

Abstract
Purpose
The Programmed Death-1 (PD-1) immune checkpoint pathway may be usurped by tumors, including diffuse large B-cell lymphoma (DLBCL), to evade immune surveillance. The reconstituting immune landscape after autologous hematopoietic stem-cell transplantation (AHSCT) may be particularly favorable for breaking immune tolerance through PD-1 blockade.
Patients and Methods
We conducted an international phase II study of pidilizumab, an anti–PD-1 monoclonal antibody, in patients with DLBCL undergoing AHSCT, with correlative studies of lymphocyte subsets. Patients received three doses of pidilizumab beginning 1 to 3 months after AHSCT.
Results
Sixty-six eligible patients were treated. Toxicity was mild. At 16 months after the first treatment, progression-free survival (PFS) was 0.72 (90% CI, 0.60 to 0.82), meeting the primary end point. Among the 24 high-risk patients who remained positive on positron emission tomography after salvage chemotherapy, the 16-month PFS was 0.70 (90% CI, 0.51 to 0.82). Among the 35 patients with measurable disease after AHSCT, the overall response rate after pidilizumab treatment was 51%. Treatment was associated with increases in circulating lymphocyte subsets including PD-L1E–bearing lymphocytes, suggesting an on-target in vivo effect of pidilizumab.
Conclusion
This is the first demonstration of clinical activity of PD-1 blockade in DLBCL. Given these results, PD-1 blockade after AHSCT using pidilizumab may represent a promising therapeutic strategy in this disease.
INTRODUCTION
PD-1 (Programmed Death-1) is a member of the B7 receptor family. Together with its ligands (PD-L1 and PD-L2), it functions as an important checkpoint in the regulation of immune responses.1 Those ligands are upregulated by the inflammatory environment and inhibit the function of PD-1–bearing lymphocytes. Thus the PD-1 immune checkpoint pathway serves to dampen peripheral lymphocyte activity in the context of inflammatory responses. This pathway seems to be co-opted by many tumors, preventing effective antitumor immunity, and therefore represents a promising therapeutic target, as demonstrated in several solid tumor subtypes.2–5 Pidilizumab (CureTech, Yavne, Israel) is an anti–PD-1 humanized immunoglobulin G1 monoclonal antibody with preclinical antitumor activity in animal models.6–8 In a phase I trial in patients with advanced hematologic malignancies, pidilizumab showed a favorable safety profile and early evidence of clinical activity.9
We conducted an international phase II study of pidilizumab in patients with diffuse large B-cell lymphoma (DLBCL) and primary mediastinal large B-cell lymphoma (PMBCL) after autologous hematopoietic stem-cell transplantation (AHSCT). PD-L1 is expressed on suppressor immune cells in the tumor microenvironment and in at least a subset of DLBCL and PMBCL tumors,10–13 where it may alter the composition and function of tumor-infiltrating lymphocytes,14 and therefore represents a valid therapeutic target.11,12 Moreover, the post-AHSCT setting may be a particularly fertile context for PD-1 blockade. This is a state of low-volume residual disease, during which there is a remodeling of the immune system. Indeed, the majority of the circulating leukocytes in the first few months after AHSCT are natural killer cells, CD45RO+ memory/effector cells, and monocytes, which comprise pidilizumab's target populations and whose presence in DLBCL tumors has been associated with a favorable prognosis.15–17 Therefore, PD-1 blockade early after AHSCT for patients with DLBCL may prevent a tumor-dependent, PD-1 driven exhaustion of antitumor lymphocytes, leading to eradication of residual disease and improvement in progression-free survival (PFS).
PATIENTS AND METHODS
Patients
Patients 18 years and older could be consented for this study if they planned or had undergone AHSCT for DLBCL, PMBCL, or transformed indolent B-cell non-Hodgkin lymphoma. Only patients with chemotherapy-sensitive disease (at least partial remission18 after salvage therapy by computed tomography [CT] scans) were eligible. Confirmatory screening was performed between 30 and 90 days after AHSCT. To enroll onto the study and receive treatment, patients had to have CT scans before first drug administration showing no evidence of progressive disease (PD) from pretransplant assessment, as well as normal hematologic, renal, hepatic, and cardiac function. Patients with type 1 diabetes, immune deficiency, active autoimmune disease, CNS involvement by lymphoma, active infection, other serious illness, concurrent investigational treatment, or performance status more than 1 were excluded, as were pregnant or nursing patients.
Patients were recruited at 30 centers in the United States, Israel, Chile, and India. All patients provided written informed consent. The study was approved by the offices for human research studies at the participating institution and conducted in accordance with the principles of the Declaration of Helsinki. The study was supported by CureTech, and the data were analyzed by three of the authors (P.A., E.A.W., and L.I.G.) and by CureTech.
Treatment and Monitoring
Patients received treatment with pidilizumab administered intravenously at a dose of 1.5 mg/kg every 42 days for three cycles, beginning 30 to 90 days from AHSCT. Premedication consisted of acetaminophen or ibuprofen, as well as diphenhydramine or promethazine. Patients were restaged with CT scans (with or without positron emission tomography [PET] scans, at the discretion of the treating clinicians) at confirmatory screening, then before the second and third cycles, and at 30, 44, and 69 weeks from the first day of treatment. Treatment was stopped if there was evidence of PD based on standard criteria.18 Patients were observed until 16 months from first pidilizumab treatment, which corresponded to approximately 18 months from AHSCT. For patients with measurable disease at post-AHSCT screening, response to pidilizumab treatment was assessed18 according to the restaging schedule described previously, using the post-AHSCT measurements as the pretreatment baseline. Toxicity was graded using National Cancer Institute Common Terminology Criteria of Adverse Events v3.0.
Correlative Studies
Blood samples collected from all treatment sites from patients treated at least once with pidilizumab were analyzed by flow cytometry at two central laboratories of Esoterix LabCorp Services (Austin, TX) using study-specific validated methodologies. Forty-one prospectively specified leukocyte subsets based on cluster of differentiation marker expression were evaluated for absolute (per microliter) and relative numbers, as well as molecules of equivalent soluble fluorochrome (MESF). Validation studies for marker stability and inter- and intra-assay precision were conducted before initiating the tests in this study.
Statistical Considerations
The primary end point of this study was the 16-month progression-free proportion from the time of first pidilizumab administration among all eligible patients who received at least one dose of pidilizumab. Secondary end points included safety and toxicity, PFS, and overall survival (OS); immunogenicity of pidilizumab; and immune subset analyses. OS was defined as the time from first treatment to death, and PFS as the time to death, relapse, or progression. OS and PFS were calculated using the Kaplan-Meier method. On the basis of data available at the time of study design, the 18-month PFS after transplantation for chemosensitive patients was estimated to be approximately 60% to 65%.19,20 This time corresponds to approximately 16 months from the planned start of pidilizumab on this trial. Because the number of patients enrolled was anticipated to range from 64 to 80 patients, an observed 16-month PFS from start of pidilizumab of at least 69% was considered to warrant further study. This design had at least 87% probability of concluding the treatment promising if the true 16-month PFS was 75% and less than 10% probability if the true 16-month PFS was 60% (given the exact binomial distribution applied to all possible sample sizes between 64 and 80 patients). Patients were eligible if they met all eligibility criteria and received at least one dose of pidilizumab.
For exploratory measurements of changes in immune subsets, we compared absolute numbers of prespecified circulating lymphocytes of a given immunophenotype before the first treatment and at 24 hours, 6, 12, and 16 weeks afterwards. We also measured MESF to assess for change in surface expression of selected markers. Pre- and post-treatment values were compared using paired Wilcoxon signed rank testing for individual time points, adjusted for multiple comparisons, as well as repeated measures analysis using a log10 transformation (with SAS proc mixed). All P values are two-tailed, using a threshold for statistical significance of .05 except as noted. The data were analyzed using SAS version 9.2 (SAS Institute, Cary, NC).
RESULTS
Patients
Patients could sign consent before or after AHSCT, but were required to pass confirmatory screening to be enrolled onto the study and treated. Among the 97 patients who gave consent, 25 were screen failures at confirmatory screening (including six with PD, five on concurrent disallowed treatment, four who withdrew consent, two with CNS disease, and two with infection). Therefore, 72 patients received at least one dose of pidilizumab (treated subset) at a median of 2.6 months after AHSCT (range, 1.1 to 4.1 months). Sixty patients (83%) completed all three cycles. Ten patients withdrew from the study before the 16-month follow-up visit for reasons other than death or progression: loss to follow-up (n = 2), investigator decision (n = 3), withdrawal of consent (n = 3), protocol violation (n = 1), and adverse event (AE; n = 1). On final review, six patients were determined ineligible (four because of refractoriness to salvage therapy, one for PD on the first day of treatment, and the other for disallowed concomitant treatments). The baseline characteristics of the 66 eligible patients are shown in Table 1. At the time of post-AHSCT restaging, 47% of patients were in complete remission (CR) by CT. Fifty-five patients had a PET scan after salvage; 31 (47%) were in PET-CR. Fifty-four patients had a post-AHSCT PET scan; 45 (68%) were in PET-CR at that time.
Table 1.
Baseline Patient Characteristics (eligible patients)
| Variable | Total No.* | % |
|---|---|---|
| No. of patients | 66 | |
| Age, years | ||
| Median | 57 | |
| Range | 19-80 | |
| Race | ||
| Asian | 7 | 11 |
| Black | 3 | 5 |
| White | 52 | 79 |
| Hispanic | 4 | 6 |
| Country | ||
| Chile | 1 | 1 |
| India | 3 | 4 |
| Israel | 7 | 11 |
| United States | 55 | 83 |
| Sex | ||
| Male | 43 | 65 |
| Female | 23 | 35 |
| Disease | ||
| De novo DLBCL | 49 | 74 |
| PMBCL | 4 | 6 |
| Transformed indolent B-NHL | 13 | 20 |
| IPI score at diagnosis | ||
| 0-1 | 15 | 23 |
| 2 | 11 | 17 |
| 3 | 7 | 11 |
| 4-5 | 7 | 11 |
| Unknown | 26 | 39 |
| Response to first-line therapy | ||
| Complete remission | 45 | 68 |
| Partial remission | 15 | 23 |
| Stable or progressive disease | 5 | 8 |
| Unknown | 1 | 1 |
| Time from diagnosis to AHSCT, months | ||
| Median | 25 | |
| Range | 8-186 | |
| Characteristics at relapse | ||
| Stage† | ||
| I | 6 | 9 |
| II | 8 | 12 |
| III | 9 | 14 |
| IV | 18 | 27 |
| Bulky‡ | 18 | 27 |
| Extranodal involvement§ | 18 | 27 |
| Marrow involvement‖ | 29 | 44 |
| IPI¶ | ||
| 0-1 | 13 | 20 |
| 2 | 7 | 11 |
| 3 | 8 | 12 |
| 4-5 | 2 | 3 |
| No. of prior treatments | ||
| 1 | 3 | 5 |
| 2 | 47 | 71 |
| 3 | 13 | 20 |
| 4 | 3 | 5 |
| Rituximab use | ||
| With first-line therapy | 56 | 85 |
| With salvage therapy | 54 | 82 |
| With conditioning | 10 | 15 |
| Radiation after transplantation | 5 | 8 |
| Response to salvage therapy by PET | ||
| Negative | 31 | 47 |
| Positive | 24 | 36 |
| PET not done | 11 | 17 |
| Status before pidilizumab treatment# | ||
| By CT imaging | ||
| CR | 31 | 47 |
| Not in CR | 35 | 53 |
| By PET imaging | ||
| Negative | 45 | 68 |
| Positive | 9 | 18 |
| PET not done/missing | 12 | 14 |
Abbreviations: AHSCT, autologous hematopoietic stem-cell transplantation; B-NHL, B-cell non-Hodgkin lymphoma; CR, complete remission; CT, computed tomography; DLBCL, diffuse large B-cell lymphoma; IPI, International Prognostic Index; PET, positron emission tomography; PMBCL, primary mediastinal B-cell lymphoma; PR, partial remission; SD, stable disease.
Percentages may not add to 100 because of rounding. Denominator used was all patients, including those with missing data.
Data missing on 25 patients.
Data missing on nine patients.
Data missing on 10 patients.
Marrow biopsy was not performed on three patients; results were determined at time of diagnosis for primary refractory patients (SD+PD) or at last relapse before transplant for the others.
Data missing on 36 patients.
CT was required per protocol and used for eligibility determination; PET was obtained at the discretion of the treating clinician and not used for eligibility determination.
Safety and Toxicity
Among all 72 treated patients, a total of 613 AEs occurred in 69 (96%) of patients (Table 2), among which 135 were considered related to treatment. The most frequently reported grade 3 to 4 AEs were neutropenia (19% of patients) and thrombocytopenia (8%). All patients with grade 4 neutropenia responded to growth factor treatment and remained asymptomatic. One patient died of disseminated herpes zoster 10 months after the third dose of pidilizumab, which was considered unrelated to study treatment. Twenty-three patients (32%) experienced at least one serious AE, and three patients (4%) experienced a related serious AE. There was no evidence of significant autoimmune toxicity, no infusion reactions, and no treatment-related mortality.
Table 2.
Adverse Events
| Event | Severity Grade |
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| All Grades |
1 |
2 |
3 |
4 |
5 |
|||||||
| No. of Patients | No. of Events | No. of Patients | No. of Events | No. of Patients | No. of Events | No. of Patients | No. of Events | No. of Patients | No. of Events | No. of Patients | No. of Events | |
| Any AE | 69 | 613 | 65 | 392 | 49 | 145 | 30 | 60 | 9 | 14 | 1* | 1 |
| Neutropenia | 19 | 25 | 3 | 3 | 7 | 7 | 9 | 10 | 5 | 5 | — | |
| Fatigue | 18 | 21 | 16 | 19 | 2 | 2 | — | — | — | |||
| Upper respiratory tract infection | 14 | 15 | 9 | 10 | 5 | 5 | — | — | — | |||
| Diarrhea | 12 | 19 | 10 | 14 | 4 | 5 | — | — | — | |||
| Cough | 12 | 14 | 11 | 13 | 1 | 1 | — | — | — | |||
| Thrombocytopenia | 10 | 15 | 5 | 6 | — | 4 | 5 | 2 | 4 | — | ||
| Hyperglycemia | 9 | 12 | 8 | 10 | 2 | 2 | — | — | — | |||
| Leukopenia | 9 | 12 | 6 | 8 | 3 | 3 | 1 | 1 | — | — | ||
| Anemia | 3 | 3 | ||||||||||
| Pyrexia | 2 | 2 | ||||||||||
| Renal failure | 2 | 2 | ||||||||||
| Vomiting | 1 | 2 | ||||||||||
| Lymphopenia | 1 | 1 | ||||||||||
| Cardiac arrest | 1 | 1 | ||||||||||
| Duodenal ulcer | 1 | 1 | ||||||||||
| GI hemorrhage | 1 | 1 | ||||||||||
| General physical health decline | 1 | 1 | ||||||||||
| Pain | 1 | 1 | ||||||||||
| Clostridium difficile colitis | 1 | 1 | ||||||||||
| Herpes zoster | 1 | 1 | 1 | 1 | ||||||||
| Lobar pneumonia | 1 | 1 | ||||||||||
| Urinary tract infection | 1 | 1 | ||||||||||
| Vascular injury | 1 | 1 | ||||||||||
| Accident | 1 | 1 | ||||||||||
| Fall | 1 | 1 | ||||||||||
| Pelvic fracture | 1 | 1 | ||||||||||
| Head injury | 1 | 1 | ||||||||||
| Facial bone fracture | 1 | 1 | ||||||||||
| aPTT prolonged | 1 | 1 | ||||||||||
| Hypophosphatemia | 1 | 1 | ||||||||||
| Bone pain | 1 | 1 | ||||||||||
| Myositis | 1 | 1 | ||||||||||
| Rhabdomyolysis | 1 | 1 | ||||||||||
| Myelodysplastic syndrome | 1† | 1 | ||||||||||
| Glioma | 1 | 1 | ||||||||||
| Intracranial hemorrhage | 1 | 1 | ||||||||||
| Subarachnoid hemorrhage | 1 | 1 | ||||||||||
| Headache | 1 | 1 | ||||||||||
| Tachypnea | 1 | 1 | ||||||||||
| COPD | 1 | 1 | ||||||||||
| ARDS | 1 | 1 | ||||||||||
| Pneumothorax | 1 | 1 | ||||||||||
| Hyperhidrosis | 1 | 1 | ||||||||||
| Cholecystectomy | 1 | 1 | ||||||||||
| DVT | 1 | 1 | ||||||||||
| Hypertension | 1 | 1 | ||||||||||
| Hypotension | 1 | 1 | ||||||||||
NOTE. Data are shown as number of patients with a given AE and number of events. Only AEs representing ≥ 2% of total events are shown for grade 1 and 2 events.
Abbreviations: AE, adverse event; aPTT, activated partial thromboplastin time; ARDS, acute respiratory distress syndrome; COPD, chronic obstructive pulmonary disorder; DVT; deep venous thrombosis.
One patient developed fatal disseminated zoster infection during the follow-up period.
One patient with pre-existing leukopenia and thrombocytopenia developed myelodysplasia 13 months after the last dose of pidilizumab. This was considered unrelated to study drug.
Clinical Outcome
Among the 66 eligible patients, 18 experienced disease progression or died before the 16-month time point. The 16-month PFS from first treatment (the primary end point) was 0.72 (90% CI, 0.60 to 0.82; Fig 1A). The study therefore met its primary end point. Nine patients died during the study period between 2.3 and 15.3 months; the cause of death was lymphoma in eight patients and disseminated herpes zoster in one patient. The 16-month OS for eligible patients was 0.85 (90% CI, 0.74 to 0.92; Fig 1A). Among the 24 patients who remained PET-positive at the conclusion of salvage therapy, 16-month PFS was 0.70 (90% CI, 0.51 to 0.82; Fig 1B). Among the 31 PET-negative patients, 16-month PFS was 0.72 (90% CI, 0.56 to 0.84); among the 11 patients who did not have a postsalvage PET scan, 16-month PFS was 0.72 (90% CI, 0.42 to 0.88). No significant difference was detected in the PFS or OS between patients when stratified by disease status assessed by CT scans after AHSCT, age, time to first relapse, time from diagnosis to AHSCT, or salvage regimen; however, there was limited power for those comparisons. We also performed an intent-to-treat analysis for the 72 treated patients. The PFS at 16 months from first pidilizumab treatment in this cohort was 0.68 (90% CI, 0.59 to 0.77), and OS was 0.84 (90% CI, 0.77 to 0.91).
Fig 1.

Progression-free survival (PFS) and overall survival (OS) after pidilizumab treatment. (A) PFS and OS of all eligible patients. (B) PFS and OS of the 24 eligible patients who remained positive on positron emission tomography after salvage therapy.
Among the 35 eligible patients with measurable disease at screening post-AHSCT and before the first dose of pidilizumab, 12 (34%) achieved a CR by CT criteria after pidilizumab treatment, and six (17%) achieved a partial remission (PR), for an overall response rate of 51%. In addition, 13 patients (37%) had stable disease, whereas four (11%) had PD. The median time to documented response was 30 weeks (range, 6 to 69 weeks). Among the nine patients who had residual disease after AHSCT and a positive PET scan, the overall response rate was 33%, and an additional 44% had stable disease.
Immune Subset Analyses
Figure 2 and Table 3 detail changes in selected lymphocyte subsets and marker expression among eligible patients. Treatment with pidilizumab resulted in a significant increase in the absolute number of PD-L1–bearing activated helper T cells (CD4+ CD25+ PD-L1+), apparent 24 hours after first treatment and sustained until at least 16 weeks (Fig 2A). There also seemed to be some changes in PD-1 ligand-bearing monocytes (CD14+ PD-L1+ and CD14+ PD-L2+ cells). For the latter subsets, the mean percentage increase was larger than the median increase at all time points (Table 3), suggesting that pidilizumab induced large increases in those cells that were early and sustained, but restricted to a subset of patients. MESF analysis excluded the possibility that these changes could be solely explained by upregulation of surface markers. There were also significant increases in the absolute number of circulating CD8+ peripheral (CD62L-CD127+) and central (CD62L+CD127+) memory T cells, as well as in CD4+ central memory T cells (Fig 2B). Finally, we found an increase in the cell surface expression of the interleukin 7α receptor (CD127) on peripheral and central memory CD4+ and CD8+ T cells (Fig 2C).
Fig 2.
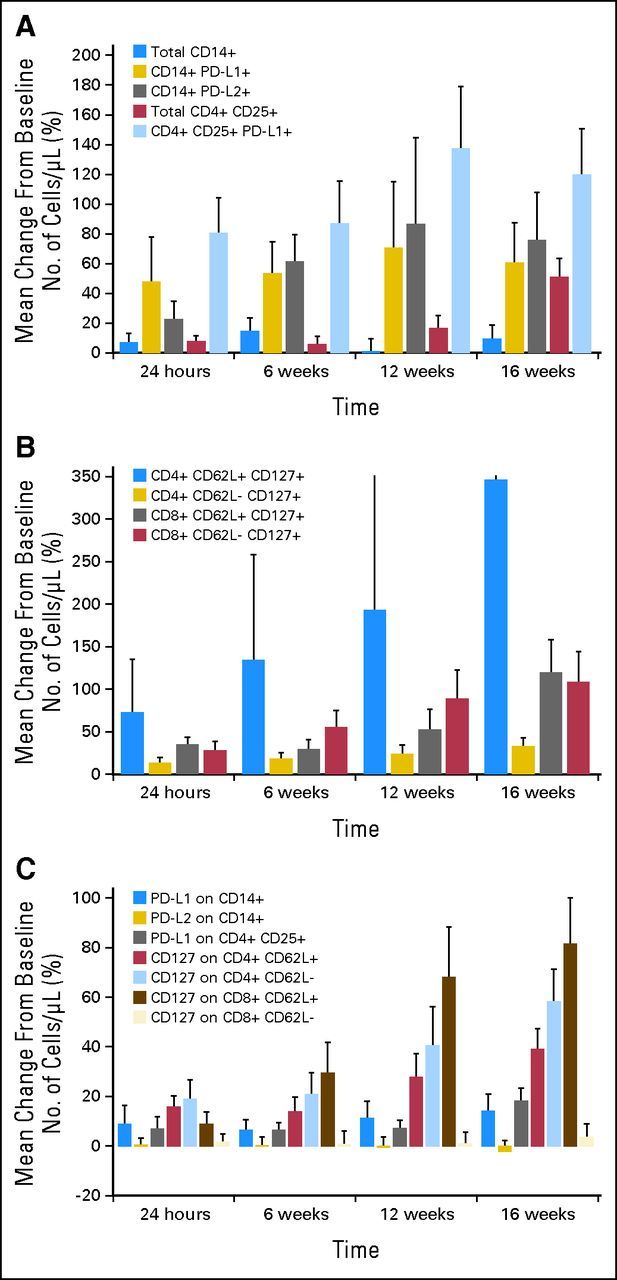
Changes in absolute number of circulating lymphocyte subsets and surface marker expression after pidilizumab administration. (A) Changes in circulating number of PD-L1 (B7-H1) and PD-L2 (B7-DC) –positive monocytes and T cells. (B) Changes in circulating number of peripheral and central memory CD8 T cells. (C) Changes in expression of selected surface markers on monocytes and T cells.
Table 3.
Changes in Selected Lymphocyte Subsets and Marker Expression
| Variable | Adjusted P | 24 Hours |
6 Weeks |
12 Weeks |
16 Weeks |
||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Mean Change | Median Change | P | Mean Change | Median Change | P | Mean Change | Median Change | P | Mean Change | Median Change | P | ||
| Lymphocyte population | |||||||||||||
| CD14+ | .2 | +6.9 | +0.00 | +14.4 | +0.0 | +1.0 | −20.2 | +9.3 | −5.3 | ||||
| CD14+ PD-L1+ | .3 | +47.6 | −9.65 | +53.3 | +10.0 | +70.7 | −31.3 | +60.5 | −6.7 | ||||
| CD14+ PD-L2+ | .3 | +22.6 | −8.82 | .015 | +61.4 | +17.3 | +86.7 | −1.6 | +75.6 | +12.1 | .09 | ||
| CD4+CD25+ | < .0001 | +7.7 | +7.77 | .041 | +5.9 | +0.0 | +16.7 | −2.6 | +50.7 | +30.0 | .0001 | ||
| CD4+CD25+ PD-L1+ | .0004 | +80.4 | +29.17 | .003 | +87.1 | +0.0 | .013 | +137.4 | +16.7 | .006 | +119.9 | +50.0 | < .0001 |
| CD4+CD62L+ CD127+ | .0025 | +72.6 | +9.97 | .02 | +134.6 | -5.5 | +192.8 | −2.9 | +346.7 | +24.2 | .0005 | ||
| CD4+CD62L– CD127+ | .07 | +12.8 | +6.83 | +18.1 | +6.5 | .02 | +23.6 | +6.3 | +32.8 | +19.8 | .002 | ||
| CD8+CD62L+ CD127+ | .003 | +34.6 | +25.93 | < .0001 | +29.4 | +9.7 | .08 | +52.2 | +9.0 | +119.8 | +37.1 | .0002 | |
| CD8+CD62L– CD127+ | .0002 | +27.9 | +11.04 | .03 | +55.3 | +2.3 | .01 | +88.8 | +54.9 | < .0001 | +108.1 | +22.5 | .001 |
| Marker expression | |||||||||||||
| PD-L1 on CD14+ | .6 | +8.8 | +0.0 | +6.5 | +2.75 | +11.4 | +1.0 | +14.1 | +4.6 | ||||
| PD-L2 on CD14+ | .2 | +0.6 | −1.0 | +0.3 | −1.14 | −0.5 | −5.1 | −2.2 | −6.8 | .09 | |||
| PD-L1 on CD4+ CD25+ | .04 | +6.9 | +5.0 | +6.5 | +4.58 | +7.3 | +8.4 | +18.2 | +15.1 | ||||
| CD127 on CD4+CD62L+ | .0001 | +15.9 | +15.9 | .0007 | +14.0 | +3.69 | .05 | +27.9 | +21.4 | .003 | +39.1 | +28.5 | < .0001 |
| CD127 on CD4+CD62L– | .0001 | +18.9 | +9.3 | .004 | +20.8 | +6.39 | .05 | +40.7 | +20.6 | .001 | +58.2 | +33.0 | < .0001 |
| CD127 on CD8+CD62L+ | < .0001 | +8.8 | +6.2 | +29.4 | +7.33 | .036 | +68.0 | +24.4 | .0007 | +81.6 | +37.0 | < .0001 | |
| CD127 on CD8+CD62L– | .7 | +1.8 | −2.9 | +0.7 | +0.04 | +1.0 | −0.9 | +3.6 | +1.5 | ||||
NOTE. Changes are reported compared with baseline values as both the mean and median percentage change in the absolute number of the selected subset or marker. Unadjusted P values are based on Wilcoxon signed rank testing; only P values < .1 are reported. Bolded P values indicate significance after adjustment for multiple comparisons (P < .0125). Adjusted P values test the significance of changes over time using repeated measurement analysis (see Patients and Methods).
DISCUSSION
Monoclonal antibody therapy as a means of targeting immune checkpoints has emerged as a viable and effective antitumor strategy.3–5,9,21,22 Hematologic malignancies may be particularly attractive targets for this type of treatment, as patients with even advanced myeloid or lymphoid tumors can be cured by adoptive immunotherapy delivered in the context of allogeneic HSCT. This raises the possibility that patients' own immune systems can be harnessed to eradicate those diseases, if the mechanisms that lead to immune tolerance of the tumor can be safely disabled. In the present trial, we show that the anti–PD-1 monoclonal antibody pidilizumab can be safely given to patients with DLBCL after AHSCT. The lack of significant autoimmune toxicity in our trial stands in contrast to the clinical experience so far with cytotoxic T-lymphocyte antigen-4 blockade.23 Treatment was associated with an apparent CR rate of 34% and overall response rate of 51% among patients with measurable disease after transplant. This suggests direct antitumor activity, although given this study's design, we cannot rule out the possibility that the residual radiologic abnormalities in some cases reflected treated disease or inflammation rather than the presence of viable lymphoma. Moreover, with a 16-month PFS of 0.72, the study met its prespecified primary end point. These results may compare favorably with those of two recent multicenter randomized clinical trials in this population, although our exclusion of patients who experienced relapse early after AHSCT precludes a direct comparison.24,25 In the rituximab era, as the prognosis of patients with relapsed or refractory disease is worse,24 new therapies are needed to increase the efficacy of salvage and to increase the success rate of AHSCT in patients without a PET-CR at transplantation26–28 who have a poorer outcome. We recently reported the outcomes of 105 patients with DLBCL who underwent transplantation in the last decade at our own institutions.29 Among this cohort, we examined (on an institutional review board–approved study) the outcome of the 46 patients who were chemosensitive but PET positive after salvage and who would have otherwise met the eligibility criteria for the present study, including no progression or relapse within 2 months of AHSCT. In this group, the 18-month post-AHSCT PFS was 0.52 (90% CI, 0.39 to 0.63). In the present study, the PFS was 0.72 among PET-positive patients treated with pidilizumab. This compares favorably with our historical experience and with other published cohorts.26–28,30–32 Although not all patients on the present study had a postsalvage PET (which was not mandated per protocol), the absence of an apparent difference in outcome between those who did and those who did not argues against a strong selection bias, again with the caveat of the small numbers involved. We emphasize that the study was not powered for a comparison among PET subgroups or with historical controls. Nonetheless, our findings support the hypothesis that PD-1 blockade may be a viable therapeutic strategy in the high-risk subset of patients with residual disease and may overcome the negative prognostic value of a pretransplant positive PET scan. This hypothesis should now be tested in a randomized clinical trial.
The increase in the number of some PD-1 ligand-bearing lymphocyte subsets within 24 hours of drug infusion is consistent with an on-target effect of pidilizumab. Indeed, the interaction of PD-1 with either of its cognate ligands seems to induce cell death and signal suppression in lymphocytes and monocytes.33 The increase in circulating levels of cells expressing PD-1 ligands may therefore reflect the reversal of PD-1's inhibition of cell survival or proliferation, although the rapidity of some of the changes occurring after treatment suggests that blockade of apoptosis is not the main or only mechanism at play and that mobilization of those cells from their reservoirs may be important. Also, our results suggest significant variability in the changes among different patients, implying additional complexity in the immune effects of PD-1 blockade in the setting of a reconstituting immune system. We also found an increase in effector and peripheral memory cell subsets, consistent with in vitro data that pidilizumab enhances the survival of human CD4+CD45RO+ cells (CureTech, unpublished data) within 72 hours of treatment. The increase in the cell surface expression of the interleukin 7α receptor CD127, pivotal for the maturation and survival of memory T cells, suggests that pidilizumab induces molecular events associated with the fate of specific memory T-cell subsets. These analyses are only exploratory, reported for hypothesis-generating purposes, and should be prospectively validated.
Because the expression of PD-L1 on DLBCL cells may be restricted to a subset of tumors,10 it may be that future selection of patients for PD-1 blockade on the basis of ligand expression in the tumor or microenvironment could lead to a greater clinical benefit in the appropriate patient subgroups; this could not be ascertained on this study because we did not have access to tumor material for most patients. Furthermore, the ability to evaluate for on-target effects that predict outcome could provide a method to adapt post-AHSCT therapy in those patients. Although the answers to those questions must await future prospective trials, the present study represents the first efficacy trial of immune checkpoint blockade in hematologic malignancies. Our rapidly expanding scientific knowledge in this area, coupled with the availability of a growing number of monoclonal antibodies targeting those pathways, will doubtlessly lead into broader investigations of this strategy in other settings.
Footnotes
See accompanying article on page 4268
P.A. was supported by an American Society of Hematology (ASH) Scholar Award and by an American Society of Clinical Oncology/Conquer Cancer Foundation Career Development Award. D.A.R. was supported by an ASH Scholar Award.
Authors' disclosures of potential conflicts of interest and author contributions are found at the end of this article.
Clinical trial information: NCT00532259.
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
Although all authors completed the disclosure declaration, the following author(s) and/or an author's immediate family member(s) indicated a financial or other interest that is relevant to the subject matter under consideration in this article. Certain relationships marked with a “U” are those for which no compensation was received; those relationships marked with a “C” were compensated. For a detailed description of the disclosure categories, or for more information about ASCO's conflict of interest policy, please refer to the Author Disclosure Declaration and the Disclosures of Potential Conflicts of Interest section in Information for Contributors.
Employment or Leadership Position: Rinat Rotem-Yehudar, Cure Tech (C) Consultant or Advisory Role: Philippe Armand, Merck (C); Arnon Nagler, CureTech (C); Edie A. Weller, CureTech (C); Yi-Bin Chen, Otsuka (C), Genzyme (C); David A. Rizzieri, Teva (C), Celgene (C), Celgene (C), Novartis (C), Spectrum (C); Leo I. Gordon, CureTech (C) Stock Ownership: None Honoraria: None Research Funding: Arnon Nagler, CureTech; David E. Avigan, CureTech; Yi-Bin Chen, Millenium, Seattle Genetics, Onyx, Otsuka; H. Kent Holland, CureTech; Chitra M. Hosing, CureTech; Hillard M. Lazarus, CureTech Expert Testimony: None Patents: None Other Remuneration: None
AUTHOR CONTRIBUTIONS
Conception and design: Philippe Armand, Arnon Nagler, Edie A. Weller, Steven M. Devine, David E. Avigan, Joseph W. Fay, David A. Rizzieri, Markus Y. Mapara, Stephanie A. Gregory, Reuven Or, Rinat Rotem-Yehudar, Leo I. Gordon
Provision of study materials or patients: Phillipe Armand, Arnon Nagler, Steven M. Devine, Jane N. Winter, Joseph W. Fay, David A. Rizzieri, Chitra M. Hosing, Edward D. Ball, Hillard M. Lazarus, Markus Y. Mapara, Stephanie A. Gregory, David Andorsky, Reuven Or, Edmund K. Waller, Leo I. Gordon
Collection and assembly of data: Philippe Armand, Arnon Nagler, Mark S. Kaminski, H. Kent Holland, Jane N. Winter, James R. Mason, David A. Rizzieri, Joseph P. Uberti, Stephanie A. Gregory, John M. Timmerman, David Andorsky, Edmund K. Waller, Rinat Rotem-Yehudar, Leo I. Gordon
Data analysis and interpretation: Philippe Armand, Edie A. Weller, Rinat Rotem-Yehudar, Leo I. Gordon
Manuscript writing: All authors
Final approval of manuscript: All authors
REFERENCES
- 1.Francisco LM, Sage PT, Sharpe AH. The PD-1 pathway in tolerance and autoimmunity. Immunol Rev. 2010;236:219–242. doi: 10.1111/j.1600-065X.2010.00923.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Topalian SL, Drake CG, Pardoll DM. Targeting the PD-1/B7-H1(PD-L1) pathway to activate anti-tumor immunity. Curr Opin Immunol. 2012;24:207–212. doi: 10.1016/j.coi.2011.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brahmer JR, Drake CG, Wollner I, et al. Phase I study of single-agent anti-programmed death-1 (MDX-1106) in refractory solid tumors: Safety, clinical activity, pharmacodynamics, and immunologic correlates. J Clin Oncol. 2010;28:3167–3175. doi: 10.1200/JCO.2009.26.7609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brahmer JR, Tykodi SS, Chow LQ, et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N Engl J Med. 2012;366:2455–2465. doi: 10.1056/NEJMoa1200694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Topalian SL, Hodi FS, Brahmer JR, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med. 2012;366:2443–2454. doi: 10.1056/NEJMoa1200690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hardy B, Indjiia L, Rodionov G, et al. Treatment with BAT monoclonal antibody decreases tumor burden in a murine model of leukemia/lymphoma. Int J Oncol. 2001;19:897–902. doi: 10.3892/ijo.19.5.897. [DOI] [PubMed] [Google Scholar]
- 7.Hardy B, Morgenstern S, Raiter A, et al. BAT monoclonal antibody immunotherapy of human metastatic colorectal carcinoma in mice. Cancer Lett. 2005;229:217–222. doi: 10.1016/j.canlet.2005.06.046. [DOI] [PubMed] [Google Scholar]
- 8.Hardy B, Yampolski I, Kovjazin R, et al. A monoclonal antibody against a human B lymphoblastoid cell line induces tumor regression in mice. Cancer Res. 1994;54:5793–5796. [PubMed] [Google Scholar]
- 9.Berger R, Rotem-Yehudar R, Slama G, et al. Phase I safety and pharmacokinetic study of CT-011, a humanized antibody interacting with PD-1, in patients with advanced hematologic malignancies. Clin Cancer Res. 2008;14:3044–3051. doi: 10.1158/1078-0432.CCR-07-4079. [DOI] [PubMed] [Google Scholar]
- 10.Andorsky DJ, Yamada RE, Said J, et al. Programmed death ligand 1 is expressed by non-Hodgkin lymphomas and inhibits the activity of tumor-associated T cells. Clin Cancer Res. 2011;17:4232–4244. doi: 10.1158/1078-0432.CCR-10-2660. [DOI] [PubMed] [Google Scholar]
- 11.Li Y, Wang J, Li C, et al. Contribution of PD-L1 to oncogenesis of lymphoma and its RNAi-based targeting therapy. Leuk Lymphoma. 2012;53:2015–2023. doi: 10.3109/10428194.2012.673228. [DOI] [PubMed] [Google Scholar]
- 12.Green MR, Monti S, Rodig SJ, et al. Integrative analysis reveals selective 9p24.1 amplification, increased PD-1 ligand expression, and further induction via JAK2 in nodular sclerosing Hodgkin lymphoma and primary mediastinal large B-cell lymphoma. Blood. 2010;116:3268–3277. doi: 10.1182/blood-2010-05-282780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rosenwald A, Wright G, Leroy K, et al. Molecular diagnosis of primary mediastinal B cell lymphoma identifies a clinically favorable subgroup of diffuse large B cell lymphoma related to Hodgkin lymphoma. J Exp Med. 2003;198:851–862. doi: 10.1084/jem.20031074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Warlick ED, Tomblyn M, Cao Q, et al. Reduced-intensity conditioning followed by related allografts in hematologic malignancies: Long-term outcomes most successful in indolent and aggressive non-Hodgkin lymphomas. Biol Blood Marrow Transplant. 2011;17:1025–1032. doi: 10.1016/j.bbmt.2010.10.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ansell SM, Stenson M, Habermann TM, et al. Cd4+ T-cell immune response to large B-cell non-Hodgkin's lymphoma predicts patient outcome. J Clin Oncol. 2001;19:720–726. doi: 10.1200/JCO.2001.19.3.720. [DOI] [PubMed] [Google Scholar]
- 16.Guillaume T, Rubinstein DB, Symann M. Immune reconstitution and immunotherapy after autologous hematopoietic stem cell transplantation. Blood. 1998;92:1471–1490. [PubMed] [Google Scholar]
- 17.Porrata LF, Litzow MR, Markovic SN. Immune reconstitution after autologous hematopoietic stem cell transplantation. Mayo Clin Proc. 2001;76:407–412. doi: 10.4065/76.4.407. [DOI] [PubMed] [Google Scholar]
- 18.Cheson BD, Pfistner B, Juweid ME, et al. Revised response criteria for malignant lymphoma. J Clin Oncol. 2007;25:579–586. doi: 10.1200/JCO.2006.09.2403. [DOI] [PubMed] [Google Scholar]
- 19.Aksentijevich I, Jones RJ, Ambinder RF, et al. Clinical outcome following autologous and allogeneic blood and marrow transplantation for relapsed diffuse large-cell non-Hodgkin's lymphoma. Biol Blood Marrow Transplant. 2006;12:965–972. doi: 10.1016/j.bbmt.2006.05.018. [DOI] [PubMed] [Google Scholar]
- 20.Philip T, Guglielmi C, Hagenbeek A, et al. Autologous bone marrow transplantation as compared with salvage chemotherapy in relapses of chemotherapy-sensitive non-Hodgkin's lymphoma. N Engl J Med. 1995;333:1540–1545. doi: 10.1056/NEJM199512073332305. [DOI] [PubMed] [Google Scholar]
- 21.Hodi FS, O'Day SJ, McDermott DF, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;363:711–723. doi: 10.1056/NEJMoa1003466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bashey A, Medina B, Corringham S, et al. CTLA4 blockade with ipilimumab to treat relapse of malignancy after allogeneic hematopoietic cell transplantation. Blood. 2009;113:1581–1588. doi: 10.1182/blood-2008-07-168468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Di Giacomo AM, Biagioli M, Maio M. The emerging toxicity profiles of anti-CTLA-4 antibodies across clinical indications. Semin Oncol. 2010;37:499–507. doi: 10.1053/j.seminoncol.2010.09.007. [DOI] [PubMed] [Google Scholar]
- 24.Gisselbrecht C, Glass B, Mounier N, et al. Salvage regimens with autologous transplantation for relapsed large B-cell lymphoma in the rituximab era. J Clin Oncol. 2010;28:4184–4190. doi: 10.1200/JCO.2010.28.1618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vose J, Carter SL, Burns LJ, et al. Randomized phase III trial of 131iodine-tositumomab (Bexxar)/carmustine, etoposide, cytarabine, melphalan (BEAM) vs rituximab/BEAM and autologous stem cell transplantation for relapsed diffuse large B-cell lymphoma (DLBCL): No difference in progression-free (PFS) or overall survival (OS) Blood. 2011;118:661. (abstr 661) [Google Scholar]
- 26.Alousi AM, Saliba RM, Okoroji GJ, et al. Disease staging with positron emission tomography or gallium scanning and use of rituximab predict outcome for patients with diffuse large B-cell lymphoma treated with autologous stem cell transplantation. Br J Haematol. 2008;142:786–792. doi: 10.1111/j.1365-2141.2008.07277.x. [DOI] [PubMed] [Google Scholar]
- 27.Dickinson M, Hoyt R, Roberts AW, et al. Improved survival for relapsed diffuse large B cell lymphoma is predicted by a negative pre-transplant FDG-PET scan following salvage chemotherapy. Br J Haematol. 2010;150:39–45. doi: 10.1111/j.1365-2141.2010.08162.x. [DOI] [PubMed] [Google Scholar]
- 28.Trneny M, Bosly A, Bouabdallah K, et al. Independent predictive value of PET-CT pre transplant in relapsed and refractory patients with CD20 diffuse large B-cell lymphoma (DLBCL) included in the CORAL study. Blood. 2009;114:881. (abstr 881) [Google Scholar]
- 29.Armand P, Welch S, Kim HT, et al. Prognostic factors for patients with diffuse large B cell lymphoma and transformed indolent lymphoma undergoing autologous stem cell transplantation in the positron emission tomography era. Br J Haematol. 2013;160:608–617. doi: 10.1111/bjh.12176. [DOI] [PubMed] [Google Scholar]
- 30.Roland V, Bodet-Milin C, Moreau A, et al. Impact of high-dose chemotherapy followed by auto-SCT for positive interim [18F] FDG-PET diffuse large B-cell lymphoma patients. Bone Marrow Transplant. 2010;46:393–399. doi: 10.1038/bmt.2010.130. [DOI] [PubMed] [Google Scholar]
- 31.Derenzini E, Musuraca G, Fanti S, et al. Pretransplantation positron emission tomography scan is the main predictor of autologous stem cell transplantation outcome in aggressive B-cell non-Hodgkin lymphoma. Cancer. 2008;113:2496–2503. doi: 10.1002/cncr.23861. [DOI] [PubMed] [Google Scholar]
- 32.Schot BW, Zijlstra JM, Sluiter WJ, et al. Early FDG-PET assessment in combination with clinical risk scores determines prognosis in recurring lymphoma. Blood. 2007;109:486–491. doi: 10.1182/blood-2005-11-006957. [DOI] [PubMed] [Google Scholar]
- 33.Keir ME, Butte MJ, Freeman GJ, et al. PD-1 and its ligands in tolerance and immunity. Annu Rev Immunol. 2008;26:677–704. doi: 10.1146/annurev.immunol.26.021607.090331. [DOI] [PMC free article] [PubMed] [Google Scholar]