms of safety profile, clinical trials have demonstrated that siRNA therapeutics are generally well-tolerated by the patients. By contrast, the clinical study of miRNA therapeutics is still in its infancy. The potential for any adverse effects may only become apparent after more clinical trials have been carried out. Since a single miRNA can affect multiple target genes, it is also difficult to predict its long-term systemic effect.
Conditions for local treatment
Apart from cancer therapy, clinical trials of siRNA therapeutics against other diseases have also been initiated, including ocular diseases, viral infections, cardiovascular and metabolic diseases, genetic disorders, as well as kidney and renal conditions (Table 5). In particular, the use of siRNAs against ocular disease has enjoyed considerable success: around one-fifth of siRNA-based therapeutics in clinical trials are indicated for the treatment of macular degeneration and related eye disorders. The relatively high proportion of siRNA therapeutics indicated for ocular diseases is largely due to the ease of delivery to the target site.196 The eyes are one of the few organs in the body where successful gene silencing can be achieved by local administration of naked siRNA, thereby minimizing systemic effects, and avoiding toxicity associated with the use of delivery vectors. The most common delivery method is by intravitreal injection of naked siRNA to the posterior segment of the eye, bypassing the corneoscleral barriers. The siRNA can also be topically administered to the ocular surface to treat conditions affecting the anterior segment of the eyes.196 Despite the promising progress of siRNA therapeutics, miRNAs have yet to enter clinical trials for the treatment of eye disorders, probably due to the uncertainty of the roles of miRNAs in the retina and other ocular tissues.136,197 With the improvement of knowledge in the field, there is no doubt that the benefits of ocular delivery of siRNAs can be applied to miRNAs for the treatment of other ocular diseases in the future.
Apart from the eyes, the lung is another site in the body where successful RNAi has been achieved in animal studies by local administration of naked siRNA.198 This effect was also demonstrated clinically by ALN-RSV01, which is a naked siRNA formulation targeting the nucleocapsid of the respiratory syncytial virus (RSV). There is no vaccine for RSV and the only approved therapy (ribavirin) is rarely used due to the risk of teratogenicity and limited antiviral effects. Two phase 2 clinical studies of ALN-RSV01 were completed. ALN-RSV01 was delivered to the lungs of subjects by either intranasal administration or nebulization, and the results of the studies showed that ALN-RSV01 was well-tolerated by patients with promising antiviral effect.5,199 Although the studies narrowly missed the primary endpoint, they established a unique proof-of-concept for RNAi therapeutics for lung infections by local administration of naked siNRA. However, the cellular uptake mechanism of naked siRNA in the airways is unclear; the presence of lung surfactants along the airways may act as natural carriers that mediate the cellular uptake of siRNA.200 It is believed that pulmonary delivery could also be applied to other siRNA and miRNA therapeutics for the treatment of other lung diseases.
Conclusions and future prospects
Synthetic siRNAs and miRNAs hold great promises as new classes of therapeutic agents by silencing the gene(s) of interest. They have been studied for the treatment of various human diseases including cancers, viral infections, ocular conditions, genetic disorders, and cardiovascular diseases. The most attractive aspect of siRNA and miRNA therapeutics is their ability to target virtually any gene(s), which may not be possible with small molecules or protein-based drugs. While the therapeutic efficacy of siRNAs and miRNAs has been successfully demonstrated in vivo, several technical barriers still need to be overcome in order for these RNA molecules to be used clinically. The experience from antisense and gene therapy has contributed to the rapid progress of siRNAs and miRNAs into clinical studies. In particular, the technologies of chemical modification and delivery of nucleic acids developed previously can be applied to both siRNAs and miRNAs. While the former possess a high specificity by targeting one single gene, the latter can target multiple related genes, often in the same cellular pathway or process, to generate pronounced therapeutic effect. Currently, the development of siRNAs is advancing ahead of miRNAs, with a larger number of candidates already entered clinical trials, possibly due to the uncertainties of the complex roles of miRNAs during the early years of their discovery. With the recent surge in intensive research concerning miRNAs, it can be expected that significant advance will be made for their future role in therapeutics.
For proper therapeutic use, the sequences of RNA must be carefully designed to avoid any specific or nonspecific unwanted effects and immune responses. The transition from bench to bedside of RNA-based therapy also depends heavily on the availability of a safe, clinically relevant delivery system that can facilitate cellular uptake of the RNA into target tissues/cells and offer protection against nuclease degradation. The use of nonviral vectors including polymer-based or lipid-based delivery systems offer the advantages of better safety profile and lower production cost over viral vectors despite the inferior delivery efficiency.
Among the various diseases being investigated, cancer is currently the major target of siRNA and miRNA therapeutics. While a large number of cancer-related genes have been identified with therapeutic potential, the duration of silencing effect has not been properly investigated or reported, and it may affect the dose interval and length of treatment. The duration of silencing effect after a single dose of siRNAs or miRNAs depends on a number of factors. These include the stability of the RNA molecules, the rate of RNA release from the delivery system, the type of target tissues, as well as the half-life and turnover rate of the target proteins. A good understanding of this area can contribute to the rational design of treatment strategy to improve clinical outcome of siRNA and miRNA therapeutics. PEGylated nanoparticles incorporated with targeting ligands are frequently employed to prolong circulation time and achieve specific targeting to tumor sites following systemic administration. However, the potential toxicity and immunogenicity effects associated with the delivery agents need to be carefully monitored.
Efficient gene silencing can also be achieved with naked RNA, but this may be limited to local administration to organs such as the eyes and the lungs. Nevertheless, chemical modification of the RNA molecule may be required to improve its stability against nuclease activity in the vitreous humor of the eye or in the airway fluid of the lungs. It is expected that by overcoming the delivery barrier, and better understanding of the effects and the duration of gene silencing, siRNAs and miRNAs will become practical therapeutics in the clinic in the near future.
Acknowledgments
J.K.W.L. is supported by the Area of Excellence Scheme of the University Grants Committee (Grant AoE/M-12/06) and Research Grant Council of Hong Kong (17110414M). M.Y.T.C. is the recipient of University Postgraduate Fellowship, The University of Hong Kong. The authors wsould like to thank Prof Paul M Vanhoutte and A James Mason for critical comments. There is no conflict of interest to disclose.
References
- Mattick, JS and Makunin, IV (2006). Non-coding RNA. Hum Mol Genet 15 Spec No 1: R17–R29. [DOI] [PubMed] [Google Scholar]
- Tabernero, J, Shapiro, GI, LoRusso, PM, Cervantes, A, Schwartz, GK, Weiss, GJ et al. (2013). First-in-humans trial of an RNA interference therapeutic targeting VEGF and KSP in cancer patients with liver involvement. Cancer Discov 3: 406–417. [DOI] [PubMed] [Google Scholar]
- Schultheis, B, Strumberg, D, Santel, A, Vank, C, Gebhardt, F, Keil, O et al. (2014). First-in-human phase I study of the liposomal RNA interference therapeutic Atu027 in patients with advanced solid tumors. J Clin Oncol 32: 4141–4148. [DOI] [PubMed] [Google Scholar]
- Bader, AG, Brown, D, Stoudemire, J and Lammers, P (2011). Developing therapeutic microRNAs for cancer. Gene Ther 18: 1121–1126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeVincenzo, J, Lambkin-Williams, R, Wilkinson, T, Cehelsky, J, Nochur, S, Walsh, E et al. (2010). A randomized, double-blind, placebo-controlled study of an RNAi-based therapy directed against respiratory syncytial virus. Proc Natl Acad Sci USA 107: 8800–8805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandra, PK, Kundu, AK, Hazari, S, Chandra, S, Bao, L, Ooms, T et al. (2012). Inhibition of hepatitis C virus replication by intracellular delivery of multiple siRNAs by nanosomes. Mol Ther 20: 1724–1736. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Sendi, H, Mehrab-Mohseni, M, Foureau, DM, Ghosh, S, Walling, TL, Steuerwald, N et al. (2015). MiR-122 decreases HCV entry into hepatocytes through binding to the 3′ UTR of OCLN mRNA. Liver Int 35: 1315–1323. [DOI] [PubMed] [Google Scholar]
- Daka, A and Peer, D (2012). RNAi-based nanomedicines for targeted personalized therapy. Adv Drug Deliv Rev 64: 1508–1521. [DOI] [PubMed] [Google Scholar]
- Behlke, MA (2006). Progress towards in vivo use of siRNAs. Mol Ther 13: 644–670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Rooij, E, Purcell, AL and Levin, AA (2012). Developing microRNA therapeutics. Circ Res 110: 496–507. [DOI] [PubMed] [Google Scholar]
- Bader, AG, Brown, D and Winkler, M (2010). The promise of microRNA replacement therapy. Cancer Res 70: 7027–7030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiler, J, Hunziker, J and Hall, J (2006). Anti-miRNA oligonucleotides (AMOs): ammunition to target miRNAs implicated in human disease? Gene Ther 13: 496–502. [DOI] [PubMed] [Google Scholar]
- Broderick, JA and Zamore, PD (2011). MicroRNA therapeutics. Gene Ther 18: 1104–1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Rooij, E and Kauppinen, S (2014). Development of microRNA therapeutics is coming of age. EMBO Mol Med 6: 851–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng, Y, Wang, CC, Choy, KW, Du, Q, Chen, J, Wang, Q et al. (2014). Therapeutic potentials of gene silencing by RNA interference: principles, challenges, and new strategies. Gene 538: 217–227. [DOI] [PubMed] [Google Scholar]
- Agrawal, N, Dasaradhi, PV, Mohmmed, A, Malhotra, P, Bhatnagar, RK and Mukherjee, SK (2003). RNA interference: biology, mechanism, and applications. Microbiol Mol Biol Rev 67: 657–685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fire, A, Xu, S, Montgomery, MK, Kostas, SA, Driver, SE and Mello, CC (1998). Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature 391: 806–811. [DOI] [PubMed] [Google Scholar]
- Elbashir, SM, Harborth, J, Lendeckel, W, Yalcin, A, Weber, K and Tuschl, T (2001). Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature 411: 494–498. [DOI] [PubMed] [Google Scholar]
- Pecot, CV, Calin, GA, Coleman, RL, Lopez-Berestein, G and Sood, AK (2011). RNA interference in the clinic: challenges and future directions. Nat Rev Cancer 11: 59–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gantier, MP and Williams, BR (2007). The response of mammalian cells to double-stranded RNA. Cytokine Growth Factor Rev 18: 363–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barik, S (2005). Silence of the transcripts: RNA interference in medicine. J Mol Med (Berl) 83: 764–773. [DOI] [PubMed] [Google Scholar]
- Sledz, CA, Holko, M, de Veer, MJ, Silverman, RH and Williams, BR (2003). Activation of the interferon system by short-interfering RNAs. Nat Cell Biol 5: 834–839. [DOI] [PubMed] [Google Scholar]
- Bridge, AJ, Pebernard, S, Ducraux, A, Nicoulaz, AL and Iggo, R (2003). Induction of an interferon response by RNAi vectors in mammalian cells. Nat Genet 34: 263–264. [DOI] [PubMed] [Google Scholar]
- Rao, DD, Vorhies, JS, Senzer, N and Nemunaitis, J (2009). siRNA vs. shRNA: similarities and differences. Adv Drug Deliv Rev 61: 746–759. [DOI] [PubMed] [Google Scholar]
- Kim, DH and Rossi, JJ (2007). Strategies for silencing human disease using RNA interference. Nat Rev Genet 8: 173–184. [DOI] [PubMed] [Google Scholar]
- Kubowicz, P, Żelaszczyk, D and Pękala, E (2013). RNAi in clinical studies. Curr Med Chem 20: 1801–1816. [DOI] [PubMed] [Google Scholar]
- Davidson, BL and McCray, PB Jr (2011). Current prospects for RNA interference-based therapies. Nat Rev Genet 12: 329–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, RC, Feinbaum, RL and Ambros, V (1993). The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell 75: 843–854. [DOI] [PubMed] [Google Scholar]
- Ha, M and Kim, VN (2014). Regulation of microRNA biogenesis. Nat Rev Mol Cell Biol 15: 509–524. [DOI] [PubMed] [Google Scholar]
- Elbashir, SM, Lendeckel, W and Tuschl, T (2001). RNA interference is mediated by 21- and 22-nucleotide RNAs. Genes Dev 15: 188–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valencia-Sanchez, MA, Liu, J, Hannon, GJ and Parker, R (2006). Control of translation and mRNA degradation by miRNAs and siRNAs. Genes Dev 20: 515–524. [DOI] [PubMed] [Google Scholar]
- Bartel, DP (2009). MicroRNAs: target recognition and regulatory functions. Cell 136: 215–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim, LP, Lau, NC, Garrett-Engele, P, Grimson, A, Schelter, JM, Castle, J et al. (2005). Microarray analysis shows that some microRNAs downregulate large numbers of target mRNAs. Nature 433: 769–773. [DOI] [PubMed] [Google Scholar]
- Shin, C, Nam, JW, Farh, KK, Chiang, HR, Shkumatava, A and Bartel, DP (2010). Expanding the microRNA targeting code: functional sites with centered pairing. Mol Cell 38: 789–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chi, SW, Hannon, GJ and Darnell, RB (2012). An alternative mode of microRNA target recognition. Nat Struct Mol Biol 19: 321–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huntzinger, E and Izaurralde, E (2011). Gene silencing by microRNAs: contributions of translational repression and mRNA decay. Nat Rev Genet 12: 99–110. [DOI] [PubMed] [Google Scholar]
- Kim, VN, Han, J and Siomi, MC (2009). Biogenesis of small RNAs in animals. Nat Rev Mol Cell Biol 10: 126–139. [DOI] [PubMed] [Google Scholar]
- Perwitasari, O, Bakre, A, Tompkins, SM and Tripp, RA (2013). siRNA Genome Screening Approaches to Therapeutic Drug Repositioning. Pharmaceuticals (Basel) 6: 124–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drosopoulos, K and Linardopoulos, S (2013). Integration of RNAi and small molecule screens to identify targets for drug development. Methods Mol Biol 986: 97–104. [DOI] [PubMed] [Google Scholar]
- Hayes, J, Peruzzi, PP and Lawler, S (2014). MicroRNAs in cancer: biomarkers, functions and therapy. Trends Mol Med 20: 460–469. [DOI] [PubMed] [Google Scholar]
- Walton, SP, Wu, M, Gredell, JA and Chan, C (2010). Designing highly active siRNAs for therapeutic applications. FEBS J 277: 4806–4813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, DH, Behlke, MA, Rose, SD, Chang, MS, Choi, S and Rossi, JJ (2005). Synthetic dsRNA Dicer substrates enhance RNAi potency and efficacy. Nat Biotechnol 23: 222–226. [DOI] [PubMed] [Google Scholar]
- Siolas, D, Lerner, C, Burchard, J, Ge, W, Linsley, PS, Paddison, PJ et al. (2005). Synthetic shRNAs as potent RNAi triggers. Nat Biotechnol 23: 227–231. [DOI] [PubMed] [Google Scholar]
- Hefner, E, Clark, K, Whitman, C, Behlke, MA, Rose, SD, Peek, AS et al. (2008). Increased potency and longevity of gene silencing using validated Dicer substrates. J Biomol Tech 19: 231–237. [PMC free article] [PubMed] [Google Scholar]
- Chaudhary, A, Srivastava, S and Garg, S (2011). Development of a software tool and criteria evaluation for efficient design of small interfering RNA. Biochem Biophys Res Commun 404: 313–320. [DOI] [PubMed] [Google Scholar]
- Zhong, R, Kim, J, Kim, HS, Kim, M, Lum, L, Levine, B et al. (2014). Computational detection and suppression of sequence-specific off-target phenotypes from whole genome RNAi screens. Nucleic Acids Res 42: 8214–8222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meister, G (2013). Argonaute proteins: functional insights and emerging roles. Nat Rev Genet 14: 447–459. [DOI] [PubMed] [Google Scholar]
- Khvorova, A, Reynolds, A and Jayasena, SD (2003). Functional siRNAs and miRNAs exhibit strand bias. Cell 115: 209–216. [DOI] [PubMed] [Google Scholar]
- Schwarz, DS, Tomari, Y and Zamore, PD (2004). The RNA-induced silencing complex is a Mg2+-dependent endonuclease. Curr Biol 14: 787–791. [DOI] [PubMed] [Google Scholar]
- Angart, P, Vocelle, D, Chan, C and Walton, SP (2013). Design of siRNA Therapeutics from the Molecular Scale. Pharmaceuticals (Basel) 6: 440–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarz, DS, Hutvágner, G, Du, T, Xu, Z, Aronin, N and Zamore, PD (2003). Asymmetry in the assembly of the RNAi enzyme complex. Cell 115: 199–208. [DOI] [PubMed] [Google Scholar]
- Petri, S and Meister, G (2013). siRNA design principles and off-target effects. Methods Mol Biol 986: 59–71. [DOI] [PubMed] [Google Scholar]
- Reynolds, A, Leake, D, Boese, Q, Scaringe, S, Marshall, WS and Khvorova, A (2004). Rational siRNA design for RNA interference. Nat Biotechnol 22: 326–330. [DOI] [PubMed] [Google Scholar]
- Wang, X, Wang, X, Varma, RK, Beauchamp, L, Magdaleno, S and Sendera, TJ (2009). Selection of hyperfunctional siRNAs with improved potency and specificity. Nucleic Acids Res 37: e152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurreck, J (2006). siRNA efficiency: structure or sequence-that is the question. J Biomed Biotechnol 2006: 83757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tafer, H, Ameres, SL, Obernosterer, G, Gebeshuber, CA, Schroeder, R, Martinez, J et al. (2008). The impact of target site accessibility on the design of effective siRNAs. Nat Biotechnol 26: 578–583. [DOI] [PubMed] [Google Scholar]
- Birmingham, A, Anderson, E, Sullivan, K, Reynolds, A, Boese, Q, Leake, D et al. (2007). A protocol for designing siRNAs with high functionality and specificity. Nat Protoc 2: 2068–2078. [DOI] [PubMed] [Google Scholar]
- Ui-Tei, K, Naito, Y, Nishi, K, Juni, A and Saigo, K (2008). Thermodynamic stability and Watson-Crick base pairing in the seed duplex are major determinants of the efficiency of the siRNA-based off-target effect. Nucleic Acids Res 36: 7100–7109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fedorov, Y, Anderson, EM, Birmingham, A, Reynolds, A, Karpilow, J, Robinson, K et al. (2006). Off-target effects by siRNA can induce toxic phenotype. RNA 12: 1188–1196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson, AL, Burchard, J, Schelter, J, Chau, BN, Cleary, M, Lim, L et al. (2006). Widespread siRNA “off-target” transcript silencing mediated by seed region sequence complementarity. RNA 12: 1179–1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson, AL and Linsley, PS (2010). Recognizing and avoiding siRNA off-target effects for target identification and therapeutic application. Nat Rev Drug Discov 9: 57–67. [DOI] [PubMed] [Google Scholar]
- Scacheri, PC, Rozenblatt-Rosen, O, Caplen, NJ, Wolfsberg, TG, Umayam, L, Lee, JC et al. (2004). Short interfering RNAs can induce unexpected and divergent changes in the levels of untargeted proteins in mammalian cells. Proc Natl Acad Sci USA 101: 1892–1897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saxena, S, Jónsson, ZO and Dutta, A (2003). Small RNAs with imperfect match to endogenous mRNA repress translation. Implications for off-target activity of small inhibitory RNA in mammalian cells. J Biol Chem 278: 44312–44319. [DOI] [PubMed] [Google Scholar]
- Doench, JG, Petersen, CP and Sharp, PA (2003). siRNAs can function as miRNAs. Genes Dev 17: 438–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snøve, O Jr and Holen, T (2004). Many commonly used siRNAs risk off-target activity. Biochem Biophys Res Commun 319: 256–263. [DOI] [PubMed] [Google Scholar]
- Lin, X, Ruan, X, Anderson, MG, McDowell, JA, Kroeger, PE, Fesik, SW et al. (2005). siRNA-mediated off-target gene silencing triggered by a 7 nt complementation. Nucleic Acids Res 33: 4527–4535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimm, D, Streetz, KL, Jopling, CL, Storm, TA, Pandey, K, Davis, CR et al. (2006). Fatality in mice due to oversaturation of cellular microRNA/short hairpin RNA pathways. Nature 441: 537–541. [DOI] [PubMed] [Google Scholar]
- Kittler, R, Surendranath, V, Heninger, AK, Slabicki, M, Theis, M, Putz, G et al. (2007). Genome-wide resources of endoribonuclease-prepared short interfering RNAs for specific loss-of-function studies. Nat Methods 4: 337–344. [DOI] [PubMed] [Google Scholar]
- Kozomara, A and Griffiths-Jones, S (2011). miRBase: integrating microRNA annotation and deep-sequencing data. Nucleic Acids Res 39(Database issue): D152–D157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naito, Y and Ui-Tei, K (2013). Designing functional siRNA with reduced off-target effects. Methods Mol Biol 942: 57–68. [DOI] [PubMed] [Google Scholar]
- Judge, AD, Sood, V, Shaw, JR, Fang, D, McClintock, K and MacLachlan, I (2005). Sequence-dependent stimulation of the mammalian innate immune response by synthetic siRNA. Nat Biotechnol 23: 457–462. [DOI] [PubMed] [Google Scholar]
- Robbins, M, Judge, A and MacLachlan, I (2009). siRNA and innate immunity. Oligonucleotides 19: 89–102. [DOI] [PubMed] [Google Scholar]
- Kleinman, ME, Yamada, K, Takeda, A, Chandrasekaran, V, Nozaki, M, Baffi, JZ et al. (2008). Sequence- and target-independent angiogenesis suppression by siRNA via TLR3. Nature 452: 591–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heil, F, Hemmi, H, Hochrein, H, Ampenberger, F, Kirschning, C, Akira, S et al. (2004). Species-specific recognition of single-stranded RNA via toll-like receptor 7 and 8. Science 303: 1526–1529. [DOI] [PubMed] [Google Scholar]
- Goodchild, A, Nopper, N, King, A, Doan, T, Tanudji, M, Arndt, GM et al. (2009). Sequence determinants of innate immune activation by short interfering RNAs. BMC Immunol 10: 40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bramsen, JB and Kjems, J (2013). Engineering small interfering RNAs by strategic chemical modification. Methods Mol Biol 942: 87–109. [DOI] [PubMed] [Google Scholar]
- Iorio, MV and Croce, CM (2012). MicroRNA dysregulation in cancer: diagnostics, monitoring and therapeutics. A comprehensive review. EMBO Mol Med 4: 143–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terasawa, K, Shimizu, K and Tsujimoto, G (2011). Synthetic Pre-miRNA-Based shRNA as Potent RNAi Triggers. J Nucleic Acids 2011: 131579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, Y, Wang, Z and Gemeinhart, RA (2013). Progress in microRNA delivery. J Control Release 172: 962–974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabbri, M, Paone, A, Calore, F, Galli, R and Croce, CM (2013). A new role for microRNAs, as ligands of Toll-like receptors. RNA Biol 10: 169–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Layzer, JM, McCaffrey, AP, Tanner, AK, Huang, Z, Kay, MA and Sullenger, BA (2004). In vivo activity of nuclease-resistant siRNAs. RNA 10: 766–771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watts, JK, Deleavey, GF and Damha, MJ (2008). Chemically modified siRNA: tools and applications. Drug Discov Today 13: 842–855. [DOI] [PubMed] [Google Scholar]
- Lima, WF, Wu, H, Nichols, JG, Sun, H, Murray, HM and Crooke, ST (2009). Binding and cleavage specificities of human Argonaute2. J Biol Chem 284: 26017–26028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, Y, Sheng, G, Juranek, S, Tuschl, T and Patel, DJ (2008). Structure of the guide-strand-containing argonaute silencing complex. Nature 456: 209–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bramsen, JB, Laursen, MB, Nielsen, AF, Hansen, TB, Bus, C, Langkjaer, N et al. (2009). A large-scale chemical modification screen identifies design rules to generate siRNAs with high activity, high stability and low toxicity. Nucleic Acids Res 37: 2867–2881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiu, YL and Rana, TM (2003). siRNA function in RNAi: a chemical modification analysis. RNA 9: 1034–1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allerson, CR, Sioufi, N, Jarres, R, Prakash, TP, Naik, N, Berdeja, A et al. (2005). Fully 2′-modified oligonucleotide duplexes with improved in vitro potency and stability compared to unmodified small interfering RNA. J Med Chem 48: 901–904. [DOI] [PubMed] [Google Scholar]
- Prakash, TP, Allerson, CR, Dande, P, Vickers, TA, Sioufi, N, Jarres, R et al. (2005). Positional effect of chemical modifications on short interference RNA activity in mammalian cells. J Med Chem 48: 4247–4253. [DOI] [PubMed] [Google Scholar]
- Amarzguioui, M, Holen, T, Babaie, E and Prydz, H (2003). Tolerance for mutations and chemical modifications in a siRNA. Nucleic Acids Res 31: 589–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Odadzic, D, Bramsen, JB, Smicius, R, Bus, C, Kjems, J and Engels, JW (2008). Synthesis of 2′-O-modified adenosine building blocks and application for RNA interference. Bioorg Med Chem 16: 518–529. [DOI] [PubMed] [Google Scholar]
- Judge, AD, Bola, G, Lee, AC and MacLachlan, I (2006). Design of noninflammatory synthetic siRNA mediating potent gene silencing in vivo. Mol Ther 13: 494–505. [DOI] [PubMed] [Google Scholar]
- Cekaite, L, Furset, G, Hovig, E and Sioud, M (2007). Gene expression analysis in blood cells in response to unmodified and 2′-modified siRNAs reveals TLR-dependent and independent effects. J Mol Biol 365: 90–108. [DOI] [PubMed] [Google Scholar]
- Hamm, S, Latz, E, Hangel, D, Müller, T, Yu, P, Golenbock, D et al. (2010). Alternating 2′-O-ribose methylation is a universal approach for generating non-stimulatory siRNA by acting as TLR7 antagonist. Immunobiology 215: 559–569. [DOI] [PubMed] [Google Scholar]
- Veedu, RN and Wengel, J (2010). Locked nucleic acids: promising nucleic acid analogs for therapeutic applications. Chem Biodivers 7: 536–542. [DOI] [PubMed] [Google Scholar]
- Elmén, J, Thonberg, H, Ljungberg, K, Frieden, M, Westergaard, M, Xu, Y et al. (2005). Locked nucleic acid (LNA) mediated improvements in siRNA stability and functionality. Nucleic Acids Res 33: 439–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mook, OR, Baas, F, de Wissel, MB and Fluiter, K (2007). Evaluation of locked nucleic acid-modified small interfering RNA in vitro and in vivo. Mol Cancer Ther 6: 833–843. [DOI] [PubMed] [Google Scholar]
- Braasch, DA, Jensen, S, Liu, Y, Kaur, K, Arar, K, White, MA et al. (2003). RNA interference in mammalian cells by chemically-modified RNA. Biochemistry 42: 7967–7975. [DOI] [PubMed] [Google Scholar]
- Grünweller, A, Wyszko, E, Bieber, B, Jahnel, R, Erdmann, VA and Kurreck, J (2003). Comparison of different antisense strategies in mammalian cells using locked nucleic acids, 2'-O-methyl RNA, phosphorothioates and small interfering RNA. Nucleic Acids Res 31: 3185–3193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hornung, V, Guenthner-Biller, M, Bourquin, C, Ablasser, A, Schlee, M, Uematsu, S et al. (2005). Sequence-specific potent induction of IFN-alpha by short interfering RNA in plasmacytoid dendritic cells through TLR7. Nat Med 11: 263–270. [DOI] [PubMed] [Google Scholar]
- Laursen, MB, Pakula, MM, Gao, S, Fluiter, K, Mook, OR, Baas, F et al. (2010). Utilization of unlocked nucleic acid (UNA) to enhance siRNA performance in vitro and in vivo. Mol Biosyst 6: 862–870. [DOI] [PubMed] [Google Scholar]
- Vaish, N, Chen, F, Seth, S, Fosnaugh, K, Liu, Y, Adami, R et al. (2011). Improved specificity of gene silencing by siRNAs containing unlocked nucleobase analogs. Nucleic Acids Res 39: 1823–1832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell, JM, Bacon, TA and Wickstrom, E (1990). Oligodeoxynucleoside phosphorothioate stability in subcellular extracts, culture media, sera and cerebrospinal fluid. J Biochem Biophys Methods 20: 259–267. [DOI] [PubMed] [Google Scholar]
- Eckstein, F (2014). Phosphorothioates, essential components of therapeutic oligonucleotides. Nucleic Acid Ther 24: 374–387. [DOI] [PubMed] [Google Scholar]
- Crooke, ST (1998). Vitravene--another piece in the mosaic. Antisense & Nucleic Acid Drug Development 8: vii–viii. [DOI] [PubMed] [Google Scholar]
- Braasch, DA, Paroo, Z, Constantinescu, A, Ren, G, Oz, OK, Mason, RP et al. (2004). Biodistribution of phosphodiester and phosphorothioate siRNA. Bioorg Med Chem Lett 14: 1139–1143. [DOI] [PubMed] [Google Scholar]
- Choung, S, Kim, YJ, Kim, S, Park, HO and Choi, YC (2006). Chemical modification of siRNAs to improve serum stability without loss of efficacy. Biochem Biophys Res Commun 342: 919–927. [DOI] [PubMed] [Google Scholar]
- Soutschek, J, Akinc, A, Bramlage, B, Charisse, K, Constien, R, Donoghue, M et al. (2004). Therapeutic silencing of an endogenous gene by systemic administration of modified siRNAs. Nature 432: 173–178. [DOI] [PubMed] [Google Scholar]
- Jahns, H, Roos, M, Imig, J, Baumann, F, Wang, Y, Gilmour, R et al. (2015). Stereochemical bias introduced during RNA synthesis modulates the activity of phosphorothioate siRNAs. Nat Commun 6: 6317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall, AH, Wan, J, Shaughnessy, EE, Ramsay Shaw, B and Alexander, KA (2004). RNA interference using boranophosphate siRNAs: structure-activity relationships. Nucleic Acids Res 32: 5991–6000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin, H, Kanasty, RL, Eltoukhy, AA, Vegas, AJ, Dorkin, JR and Anderson, DG (2014). Non-viral vectors for gene-based therapy. Nat Rev Genet 15: 541–555. [DOI] [PubMed] [Google Scholar]
- Couto, LB and High, KA (2010). Viral vector-mediated RNA interference. Curr Opin Pharmacol 10: 534–542. [DOI] [PubMed] [Google Scholar]
- Kasar, S, Salerno, E, Yuan, Y, Underbayev, C, Vollenweider, D, Laurindo, MF et al. (2012). Systemic in vivo lentiviral delivery of miR-15a/16 reduces malignancy in the NZB de novo mouse model of chronic lymphocytic leukemia. Genes Immun 13: 109–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar, MS, Erkeland, SJ, Pester, RE, Chen, CY, Ebert, MS, Sharp, PA et al. (2008). Suppression of non-small cell lung tumor development by the let-7 microRNA family. Proc Natl Acad Sci USA 105: 3903–3908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, YP, Vink, MA, Westerink, JT, Ramirez de Arellano, E, Konstantinova, P, Ter Brake, O et al. (2010). Titers of lentiviral vectors encoding shRNAs and miRNAs are reduced by different mechanisms that require distinct repair strategies. RNA 16: 1328–1339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandt, MR, Kirste, AG, Pozzuto, T, Schubert, S, Kandolf, R, Fechner, H et al. (2013). Adenovirus vector-mediated RNA interference for the inhibition of human parvovirus B19 replication. Virus Res 176: 155–160. [DOI] [PubMed] [Google Scholar]
- Esquela-Kerscher, A and Slack, FJ (2006). Oncomirs - microRNAs with a role in cancer. Nat Rev Cancer 6: 259–269. [DOI] [PubMed] [Google Scholar]
- Lou, W, Chen, Q, Ma, L, Liu, J, Yang, Z, Shen, J et al. (2013). Oncolytic adenovirus co-expressing miRNA-34a and IL-24 induces superior antitumor activity in experimental tumor model. J Mol Med (Berl) 91: 715–725. [DOI] [PubMed] [Google Scholar]
- Borel, F, Kay, MA and Mueller, C (2014). Recombinant AAV as a platform for translating the therapeutic potential of RNA interference. Mol Ther 22: 692–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kota, J, Chivukula, RR, O'Donnell, KA, Wentzel, EA, Montgomery, CL, Hwang, HW et al. (2009). Therapeutic microRNA delivery suppresses tumorigenesis in a murine liver cancer model. Cell 137: 1005–1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanek, LM, Sardi, SP, Mastis, B, Richards, AR, Treleaven, CM, Taksir, T et al. (2014). Silencing mutant huntingtin by adeno-associated virus-mediated RNA interference ameliorates disease manifestations in the YAC128 mouse model of Huntington's disease. Hum Gene Ther 25: 461–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendrickx, R, Stichling, N, Koelen, J, Kuryk, L, Lipiec, A and Greber, UF (2014). Innate immunity to adenovirus. Hum Gene Ther 25: 265–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baum, C, Kustikova, O, Modlich, U, Li, Z and Fehse, B (2006). Mutagenesis and oncogenesis by chromosomal insertion of gene transfer vectors. Hum Gene Ther 17: 253–263. [DOI] [PubMed] [Google Scholar]
- Pauwels, K, Gijsbers, R, Toelen, J, Schambach, A, Willard-Gallo, K, Verheust, C et al. (2009). State-of-the-art lentiviral vectors for research use: risk assessment and biosafety recommendations. Curr Gene Ther 9: 459–474. [DOI] [PubMed] [Google Scholar]
- Seow, Y and Wood, MJ (2009). Biological gene delivery vehicles: beyond viral vectors. Mol Ther 17: 767–777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Movahedi, F, Hu, RG, Becker, DL and Xu, C (2015). Stimuli-responsive liposomes for the delivery of nucleic acid therapeutics. Nanomedicine 11: 1575–1584. [DOI] [PubMed] [Google Scholar]
- Videira, M, Arranja, A, Rafael, D and Gaspar, R (2014). Preclinical development of siRNA therapeutics: towards the match between fundamental science and engineered systems. Nanomedicine 10: 689–702. [DOI] [PubMed] [Google Scholar]
- Williford, JM, Wu, J, Ren, Y, Archang, MM, Leong, KW and Mao, HQ (2014). Recent advances in nanoparticle-mediated siRNA delivery. Annu Rev Biomed Eng 16: 347–370. [DOI] [PubMed] [Google Scholar]
- Lv, H, Zhang, S, Wang, B, Cui, S and Yan, J (2006). Toxicity of cationic lipids and cationic polymers in gene delivery. J Control Release 114: 100–109. [DOI] [PubMed] [Google Scholar]
- Neuberg, P and Kichler, A (2014). Recent developments in nucleic acid delivery with polyethylenimines. Adv Genet 88: 263–288. [DOI] [PubMed] [Google Scholar]
- Höbel, S and Aigner, A (2013). Polyethylenimines for siRNA and miRNA delivery in vivo. Wiley Interdiscip Rev Nanomed Nanobiotechnol 5: 484–501. [DOI] [PubMed] [Google Scholar]
- Urban-Klein, B, Werth, S, Abuharbeid, S, Czubayko, F and Aigner, A (2005). RNAi-mediated gene-targeting through systemic application of polyethylenimine (PEI)-complexed siRNA in vivo. Gene Ther 12: 461–466. [DOI] [PubMed] [Google Scholar]
- Dufès, C, Uchegbu, IF and Schätzlein, AG (2005). Dendrimers in gene delivery. Adv Drug Deliv Rev 57: 2177–2202. [DOI] [PubMed] [Google Scholar]
- Shcharbin, D, Shakhbazau, A and Bryszewska, M (2013). Poly(amidoamine) dendrimer complexes as a platform for gene delivery. Expert Opin Drug Deliv 10: 1687–1698. [DOI] [PubMed] [Google Scholar]
- Kala, S, Mak, AS, Liu, X, Posocco, P, Pricl, S, Peng, L et al. (2014). Combination of dendrimer-nanovector-mediated small interfering RNA delivery to target Akt with the clinical anticancer drug paclitaxel for effective and potent anticancer activity in treating ovarian cancer. J Med Chem 57: 2634–2642. [DOI] [PubMed] [Google Scholar]
- Liu, C, Liu, X, Rocchi, P, Qu, F, Iovanna, JL and Peng, L (2014). Arginine-terminated generation 4 PAMAM dendrimer as an effective nanovector for functional siRNA delivery in vitro and in vivo. Bioconjug Chem 25: 521–532. [DOI] [PubMed] [Google Scholar]
- Liu, X, Li, G, Su, Z, Jiang, Z, Chen, L, Wang, J et al. (2013). Poly(amido amine) is an ideal carrier of miR-7 for enhancing gene silencing effects on the EGFR pathway in U251 glioma cells. Oncol Rep 29: 1387–1394. [DOI] [PubMed] [Google Scholar]
- Shah, V, Taratula, O, Garbuzenko, OB, Taratula, OR, Rodriguez-Rodriguez, L and Minko, T (2013). Targeted nanomedicine for suppression of CD44 and simultaneous cell death induction in ovarian cancer: an optimal delivery of siRNA and anticancer drug. Clin Cancer Res 19: 6193–6204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siu, KS, Chen, D, Zheng, X, Zhang, X, Johnston, N, Liu, Y et al. (2014). Non-covalently functionalized single-walled carbon nanotube for topical siRNA delivery into melanoma. Biomaterials 35: 3435–3442. [DOI] [PubMed] [Google Scholar]
- Chiou, GY, Cherng, JY, Hsu, HS, Wang, ML, Tsai, CM, Lu, KH et al. (2012). Cationic polyurethanes-short branch PEI-mediated delivery of Mir145 inhibited epithelial-mesenchymal transdifferentiation and cancer stem-like properties and in lung adenocarcinoma. J Control Release 159: 240–250. [DOI] [PubMed] [Google Scholar]
- Yang, YP, Chien, Y, Chiou, GY, Cherng, JY, Wang, ML, Lo, WL et al. (2012). Inhibition of cancer stem cell-like properties and reduced chemoradioresistance of glioblastoma using microRNA145 with cationic polyurethane-short branch PEI. Biomaterials 33: 1462–1476. [DOI] [PubMed] [Google Scholar]
- Yhee, JY, Song, S, Lee, SJ, Park, SG, Kim, KS, Kim, MG et al. (2015). Cancer-targeted MDR-1 siRNA delivery using self-cross-linked glycol chitosan nanoparticles to overcome drug resistance. J Control Release 198: 1–9. [DOI] [PubMed] [Google Scholar]
- Deng, X, Cao, M, Zhang, J, Hu, K, Yin, Z, Zhou, Z et al. (2014). Hyaluronic acid-chitosan nanoparticles for co-delivery of MiR-34a and doxorubicin in therapy against triple negative breast cancer. Biomaterials 35: 4333–4344. [DOI] [PubMed] [Google Scholar]
- Yuan, Y, Makita, N, Cao, D, Mihara, K, Kadomatsu, K and Takei, Y (2015). Atelocollagen-mediated intravenous siRNA delivery specific to tumor tissues orthotopically xenografted in prostates of nude mice and its anticancer effects. Nucleic Acid Ther 25: 85–94. [DOI] [PubMed] [Google Scholar]
- Kawakami, E, Kawai, N, Kinouchi, N, Mori, H, Ohsawa, Y, Ishimaru, N et al. (2013). Local applications of myostatin-siRNA with atelocollagen increase skeletal muscle mass and recovery of muscle function. PLoS One 8: e64719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeshita, F, Patrawala, L, Osaki, M, Takahashi, RU, Yamamoto, Y, Kosaka, N et al. (2010). Systemic delivery of synthetic microRNA-16 inhibits the growth of metastatic prostate tumors via downregulation of multiple cell-cycle genes. Mol Ther 18: 181–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis, ME and Brewster, ME (2004). Cyclodextrin-based pharmaceutics: past, present and future. Nat Rev Drug Discov 3: 1023–1035. [DOI] [PubMed] [Google Scholar]
- Bartlett, DW and Davis, ME (2008). Impact of tumor-specific targeting and dosing schedule on tumor growth inhibition after intravenous administration of siRNA-containing nanoparticles. Biotechnol Bioeng 99: 975–985. [DOI] [PubMed] [Google Scholar]
- Hu-Lieskovan, S, Heidel, JD, Bartlett, DW, Davis, ME and Triche, TJ (2005). Sequence-specific knockdown of EWS-FLI1 by targeted, nonviral delivery of small interfering RNA inhibits tumor growth in a murine model of metastatic Ewing's sarcoma. Cancer Res 65: 8984–8992. [DOI] [PubMed] [Google Scholar]
- Murata, N, Takashima, Y, Toyoshima, K, Yamamoto, M and Okada, H (2008). Anti-tumor effects of anti-VEGF siRNA encapsulated with PLGA microspheres in mice. J Control Release 126: 246–254. [DOI] [PubMed] [Google Scholar]
- Mountziaris, PM, Tzouanas, SN, Sing, DC, Kramer, PR, Kasper, FK and Mikos, AG (2012). Intra-articular controlled release of anti-inflammatory siRNA with biodegradable polymer microparticles ameliorates temporomandibular joint inflammation. Acta Biomater 8: 3552–3560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das, J, Das, S, Paul, A, Samadder, A, Bhattacharyya, SS and Khuda-Bukhsh, AR (2014). Assessment of drug delivery and anticancer potentials of nanoparticles-loaded siRNA targeting STAT3 in lung cancer, in vitro and in vivo. Toxicol Lett 225: 454–466. [DOI] [PubMed] [Google Scholar]
- Bitar, A, Ahmad, NM, Fessi, H and Elaissari, A (2012). Silica-based nanoparticles for biomedical applications. Drug Discov Today 17: 1147–1154. [DOI] [PubMed] [Google Scholar]
- Steinbacher, JL and Landry, CC (2014). Adsorption and release of siRNA from porous silica. Langmuir 30: 4396–4405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, Y, Wang, X, Liu, T, Zhang, DS, Wang, Y, Gu, H et al. (2015). Highly effective antiangiogenesis via magnetic mesoporous silica-based siRNA vehicle targeting the VEGF gene for orthotopic ovarian cancer therapy. Int J Nanomedicine 10: 2579–2594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tivnan, A, Orr, WS, Gubala, V, Nooney, R, Williams, DE, McDonagh, C et al. (2012). Inhibition of neuroblastoma tumor growth by targeted delivery of microRNA-34a using anti-disialoganglioside GD2 coated nanoparticles. PLoS One 7: e38129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, Y, Gu, H, Zhang, DS, Li, F, Liu, T and Xia, W (2014). Highly effective inhibition of lung cancer growth and metastasis by systemic delivery of siRNA via multimodal mesoporous silica-based nanocarrier. Biomaterials 35: 10058–10069. [DOI] [PubMed] [Google Scholar]
- Ozpolat, B, Sood, AK and Lopez-Berestein, G (2014). Liposomal siRNA nanocarriers for cancer therapy. Adv Drug Deliv Rev 66: 110–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu, SY and McMillan, NA (2009). Lipidic systems for in vivo siRNA delivery. AAPS J 11: 639–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang, W and Lam, JKW. Endosomal escape pathways for non-viral nucleic acid delivery systems. In: Ceresa B (ed.). Molecular Regulation of Endocytosis. InTech, 2012. pp. 429–456. [Google Scholar]
- Wu, Y, Crawford, M, Mao, Y, Lee, RJ, Davis, IC, Elton, TS et al. (2013). Therapeutic Delivery of MicroRNA-29b by Cationic Lipoplexes for Lung Cancer. Mol Ther Nucleic Acids 2: e84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, Z, Tan, S and Feng, SS (2012). Vitamin E TPGS as a molecular biomaterial for drug delivery. Biomaterials 33: 4889–4906. [DOI] [PubMed] [Google Scholar]
- Kenjo, E, Asai, T, Yonenaga, N, Ando, H, Ishii, T, Hatanaka, K et al. (2013). Systemic delivery of small interfering RNA by use of targeted polycation liposomes for cancer therapy. Biol Pharm Bull 36: 287–291. [DOI] [PubMed] [Google Scholar]
- Yang, ZZ, Li, JQ, Wang, ZZ, Dong, DW and Qi, XR (2014). Tumor-targeting dual peptides-modified cationic liposomes for delivery of siRNA and docetaxel to gliomas. Biomaterials 35: 5226–5239. [DOI] [PubMed] [Google Scholar]
- Buyens, K, De Smedt, SC, Braeckmans, K, Demeester, J, Peeters, L, van Grunsven, LA et al. (2012). Liposome based systems for systemic siRNA delivery: stability in blood sets the requirements for optimal carrier design. J Control Release 158: 362–370. [DOI] [PubMed] [Google Scholar]
- Shi, S, Han, L, Deng, L, Zhang, Y, Shen, H, Gong, T et al. (2014). Dual drugs (microRNA-34a and paclitaxel)-loaded functional solid lipid nanoparticles for synergistic cancer cell suppression. J Control Release 194: 228–237. [DOI] [PubMed] [Google Scholar]
- Lobovkina, T, Jacobson, GB, Gonzalez-Gonzalez, E, Hickerson, RP, Leake, D, Kaspar, RL et al. (2011). In vivo sustained release of siRNA from solid lipid nanoparticles. ACS Nano 5: 9977–9983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, Y, Zhu, X, Zhang, X, Liu, B and Huang, L (2010). Nanoparticles modified with tumor-targeting scFv deliver siRNA and miRNA for cancer therapy. Mol Ther 18: 1650–1656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chono, S, Li, SD, Conwell, CC and Huang, L (2008). An efficient and low immunostimulatory nanoparticle formulation for systemic siRNA delivery to the tumor. J Control Release 131: 64–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramezani, M, Khoshhamdam, M, Dehshahri, A and Malaekeh-Nikouei, B (2009). The influence of size, lipid composition and bilayer fluidity of cationic liposomes on the transfection efficiency of nanolipoplexes. Colloids Surf B Biointerfaces 72: 1–5. [DOI] [PubMed] [Google Scholar]
- Schäfer, J, Höbel, S, Bakowsky, U and Aigner, A (2010). Liposome-polyethylenimine complexes for enhanced DNA and siRNA delivery. Biomaterials 31: 6892–6900. [DOI] [PubMed] [Google Scholar]
- Alshamsan, A, Hamdy, S, Samuel, J, El-Kadi, AO, Lavasanifar, A and Uludağ, H (2010). The induction of tumor apoptosis in B16 melanoma following STAT3 siRNA delivery with a lipid-substituted polyethylenimine. Biomaterials 31: 1420–1428. [DOI] [PubMed] [Google Scholar]
- Jang, YL, Yun, UJ, Lee, MS, Kim, MG, Son, S, Lee, K et al. (2012). Cell-penetrating peptide mimicking polymer-based combined delivery of paclitaxel and siRNA for enhanced tumor growth suppression. Int J Pharm 434: 488–493. [DOI] [PubMed] [Google Scholar]
- Hong, J, Ku, SH, Lee, MS, Jeong, JH, Mok, H, Choi, D et al. (2014). Cardiac RNAi therapy using RAGE siRNA/deoxycholic acid-modified polyethylenimine complexes for myocardial infarction. Biomaterials 35: 7562–7573. [DOI] [PubMed] [Google Scholar]
- Kim, WJ, Chang, CW, Lee, M and Kim, SW (2007). Efficient siRNA delivery using water soluble lipopolymer for anti-angiogenic gene therapy. J Control Release 118: 357–363. [DOI] [PubMed] [Google Scholar]
- Ozcan, G, Ozpolat, B, Coleman, RL, Sood, AK and Lopez-Berestein, G (2015). Preclinical and clinical development of siRNA-based therapeutics. Adv Drug Deliv Rev 87: 108–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Junn, E and Mouradian, MM (2012). MicroRNAs in neurodegenerative diseases and their therapeutic potential. Pharmacol Ther 133: 142–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mack, GS (2007). MicroRNA gets down to business. Nat Biotechnol 25: 631–638. [DOI] [PubMed] [Google Scholar]
- Li, T, Wu, M, Zhu, YY, Chen, J and Chen, L (2014). Development of RNA interference-based therapeutics and application of multi-target small interfering RNAs. Nucleic Acid Ther 24: 302–312. [DOI] [PubMed] [Google Scholar]
- Obici, L and Merlini, G (2014). An overview of drugs currently under investigation for the treatment of transthyretin-related hereditary amyloidosis. Expert Opin Investig Drugs 23: 1239–1251. [DOI] [PubMed] [Google Scholar]
- Sehgal, A, Barros, S, Ivanciu, L, Cooley, B, Qin, J, Racie, T et al. (2015). An RNAi therapeutic targeting antithrombin to rebalance the coagulation system and promote hemostasis in hemophilia. Nat Med 21: 492–497. [DOI] [PubMed] [Google Scholar]
- Barr, FA, Silljé, HH and Nigg, EA (2004). Polo-like kinases and the orchestration of cell division. Nat Rev Mol Cell Biol 5: 429–440. [DOI] [PubMed] [Google Scholar]
- Liu, X and Erikson, RL (2003). Polo-like kinase (Plk)1 depletion induces apoptosis in cancer cells. Proc Natl Acad Sci USA 100: 5789–5794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao, J, Zhou, B, Chu, B and Yen, Y (2006). Ribonucleotide reductase inhibitors and future drug design. Curr Cancer Drug Targets 6: 409–431. [DOI] [PubMed] [Google Scholar]
- Heidel, JD, Liu, JY, Yen, Y, Zhou, B, Heale, BS, Rossi, JJ et al. (2007). Potent siRNA inhibitors of ribonucleotide reductase subunit RRM2 reduce cell proliferation in vitro and in vivo. Clin Cancer Res 13: 2207–2215. [DOI] [PubMed] [Google Scholar]
- Davis, ME, Zuckerman, JE, Choi, CH, Seligson, D, Tolcher, A, Alabi, CA et al. (2010). Evidence of RNAi in humans from systemically administered siRNA via targeted nanoparticles. Nature 464: 1067–1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuckerman, JE, Gritli, I, Tolcher, A, Heidel, JD, Lim, D, Morgan, R et al. (2014). Correlating animal and human phase Ia/Ib clinical data with CALAA-01, a targeted, polymer-based nanoparticle containing siRNA. Proc Natl Acad Sci USA 111: 11449–11454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hicklin, DJ and Ellis, LM (2005). Role of the vascular endothelial growth factor pathway in tumor growth and angiogenesis. J Clin Oncol 23: 1011–1027. [DOI] [PubMed] [Google Scholar]
- Tao, W, South, VJ, Zhang, Y, Davide, JP, Farrell, L, Kohl, NE et al. (2005). Induction of apoptosis by an inhibitor of the mitotic kinesin KSP requires both activation of the spindle assembly checkpoint and mitotic slippage. Cancer Cell 8: 49–59. [DOI] [PubMed] [Google Scholar]
- Santel, A, Aleku, M, Röder, N, Möpert, K, Durieux, B, Janke, O et al. (2010). Atu027 prevents pulmonary metastasis in experimental and spontaneous mouse metastasis models. Clin Cancer Res 16: 5469–5480. [DOI] [PubMed] [Google Scholar]
- Aleku, M, Schulz, P, Keil, O, Santel, A, Schaeper, U, Dieckhoff, B et al. (2008). Atu027, a liposomal small interfering RNA formulation targeting protein kinase N3, inhibits cancer progression. Cancer Res 68: 9788–9798. [DOI] [PubMed] [Google Scholar]
- Leenders, F, Möpert, K, Schmiedeknecht, A, Santel, A, Czauderna, F, Aleku, M et al. (2004). PKN3 is required for malignant prostate cell growth downstream of activated PI 3-kinase. EMBO J 23: 3303–3313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pereira, DM, Rodrigues, PM, Borralho, PM and Rodrigues, CM (2013). Delivering the promise of miRNA cancer therapeutics. Drug Discov Today 18: 282–289. [DOI] [PubMed] [Google Scholar]
- Bouchie, A (2013). First microRNA mimic enters clinic. Nat Biotechnol 31: 577. [DOI] [PubMed] [Google Scholar]
- Bader, AG (2012). miR-34 - a microRNA replacement therapy is headed to the clinic. Front Genet 3: 120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aqeilan, RI, Calin, GA and Croce, CM (2010). miR-15a and miR-16-1 in cancer: discovery, function and future perspectives. Cell Death Differ 17: 215–220. [DOI] [PubMed] [Google Scholar]
- Guzman-Aranguez, A, Loma, P and Pintor, J (2013). Small-interfering RNAs (siRNAs) as a promising tool for ocular therapy. Br J Pharmacol 170: 730–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu, S (2009). microRNA expression in the eyes and their significance in relation to functions. Prog Retin Eye Res 28: 87–116. [DOI] [PubMed] [Google Scholar]
- Lam, JK, Liang, W and Chan, HK (2012). Pulmonary delivery of therapeutic siRNA. Adv Drug Deliv Rev 64: 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamora, MR, Budev, M, Rolfe, M, Gottlieb, J, Humar, A, Devincenzo, J et al. (2011). RNA interference therapy in lung transplant patients infected with respiratory syncytial virus. Am J Respir Crit Care Med 183: 531–538. [DOI] [PubMed] [Google Scholar]
- Chow, MYT and Lam, JKW. Dry powder formulation of plasmid DNA and siRNA for inhalation. Current Pharmaceutical Design, in press. [DOI] [PubMed]
- Bumcrot, D, Manoharan, M, Koteliansky, V and Sah, DW (2006). RNAi therapeutics: a potential new class of pharmaceutical drugs. Nat Chem Biol 2: 711–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lively, TN, Kossen, K, Balhorn, A, Koya, T, Zinnen, S, Takeda, K et al. (2008). Effect of chemically modified IL-13 short interfering RNA on development of airway hyperresponsiveness in mice. J Allergy Clin Immunol 121: 88–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mostafa Anower, AK, Shim, JA, Choi, B and Sohn, S (2012). Pretreatment with interleukin-6 small interfering RNA can improve the survival rate of polymicrobial cecal ligation and puncture mice by down regulating interleukin-6 production. Eur J Pharmacol 688: 76–83. [DOI] [PubMed] [Google Scholar]
- Ibrahim, AF, Weirauch, U, Thomas, M, Grünweller, A, Hartmann, RK and Aigner, A (2011). MicroRNA replacement therapy for miR-145 and miR-33a is efficacious in a model of colon carcinoma. Cancer Res 71: 5214–5224. [DOI] [PubMed] [Google Scholar]
- Lin, D, Cheng, Q, Jiang, Q, Huang, Y, Yang, Z, Han, S et al. (2013). Intracellular cleavable poly(2-dimethylaminoethyl methacrylate) functionalized mesoporous silica nanoparticles for efficient siRNA delivery in vitro and in vivo. Nanoscale 5: 4291–4301. [DOI] [PubMed] [Google Scholar]
- Shim, G, Choi, HW, Lee, S, Choi, J, Yu, YH, Park, DE et al. (2013). Enhanced intrapulmonary delivery of anticancer siRNA for lung cancer therapy using cationic ethylphosphocholine-based nanolipoplexes. Mol Ther 21: 816–824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakurai, Y, Hatakeyama, H, Akita, H and Harashima, H (2014). Improvement of doxorubicin efficacy using liposomal anti-polo-like kinase 1 siRNA in human renal cell carcinomas. Mol Pharm 11: 2713–2719. [DOI] [PubMed] [Google Scholar]
- Akao, Y, Nakagawa, Y, Hirata, I, Iio, A, Itoh, T, Kojima, K et al. (2010). Role of anti-oncomirs miR-143 and -145 in human colorectal tumors. Cancer Gene Ther 17: 398–408. [DOI] [PubMed] [Google Scholar]
- Trang, P, Wiggins, JF, Daige, CL, Cho, C, Omotola, M, Brown, D et al. (2011). Systemic delivery of tumor suppressor microRNA mimics using a neutral lipid emulsion inhibits lung tumors in mice. Mol Ther 19: 1116–1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geisbert, TW, Hensley, LE, Kagan, E, Yu, EZ, Geisbert, JB, Daddario-DiCaprio, K et al. (2006). Postexposure protection of guinea pigs against a lethal ebola virus challenge is conferred by RNA interference. J Infect Dis 193: 1650–1657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi, S, Han, L, Gong, T, Zhang, Z and Sun, X (2013). Systemic delivery of microRNA-34a for cancer stem cell therapy. Angew Chem Int Ed Engl 52: 3901–3905. [DOI] [PubMed] [Google Scholar]
- DeVincenzo, J, Cehelsky, JE, Alvarez, R, Elbashir, S, Harborth, J, Toudjarska, I et al. (2008). Evaluation of the safety, tolerability and pharmacokinetics of ALN-RSV01, a novel RNAi antiviral therapeutic directed against respiratory syncytial virus (RSV). Antiviral Res 77: 225–231. [DOI] [PubMed] [Google Scholar]
- Nguyen, QD, Schachar, RA, Nduaka, CI, Sperling, M, Basile, AS, Klamerus, KJ et al.; PF-04523655 Study Group. (2012). Phase 1 dose-escalation study of a siRNA targeting the RTP801 gene in age-related macular degeneration patients. Eye (Lond) 26: 1099–1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen, QD, Schachar, RA, Nduaka, CI, Sperling, M, Klamerus, KJ, Chi-Burris, K et al.; MONET Clinical Study Group. (2012). Evaluation of the siRNA PF-04523655 versus ranibizumab for the treatment of neovascular age-related macular degeneration (MONET Study). Ophthalmology 119: 1867–1873. [DOI] [PubMed] [Google Scholar]
- Nguyen, QD, Schachar, RA, Nduaka, CI, Sperling, M, Basile, AS, Klamerus, KJ et al.; DEGAS Clinical Study Group. (2012). Dose-ranging evaluation of intravitreal siRNA PF-04523655 for diabetic macular edema (the DEGAS study). Invest Ophthalmol Vis Sci 53: 7666–7674. [DOI] [PubMed] [Google Scholar]
- Fitzgerald, K, Frank-Kamenetsky, M, Shulga-Morskaya, S, Liebow, A, Bettencourt, BR, Sutherland, JE et al. (2014). Effect of an RNA interference drug on the synthesis of proprotein convertase subtilisin/kexin type 9 (PCSK9) and the concentration of serum LDL cholesterol in healthy volunteers: a randomised, single-blind, placebo-controlled, phase 1 trial. Lancet 383: 60–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coelho, T, Adams, D, Silva, A, Lozeron, P, Hawkins, PN, Mant, T et al. (2013). Safety and efficacy of RNAi therapy for transthyretin amyloidosis. N Engl J Med 369: 819–829. [DOI] [PubMed] [Google Scholar]
- Leachman, SA, Hickerson, RP, Schwartz, ME, Bullough, EE, Hutcherson, SL, Boucher, KM et al. (2010). First-in-human mutation-targeted siRNA phase Ib trial of an inherited skin disorder. Mol Ther 18: 442–446. [DOI] [PMC free article] [PubMed] [Google Scholar]