

Abstract
Cisplatin, a commonly used cancer chemotherapeutic, has a dose-limiting side effect of nephrotoxicity. Approximately 30% of patients administered cisplatin suffer from kidney injury, and there are limited treatment options for the treatment of cisplatin-induced kidney injury. Suramin, which is Federal Drug Administration-approved for the treatment of trypanosomiasis, improves kidney function after various forms of kidney injury in rodent models. We hypothesized that suramin would attenuate cisplatin-induced kidney injury. Suramin treatment before cisplatin administration reduced cisplatin-induced decreases in kidney function and injury. Furthermore, suramin attenuated cisplatin-induced expression of inflammatory cytokines and chemokines, endoplasmic reticulum stress, and apoptosis in the kidney cortex. Treatment of mice with suramin 24 h after cisplatin also improved kidney function, suggesting that the mechanism of protection is not by inhibition of tubular cisplatin uptake or its metabolism to nephrotoxic species. If suramin is to be used in the context of cancer, then it cannot prevent cisplatin-induced cytotoxicity of cancer cells. Suramin did not alter the dose-response curve of cisplatin in lung adenocarcinoma cells in vitro. In addition, suramin pretreatment of mice harboring lung adenocarcinomas did not alter the initial cytotoxic effects of cisplatin (DNA damage and apoptosis) on tumor cells. These results provide evidence that suramin has potential as a renoprotective agent for the treatment/prevention of cisplatin-induced acute kidney injury and justify future long-term preclinical studies using cotreatment of suramin and cisplatin in mouse models of cancer.
Keywords: cisplatin, acute kidney injury, suramin, renoprotective strategies
cisplatin is a commonly used chemotherapeutic in the treatment of many solid tumor cancer types, such as those of the head and neck, bladder, ovary, lung, and testicule (10, 28, 33). However, acute kidney injury (AKI) is the most critical dose-limiting toxicity in cancer patients treated with cisplatin. Unfortunately, treatment options for cisplatin-induced kidney injury are limited. Cisplatin must be withheld from patients that have confounding factors predisposing them to an increased risk for kidney injury, including advanced age, smoking, hypoalbuminemia, or known chronic kidney disease (26). In fact, even without one of these comorbidities, 30% of patients administered cisplatin will develop kidney injury, according to the Risk, Injury, Failure, Loss of Function, and End-Stage Renal Disease (RIFLE) criteria of kidney injury (26, 28, 38, 43), requiring that the patient be switched to a less effective therapeutic regimen.
Due to the complexity of cisplatin's nephrotoxic mechanism of action, the development of renoprotective agents remains a challenge. In the kidney, cisplatin is metabolized to its nephrotoxic metabolite, which can induce AKI and kidney failure (22, 26–28, 43). Cisplatin-induced AKI involves several biological responses, including mitochondrial dysfunction, activation of cell death pathways, generation of ROS, and a robust inflammatory response (26, 28). The inflammatory response associated with cisplatin-induced AKI is known to have many components, such as the production of proinflammatory cytokines and chemokines, activation of resident macrophages, and infiltration of immune cells into the kidney (26, 27).
Suramin is a polysulfonated napthylurea compound originally designed for the treatment of trypanosomiasis (also known as African sleeping sickness). Interestingly, suramin has been shown to speed recovery from ischemia-reperfusion and glycerol-induced AKI in rodents, in part by dampening the inflammatory response and apoptosis and promoting renal proximal tubule cell proliferation (18–20, 23–25, 45). In models of chronic kidney disease such as diabetic nephropathy, unilateral ureteral obstruction, and the remnant kidney model, suramin treatment has been shown to decrease the inflammatory response and reverse the associated fibrotic phenotypes (18–20, 23–25, 45); however, a role for the use of suramin as a renoprotective agent in cisplatin-induced AKI has not been examined. We hypothesized that suramin treatment would attenuate the inflammatory response after cisplatin administration and protect the kidney from injury. To test this hypothesis, we pretreated mice with suramin before cisplatin treatment.
MATERIALS AND METHODS
The following antibodies were purchased from Cell Signaling (Beverly, MA) unless otherwise noted: inositol-requiring enzyme (IRE)-1α (no. 3294), cleaved caspase-3 (no. 9664), cleaved caspase-8 (no. 8592), C/EBP homologous protein (CHOP; no. 2895), JNK (no. 9258), phosphorylated (p-)JNK (no. 4668), p-histone H2Ax (no. 9718), and β-actin (catalog no. A5441, Sigma-Aldrich, St. Louis, MO). Suramin (catalog no. S2671) and cisplatin (catalog no. P4394) were purchased from Sigma-Aldrich. Pharmacy grade cisplatin (1 mg/ml) was obtained from the University of Louisville Hospital pharmacy for FVB experiments and suramin posttreatment experiments.
C57BL/6J mice (male, 8 wk old) were purchased from The Jackson Laboratory (Bar Harbor, ME) and acclimated for 1 wk before the initiation of experiments. Mice were maintained on a 12:12-h light-dark cycle and provided food and water ad libitum. All animal procedures were approved by the Institutional Animal Care and Use Committee and followed the guidelines of the American Veterinary Medical Association. For pretreatment experiments, suramin (1 or 10 mg/kg) in PBS (200 μl/animal) was administered by tail vein injection 72 h before cisplatin. Cisplatin at 20 mg/kg in water (200 μl/animal) was administered by intraperitoneal injection. Forty-eight hours after cisplatin injection, mice were put into metabolic cages for urine collection for 24 h and then euthanized. For posttreatment experiments, suramin (10 mg/kg) in PBS (200 μl/animal) was administered by tail vein injection 24 h after cisplatin. Cisplatin at 25 mg/kg in normal saline (1 mg/ml) was administered by intraperitoneal injection. Blood was collected, and kidneys were flash frozen or fixed in 10% neutral buffered formalin. Eight- to ten-mo-old FVB mice expressing mutant Kirsten rat sarcoma viral oncogene homolog (KRAS)-driven lung tumors were obtained as a gift from L. J. Beverly. These bitransgenic mice were made by crossing secretoglobin family 1A member 1 (uteroglobin)-reverse tetracycline-controlled transactivator mice to Tet-op-K-rasG12D mice. Genotyping was performed via PCR as previously described (14). These mice were pretreated with 10 mg/kg cisplatin 72 h before cisplatin treatment (20 mg/kg ip) followed by euthanization 48 h after cisplatin administration. Blood was collected, and kidneys and lungs were flash frozen in liquid nitrogen or fixed in 10% neutral buffered formalin.
Blood urea nitrogen (BUN; DIUR-500) and serum creatinine (C7548-120), were determined using kits from Bioassay Systems (Hayward, CA) and Point Scientific (Canton, MI), respectively, following the manufacturers' instructions. ELISAs for kidney injury molecule (KIM)-1 (DT1817, R&D Systems, Minneapolis, MN) and neutrophil gelatinase-associated lipocalin (NGAL; DY1857, R&D Systems) were performed on the urine as directed by the manufacturer. Protein quantification and Western blot analysis were performed as previously described (32). RNA was isolated using RNA-STAT 60 (TEL-TEST, Friendswood, TX) using mini-Bead-beater glass beads and a mini-Bead Beater machine (Cole-Palmer, Vernon Hills, IL) per the manufacturer's protocol. cDNA was synthesized with an iScript cDNA Kit (Bio-Rad) per the manufacturer's instructions. The transcripts quantitated using SYBR green included TNF-α (5′-AATGGCCTCCCTCTCATCAGTT-3′ and 5′-CCACTTGGTGGTTTGCTACGA-3′) and the housekeeping gene for normalization β2-microglobulin (B2M; 5′-TTCTGGTGCTTGTCTCACTGA-3′ and 5′-CAGTATGTTCGGCTTCCCATTC-3′) using Sso Fast Evagreen Supermix (Bio-Rad) per the manufacturers' instructions. Chemokine (C-X-C) ligand (CXCL)1 (Mm04207460_m1), monocyte chemotactic protein (MCP)-1 (Mm00441242_m1), IL-1β (Mm0043228_m1), and IL-6 (Mm00446190_m1, Life Technologies, Grand Island, NY) were used in combination with 2× gene expression Master Mix (Life Technologies). KIM-1 (Bio-Rad) was used in combination with Sso Advance Universal Probe Master Mix (Bio-Rad) per the manufacturer's instructions. Kidney and lung histology and immunohistochemistry were performed as previously described (20, 34). pH2A.x staining was quantified by image analysis. Sections were visualized via a Nikon Eclipse E600 microscope (Nikon, Tokyo, Japan), and Metamorph software was used to capture and analyze each tumor per section (×200 magnification). Image analysis was performed using modifications of previously described techniques (2). Detection thresholds were set for the brown (pH2A.x) and blue (hematoxylin) colors based on an intensely labeled point and a default color threshold. The degree of labeling in each section was determined from the area within the brown color range divided by the total area within the blue color range. Cell viability was performed as previously described (35). TUNEL assays were performed on paraffin-embedded tissues with an Apoptag red in situ apoptosis detection kit per the manufacturer's instructions (catalog no. S7165, Millipore, Temecula, CA). Sections were counterstained and mounted with Vectashield Antifade Mounting Medium with 4′,6-diamidino-2-phenylindole. Slides were visualized via immunofluorescent microscopy with a Nikon Eclipse Ti-E microscope using Nikon NIS Elements software (Nikon). For quantification of TUNEL positivity in the kidneys or lung tumors, TUNEL foci were enumerated and summed. In the lung tumors, TUNEL foci were summed per micrometer squared of tumor per ×200 magnification field.
Data are expressed as means ± SE for all experiments. Multiple comparisons of normally distributed data were analyzed by two-way ANOVA as appropriate, and group means were compared using Bonferonni posttests. For Western blot analysis, multiple comparisons of normally distributed data were analyzed by one-way ANOVA as appropriate, and group means were compared using Tuckey posttests. The criterion for statistical differences was P < 0.05 for all comparisons.
RESULTS
Effect of suramin pretreatment on serum and urinary markers of kidney function and injury in cisplatin-treated mice.
Mice were pretreated with suramin (1 mg/kg and 10 mg/kg iv) 72 h before cisplatin (20 mg/kg ip) administration and then euthanized 72 h after cisplatin treatment. Cisplatin-treated mice had increased BUN (6-fold), serum creatinine (10-fold), urinary NGAL (4,100-fold), urinary Kim-1 (302-fold), and renal Kim-1 mRNA expression (4,000-fold) compared with vehicle-treated animals (Fig. 1). Mice pretreated with 10 mg/kg suramin before cisplatin had decreased BUN (3-fold), serum creatinine (3-fold), urinary NGAL (1,500-fold), urinary Kim-1 (126-fold), and renal Kim-1 mRNA expression (1,700-fold) compared with control values (Fig. 1). However, mice pretreated with 1 mg/kg suramin before cisplatin did not show a decrease in the aforementioned kidney markers (Fig. 1). These data reveal that pretreatment of mice with 10 mg/kg suramin reduced cisplatin-induced AKI, whereas pretreatment of mice with 1 mg/kg suramin did not.
Fig. 1.

Pretreatment with high-dose suramin improves markers of kidney function and injury. C57BL/6J mice were pretreated with suramin [at 1 mg/kg (S1) or 10 mg/kg (S10)] or vehicle control (V) via an intravenous injection through the tail vein 72 h before cisplatin administration (C; 20 mg/kg ip), and mice were euthanized 72 h after cisplatin administration. Levels of blood urea nitrogen (BUN; A) and serum creatinine (SCr; B) were assessed via colorimetric and enzymatic assays, respectively. Urinary neutrophil gelatinase-associated lipocalin (NGAL; C) and kidney injury molecule (KIM)-1 (D) levels were assessed via ELISA. E: mRNA expression of KIM-1 in the kidney cortex was assessed via qRT-PCR. Data are means ± SE; n = 5–10. *P ≤ 0.05 compared with the cisplatin treated group as determined by two-way ANOVA.
Effect of suramin pretreatment on pathological damage to proximal tubules in cisplatin-treated mice.
To determine if the suramin-induced changes in kidney function and injury markers were associated with improved kidney pathology, we assessed renal histology on hematoxylin and eosin- and periodic acid-Schiff-stained paraffin-embedded sections collected 72 h after cisplatin administration (Fig. 2A). Blinded scoring by a renal pathologist indicated that markers of proximal tubule histological damage, such as acute tubular necrosis, loss of the brush border, proximal tubular cast formation, tubule dilation, and degeneration (Fig. 2, B–F), were increased in cisplatin-treated mice. All of these histological changes were attenuated in mice pretreated with suramin at 10 mg/kg (Fig. 2, B–F). Thus, suramin protected mice from histological damage resulting from cisplatin treatment.
Fig. 2.
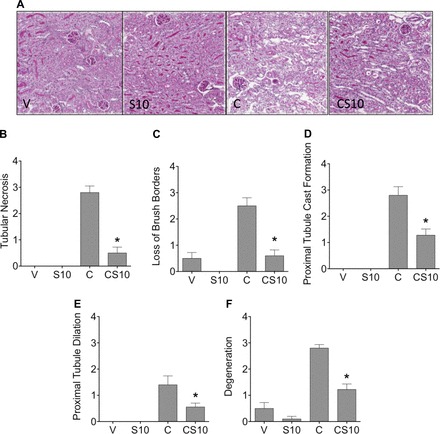
Suramin attenuates cisplatin-induced deterioration in kidney pathology. Renal histological changes were assessed on hematoxylin and eosin- and periodic acid-Schiff-stained sections (5 μm thick). A: representative images of renal histology at ×200 magnification. B–F: tubule necrosis (B), loss of proximal tubule brush borders (C), proximal tubule cast formation (D), proximal tubule dilation (E), and degradation (F) were assessed as markers of histological changes. In B–F, scoring of the sections was performed in a blinded manner by a renal pathologist (J. Megyesi) using a scale of 0–4 (where 0 = not present, 1 = mild, 2 = moderate, 3 = severe, and 4 = very severe renal histological changes in proximal tubules). V indicates vehicle control, S10 indicates suramin iv pretreatment at 10 mg/kg, C indicates cisplatin ip at 20 mg/kg, and CS10 indicates suramin iv pretreatment at 10 mg/kg followed by cisplatin ip at 20 mg/kg 72 h later. Data are means ± SE; n = 5–10. *P ≤ 0.05 compared with the cisplatin-treated group as determined by two-way ANOVA.
Effect of suramin pretreatment on inflammatory cytokines and chemokines in cisplatin-treated mice.
Treatment with cisplatin has been shown to induce the expression of many proinflammatory cytokines and chemokines in the kidney (1, 13, 27). Expression of TNF-α, IL-6, CXCL1, MCP-1, and IL-1β were increased 11-, 66-, 144-, 15-, and 3-fold, respectively, after cisplatin treatment; however, mice pretreated with suramin (10 mg/kg) 72 h before cisplatin treatment had significantly reduced mRNA expression of TNF-α, IL-6, CXCL1, MCP-1, and IL-1β to 3-, 10-, 20-, 3-, and 0-fold over control values (Fig. 3). These data reveal that pretreatment of mice with suramin reduced cisplatin-induced expression of proinflammatory cytokines and chemokines.
Fig. 3.
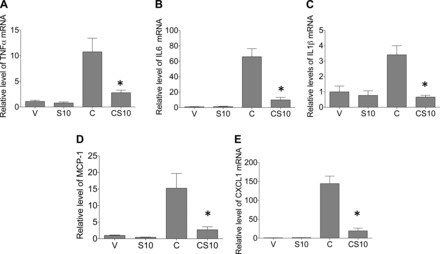
Suramin pretreatment reduces inflammation in the kidney after cisplatin treatment. C57BL/6J mice were pretreated with suramin (10 mg/kg) or an equal volume of vehicle via tail vein injection 72 h before cisplatin administration (20 mg/kg ip). Mice were euthanized 72 h after cisplatin administration, and the relative expression of TNF-α (A), IL-6 (B), chemokine (C-X-C) ligand (CXCL)1 (C), monocyte chemotactic protein (MCP)-1 (D), and IL-1β (E) was measured via qRT-PCR. V indicates vehicle control, S10 indicates suramin iv pretreatment at 10 mg/kg, C indicates cisplatin ip at 20 mg/kg, and CS10 indicates suramin iv pretreatment at 10 mg/kg followed by cisplatin ip at 20 mg/kg 72 h later. Data are means ± SE. *P ≤ 0.05 compared with the cisplatin-treated group as determined by two-way ANOVA.
Effect of suramin pretreatment on markers of cell stress and proliferation in cisplatin- treated mice.
A number of pathways are known to be activated in response to cisplatin-induced AKI, including cell stress and proliferation signaling pathways (27). Thus, we assessed markers of these signaling pathways in cisplatin-induced AKI in the presence and absence of suramin pretreatment. Cisplatin-treated mice had increased levels of the activated form of JNK protein (p-JNK) compared with vehicle treatment, whereas pretreatment with suramin before cisplatin significantly decreased levels of p-JNK (5-fold) compared with cisplatin treatment alone (Fig. 4, A and B). We also assessed markers of endoplasmic reticulum (ER) stress, as it has previously been implicated in cisplatin-induced AKI (28). Cisplatin treatment increased both IRE-1α protein and CHOP protein levels, whereas suramin pretreatment before cisplatin decreased IRE-1α levels threefold (Fig. 4, A and C). Suramin treatment before cisplatin decreased CHOP expression, albeit not significantly (Fig. 4, A and D). Taken together, these data indicate that suramin pretreatment decreased the cisplatin-induced increase in levels of markers of cell stress and proliferation in the kidney cortex.
Fig. 4.

Suramin reduces cisplatin-induced increases in markers of cell stress responses and proliferation. A: Western blot analysis was performed to assess relative protein levels of the indicated proteins in the renal cortex of mice. Samples from the kidney cortex were prepared from mice euthanized 72 h after cisplatin administration. p-JNK, phosphorylated JNK; IRE-1α, inositol-requiring enzyme-1α; CHOP, C/EBP homologous protein. In B–D, densitometry was performed to assess relative protein levels normalized to actin. B: p-JNK/JNK; C: IRE-1α; D: CHOP. V indicates vehicle control, C indicates cisplatin ip at 20 mg/kg, and CS10 indicates suramin iv pretreatment at 10 mg/kg followed by cisplatin ip at 20 mg/kg 72 h later. Data are mean ± SE; n = 5. *Statistically significant difference compared with the cisplatin-treated group as determined by one-way ANOVA.
Effect of suramin pretreatment on markers of apoptosis in cisplatin-treated mice.
Cisplatin treatment is known to induce apoptosis in the kidney (27). Thus, we assessed markers of apoptosis in the kidney cortex. Apoptosis is dependent on the activation and cleavage of cysteine and aspartate proteases (caspases). Cisplatin increased protein levels of cleaved caspase-8 and cleaved caspase-3, respectively (Fig. 5, A–C). Suramin treatment before cisplatin significantly reduced levels of cleaved caspase-8 and cleaved caspase-3 in the kidney cortex by 10- and 3-fold, respectively, compared with cisplatin alone-treated mice (Fig. 5, A–C). TUNEL assays were also performed on paraffin-embedded kidney sections for the detection of apoptotic cells in the kidney cortex (Fig. 5D). Quantification of TUNEL-stained kidneys showed that cisplatin treatment alone led to a significant increase in TUNEL-positive foci in the kidney cortex, whereas suramin pretreatment before cisplatin significantly decreased the number of TUNEL-positive foci compared with cisplatin alone-treated animals (Fig. 5, D and E). These data indicate that suramin pretreatment protects mice from cisplatin-induced kidney cell apoptosis.
Fig. 5.
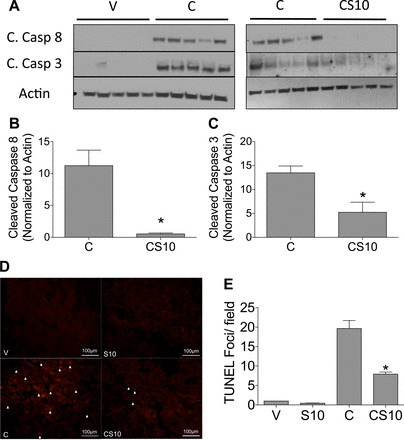
Pretreatment with suramin inhibits cisplatin-induced cell death in the kidney. A: Western blot analysis of cleaved (C.) caspase-8 and cleaved caspase-3 from homogenates of the kidney cortex for the indicated treatment groups. In B and C, densitometry was performed to assess relative protein levels normalized to actin. B: cleaved caspase-8; C: cleaved caspase-3. D: representative photomicrographs of TUNEL immunofluorescence (final magnification: ×200). Arrows indicate TUNEL-positive foci. E: quantification of TUNEL immunofluorescence. Data are means ± SE; n = 5. V indicates vehicle control, S10 indicates suramin iv pretreatment at 10 mg/kg, C indicates cisplatin ip at 20 mg/kg, and CS10 indicates suramin iv pretreatment at 10 mg/kg followed by cisplatin ip at 20 mg/kg 72 h later. *Statistically significant difference compared with the cisplatin-treated group as determined by one-way ANOVA.
Effect of suramin posttreatment on markers of kidney function, injury, cell death, and ER stress after cisplatin treatment.
Our data indicate that suramin pretreatment before cisplatin protects from cisplatin-induced AKI. The protective effects of suramin may result from decreased cisplatin uptake into proximal tubule cells or its metabolism. Cisplatin is concentrated in the kidney, reaching a peak renal concentrations within 1–6 h after cisplatin administration, and ∼80% of cisplatin is excreted within 24 h after cisplatin administration (9, 21, 38). If suramin inhibited cisplatin uptake or its metabolism in proximal tubules, then suramin treatment 24 h after cisplatin would not be protective. Mice were treated with cisplatin (25 mg/kg) and then treated with suramin (10 mg/kg) 24 h after cisplatin. Kidney function was assessed 72 h after cisplatin treatment. Cisplatin treatment increased levels of BUN and serum creatinine nine- and sevenfold, respectively, whereas mice treated with cisplatin and then treated with suramin 24 h later had significantly decreased BUN and serum creatinine levels of seven- and fourfold, respectively (Fig. 6, A and B). We also assessed markers of ER stress and apoptosis, as they have been implicated in cisplatin-induced AKI (28). Cisplatin-treated mice had increased levels of p-JNK protein compared with vehicle and suramin alone treatment, whereas suramin posttreatment followed by cisplatin administration showed decreased levels of p-JNK compared with cisplatin treatment alone (Fig. 6C). Cisplatin treatment increased both IRE-1α protein and CHOP protein levels, whereas suramin posttreatment after cisplatin decreased both IRE-1α and CHOP expression (Fig. 6C). Markers of apoptosis were increased after cisplatin treatment, as evidenced by increases in cleaved caspase-8 and cleaved caspase-3 protein levels compared with vehicle- or suramin alone-treated mice (Fig. 6C). Suramin posttreatment followed by cisplatin administration showed reduced levels of cleaved caspase-8 and cleaved caspase-3 in the kidney cortex compared with cisplatin-treated mice (Fig. 6C). To further assess apoptosis, TUNEL assays were performed on paraffin-embedded tissues. Quantification of TUNEL-stained kidneys showed that cisplatin treatment alone led to a significant increase in TUNEL-positive foci in the kidney cortex, whereas suramin posttreatment 24 h after cisplatin significantly decreased the number of TUNEL-positive foci compared with cisplatin alone-treated animals (Fig. 6D). These data reveal that posttreatment of suramin 24 h after cisplatin partially restored kidney function and reduced markers of ER stress and apoptosis in the renal cortex.
Fig. 6.
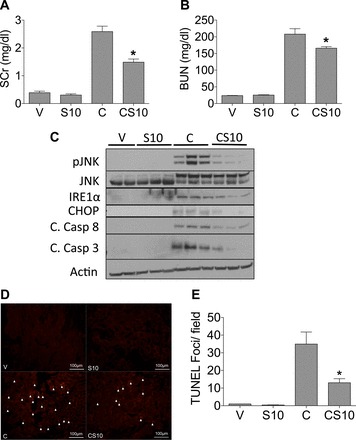
Suramin posttreatment preserves markers of kidney function and reduces markers of cisplatin-induced cell death and stress. C57BL/6J mice were posttreated with suramin (10 mg/kg) or vehicle control via an intravenous injection through the tail vein 24 h after cisplatin administration (25 mg/kg ip), and mice were euthanized 72 h after cisplatin administration. Levels of SCr (A) and BUN (B) were assessed via colorimetric and enzymatic assays, respectively. C: Western blot analysis was performed to assess relative protein levels of the indicated proteins in the renal cortex of mice. D: representative photomicrographs of TUNEL immunofluorescence (final magnification: ×200). Arrows indicate TUNEL-positive foci. E: quantification of TUNEL immunofluorescence (n = 5). In A and B, n = 8; in C, n = 3; in D, n = 5 with representative images provided. V indicates vehicle control, S10 indicates suramin iv posttreatment at 10mg/kg, C indicates cisplatin ip at 25 mg/kg, and CS10 indicates suramin iv posttreatment at 10 mg/kg 24 h following cisplatin ip at 20 mg/kg. Data are means ± SE. *P ≤ 0.05 compared with the cisplatin-treated group as determined by two-way ANOVA.
Effect of suramin on cisplatin-induced cytotoxicity in mutant KRAS-driven lung cancer.
The cisplatin-induced reduction in tumor mass is dependent on its ability to induce DNA damage and apoptosis in proliferating tumor cells. If suramin is to be used with cisplatin as part of a treatment protocol, then it cannot reduce the antitumor efficacy of cisplatin. We performed experiments to assess the effects of suramin on cisplatin-induced decreases in lung cancer cell viability in vitro and the initial ability of cisplatin to induced DNA damage and apoptosis within lung tumors in vivo. Zhuang et al. (46) showed that 50 μM suramin promotes renal proximal tubule cell scattering and proliferation. Thus, we chose 50 μM suramin for our in vitro experiments. A549 cells, a non-small cell lung cancer cell line expressing mutant KRAS, were pretreated with 50 μM suramin or diluent for 1 h and then treated with cisplatin (0–135 μM, 48 h), and relative cell numbers were determined by Alamar blue fluorescence. There were no differences in the dose-response curves of cisplatin-treated A549 cells with or without suramin pretreatment (Fig. 7A). A dose-response curve of A549 cells treated only with suramin (0–135 μM, 48 h) indicated that suramin alone had no impact on the relative numbers of A549 cells (data not shown). To examine the in vivo effects of suramin, FVB mice expressing doxycline-inducible mutant KRAS-driven lung adenocarcinoma were treated with cisplatin alone or pretreated with suramin 72 h before cisplatin. Levels of BUN were measured as markers of kidney function. Similar to the above experiments, BUN levels were greater in mice treated with cisplatin than in mice pretreated with suramin (10 mg/kg) before cisplatin (Fig. 7B). The ability of cisplatin to initially induce DNA damage in lung adenocarcinomas in the presence and absence of suramin was evaluated by immunohistochemistry of pH2A.x staining in lung sections. As shown in Fig. 7C, we observed an increase in pH2A.x staining in lung nodules of cisplatin-treated mice compared with vehicle-treated mice. Lung nodules from mice that received suramin pretreatment before cisplatin showed a trending increase in pH2A.x staining compared with pH2A.x staining in cisplatin only-treated mice; however, this increase was not statistically significant (Fig. 7, C and E). Cisplatin-induced DNA damage is known to lead to the cell's demise via the activation of apoptotic pathways in tumor cells. Thus, we examined apoptosis levels via TUNEL immunofluorescence on lung nodules. We observed a trending increase in TUNEL-positive foci in the suramin alone-treated group compared with the vehicle-treated group, whereas cisplatin treatment showed an even greater increase in TUNEL-positive foci in lung nodules compared with the vehicle-treated group. More importantly, suramin pretreatment before cisplatin did not decrease the ability of cisplatin to induce apoptosis in lung nodules (Fig. 7, D and F). Taken together, these data indicate that suramin pretreatment protects mice from cisplatin-induced AKI without inhibiting the initial ability of cisplatin to induce both DNA damage and apoptosis in lung adenocarcinomas.
Fig. 7.
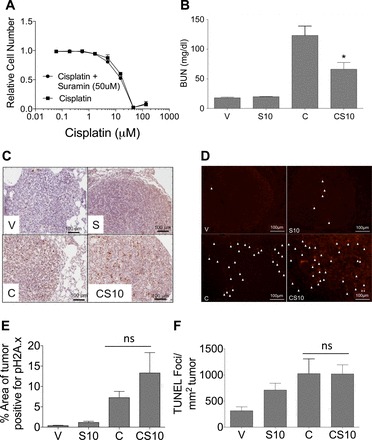
Suramin treatment does not inhibit the antitumor efficacy of cisplatin. A: A549 cells were treated with 0–135 μM cisplatin in the presence and absence of 50μM suramin for 48 h. Cell viability was assessed via an Alamar blue assay. B: BUN levels of 10-mo-old FVB mice harboring mutant Kirsten rat sarcoma viral oncogene homolog-driven lung tumors treated with 10 mg/kg suramin 72 h before cisplatin treatment (20 mg/kg, n = 5–10). Mice were euthanized 48 h after cisplatin administration. C: representative photomicrographs of pH2A.x immunohistochemistry (final magnification: ×200). Arrows indicate TUNEL-positive foci. D: representative photomicrographs of TUNEL immunofluorescence (final magnification: ×200). E: quantification of pH2A.x immunohistochemistry (n = 3). F: quantification of TUNEL immunofluorescence (n = 5). V indicates vehicle control, S10 indicates suramin iv pretreatment at 10 mg/kg, C indicates cisplatin ip at 20 mg/kg, and CS10 indicates suramin iv pretreatment at 10 mg/kg followed by cisplatin ip at 20 mg/kg 72 h later. Data are means ± SE. *P ≤ 0.05 compared with the cisplatin-treated group; ns indicates P > 0.05 compared with cisplatin-treated group as determined by two-way ANOVA.
DISCUSSION
Studies have shown that treatment with suramin after the onset of AKI improved recovery of renal function after renal ischemia-reperfusion and glycerol-induced rhabdomyolysis (20, 25, 45). For many forms of AKI, therapeutics aimed at preventing kidney injury are not useful due to the unpredictable nature of the insult. On the other hand, cisplatin is a known nephrotoxic agent whose administration is scheduled and thus the development of renoprotective strategies is feasible. Suramin has not been pursued as a renoprotective agent or in the context of cisplatin-induced AKI. Thus, in our study, we examined whether the prophylactic treatment of suramin would protect the kidney from the nephrotoxic side effects of cisplatin. Our data indicate that pretreatment of mice with suramin at a higher dose of 10 mg/kg but not a lower dose of 1 mg/kg protected from the cisplatin-induced decline in kidney function and increase in markers of kidney injury. Suramin pretreatment before cisplatin also attenuated the expression of proinflammatory chemokines and cytokines, suggesting that suramin is dampening the inflammatory response associated with cisplatin-induced AKI. The data also indicate that suramin pretreatment before cisplatin administration attenuated markers of both ER stress and apoptosis. Furthermore, posttreatment of mice with suramin after cisplatin administration decreased the cisplatin-induced decline in kidney function and attenuated markers of both ER stress and apoptosis in the renal cortex, suggesting that the renoprotective effects of suramin are not via the inhibition of uptake or metabolism of cisplatin in the kidney. In addition, our in vitro data suggest that the protection observed in vivo is not mediated by suramin directly blocking cell death as pretreatment of A549 lung cancer cells with 50 μM suramin did not alter the dose-response curve of cisplatin. Finally, suramin pretreatment of FVB mice harboring mutant KRAS-driven lung adenocarcinomas protected from the cisplatin-induced increase in BUN levels; however, suramin did not block the ability of cisplatin to initially induce DNA damage and apoptosis in lung adenocarcinomas. Taken together, we suggest that suramin may be viable for development as a renoprotective agent as our data suggest that suramin does not inhibit the initial ability of cisplatin to induce DNA damage and apoptosis in lung tumors. As such, suramin may be efficacious for widening the therapeutic window of cisplatin for the treatment of lung and other cancers.
Suramin is a pan P2 receptor antagonist and an inhibitor of growth factor receptors (17). Multiple P2 receptors are expressed on both proximal tubule cells (3, 39) and leukocytes (7) involved in the pathogenesis of cisplatin-induced AKI. P2 receptors play a crucial role in the mounting of an inflammatory response through the recognition of danger signals, which are released by dying cells or damaged tissue (7). In addition, P2 receptors are known to play a regulatory role in the release of proinflammatory cytokines and chemokines such as MCP-1 and CXCL1 (4, 16, 36). Suramin has been shown to attenuate the inflammatory response associated with pathogenesis of multiple models of AKI and chronic kidney disease (18, 20, 23, 45). In our study, we found that expression of both MCP-1 and CXCL1 were decreased after cisplatin treatment in mice that had been pretreated with suramin, which is important as CXCL1 receptor-deficient mice are protected from cisplatin-induced AKI (1).
Our data also indicate that suramin pretreatment decreases the renal expression of other proinflammatory chemokines and cytokines, namely, TNF-α, IL-1β, and IL-6. Proinflammatory cytokines such as IL-6, IL-1β, and IL-18 have all been shown to be involved in cisplatin-induced AKI; however, their exact role in cisplatin-induced AKI remains unknown. Faubel et al. (13) showed that mice administered cisplatin have increased IL-6, IL-1β, and IL-18 levels, but inhibition of these proinflammatory cytokines did not attenuate cisplatin-induced AKI; however, it is known that decreased levels of IL-6, IL-1β, and IL-18 are associated with less severe injury resulting from cisplatin administration (13).
The release of proinflammatory cytokines such as TNF-α, IL-1β, and IL-6 have been implicated in cisplatin-induced AKI and are known to be released from mast cells upon degranulation (27, 37). P2 receptors are known to be expressed on multiple inflammatory cell types, such as mast cells, T cells, macrophages, and neutrophils (7), all of which are known to infiltrate the kidney during cisplatin-induced AKI (26, 27). Mast cells are known to release proinflammatory cytokines upon degranulation, including TNF-α, IL-1β, and IL-6 (15, 37). After cisplatin treatment, mast cell degranulation within the kidney induces TNF-α production, and inhibition of mast cell degranulation protects from cisplatin-induced AKI (37). Importantly, suramin is known to inhibit mast cell degranulation, thus decreasing TNF-α (15). Interestingly, Ramesh and Reeves (29, 30) found that inhibition of TNF-α synthesis or a neutralizing TNF-α antibody attenuated cisplatin-induced AKI in mice. Likewise, mice deficient in TNF-α as well as TNF-α receptor (TNFR)1-deficient mice are protected from cisplatin-induced kidney injury (29, 30). Thus, a potential mechanism by which suramin treatment provides protection from cisplatin-induced AKI could be through the inhibition of mast cell degranulation and TNF-α release, which would decrease the production of additional TNF-α by proximal tubule cells and further dampen the proinflammatory response.
After cisplatin treatment, the binding of TNF-α to TNFR is known to activate many cell stress and cell death pathways during cisplatin-induced AKI (26, 27, 29, 30). TNF-α signaling through TNFR has been shown to induce phosphorylation of JNK, which participates in both ER stress and apoptosis pathways (5, 11, 40). Cisplatin is known to induce ER stress during kidney injury (28). IRE-1α is a serine-threonine kinase that plays an important role in the unfolded protein response during ER stress and signals through JNK for cells to undergo ER stress-induced apoptosis (8, 40). Our results indicate that cisplatin treatment increases IRE-1α protein levels and JNK activation, which are reduced in mice either pre- or posttreated with suramin. More importantly, this is not the only pathway of ER stress that induces apoptosis. CHOP is a transcription factor associated with the activation of ER stress-induced apoptosis independently of IRE-1α and JNK activation (8). We found that CHOP protein levels were decreased with suramin treatment when suramin was administered either before or after cisplatin. These results suggest that suramin reduces from ER stress-induced apoptosis as a result of cisplatin treatment; however, there are many other pathways of apoptosis that have been implicated in cisplatin-induced AKI.
Cisplatin administration has been shown to induce cell death through both apoptosis and necrosis (26–28). During cisplatin-induced AKI, many pathways of apoptosis are activated, such as the death receptor-mediated pathway of apoptosis (26–28). In this pathway of apoptosis, TNF-α or other death receptor ligands can activate death receptors on the proximal tubule cell surface leading to caspase-8 activation followed by the initiation of downstream effector caspases such as caspase-3, leading to apoptosis (5). Suramin has been shown to inhibit TNF-α from binding with the death receptor (12). Interestingly, our results show that pretreatment with suramin before cisplatin significantly decreased both caspase-8 and caspase-3 cleavage, suggesting that suramin is decreasing cisplatin-induced death receptor-mediated apoptosis in the kidney. Another potential mechanism by which suramin may be protective of cisplatin-induced AKI is via inhibition of receptor-interacting protiein (RIP) kinase-mediated cell death. It is known that the interaction of TNF-α with TNFR can lead to the activation of RIP kinases for the activation of necroptosis (41). Necroptosis a programmed form cell death that occurs independently of caspase activation (41). Thus, based on the current literature, it could be hypothesized that suramin would reduce necroptosis mediated by RIP kinases, and this will be examined in future studies.
Cisplatin is known to reach peak renal concentrations 1–6 h after cisplatin administration (9, 38), and ∼80% of cisplatin is excreted from the body within the first 24 h after cisplatin administration (21). Posttreatment of suramin restored kidney function after cisplatin injury, as evidenced by decreased BUN levels. These data also showed that posttreatment of suramin after cisplatin administration decreased markers of ER stress and apoptosis, as evidenced by decreased JNK activation, IRE-1α protein expression, CHOP protein expression, cleaved caspase-8 and cleaved caspase-3 levels, and TUNEL-positive foci in the renal cortex. Thus, these data suggest that suramin protects from cisplatin-induced AKI through mechanisms that do not interfere with the uptake and/or metabolism of cisplatin in the kidney.
Cisplatin is still routinely used to treat a number of cancers, and nephrotoxicity is the primary reason the use of cisplatin is limited. Suramin is currently being pursued in drug trials for the treatment of many cancer types as a combination therapy with a number of chemotherapeutics, including cisplatin (6, 31, 42, 44). Our data indicate that suramin did not inhibit cisplatin-induced cell death of lung cancer cells grown in culture. In addition, suramin did not decrease the initial ability of cisplatin to induce DNA damage or apoptosis in a mouse model of lung cancer. Future preclinical studies using a variety of mouse models of cancer will be necessary to truly determine the translational potential of suramin as a renoprotective agent in the context of cancer.
GRANTS
This work was supported by National Institutes of Health (NIH) Grant R01-DK-093462 (to L. J. Siskind). This work was also supported by NIH Grants R01-GM-084147 (to R. G. Schnellmann), Grant UL1-RR-029882 (to R. G. Schnellmann), and Small Business Innovation Research/Small Business Technology Transfer Grant ES-023767 (to R. G. Schnellmann), Biomedical Laboratory Research and Development Program of the Department of Veterans Affairs Grant 1BX000851 (to R. G. Schnellmann), and the South Carolina Clinical and Translational Research Institute, with an academic home at the Medical University of South Carolina (to R. G. Schnellmann).
DISCLOSURES
The Medical University of South Carolina has a patent on the use of suramin for cell repair and regeneration, and this patent has been licensed to Schnellgen, a company owned by R. G. Schnellmann. The University of Louisville has filed a provisional patent (United States Application No. 62/234,427, filing date: 09/29/2015, on behalf of L. J. Siskind, T. V. Dupre, L. J. Beverly, R. G. Schnellmann, and M. A. Doll) for the use of suramin as a combinatorial treatment with standard of care chemotherapeutics for the prevention of nephrotoxicity.
AUTHOR CONTRIBUTIONS
Author contributions: T.V.D., R.G.S., L.J.B., and L.J.S. conception and design of research; T.V.D., C.N.S., A.K., M.T.S., K.S., and L.J.S. performed experiments; T.V.D., M.A.D., P.P.S., D. Saforo, D. Siow, L.C., G.E.A., A.B.J., J.M., and L.J.S. analyzed data; T.V.D., P.P.S., L.J.B., and L.J.S. interpreted results of experiments; T.V.D., M.A.D., J.M., and L.J.S. prepared figures; T.V.D., M.A.D., and L.J.S. drafted manuscript; T.V.D., M.A.D., J.M., R.G.S., L.J.B., and L.J.S. edited and revised manuscript; T.V.D., M.A.D., P.P.S., C.N.S., A.K., M.T.S., K.S., D. Saforo, D. Siow, L.C., G.E.A., A.B.J., J.M., R.G.S., L.J.B., and L.J.S. approved final version of manuscript.
REFERENCES
- 1.Akcay A, Nguyen Q, He Z, Turkmen K, Won Lee D, Hernando AA, Altmann C, Toker A, Pacic A, Ljubanovic DG, Jani A, Faubel S, Edelstein CL. IL-33 exacerbates acute kidney injury. J Am Soc Nephrol 22: 2057–2067, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Arteel GE, Iimuro Y, Yin M, Raleigh JA, Thurman RG. Chronic enteral ethanol treatment causes hypoxia in rat liver tissue in vivo. Hepatology 25: 920–926, 1997. [DOI] [PubMed] [Google Scholar]
- 3.Bailey MA, Imbert-Teboul M, Turner C, Srai SK, Burnstock G, Unwin RJ. Evidence for basolateral P2Y6 receptors along the rat proximal tubule: functional and molecular characterization. J Am Soc Nephrol 12: 1640–1647, 2001. [DOI] [PubMed] [Google Scholar]
- 4.Braganhol E, Kukulski F, Levesque SA, Fausther M, Lavoie EG, Zanotto-Filho A, Bergamin LS, Pelletier J, Bahrami F, Ben Yebdri F, Fonseca Moreira JC, Battastini AM, Sevigny J. Nucleotide receptors control IL-8/CXCL8 and MCP-1/CCL2 secretions as well as proliferation in human glioma cells. Biochim Biophys Acta 1852: 120–130, 2015. [DOI] [PubMed] [Google Scholar]
- 5.Brenner D, Blaser H, Mak TW. Regulation of tumour necrosis factor signalling: live or let die. Nat Rev Immunol 15: 362–374, 2015. [DOI] [PubMed] [Google Scholar]
- 6.Chahinian AP, Mandeli JP, Gluck H, Naim H, Teirstein AS, Holland JF. Effectiveness of cisplatin, paclitaxel, and suramin against human malignant mesothelioma xenografts in athymic nude mice. J Surg Oncol 67: 104–111, 1998. [DOI] [PubMed] [Google Scholar]
- 7.Chen J, Zhao Y, Liu Y. The role of nucleotides and purinergic signaling in apoptotic cell clearance–implications for chronic inflammatory diseases. Front Immunol 5: 656, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen Y, Brandizzi F. IRE1: ER stress sensor and cell fate executor. Trends Cell Biol 23: 547–555, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Choie DD, del Campo AA, Guarino AM. Subcellular localization of cis-dichlorodiammineplatinum(II) in rat kidney and liver. Toxicol Appl Pharmacol 55: 245–252, 1980. [DOI] [PubMed] [Google Scholar]
- 10.Cohen SM, Lippard SJ. Cisplatin: from DNA damage to cancer chemotherapy. Prog Nucleic Acid Res Mol Biol 67: 93–130, 2001. [DOI] [PubMed] [Google Scholar]
- 11.Dhanasekaran DN, Reddy EP. JNK signaling in apoptosis. Oncogene 27: 6245–6251, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Eichhorst ST, Krueger A, Muerkoster S, Fas SC, Golks A, Gruetzner U, Schubert L, Opelz C, Bilzer M, Gerbes AL, Krammer PH. Suramin inhibits death receptor-induced apoptosis in vitro and fulminant apoptotic liver damage in mice. Nat Med 10: 602–609, 2004. [DOI] [PubMed] [Google Scholar]
- 13.Faubel S, Lewis EC, Reznikov L, Ljubanovic D, Hoke TS, Somerset H, Oh DJ, Lu L, Klein CL, Dinarello CA, Edelstein CL. Cisplatin-induced acute renal failure is associated with an increase in the cytokines interleukin (IL)-1β, IL-18, IL-6, and neutrophil infiltration in the kidney. J Pharmacol Exp Ther 322: 8–15, 2007. [DOI] [PubMed] [Google Scholar]
- 14.Fisher GH, Wellen SL, Klimstra D, Lenczowski JM, Tichelaar JW, Lizak MJ, Whitsett JA, Koretsky A, Varmus HE. Induction and apoptotic regression of lung adenocarcinomas by regulation of a K-Ras transgene in the presence and absence of tumor suppressor genes. Genes Dev 15: 3249–3262, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ganapaty S, Chandrashekhar VM, Narsu ML. Evaluation of anti-allergic activity of gossypin and suramin in mast cell-mediated allergy model. Indian J Biochem Biophys 47: 90–95, 2010. [PubMed] [Google Scholar]
- 16.Grbic DM, Degagne E, Larrivee JF, Bilodeau MS, Vinette V, Arguin G, Stankova J, Gendron FP. P2Y6 receptor contributes to neutrophil recruitment to inflamed intestinal mucosa by increasing CXC chemokine ligand 8 expression in an AP-1-dependent manner in epithelial cells. Inflamm Bowel Dis 18: 1456–1469, 2012. [DOI] [PubMed] [Google Scholar]
- 17.Hoyle CH, Knight GE, Burnstock G. Suramin antagonizes responses to P2-purinoceptor agonists and purinergic nerve stimulation in the guinea-pig urinary bladder and taenia coli. Br J Pharmacol 99: 617–621, 1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Korrapati MC, Howell LA, Shaner BE, Megyesi JK, Siskind LJ, Schnellmann RG. Suramin: a potential therapy for diabetic nephropathy. PLos One 8: e73655, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Korrapati MC, Shaner BE, Neely BA, Alge JL, Arthur JM, Schnellmann RG. Diabetes-induced renal injury in rats is attenuated by suramin. J Pharmacol Exp Ther 343: 34–43, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Korrapati MC, Shaner BE, Schnellmann RG. Recovery from glycerol-induced acute kidney injury is accelerated by suramin. J Pharmacol Exp Ther 341: 126–136, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lange RC, Spencer RP, Harder HC. Synthesis and distribution of a radiolabeled antitumor agent: cis-diamminedichloroplatinum II. J Nucl Med 13: 328–330, 1972. [PubMed] [Google Scholar]
- 22.Lieberthal W, Triaca V, Levine J. Mechanisms of death induced by cisplatin in proximal tubular epithelial cells: apoptosis vs. necrosis. Am J Physiol Renal Fluid Electrolyte Physiol 270: F700–F708, 1996. [DOI] [PubMed] [Google Scholar]
- 23.Liu N, He S, Tolbert E, Gong R, Bayliss G, Zhuang S. Suramin alleviates glomerular injury and inflammation in the remnant kidney. PLos One 7: e36194, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liu N, Tolbert E, Pang M, Ponnusamy M, Yan H, Zhuang S. Suramin inhibits renal fibrosis in chronic kidney disease. J Am Soc Nephrol 22: 1064–1075, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liu N, Tolbert E, Ponnusamy M, Yan H, Zhuang S. Delayed administration of suramin attenuates the progression of renal fibrosis in obstructive nephropathy. J Pharmacol Exp Ther 338: 758–766, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Miller RP, Tadagavadi RK, Ramesh G, Reeves WB. Mechanisms of cisplatin nephrotoxicity. Toxins 2: 2490–2518, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ozkok A, Edelstein CL. Pathophysiology of cisplatin-induced acute kidney injury. Biomed Res Int 2014: 967826, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pabla N, Dong Z. Cisplatin nephrotoxicity: mechanisms and renoprotective strategies. Kidney Int 73: 994–1007, 2008. [DOI] [PubMed] [Google Scholar]
- 29.Ramesh G, Reeves WB. TNF-α mediates chemokine and cytokine expression and renal injury in cisplatin nephrotoxicity. J Clin Invest 110: 835–842, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ramesh G, Reeves WB. TNFR2-mediated apoptosis and necrosis in cisplatin-induced acute renal failure. Am J Physiol Renal Physiol 285: F610–F618, 2003. [DOI] [PubMed] [Google Scholar]
- 31.Safarinejad MR. Combination chemotherapy with docetaxel, estramustine and suramin for hormone refractory prostate cancer. Urol Oncol 23: 93–101, 2005. [DOI] [PubMed] [Google Scholar]
- 32.Shah PP, Beverly LJ. Regulation of VCP/p97 demonstrates the critical balance between cell death and epithelial-mesenchymal transition (EMT) downstream of ER stress. Oncotarget 6: 17725–17737, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Siddik ZH. Cisplatin: mode of cytotoxic action and molecular basis of resistance. Oncogene 22: 7265–7279, 2003. [DOI] [PubMed] [Google Scholar]
- 34.Stallons LJ, Whitaker RM, Schnellmann RG. Suppressed mitochondrial biogenesis in folic acid-induced acute kidney injury and early fibrosis. Toxicol Lett 224: 326–332, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Stathem M, Marimuthu S, O'Neal J, Rathmell JC, Chesney JA, Beverly LJ, Siskind LJ. Glucose availability and glycolytic metabolism dictate glycosphingolipid levels. J Cell Biochem 116: 67–80, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Stokes L, Surprenant A. Purinergic P2Y2 receptors induce increased MCP-1/CCL2 synthesis and release from rat alveolar and peritoneal macrophages. J Immunol 179: 6016–6023, 2007. [DOI] [PubMed] [Google Scholar]
- 37.Summers SA, Chan J, Gan PY, Dewage L, Nozaki Y, Steinmetz OM, Nikolic-Paterson DJ, Kitching AR, Holdsworth SR. Mast cells mediate acute kidney injury through the production of TNF. J Am Soc Nephrol 22: 2226–2236, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Townsend DM, Deng M, Zhang L, Lapus MG, Hanigan MH. Metabolism of cisplatin to a nephrotoxin in proximal tubule cells. J Am Soc Nephrol 14: 1–10, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Unwin RJ, Bailey MA, Burnstock G. Purinergic signaling along the renal tubule: the current state of play. News Physiol Sci 18: 237–241, 2003. [DOI] [PubMed] [Google Scholar]
- 40.Urano F, Wang X, Bertolotti A, Zhang Y, Chung P, Harding HP, Ron D. Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1. Science 287: 664–666, 2000. [DOI] [PubMed] [Google Scholar]
- 41.Vanden Berghe T, Linkermann A, Jouan-Lanhouet S, Walczak H, Vandenabeele P. Regulated necrosis: the expanding network of non-apoptotic cell death pathways. Nat Rev Mol Cell Biol 15: 135–147, 2014. [DOI] [PubMed] [Google Scholar]
- 42.Vogelzang NJ, Karrison T, Stadler WM, Garcia J, Cohn H, Kugler J, Troeger T, Giannone L, Arrieta R, Ratain MJ, Vokes EE. A Phase II trial of suramin monthly × 3 for hormone-refractory prostate carcinoma. Cancer 100: 65–71, 2004. [DOI] [PubMed] [Google Scholar]
- 43.Yao X, Panichpisal K, Kurtzman N, Nugent K. Cisplatin nephrotoxicity: a review. Am J Med Sci 334: 115–124, 2007. [DOI] [PubMed] [Google Scholar]
- 44.Zhang P, He JB, Ou LW, Wang XH. [Effects of suramin in combination with cisplatin on growth and metastasis of lung adenocarcinoma xenografts in mice]. Ai Zheng 25: 409–413, 2006. [PubMed] [Google Scholar]
- 45.Zhuang S, Lu B, Daubert RA, Chavin KD, Wang L, Schnellmann RG. Suramin promotes recovery from renal ischemia/reperfusion injury in mice. Kidney Int 75: 304–311, 2009. [DOI] [PubMed] [Google Scholar]
- 46.Zhuang S, Schnellmann RG. Suramin promotes proliferation and scattering of renal epithelial cells. J Pharmacol Exp Ther 314: 383–390, 2005. [DOI] [PubMed] [Google Scholar]