

Abstract
The importance of wound healing to medicine and biology has long been evident, and consequently, wound healing has been the subject of intense investigation for many years. However, several relatively recent developments have added new impetus to wound repair research: the increasing application of model systems; the growing recognition that single cells have a robust, complex, and medically relevant wound healing response; and the emerging recognition that different modes of wound repair bear an uncanny resemblance to other basic biological processes such as morphogenesis and cytokinesis. In this review, each of these developments is described, and their significance for wound healing research is considered. In addition, overlapping mechanisms of single-cell and multicellular wound healing are highlighted, and it is argued that they are more similar than is often recognized. Based on this and other information, a simple model to explain the evolutionary relationships of cytokinesis, single-cell wound repair, multicellular wound repair, and developmental morphogenesis is proposed. Finally, a series of important, but as yet unanswered, questions is posed.
Keywords: Rho GTPases, actin, myosin, muscular dystrophy, injury, damage, regeneration, morphogenesis, cell migration
INTRODUCTION
Understanding wounds and wound healing is arguably one of the most important challenges faced by biomedical researchers. Although the significance of wounding is inherently obvious, its frequency may not be: Wounds result not only from accidental trauma or surgery but also from a variety of pathological conditions ranging from cancer to infection (Crosby & Waters 2010, Gurtner et al. 2008, Singer & Clark 1999, Velnar et al. 2009). Wounds are also a consequence of biological activities that are otherwise considered normal, such as muscle contraction (McNeil & Khakee 1992). Furthermore, a remarkably large number of mutually negative interactions occur between wounds, wound healing, and other pathologies. Diabetes, for example, impairs healing (Velnar et al. 2009). Conversely, wounds are a primary source of infection, and wounding can promote tumor initiation (Schuh et al. 1990) and metastasis (Walter et al. 2011). In addition, excessive or abnormal wound repair results in inflammation and fibrosis, which initiate and/or exacerbate a whole host of diseases (Furie & Furie 2008).
As important as wound healing is to medicine, it is no less so to biology. Indeed, although wound healing is not considered one of the basic attributes of living systems, it certainly should be. Efficient self-repair is evident in all fundamental levels of biological organization including cells, cell layers, tissues, organs, and organisms. Moreover, self-repair is evident in every organism studied: Bacteria heal (Hambleton 1971), yeast heal (Levin 2005), protists heal (Szubinska 1971), plants heal (Volkmann & Baluska 1999), and of course animals heal. Thus, as a criterion for life, self-repair has the virtue of being appropriately inclusive and, in contrast to some of the other standard criteria such as movement or complexity, is also appropriately exclusive.
If the medical and biological importance of wound healing compels our interest, so should the process itself. By any standards, it is utterly fascinating. The crudest of all possible stimuli—damage—unleashes a cryptic pattern formation program. The program unfolds in a manner that is scaled to match the size of the wound. It triggers local changes and, if needed, long-range changes. Step by step, the program deals with the challenge posed by the wound: The hole is plugged, unwanted material is destroyed or expelled, the material lost due to wounding is regenerated and repositioned, and finally much or all of the evidence of the wound and the wound response is removed.
Wound repair research has a history going back at least two millennia (Sipos et al. 2004), but three recent developments have spurred increased interest in the field. The first is the rapidly growing application of model systems for understanding of wound repair. Although decades of wound healing studies using a variety of mammalian models have provided much of our basic repair knowledge, it is increasingly common to utilize models that provide advantages for imaging, genetic or molecular genetic analyses, molecular and cellular manipulations, or combinations of all of these strengths.
With the development of RNA interference, expression profiling, and other techniques, cultured human cells can have many of the above strengths, but they lack one feature essential for wound healing studies: context. The typical epidermal wound, for example, involves multiple cell types of several distinct lineages, each responding to and generating different signals at different times (Shaw & Martin 2009, Singer and Clark, 1999). It is impossible even to approximate this complexity in vitro.
This is not to say that the most complex model system should always be chosen, as one of the most successful strategies in areas such as cell cycle regulation, membrane trafficking, and development has been to use simple model organisms to help identify the most conserved essential players and processes and then build from there. In addition, the qualitative differences apparent in model systems can be particularly useful. Again, this is particularly true for wound healing, as simpler model organisms are typically better healers than complex organisms even though the same general mechanisms and molecules are often used in both. Thus, to understand how to make healing more rapid or more complete in humans, there is good reason to study healing in invertebrates, fish, and amphibians.
The second major development in wound healing research is the increasing appreciation that single cells have a wound response (McNeil & Steinhardt 2003). That cells repair even serious plasma membrane wounds has been apparent for at least a century (e.g., Kite 1913). However, systematic analysis of single-cell wound repair did not occupy much research effort until relatively recently. This is due largely to the efforts of McNeil and collaborators (McNeil & Ito 1989, 1990; McNeil & Khakee 1992; McNeil et al. 1989), who showed that plasma membrane damage and repair are common features of many different mammalian cell types in vivo and demonstrated that healing of plasma membrane wounds is an active cellular process rather than a passive reorganization of membrane lipids as was commonly assumed (McNeil et al. 2003). Further excitement was generated by studies showing that basic mechanisms of cellular repair are conserved and similar to those involved in regulated exocytosis (Steinhardt et al. 1994). In parallel, it has become evident that cellular damage provides a powerful experimental model for understanding how rapid changes in the cortical cytoskeleton are controlled (Bement & Capco 1991, Chowdhury et al. 1992, Levin 2005). And, with the recent discovery that deficient cell repair underlies some forms of muscular dystrophy (Bansal et al. 2003) and the growing appreciation that understanding single-cell damage and healing is essential for understanding diseases of the heart (Wang et al. 2010), lung (Oeckler & Hubmayr 2008), and nervous system (Fishman & Bittner 2003), single-cell wound repair has become, if not quite mainstream, at least respectable.
The third major development is the emerging recognition of what might be called the suspicious similarity of wound healing to other basic biological processes. For example, multicellular wound healing of embryos (Martin & Lewis 1992) and small wounds in epithelia (Bement et al. 1993) by a multicellular “purse string” composed of actin filaments (F-actin) and myosin-2 bears a remarkable resemblance to Drosophila dorsal closure (Wood et al. 2002; Young et al. 1991, 1993). Likewise, single-cell wound healing entails formation and closure of a plasma membrane–associated F-actin and myosin-2 purse string (Bement et al. 1999, Mandato & Bement 2001) that bears a remarkable resemblance to the plasma membrane–associated purse string that drives cytokinesis in a variety of organisms (Pollard 2010). In addition, the single-cell wound purse string both resembles and transitions seamlessly into a multicellular wound purse string during development (Clark et al. 2009). Collectively, these and other observations suggest that these various processes are evolutionarily linked.
A comprehensive review of wound healing, even if limited to studies from model systems, is well beyond the scope of a single review. Instead, we focus on the following objectives: first, to consider basic, conserved features of both multicellular wound healing and single-cell wound healing; second, to discuss recent advances made in multicellular and single-cell wound healing research with an emphasis on contributions from model systems where appropriate; third, to explore the suspicious similarities referred to above; and finally, to highlight some of the more pressing unanswered questions in the field of wound healing. As a more general goal, we aim to convince the reader that there is enormous value in obtaining detailed information from wound healing in relatively simple model systems and from considering the connections and relationships between different kinds of wound healing rather than treating each example as if it is completely unique.
MULTICELLULAR WOUND REPAIR
Adult Wound Healing
Most of our information concerning multicellular wound repair comes from studies of epidermal (or cutaneous) healing. However, the sequence of events elicited by wounding, although varied in some details, is nevertheless conserved among different tissues (Gurtner et al. 2008). This makes sense because most multicellular wounds challenge the wounded organism with the same fundamental tasks: (a) plugging of the hole created by the wound, (b) destruction of any potentially damaging material that has entered the tissue through the wound, (c) replenishment and restoration of the cells destroyed by the wound, and (d ) removal of any structures developed to facilitate the first three steps (e.g., the clot).
Traditionally, epidermal wound healing is broken down into four steps that correspond roughly to the tasks above: hemostasis, inflammation, proliferation and migration, and resolution and remodeling (Figure 1; Janis et al. 2010, Shaw & Martin 2009, Singer & Clark 1999). Although it is conceptually useful to recognize such discrete steps, healing is of course an interconnected continuum such that the early processes overlap with and promote later processes, and the later processes positively or negatively influence the processes begun earlier. Considerable variation also exists in the time taken by each of these steps as well as the healing process as a whole, which may require days, months, or even years to complete depending on the size and type of wound as well as the presence or absence of confounding factors such as other diseases.
Figure 1.

Stages of multicellular adult wound healing. (a) An intact epidermis with an underlying capillary in the prewound state. (b) Hemostasis: plugging the hole. Immediately after wounding, vasoconstriction commences while platelets emerge from the damaged vasculature and aggregate to dam the wound. The platelets also release cytokines and other inflammatory agents to recruit inflammatory cells to the wound. (c) Inflammation: killing your enemies. Neutrophils provide the initial inflammatory response, arriving within an hour after wounding to clear the site of pathogens while also releasing additional inflammatory cytokines. Within 24–48 h after wounding, monocytes arrive, mature into macrophages, and continue clearing out wound debris and expired neutrophils by phagocytosis. The macrophages also secrete cytokines both to stimulate angiogenesis and to recruit fibroblasts to the wound. (d ) Proliferation and migration: bringing back your friends. Fibroblasts from undamaged tissue migrate to the wound, lay down a collagen-rich matrix, and through the upregulation of muscle-specific genes, take on a contractile phenotype. Epidermal cells at the wound edge proliferate and migrate into the wound; they are assisted by the new collagen matrix and the pulling forces produced by the contractile myofibroblasts in forming a new epithelial layer over the wound. (e) Resolution and remodeling: removal of wound-specific structures. The remaining fibroblasts and inflammatory cells secrete matrix metalloproteinases to assist in the realignment and cross-linking of the collagen matrix.
Hemostasis: plugging the hole
Hemostasis is the process whereby blood loss and pathogen incursion are limited physically and is based on constriction of the damaged vessels and clot formation. Vessel constriction results from contraction of the vascular smooth muscle cells, whereas platelets that aggregate at the wound site control clot formation. The platelets initially form a loose plug and release a variety of factors that initiate formation of a network of fibrin fibrils as well as promote the recruitment of other cells in the subsequent inflammatory phase. The fibrin fibrils, which are generated via thrombin-mediated cleavage of fibrinogen, are highly insoluble and convert the platelet plug into a clot both by stabilizing the plug and by ensnaring other cells. Although the clot essentially plugs the hole, it also acts as a scaffold to which growth factors bind, thereby promoting the next stage of wound healing—inflammation.
Inflammation: killing your enemies
Inflammation refers to the accumulation and activation of leukocytes at the wound. Neutrophils, which are among the first leukocytes recruited during inflammation, migrate from the damaged vasculature. Once at the wound site, neutrophils destroy bacteria and other pathogenic organisms by (among other activities) production of reactive oxygen species (ROS). Monocytes typically are recruited to wounds later than neutrophils; once there, they differentiate to become macrophages, which remove debris as well as moribund neutrophils by phagocytosis.
Inflammation is also typically accompanied by angiogenesis, as vessels bordering the wound generate branches that subsequently invade the wound area. The newly formed vasculature not only provides oxygen and nutrients to the wound but also serves as a conduit that other cells involved in healing can use to migrate to the wound.
Proliferation and migration: bringing back your friends
The proliferation stage of healing entails a local increase in epidermal cell division to replace those cells destroyed by wounding. It involves migration of epidermal cells over the wound site (reepithelialization), recruitment of other cell types such as fibroblasts to the wound, and generation of the so-called granulation tissue. Epidermal cells near the wound edge undergo a transient dedifferentiation; assume a more flattened, elongate phenotype; and migrate into the wound area. During the migration, these cells maintain contacts with their neighbors not immediately bordering the wound; thus, the epidermis moves inward over the wound area as a coherent sheet.
Fibroblasts are recruited to the wound from border regions, whereupon they contribute to closure of the hole by a mechanism that is distinct from but complementary to epidermal cell migration. Specifically, the fibroblasts can differentiate into myofibroblasts as a result of exposure to growth factors in the wound. The fibroblasts and myofibroblasts secrete a large amount of collagen and other extracellular matrix (ECM) proteins into the wound area. This results in formation of a relatively disorganized ECM that, together with the fibroblasts, myofibroblasts, and new vasculature, forms the basis of the so-called granulation tissue. The myofibroblasts contract the granulation tissue and the cells associated with it, thereby pulling the edges of the wound closer to each other and, simultaneously, aligning the collagen fibers that compose the ECM.
Resolution and remodeling: removal of wound-specific structures
During resolution, the processes initiated during the first three stages culminate, and the structures assembled during those stages are removed or remodeled. Epidermal cell migration ceases as the edges of the migrating sheets come together, and epidermal proliferation decreases. The leukocytes remaining in the wound area either emigrate or undergo programmed cell death, the ECM is remodeled, and the granulation tissue is removed. By the end of resolution, only the scar—the aligned ECM filaments—remains.
Although failure of one or more of the above processes obviously has the potential to be catastrophic as a result of hemorrhaging or infection, if the processes occur in excess, or for longer than needed, a variety of very serious problems also may ensue. Excessive production of thrombin, for example, can result in clot formation inside blood vessels, which can lead to cardiac arrest or stroke (Furie & Furie 2008). Excessive or prolonged inflammation can actually slow the healing process or even exacerbate the original wound, as high levels of ROS production by neutrophils cause collateral damage (Novo & Parola 2008). Thus, the possibility that otherwise normal healing could occur with limited or even no clotting or inflammation is an attractive one. As described next, embryos manage healing in this fashion.
Embryonic Wound Healing
Healing of cutaneous wounds in embryos is distinctly different from that in adults even though the same four tasks must be mastered. Note that the adult-versus-embryo healing dichotomy used here and elsewhere is based more on convenience than precision, in that the embryonic (also referred to as fetal) response is thought to be progressively lost in most tissues while the organism is still an embryo (or fetus); conversely, a mode of healing that closely resembles the embryonic response is observed in some epithelial wounds in adults.
The first major difference between embryonic and adult healing is that the former is far simpler because many of the basic components of the adult response are missing or greatly attenuated. These include the clotting response (Hopkinson-Woolley et al. 1994), which presumably does not occur simply because platelets are derived from megakaryocytes, which do not differentiate until later in development. In addition, the inflammatory response is absent or minimal, as leukocytes, even though present in the embryos, fail to accumulate significantly at wounds until the healing process is almost finished (Adzick et al. 1985, Armstrong & Ferguson 1995, Cowin et al. 1998, Hopkinson-Woolley et al. 1994, McCluskey & Martin 1995). This may occur because fewer recruitment signals are generated, the leukocytes are less sensitive to the signals that are generated, or both (Liechty et al. 1998, Naik-Mathuria et al. 2007, Olutoye et al. 1996). Furthermore, the extent of ECM deposition and granulation tissue formation is greatly reduced in embryonic wounds, possibly because although fibroblasts accumulate in embryonic wounds, they do not differentiate into myofibroblasts (McCluskey & Martin 1995).
The second difference between embryonic and adult healing is that the mode of reepithelialization is quite different during embryonic wound healing. Rather than undergoing partial dedifferentiation; assuming a classically flattened, motile morphology; and then crawling into the wound, epidermal cells at the edge of embryonic wounds assemble a continuous multicellular purse string of F-actin and myosin-2 that encircles the wound (Figure 2; Martin & Lewis 1992, McCluskey & Martin 1995). The purse string is assembled within a few minutes of wounding and then undergoes progressive contraction that drives inward translocation of the leading edge cells as well as their neighbors, which remain firmly anchored to the leading edge cells. Few leading edge cell lamellipodia are seen; rather, the contraction of the purse string powers the epidermal sheet movement.
Figure 2.
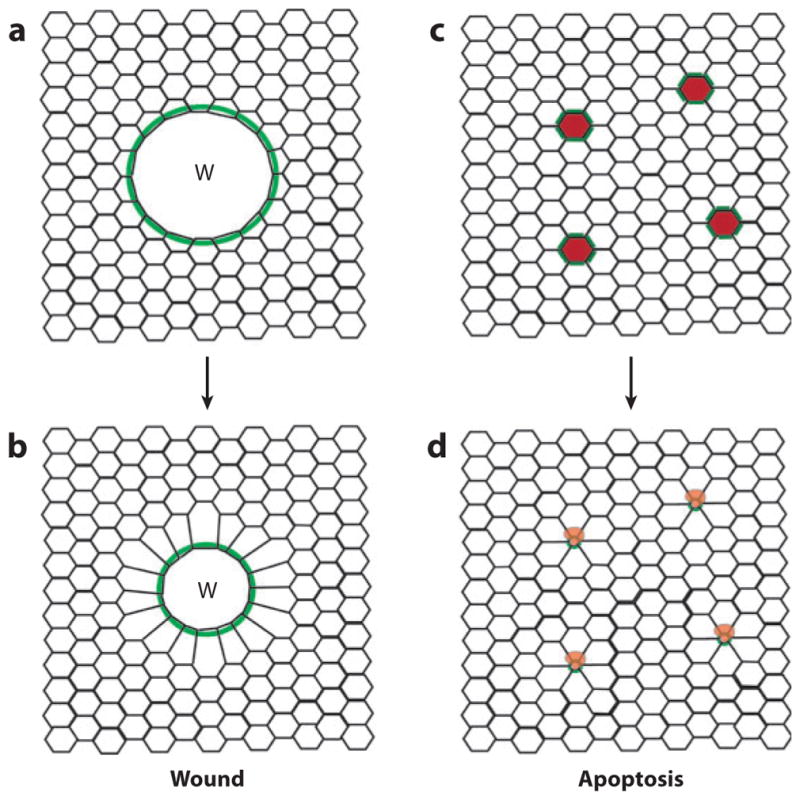
The embryonic multicellular wound response. (a) A wound (W) made in the epithelium triggers formation of a multicellular purse string of F-actin and myosin-2 ( green) that runs from cell to cell in the cells bordering the wound. (b) The purse string contracts over time to drive reepithelialization. (c) Cells in epithelium undergoing programmed cell death (red ) initiate formation of a multicellular purse string of F-actin and myosin-2 ( green) in their neighbors. (d ) These purse strings contract over time, expelling the dying cells from the epithelium.
Purse string wound closure was first described nearly 20 years ago (Bement et al. 1993, Martin & Lewis 1992) and has since been observed in the embryos of mice (McCluskey & Martin 1995), frogs (Bement et al. 1999), and flies (Kiehart et al. 2000, Wood et al. 2002); chick inner ear (Bird et al. 2010); and several epithelial cell lines (Florian et al. 2002, Lotz et al. 2000, Russo et al. 2005, Tamada et al. 2007). However, it is not limited to embryos or cultured cells, as it also has been observed in adult mouse cornea (Danjo & Gipson 1998) and human intestine (Bullen et al. 2006, Russo et al. 2005). In addition, extrusion of apoptotic cells from epithelia is accompanied by formation of an actomyosin purse string around the dying cells in Drosophila embryos (Rosenblatt et al. 2001), cultured mammalian epithelial cells (Rosenblatt et al. 2001, Slattum et al. 2009), and mammalian intestine (Bullen et al. 2006, Marchiando et al. 2011).
Besides providing motive force for reepithelialization, the purse string has other potential roles. It is associated with a local accumulation of adherens and tight junction proteins that may help maintain the polarity of the cells as they migrate into the wound (Bement et al. 1993, Bullen et al. 2006, Danjo & Gipson 1998, Florian et al. 2002, Tamada et al. 2007). Furthermore, in cases in which the wound does not completely destroy the cells within it, or when the wound results from apoptosis, accumulation of junctional proteins combined with purse string contraction may allow the epithelium to maintain nearly complete integrity (Florian et al. 2002, Madara 1990, Marchiando et al. 2011, Rosenblatt et al. 2001).
The third difference between embryonic and adult healing is speed: Embryonic wounds heal much faster than adult wounds. For example, mouse limb bud wounds made at embryonic day (E)11.5 healed completely within 24 h, whereas a wound of half the size took twice as long at E14.5 (McCluskey & Martin 1995). In sheep, reepithelialization of a trunk wound took 24 h for wounds made at day 100 of gestation, 48 h at day 120, and ~7 days in adults (Whitby et al. 1991).
The fourth difference between embryonic and adult healing is that embryonic wounds heal without scarring (Ferguson & O’Kane 2004, Larson et al. 2010, Martin 1997). This is true not only in lower organisms but also in humans, where scarless healing is evident in fetuses during the first two trimesters (Larson et al. 2010). Because scars inherently lack the complete function of unscarred tissue, considerable benefit would be derived from the ability to make adult wounds heal in the embryonic manner.
The speed and scarlessness of embryonic healing have prompted much effort to determine what, exactly, accounts for the differences between the two (Ferguson & O’Kane 2004, Larson et al. 2010, Martin 1997). Two likely, nonexclusive factors are the lack of inflammatory response and differences in ECM isoforms and deposition. There is as yet no consensus, but studies in model systems are providing important insights.
Insights from Model Systems into Adult Multicellular Wound Healing
Our understanding of multicellular wound repair—both adult and embryonic—has benefited greatly from the study of model systems. And, although in some systems the results could be considered confirmatory of existing models, other discoveries have both challenged long-standing assumptions and generated completely new ideas.
Inflammation
Several recent inflammation studies have been based on zebrafish and Drosophila, which have the virtues of being relatively transparent and amenable to a variety of molecular and genetic manipulations. Despite the open nature of the Drosophila circulatory system, healing bears many similarities to vertebrate inflammation including recruitment of blood cells to wound sites via both directed migration and local adhesion, albeit at different stages of development. That is, hemocytes—the leukocyte equivalent in insects—undergo directed migration toward wounds in embryos (Stramer et al. 2005, Wood et al. 2006), whereas in larvae (and, presumably, adults) they accumulate at wounds via adhesion (Babcock et al. 2008). This reflects the fact that prior to the larval stage, hemolymph flow (powered at least in part by peristalsis) does not occur.
An enormous amount of information has been amassed concerning the signals generated and released during the inflammatory response, but surprisingly, the signals that actually direct leukocytes to wound sites have been mysterious. However, a recent study of wounding in zebrafish (Niethammer et al. 2009) obtained compelling evidence that hydrogen peroxide (H2O2) is one such signal. Using a fluorescent protein reporter for H2O2, the authors found that within minutes of wounding, a H2O2 gradient develops around the wound, with peak levels present at the wound site itself. Epithelial cells at or near the wound site mediated H2O2 production, and formation of the gradient coincided with the onset of leukocyte migration toward the wound. Most convincingly, pharmacological- or antisense-mediated suppression of H2O2 production sharply suppressed leukocyte recruitment to wounds. H2O2 was also shown to be required for hemocyte recruitment to wounds in Drosophila (Moreira et al. 2010), which indicates that it is likely a key signal for leukocyte wound recruitment in most if not all other animals.
Directed migration of cells requires polarization of the cytoskeleton of the migrating cells. The Rho GTPases—Rho, Rac and Cdc42—are well-known regulators of cytoskeleton polarity within cells: Active (i.e., GTP-bound) Rho stimulates contractility by activating myosin-2 and promoting unbranched actin filament assembly; active Rac stimulates formation of lamellipodia by promotion of branched networks of actin filaments; and active Cdc42 is associated with cell polarization and formation of filopodia (Heasman & Ridley 2008). Although both in vitro and mouse knockout studies suggested that the Rho GTPases participate in cell locomotion (Heasman & Ridley 2008), the wound healing response of Rho GTPase leukocyte-specific knockout mice has not yet been tested. However, the role of the Rho GTPases has been assessed in the recruitment of hemocytes to Drosophila embryo wounds (Stramer et al. 2005). Consistent with the in vitro analyses, Rac mutant hemocytes have impaired lamellipodial extension and fail to recruit to wounds. Rho mutant hemocytes, although able to polarize toward the wound, have difficulty retracting their trailing edges, as expected if contraction is impaired. No overall difference in recruitment was seen between controls and Cdc42 mutant hemocytes, which is surprising in light of previous in vitro results (Weber et al. 1998). Rather, hemocyte navigation was impaired but was compensated for by an increase in the rate of migration (Stramer et al. 2005).
The phosphoinositide kinase PI(3)K, which generates the second messengers PI(3,4,5)P3 and PI(3,4)P2, has also been implicated in leukocyte motility in vitro (Chen et al. 2007). However, results obtained with different cell types under different culture conditions have produced conflicting results (Andrew & Insall 2007). Consistent with PI(3)K being essential for inflammatory migration, genetic or pharmacological inhibition of PI(3)K largely abrogated recruitment of hemocytes to wounds in embryos (Wood et al. 2006). These findings were corroborated and extended in a study of neutrophil migration in zebrafish embryos, where disrupting PI(3)K function inhibited recruitment of leukocytes to tail fin wounds (Yoo et al. 2010). Moreover, imaging of the PI(3)K products PI(3,4,5)P3 and PI(3,4)P2 with a fluorescent reporter showed that they concentrated at the leading edge of locomoting leukocytes in a manner consistent with the products regulating cell polarity. Furthermore, local photoactivation of Rac—a putative target of PI(3,4,5)P3 and PI(3,4)P2—could be used to steer the migrating leukocytes in any direction desired (Yoo et al. 2010), and local activation of Rac resulted in a corresponding accumulation of PI(3,4,5)P3 and PI(3,4)P2, which suggests positive feedback between Rac and PI(3)K signaling.
In vivo imaging of wound healing has also revealed important features of other suspected players in directed cell locomotion as well as details about the locomotion process itself. For example, depolymerization of microtubules prevents locomotion of macrophages to wound sites in zebrafish, but remarkably, simultaneous inhibition of the Rho target, Rho kinase, can rescue this defect (Redd et al. 2006). WASp, a regulator of actin assembly, is essential for proper directional migration in zebrafish embryos but not for polarization or migration per se (Cvejic et al. 2008). Conversely, in Drosophila embryos, the F-actin regulatory protein Ena controls the rate of migration, rather than steering the cells (Tucker et al. 2011).
Proliferation and migration
What are the upstream signals that control reepithelialization? Ideally, one would like to trace a path from wounding to a signal or ligand, to a receptor, to the targets of the receptor, through the cytoplasm (assuming the receptor is at the plasma membrane) to the nucleus, and so forth. Although this is not yet possible, pieces of such potential signal transduction pathways have been identified. For example, several different ligands that bind to receptor tyrosine kinases (RTKs) are released at cutaneous wounds (Martin 1997), and knockout of the receptors for at least two of these, the epidermal growth factor (EGF) receptor and the keratinocyte growth factor (KGF) receptor, reduce the rate of epidermal migration and impair proliferation and other features of the healing process including inflammation (Repertinger et al. 2004, Werner et al. 1994). A specific role in reepithelialization for the hepatocyte growth factor (HGF) receptor, c-Met, was recently demonstrated via knockout in mouse skin (Chmielowiec et al. 2007). In particular, targeted knockout of c-Met in epidermal cells did not impair cell division but did delay reepithelialization. Remarkably, further analysis showed that the only epidermal cells in the wound were those ~5% that escaped ablation of the c-Met receptor. Chmielowiec et al. (2007) also found that in controls, both HGF and c-Met expression are upregulated in epidermal cells bordering wounds, thus revealing the existence of an autocrine signaling pathway for regulation of reepithelialization. The extent to which HGF–c-Met signaling controls wound repair in other organs remains to be seen, but targeted ablation of c-Met in liver grossly impairs healing (Huh et al. 2004), consistent with a general role for c-Met signaling in wound repair.
Another signaling pathway based on an epidermal RTK was found in Drosophila larval wound repair (adult healing). Pvr [a member of the platelet-derived growth factor (PDGF) receptor family] is concentrated at the lateral domains of intact epidermal cells and is essential for reepithelialization (Wu et al. 2009). The ligand for Pvr, Pvf1, is normally secreted into the hemolymph by the epidermal cells, which suggests that wounding could trigger Pvr-Pvf1 signaling simply by allowing Pvf1 access to Pvr exposed at the wound edge. Stitcher (Stit) is another recently identified RTK involved in Drosophila healing, but in embryos rather than larva (Wang et al. 2009). Stit is a member of the Ret RTK family; its ligand in Drosophila is unknown. Upon wounding, Stit becomes concentrated at the wound edge, and Stit mutants show delayed wound healing and loss of the actin purse string.
RTKs often signal via kinase relays and, indeed, Stit is upstream of the ERK (MAPK) kinase relay during Drosophila embryonic reepithelialization (Wang et al. 2009). It is also required for normal reepithelialization in C. elegans embryos (Pujol et al. 2008) and adult mouse cutaneous wounds (Deng et al. 2006). The Jun kinase relay is known to be required for epidermal healing in Drosophila embryos (Campos et al. 2010), larvae (Galko & Krasnow 2004, Kwon et al. 2010, Lesch et al. 2010), and adults (Ramet et al. 2002); its trigger is unknown, but apparently it is not Pvl (Wu et al. 2009). Although Jun kinase signaling has not yet been linked to wound repair in mammals via targeted knockout, a growing body of evidence from other approaches supports this connection (Deng et al. 2006).
Multiple transcription factors have been implicated in wound healing (Schafer & Werner 2007), including those of the AP-1 family such as c-fos, Jun, and CREB/ATF (Martin & Nobes 1992), but how they are activated and the identity of their targets are unclear. One exciting exception is grainy head (Grh), a transcription factor that controls healing of the integument (the outermost layer of the epidermis) in both Drosophila larvae and mouse embryos following wounding (Mace et al. 2005, Ting et al. 2005). In Drosophila embryos, Grh activation results from Stit-dependent activation of the ERK pathway (Mace et al. 2005, Wang et al. 2009). In both Drosophila and mice, Grh binds to and activates the transcription of genes involved in the repair of the integument (Mace et al. 2005, Ting et al. 2005).
In mice, Grh also targets the RhoGEF19 gene (Caddy et al. 2010). RhoGEF19 is a Rho activator and is required for normal purse string wound repair in mouse embryos and actin organization in later embryos undergoing adult repair (Caddy et al. 2010). Thus, even though the process of epithelial migration is morphologically different in embryonic and adult wounds, the upstream control mechanisms are the same. The same study found that epidermal wound healing is under the control of the planar cell polarity (PCP) pathway, an evolutionarily ancient signaling system that is well known to regulate developmental processes involving epithelia and the actomyosin cytoskeleton (Zallen 2007). In PCP signaling, a conserved cassette of proteins including Wnt, the Wnt receptor Frizzled, and the target of Frizzled, Dishevelled, essentially distinguishes one side of the apical domain of an epithelial cell from the other.
Ultimately, the signals initiated during adult reepithelialization are likely to converge on the Rho GTPases (see above). The Grh-RhoGEF19 results are consistent with this, as are the findings of Lesch et al. (2010), who found a requirement for Rac and Cdc42 but not Rho in Drosophila larval wound repair.
The other major physical contributor to reepithelialization is contraction of the granulation tissue, which is powered by myofibroblast contraction. As myofibroblasts are recruited to wound sites, there has been considerable interest in understanding their source. For cutaneous wounds, fibroblasts come from the dermis at the wound margin or the vascular system (Shaw & Martin 2009). However, in wounds of other organs, the source has been less clear. In kidney and liver, for example, at least some of the myofibroblasts might be derived from the wounded epithelial cells, which are proposed to undergo an epithelial to mesenchymal transition, migrate into the wound area, and then differentiate into myofibroblasts (Duffield 2010). This model was based on both the well-documented ability of epithelial cells to undergo partial dedifferentiation in response to wounding or growth factor application as well as some early lineage tracing studies of wounded liver and kidney. However, two recent studies applying newer approaches that afford greater signal-to-noise fluorescence imaging of marked epithelial cells in mouse models show quite clearly that in liver wounds, hepatocyte markers and myofibroblast markers are not observed in the same cells (Taura et al. 2010). Similarly, analysis of lineage-traced kidney epithelial cells indicated that none of these migrate out of the epithelium into the wound and that the myofibroblasts are derived largely if not exclusively from the mesenchyme (Humphreys et al. 2010). Strikingly, in both studies, the ability of the epithelial cells in question to undergo at least a partial transition in morphology and/or expression pattern that mimicked myofibroblasts was confirmed, demonstrating once again the potential difficulties with drawing too many conclusions strictly on the basis of in vitro data.
Thus, although the diversity and interconnections among different wound-triggered signaling pathways are only beginning to be uncovered, multiple signaling pathways clearly are used—at least three RTKs in mammals, for example, to say nothing of the many other signals, both mechanical and chemical, are likely to be triggered by wounding. Why so many? We think it likely that this abundance imposes a simple pattern formation program at the wound site that is analogous to the pattern formation programs of early development but with the function of telling cells where they are with respect to the wound (Figure 3). That is, assuming that different upstream signals can travel different distances from the wound site, cells at different locations will receive different combinations of signals. Broad gradients of the initial signals could then be converted to sharper boundaries by way of cross talk and signaling to downstream targets. Consistent with this hypothesis, epidermal cells apparently do know where they are with respect to the wound: Cells closest to the wound preferentially migrate, whereas those more distant preferentially proliferate (Singer & Clark 1999). Moreover, directed knockout of different candidate genes for Drosophila larval wound healing followed by a detailed phenotypic analysis of cell behavior showed that non-leading edge cells could migrate toward the wound even when leading edge cells could not, which implies that each group of cells was receiving qualitatively different information (Lesch et al. 2010). In addition, analysis of DNA regulatory elements required for transcription of wound-induced genes in Drosophila revealed that the elements generally require input from at least two transcription factors targeted by different upstream signaling pathways (Pearson et al. 2009). Even more significantly, several distinct wound response elements were apparent, and the genes controlled by these were activated at different distances from the wound (Pearson et al. 2009).
Figure 3.

Pattern formation around multicellular wounds. (Top) Initial signals (orange stars, yellow moons, and green clovers) are imprecisely distributed around the wound (W) as gradients extending different distances from the wound. (Bottom) Over time, the downstream signals ( purple horseshoes, blue diamonds, and red balloons) elicited by the initial signals are expressed in a relatively precise manner, such that cells at different distances from the wound display qualitatively different responses.
Resolution
Careful cell tracking studies in zebrafish have also contributed surprising information to our understanding of resolution. In contrast to the assumption that most neutrophils remain at wounds until cleared by macrophages, neutrophils undergo frequent retrograde chemotaxis (i.e., movement away from wounds) (Mathias et al. 2006). This motility is nonrandom and implies that neutrophils undergo some kind of signaling switch that converts chemoattraction to chemorepulsion. Retrograde chemotaxis was also observed in Drosophila embryos (Stramer et al. 2005), which indicates that this is a broadly conserved feature of resolution. Even more remarkably, the recent use of a photoconvertible fluorescent protein that allowed the tracking of specific fluorescent neutrophils showed that a given neutrophil can undergo repeated cycles of migration to and from the wound (Yoo & Huttenlocher 2011). These findings not only reveal far more complexity in leukocyte behavior than previously envisioned, they also suggest that retrograde chemotaxis may make a significant contribution to the resolution phase of healing.
Insights from Model Systems into Embryonic Multicellular Wound Healing
Detailed analysis of embryonic wound healing in Drosophila embryos revealed that in addition to purse string contraction, wound closure was also dependent on interaction of filopodia and lamellipodia extending from epidermal cells on opposing sides of the wound (Wood et al. 2002). Following interaction, the filopodia and lamellipodia shortened, pulling the epidermal cells closer to each other. Subsequent analysis of wound-induced purse strings in Drosophila using a combination of imaging, genetic manipulation, and biophysical analyses directly demonstrated that the purse strings contain myosin-2 and are under tension (Rodriguez-Diaz et al. 2008), an essential requirement for the purse string contraction model.
The dynamics of signaling events in embryonic healing have been studied in laser-wounded Xenopus embryos, which permit expression of multiple fluorescent probes combined with high-resolution live-cell imaging of wound repair in a vertebrate model. Wounding triggered rapid (within 1 s) calcium elevation not only at the wound site but also at cell-cell junctions in the wounded cell and in neighboring cells (Clark et al. 2009). Rho and Cdc42 were subsequently activated (within 12–18 s) at sites of calcium elevation, which was followed, in turn, by local activation of myosin-2 and actin assembly that resulted in formation of the purse string. The timescale of the signaling events and other factors suggest that extremely rapid, transcription-independent signals such as tension changes (Clark et al. 2009) or paracellular signaling via material released from the damaged cell ( Joshi et al. 2010) can trigger the initial embryonic healing response. This likely holds true for some of the other examples of embryonic healing, as damage is followed by assembly of a purse string within 1–3 min (Florian et al. 2002, Russo et al. 2005).
With the exception of RhoGEF19 transcription (see above), the immediate signal transduction mechanisms that lead to purse string assembly (i.e., the activators of Rho and Cdc42) are not well understood, with one exception—extrusion of apoptotic cells in simple epithelia. Here, a key player is GEF-H1, a Rho GEF that is targeted to the basolateral domain of neighboring cells on the sides where they contact the dying cell (Slattum et al. 2009). Targeting of GEF-H1 to this site is dependent on microtubules, and recent work in zebrafish and cultured cells shows that the signal provided by the dying cell is sphingosine-1-phosphate (S1P), which binds to S1P receptors on the neighboring cells (Gu et al. 2011).
Relationships Between Different Steps in Wound Repair
The use of model systems also permits direct assessment of the relationships between different steps of wound healing. This is critically important because, as noted above, the four steps of adult wound healing are considered to be interconnected and interdependent. Furthermore, some of the processes, such as clot formation and inflammation, lead to serious medical problems if excessive or extended. Thus, knowing exactly what happens to the other steps of healing when they are limited or eliminated would be useful. One of the most important examples of this dependency is the relationship between inflammation and the rest of healing. It seems self-evident that inflammation is a good thing for healing in that it limits the potential for infection. Furthermore, older studies provided evidence that experimental suppression of inflammation by pharmacological and other means results in impaired healing (Martin & Leibovich 2005).
However, several studies of cutaneous healing in different mouse models suggest that not only is inflammation not strictly needed for proper healing, it may actually retard healing and promote scarring. Knockout of the transforming growth factor (TGF) β1 receptor SMAD3 sharply attenuated the inflammatory response and resulted in faster healing with less scarring (Ashcroft et al. 1999). A more direct test was provided by eliminating the macrophage and neutrophil lineages via targeted knockdown of a transcription factor (PU.1) required for their differentiation (Martin et al. 2003). This resulted in the complete loss of these cell types, and yet wounds not only healed but, again, did so with little scarring. In addition, depletion of the gap junction protein connexin43 (Mori et al. 2006) or osteopontin, a protein upregulated during inflammation (Mori et al. 2008), caused a suppressed inflammatory response with an accompanying acceleration of the healing process and, in the case of osteopontin depletion, reduced scarring. These results indicate that rapid and complete defense against bacteria and pathogens was much more important in the past than it is now, particularly when wounds and healing often occur in a sterile environment and with the aid of antibiotics. More importantly, these results prompt the simple notion that deliberate suppression of inflammation might facilitate rapid, scarless wound repair.
SINGLE-CELL WOUND HEALING
Basic Features of the Single-Cell Wound Response
As with multicellular wound healing, although there are important differences between cell types, nevertheless a series of responses clearly are conserved across different cell types and organisms (Bement et al. 2007, McNeil & Steinhardt 2003, Spaeth et al. 2010). The analogy to multicellular wound healing also can be extended from the standpoint of the tasks that the wounded cell faces: It must plug the hole generated by wounding to limit the influx of compounds that would otherwise damage or kill the cell and prevent the loss of the cell contents, it must deal with whatever has entered the cell prior to plugging the hole, it must regenerate and reposition the players destroyed by wounding, and finally, it must remove any special structures generated as a result of the initial steps of the wound response to restore full function of the cell surface.
Membrane patching: plugging the hole
The immediate response to plasma membrane damage is rapid membrane fusion at the site of damage, triggered by oxidation and exposure of the cytoplasm to the high levels of calcium in the extracellular medium (Figure 4). Fusion of membranous organelles at the site of damage is thought to create a patch that is sealed to the undamaged plasma membrane flanking the hole by exocytosis (McNeil et al. 2000) and that prevents the cell from being killed as a result of either continuous calcium influx or the loss of cytoplasm. It has been argued energetically that lysosomes are the major if not the only organelles that contribute to the membrane patch ( Jaiswal et al. 2002, Reddy et al. 2001), but this notion has been disputed (Borgonovo et al. 2002, Shen et al. 2005). On the basis of a variety of considerations, we think it likely that most of the membranous compartments near the wound site would be likely to fuse with the plasma membrane upon exposure to the millimolar level of calcium in the extracellular medium (Bement et al. 2007). Consistent with this expectation, proteomic analysis of plasma membrane wounds has revealed several components of the endoplasmic reticulum (Mellgren 2010) in membrane patches.
Figure 4.

Stages of single-cell wound healing. (a) An undamaged plasma membrane with organized cortical actin cytoskeleton and cytoplasmic vesicles. Dysferlin and MG53 each localize to both the plasma membrane and vesicles owing to either a transmembrane domain (dysferlin) or lipid binding (MG53). (b) Contraction-induced stress causes membrane damage that allows the efflux of cytoplasm and the influx of extracellular milieu. Calcium flows down its concentration gradient and into the cell, activating calcium-dependent proteases that are thought to degrade and depolymerize the cortical actin cytoskeleton. Vesicles rush to the wound site and interact via both calcium-dependent (dysferlin) and oxidation-dependent (MG53) mechanisms. (c) Neighboring vesicles fuse and form a vesicle “patch” that serves to plug the hole and prevent further influx/efflux. (d ) As the patch reaches sufficient size, peripheral regions of the patch interact and fuse to the wound boundary of the plasma membrane to restore a continuous bilayer. (The mechanics of vesicle-vesicle and vesicle-membrane fusion have not yet been elucidated in muscle membrane repair but are thought to be regulated by SNARE proteins.) Rho GTPases (not shown) direct the cortical actomyosin-mediated contraction of the repaired membrane and restoration of the cortical actin cytoskeleton (e) to structurally reinforce the repair.
Exactly how the fusion is controlled is unclear, although it is sensitive to toxins known to target SNARE proteins (Steinhardt et al. 1994) and to dominant negative synaptotagmin constructs (Shen et al. 2005). In plants, the relevant synaptotagmin is syt-1 (Schapire et al. 2008); in animals it has yet to be definitively identified. Furthermore, if multiple organelles contribute to the patch, more than one synaptotagmin is likely involved, and it may vary between distinct cell types. Other potential regulators of membrane fusion during the patch response include the annexins (McNeil et al. 2006), dysferlin (Bansal et al. 2003), and MG53 (Cai et al. 2009), although it is not yet known whether these proteins are required for linking vesicles to each other and the plasma membrane at the wound site, for promoting fusion, or for both (see also below).
The patching and sealing responses are abetted by transient, local disassembly of cortical actin around the wound, which is thought to promote local exocytosis and lateral plasma membrane lipid mobility (Godin et al. 2011, Miyake et al. 2001, Togo et al. 2000). Although the means by which local actin disassembly is controlled is not well characterized, it may involve calcium-dependent actin-severing proteins such as gelsolin and calpain (Godell et al. 1997).
Expulsion and endocytosis: killing your enemies
The major external threat to wounded cells is calcium, the unrestrained influx of which would kill cells rapidly. Calcium does indeed enter damaged cells, but only transiently, unless the patching process is compromised (Terasaki et al. 1997). The basic pumping mechanisms responsible for intracellular free calcium homeostasis might be responsible for coping with calcium that enters the cell before patching is complete, but this has not been tested. An alternative mechanism is that excess calcium is trapped in membranous vesicles that are extruded into the intracellular space from the plasma membrane (Babiychuk et al. 2009). A similar mechanism for the expulsion of agents that permeabilize or damage the plasma membrane, which trigger the single-cell wound response and, similar to calcium, are potentially deadly enemies of the cell, has been observed directly (Morgan et al. 1987). Toxins are also internalized via endocytosis (Idone et al. 2008), which is commonly triggered by wounding (see below).
Membrane and cytoskeleton repair: bringing back your friends
The patching response essentially leaves the cell in the same position as the epithelial layer following the clotting response: The hole is plugged, but the plug is only temporary. That is, the patching membrane differs in lipid and protein composition from the plasma membrane because it is derived from intracellular organelles. Furthermore, both the initial wound and the subsequent depolymerization of F-actin leave the area beneath and flanking the patch denuded of the cortical cytoskeleton that normally provides essential support for the plasma membrane.
The means by which new membrane is restored to the wound site are not well understood but may involve microtubule-dependent membrane trafficking. Wounding initially triggers local, calcium-dependent depolymerization of microtubules, but within approximately a minute of wounding, microtubules polymerize and are transported to the wound (via actomyosin-powered contraction) such that the wound becomes surrounded by a radial array of microtubules (Mandato & Bement 2003, Togo 2006). Golgi complex–derived membranous compartments are transported along the microtubules to the wound site (Togo 2006), and as Golgi complex–derived vesicles are thought normally to be responsible for replenishing plasma membrane lipids and proteins during constitutive exocytosis ( Jaiswal et al. 2009), it is reasonable to surmise that this transport contributes to plasma membrane repair.
Cytoskeletal repair entails local accumulation of F-actin and myosin-2 at the wound site (Bement et al. 1999, Godin et al. 2011, Miyake et al. 2001). When the initial wound is sufficiently large, in both plant (La Claire 1983) and vertebrate cells (Bement & Capco 1991, Bement et al. 1999, Gingell 1970) the F-actin and myosin-2 assemble into a purse string around the wound and close inward. This purse string closure pulls unwounded plasma membrane inward, facilitating membrane repair.
Cytoskeletal restoration has been studied predominantly in frog oocytes, which have an extraordinary healing capacity and permit high-resolution live-cell imaging. F-actin and myosin-2 accumulate as a result of both local assembly and contraction-powered transport (Mandato & Bement 2001). The assembly is directed by Rho and Cdc42, which segregate into concentric zones of activity around the wound (Figure 5; Benink & Bement 2005). This pattern of activity is dependent on Abr, a dual GEF and GAP that locally activates Rho and inactivates Cdc42 near the wound edge (Vaughan et al. 2011). Analogous to multicellular wounds, the end result of this process is that different regions of the plasma membrane and cortex know where they are with respect to the wound; active Rho directs myosin-2 accumulation at the immediate edge of the wound, and Cdc42 directs dynamic actin assembly several micrometers away.
Figure 5.
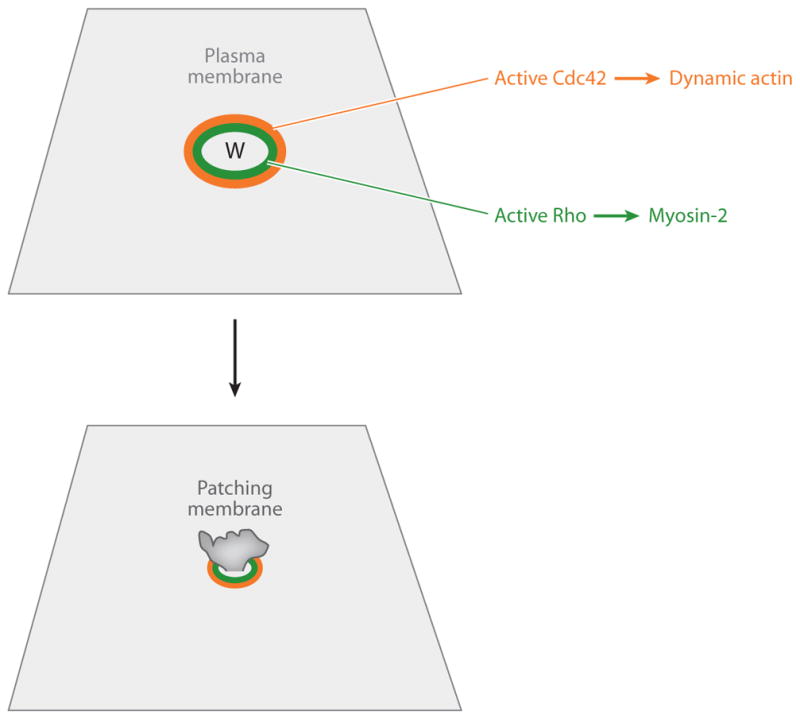
Cytoskeletal signaling during the single-cell wound response. (Top) Wounding elicits local activation of Rho ( green) and Cdc42 (orange) in concentric zones around the wound (W). Active Rho directs accumulation of myosin-2 near the wound edge; active Cdc42 directs accumulation of dynamic actin farther from the wound edge. (Bottom) Over time, the contracting purse string, in association with the Rho and Cdc42 zones, closes over the wound site, which helps to expel the patching membrane.
Resolution and remodeling: removal of wound-specific structures
Ultimately, it is not enough simply to add new membrane and membrane proteins to the site of damage. The patching membrane must be removed, just as the clot has to be removed. This is likely accomplished either via endocytosis or expulsion of plasma membrane extensions. Consistent with a role for endocytosis, wounding triggers not only local exocytosis but also local endocytosis (Bi et al. 1995, Idone et al. 2008). However, it is not clear that this is sufficient to account for retrieval of the patching membrane, particularly as the resultant endosomes may form from vesicles that fuse with the plasma membrane in response to calcium elevation but do not actually participate in patching. Consistent with a role for extrusion/excision, wounding triggers extensive formation of blebs and thin tubular extensions (Figure 6; Geuskens & Tencer 1979, Morgan et al. 1987).
Figure 6.
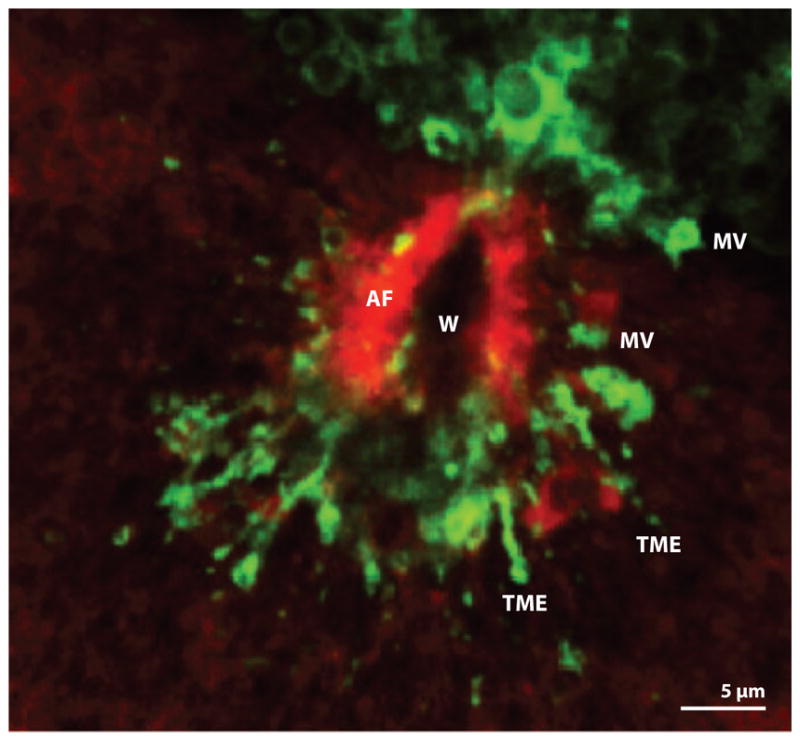
Fluorescence micrograph of late stages of wound healing in a Xenopus oocyte. Actin filaments (AF) have almost completely closed over the original wound (W). A marker for membrane ( green) reveals the existence of extracellular microvesicles (MVs) and tubular membrane extensions (TMEs) apparently derived from the patching membrane.
Generality and Specificity of Single-Cell Wound Repair
It is now generally recognized that single-cell wound repair is a conserved feature of the Metazoa (Bement et al. 2007, McNeil & Steinhardt 2003, Schapire et al. 2009). However, it is almost certainly even more ancient, as amoebae and budding yeast heal single-cell wounds in a manner that resembles metazoan cellular wound repair. In Amoeba proteus, plasma membrane damage is healed initially in a calcium-dependent manner by formation of new membrane over the site of damage and/or around extruding cytoplasm (Szubinska 1971), exactly as described for the vertebrate cell patching response. Moreover, the cytoskeletal response apparently is also conserved in these organisms in that actin accumulates around plasma membrane wounds (Szubinska 1978, Taylor et al. 1980), and if the wound is large enough, a contractile purse string containing F-actin assembles and closes around its edges ( Jeon & Jeon 1975). In budding yeast subject to cell wall (and presumptive plasma membrane) damage, rapid cytoskeletal remodeling ensues (Levin 2005). Similar to vertebrates, this remodeling entails rapid recruitment of active Rho to the plasma membrane and resultant actin assembly at sites of Rho activation (Andrews & Stark 2000, Delley & Hall 1999, Levin 2005). Given the conservation of the signaling systems and mechanisms involved (see also below), and that fungi, amoebae, and animals diverged from each other well before the development of multicellularity, it is difficult to escape the conclusion that an ancient single-cell repair system developed before the Metazoa arose.
Although the single-cell wound repair response is highly conserved, in no cell type is the need for an efficient and robust repair response more evident than in striated muscle cells. The contractile demands of everyday life dictate that the muscle cell membrane is under incessant and intense physical stress and thus susceptible to contraction-induced damage. Membrane damage has long been correlated with both strenuous eccentric exercise and various forms of muscular dystrophy, but only recently have the molecular players involved in muscle membrane repair begun to be elucidated. Positional cloning efforts identified the dysferlin gene as the genetic basis of the recessive limb girdle muscular dystrophy type 2B (Bashir et al. 1998), and dysferlin mutations were subsequently shown to cause Myoshi myopathy (Liu et al. 1998) and distal anterior compartment myopathy (Illa et al. 2001). Dysferlin knockout mice present with a progressive muscle phenotype (Bansal et al. 2003, Ho et al. 2004), and laser-induced wounding assays in isolated muscle fibers demonstrated that dysferlin facilitates calcium-dependent repair in both skeletal (Bansal et al. 2003, Ho et al. 2004) and cardiac muscle (Han et al. 2007). Although exactly how dysferlin participates in membrane repair is unclear, its amino-terminal C2 domain (C2A) binds phosphatidylserine (Davis et al. 2002, Therrien et al. 2009), and vesicle-anchored dysferlin translocates to sites of damage (Bansal et al. 2003, Klinge et al. 2007), where it binds the repair proteins annexins A1 and A2 (Lennon et al. 2003), all in a calcium-sensitive manner. These results have led to a model in which the damage-induced influx of calcium prompts the aggregation of subsarcolemmal vesicles and ultimately dysferlin/annexin-mediated vesicle-vesicle and vesicle-membrane fusion to repair the site of injury.
Although the influx of calcium is a universal initiator of membrane repair, recent studies have indicated that the influx of oxidative species may be just as important for proper membrane resealing in striated muscle. Similar to dysferlin knockout mice, MG53 (a TRIM family protein) knockout mice demonstrate defective membrane repair in both skeletal (Cai et al. 2009) and cardiac muscle (Wang et al. 2010). Although MG53 binds phosphatidylserine and translocates to sites of damage in isolated muscle fibers, it does so independently of calcium. Instead, the function of MG53 in mediating repair is dependent on an oxidative environment: In the oxidative cytosol immediately underlying the site of damage, vesicle-localized MG53 forms intermolecular disulfide bonds with MG53 molecules on neighboring vesicles and/or membrane, thereby linking vesicles to one another and to the membrane and facilitating fusion (Cai et al. 2009, Wang et al. 2010).
SUSPICIOUS SIMILARITIES AND POTENTIAL EVOLUTIONARY RELATIONSHIPS BETWEEN WOUND HEALING AND OTHER BASIC BIOLOGICAL PROCESSES
It was noted some time ago that the purse string wound response resembles the epithelial sheet migration observed during morphogenetic processes such as dorsal closure in Drosophila (Bement et al. 1993), in which converging sheets of epithelial cells close over the underlying amnioserosa with the aid of a multicellular purse string (Young et al. 1991, 1993). A beautiful series of studies on dorsal closure and healing of Drosophila embryos has confirmed this observation. At the level of morphology, both embryonic wound healing and dorsal closure entail the assembly and closure of a multicellular contractile ring of F-actin and myosin-2 (Figure 7), and in both cases filopodia contact and contraction abet the process ( Jacinto et al. 2000, 2002; Wood et al. 2002). Furthermore, in both cases, Rho and Cdc42 play analogous roles: Rho controls purse string formation, whereas Cdc42 controls filopodia formation ( Jacinto et al. 2000, 2002; Wood et al. 2002; Woolner et al. 2005). Moreover, upstream signaling is apparently conserved in that both dorsal closure and embryonic wound healing in Drosophila require signaling by the Jnk pathway (Campos et al. 2010, Jacinto et al. 2000, Riesgo-Escovar et al. 1996, Woolner et al. 2005).
Figure 7.

Suggested scheme for the evolutionary relationship between cytokinesis, wound healing, and morphogenesis. Cytokinesis: A single-cell purse string of actin filaments and myosin-2 ( green) drives cell division. Cellular wound repair: A single-cell purse string of actin filaments and myosin-2 ( green) closes over a single-cell wound. Multicellular wound healing: A multicellular purse string of actin filaments and myosin-2 drives reepithelialization. Morphogenesis: A multicellular purse string of actin filaments and myosin-2 drives epithelialization. In each case, filopod-based contraction and Rho GTPase signaling complement purse string closure (not shown).
The purse strings observed in embryonic wound healing and morphogenesis are strikingly similar to the F-actin and myosin-2 purse strings assembled during single-cell repair (see above) except that the former are multicellular and the latter are contained within single cells. This similarity includes not only the purse string component but also filopodial interaction and contraction (Mandato & Bement 2001). Moreover, the single-cell purse string response is controlled by calcium, Rho, and Cdc42, as is the embryonic purse string response (Clark et al. 2009). And although there are relatively few studies on the transcriptional response to plasma membrane damage, such damage is known to initiate activation of ERK signaling (Togo 2004) and AP-1 transcription factors (Grembowicz et al. 1999, Takada et al. 2007), which are conserved features of the embryonic multicellular wound healing response (see above).
The actomyosin rings that close over wound sites in damaged cells are remarkably similar to the actomyosin rings involved in cytokinesis in organisms ranging from fungi to mammals (Pollard 2010). Again, this similarity extends beyond morphology to regulation by Rho and/or Cdc42 (Bement et al. 2006). In addition, rapid local exocytosis and endocytosis is a conserved feature of cytokinesis (Echard 2008, Montagnac et al. 2008) just as it is for single-cell wound repair (see above). Moreover, genetic overlap between cytokinesis and cell repair systems in yeast is considerable (Levin 2005, Wu et al. 2010). Finally, although not widely recognized, a variety of cell types extend filopodia in the wake of the closing cytokinetic furrow (Danilchik & Brown 2008, Vacquier 1968). These filopodia play an analogous role to those discussed above for wound healing by pulling the daughter cells into tighter contact.
Where do all of these observations leave us? The similarity between dorsal closure and embryonic wound healing could reflect an evolutionary relationship in which developmental morphogenetic mechanisms were co-opted by evolution to drive wound repair. However, we think it more likely occurred the other way around. Given the information presented above, it seems probable that the single-cell wound response arose before multicellularity via the co-option of the cell polarity machinery used to drive cytokinesis or vice versa (Figure 7). Subsequently, with the development of multicellularity, the actomyosin purse string could be employed to close holes created in a cell layer as long as cell-cell junctions were present and capable of regulating the same signaling mechanisms used for the single-cell purse string (Clark et al. 2009). The co-option of this mechanism for morphogenesis would then entail a switch from damage-induced signaling to developmentally regulated signaling. From this perspective, the proposed patterning of cells around the wound by wound-induced signals would represent the evolutionary precursor of embryonic pattern formation mechanisms.
Although this view might seem unduly influenced by our enthusiasm for wound healing, it has two virtues. First, it is far easier to imagine evolution of the complex morphogenetic programs of complex organisms if the basic machinery and control mechanisms for such movements were already in place for an older, more basic role (wound repair). Second, assuming an evolutionary link between these various processes can suggest useful lines of inquiry. To a certain extent, such inquiries have already begun with the discovery that the transcription factor Grh is a component of the planar cell polarity pathway (Caddy et al. 2010).
FUTURE PROSPECTS AND QUESTIONS
The increasing adoption of model systems for the study of multicellular wound repair has already paid major dividends in our understanding of healing mechanisms and will continue to do so. This is fortunate, as several essential questions remain unanswered, and recent advances have prompted a series of new questions.
One long-standing and fundamentally important issue likely to attract further attention is the relationship between inflammation, the speed of wound healing, and the extent of scarring. Is it possible to convert an adult wound response to an embryonic one by limiting or eliminating inflammation? The studies so far provide exciting hints that this may be the case, but far more work needs to be done. It is also unclear to what extent the embryonic healing response and the purse string response are aligned. Does an embryonic healing response always come with the purse string? With respect to signals, not only are we likely missing at least some of the key signaling pathways for multicellular wound repair, but for those pathways we have identified, the downstream targets need to be characterized. This is true both for inflammation, wherein the means by which the H2O2 gradient is converted to directed leukocyte migration is unknown, as well as for the RTK pathways involved in reepithelialization. We also do not understand the basis of cell behaviors observed in recent experiments. Why, for example, do leukocytes make repeated trips to and from the wound?
To what extent does multicellular wound repair resemble pattern formation? That is, do cells at different distances from the wound display progressively narrower patterns of expression of specific wound-induced gene products? Determining this will require further inquiry into those genes specifically turned on or off in response to wounding.
For single-cell wound repair, how, exactly, does patching work? Which proteins regulate fusion versus aggregation, and how is the patch sealed to the unwounded plasma membrane at the wound periphery? Is there a broadly conserved “woundcyst” complex containing dysferlin and annexin (or related proteins) found in cell types other than muscle? How are the initial signals generated by plasma membrane damage—calcium inrush and oxidation—coupled to the patching and cytoskeletal responses? The responses appear to be far more precise than the initial signals, again suggesting a kind of pattern formation system, but the details of how this might work are completely mysterious.
Finally, how are the single- and the multicellular wound responses coordinated? What are the essential signals produced by damaged cells that allow them to communicate with their neighbors? Presumably, both rapid responses such as paracrine, tension, or electrically mediated signals as well as long-term responses involving differential transcription and translation are generated, but at present almost nothing is understood about how they might be coupled.
Acknowledgments
We are grateful to our labmates for support and advice. We are also thankful to all of our colleagues in the wound healing field, and we apologize to those whose work we did not discuss. Many thanks to the National Institutes of Health (GMO52932) and the Jain Foundation for support. This work is dedicated to the memory of Leslie Nielsen (February 26, 1926–November 28, 2010). He will be sorely missed.
Footnotes
DISCLOSURE STATEMENT
The authors are not aware of any affiliations, memberships, funding, or financial holdings that might be perceived as affecting the objectivity of this review.
LITERATURE CITED
- Adzick NS, Harrison MR, Glick PL, Beckstead JH, Villa RL, et al. Comparison of fetal, newborn, and adult wound healing by histologic, enzyme-histochemical, and hydroxyproline determinations. J Pediatr Surg. 1985;20:315–19. doi: 10.1016/s0022-3468(85)80210-4. [DOI] [PubMed] [Google Scholar]
- Andrew N, Insall RH. Chemotaxis in shallow gradients is mediated independently of PtdIns 3-kinase by biased choices between random protrusions. Nat Cell Biol. 2007;9:193–200. doi: 10.1038/ncb1536. [DOI] [PubMed] [Google Scholar]
- Andrews PD, Stark MJ. Dynamic, Rho1p-dependent localization of Pkc1p to sites of polarized growth. J Cell Sci. 2000;113(Pt. 15):2685–93. doi: 10.1242/jcs.113.15.2685. [DOI] [PubMed] [Google Scholar]
- Armstrong JR, Ferguson MW. Ontogeny of the skin and the transition from scar-free to scarring phenotype during wound healing in the pouch young of a marsupial, Monodelphis domestica. Dev Biol. 1995;169:242–60. doi: 10.1006/dbio.1995.1141. [DOI] [PubMed] [Google Scholar]
- Ashcroft GS, Yang X, Glick AB, Weinstein M, Letterio JL, et al. Mice lacking Smad3 show accelerated wound healing and an impaired local inflammatory response. Nat Cell Biol. 1999;1:260–66. doi: 10.1038/12971. [DOI] [PubMed] [Google Scholar]
- Babcock DT, Brock AR, Fish GS, Wang Y, Perrin L, et al. Circulating blood cells function as a surveillance system for damaged tissue in Drosophila larvae. Proc Natl Acad Sci USA. 2008;105:10017–22. doi: 10.1073/pnas.0709951105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babiychuk EB, Monastyrskaya K, Potez S, Draeger A. Intracellular Ca2+ operates a switch between repair and lysis of streptolysin O-perforated cells. Cell Death Differ. 2009;16:1126–34. doi: 10.1038/cdd.2009.30. [DOI] [PubMed] [Google Scholar]
- Bansal D, Miyake K, Vogel SS, Groh S, Chen CC, et al. Defective membrane repair in dysferlin-deficient muscular dystrophy. Nature. 2003;423:168–72. doi: 10.1038/nature01573. [DOI] [PubMed] [Google Scholar]
- Bashir R, Britton S, Strachan T, Keers S, Vafiadaki E, et al. A gene related to Caenorhabditis elegans spermatogenesis factor fer-1 is mutated in limb-girdle muscular dystrophy type 2B. Nat Genet. 1998;20:37–42. doi: 10.1038/1689. [DOI] [PubMed] [Google Scholar]
- Bement WM, Capco DG. Analysis of inducible contractile rings suggests a role for protein kinase C in embryonic cytokinesis and wound healing. Cell Motil Cytoskelet. 1991;20:145–57. doi: 10.1002/cm.970200207. [DOI] [PubMed] [Google Scholar]
- Bement WM, Forscher P, Mooseker MS. A novel cytoskeletal structure involved in purse string wound closure and cell polarity maintenance. J Cell Biol. 1993;121:565–78. doi: 10.1083/jcb.121.3.565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bement WM, Mandato CA, Kirsch MN. Wound-induced assembly and closure of an actomyosin purse string in Xenopus oocytes. Curr Biol. 1999;9:579–87. doi: 10.1016/s0960-9822(99)80261-9. [DOI] [PubMed] [Google Scholar]
- Bement WM, Miller AL, Von Dassow G. Rho GTPase activity zones and transient contractile arrays. BioEssays. 2006;28:983–93. doi: 10.1002/bies.20477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bement WM, Yu HY, Burkel BM, Vaughan EM, Clark AG. Rehabilitation and the single cell. Curr Opin Cell Biol. 2007;19:95–100. doi: 10.1016/j.ceb.2006.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benink HA, Bement WM. Concentric zones of active RhoA and Cdc42 around single cell wounds. J Cell Biol. 2005;168:429–39. doi: 10.1083/jcb.200411109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bi GQ, Alderton JM, Steinhardt RA. Calcium-regulated exocytosis is required for cell membrane resealing. J Cell Biol. 1995;131:1747–58. doi: 10.1083/jcb.131.6.1747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bird JE, Daudet N, Warchol ME, Gale JE. Supporting cells eliminate dying sensory hair cells to maintain epithelial integrity in the avian inner ear. J Neurosci. 2010;30:12545–56. doi: 10.1523/JNEUROSCI.3042-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borgonovo B, Cocucci E, Racchetti G, Podini P, Bachi A, et al. Regulated exocytosis: a novel, widely expressed system. Nat Cell Biol. 2002;4:955–62. doi: 10.1038/ncb888. [DOI] [PubMed] [Google Scholar]
- Bullen TF, Forrest S, Campbell F, Dodson AR, Hershman MJ, et al. Characterization of epithelial cell shedding from human small intestine. Lab Investig. 2006;86:1052–63. doi: 10.1038/labinvest.3700464. [DOI] [PubMed] [Google Scholar]
- Caddy J, Wilanowski T, Darido C, Dworkin S, Ting SB, et al. Epidermal wound repair is regulated by the planar cell polarity signaling pathway. Dev Cell. 2010;19:138–47. doi: 10.1016/j.devcel.2010.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai C, Masumiya H, Weisleder N, Matsuda N, Nishi M, et al. MG53 nucleates assembly of cell membrane repair machinery. Nat Cell Biol. 2009;11:56–64. doi: 10.1038/ncb1812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campos I, Geiger JA, Santos AC, Carlos V, Jacinto A. Genetic screen in Drosophila melanogaster uncovers a novel set of genes required for embryonic epithelial repair. Genetics. 2010;184:129–40. doi: 10.1534/genetics.109.110288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Iijima M, Tang M, Landree MA, Huang YE, et al. PLA2 and PI3K/PTEN pathways act in parallel to mediate chemotaxis. Dev Cell. 2007;12:603–14. doi: 10.1016/j.devcel.2007.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chmielowiec J, Borowiak M, Morkel M, Stradal T, Munz B, et al. c-Met is essential for wound healing in the skin. J Cell Biol. 2007;177:151–62. doi: 10.1083/jcb.200701086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chowdhury S, Smith KW, Gustin MC. Osmotic stress and the yeast cytoskeleton: phenotype-specific suppression of an actin mutation. J Cell Biol. 1992;118:561–71. doi: 10.1083/jcb.118.3.561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark AG, Miller AL, Vaughan E, Yu HY, Penkert R, et al. Integration of single and multicellular wound responses. Curr Biol. 2009;19:1389–95. doi: 10.1016/j.cub.2009.06.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowin AJ, Brosnan MP, Holmes TM, Ferguson MW. Endogenous inflammatory response to dermal wound healing in the fetal and adult mouse. Dev Dyn. 1998;212:385–93. doi: 10.1002/(SICI)1097-0177(199807)212:3<385::AID-AJA6>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- Crosby LM, Waters CM. Epithelial repair mechanisms in the lung. Am J Physiol Lung Cell Mol Physiol. 2010;298:L715–31. doi: 10.1152/ajplung.00361.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cvejic A, Hall C, Bak-Maier M, Flores MV, Crosier P, et al. Analysis of WASp function during the wound inflammatory response—live-imaging studies in zebrafish larvae. J Cell Sci. 2008;121:3196–206. doi: 10.1242/jcs.032235. [DOI] [PubMed] [Google Scholar]
- Danilchik MV, Brown EE. Membrane dynamics of cleavage furrow closure in Xenopus laevis. Dev Dyn. 2008;237:565–79. doi: 10.1002/dvdy.21442. [DOI] [PubMed] [Google Scholar]
- Danjo Y, Gipson IK. Actin “purse string” filaments are anchored by E-cadherin-mediated adherens junctions at the leading edge of the epithelial wound, providing coordinated cell movement. J Cell Sci. 1998;111(Pt. 22):3323–32. doi: 10.1242/jcs.111.22.3323. [DOI] [PubMed] [Google Scholar]
- Davis DB, Doherty KR, Delmonte AJ, McNally EM. Calcium-sensitive phospholipid binding properties of normal and mutant ferlin C2 domains. J Biol Chem. 2002;277:22883–88. doi: 10.1074/jbc.M201858200. [DOI] [PubMed] [Google Scholar]
- Delley PA, Hall MN. Cell wall stress depolarizes cell growth via hyperactivation of RHO1. J Cell Biol. 1999;147:163–74. doi: 10.1083/jcb.147.1.163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng M, Chen WL, Takatori A, Peng Z, Zhang L, et al. A role for the mitogen-activated protein kinase kinase kinase 1 in epithelial wound healing. Mol Biol Cell. 2006;17:3446–55. doi: 10.1091/mbc.E06-02-0102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duffield JS. Epithelial to mesenchymal transition in injury of solid organs: fact or artifact? Gastroenterology. 2010;139:1081–83. doi: 10.1053/j.gastro.2010.08.017. [DOI] [PubMed] [Google Scholar]
- Echard A. Membrane traffic and polarization of lipid domains during cytokinesis. Biochem Soc Trans. 2008;36:395–99. doi: 10.1042/BST0360395. [DOI] [PubMed] [Google Scholar]
- Ferguson MWJ, O’Kane S. Scar-free healing: from embryonic mechanisms to adult therapeutic intervention. Philos Trans R Soc Lond B. 2004;359:839–50. doi: 10.1098/rstb.2004.1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fishman HM, Bittner GD. Vesicle-mediated restoration of a plasmalemmal barrier in severed axons. News Physiol Sci. 2003;18:115–18. doi: 10.1152/nips.01429.2002. [DOI] [PubMed] [Google Scholar]
- Florian P, Schoneberg T, Schulzke JD, Fromm M, Gitter AH. Single-cell epithelial defects close rapidly by an actinomyosin purse string mechanism with functional tight junctions. J Physiol. 2002;545:485–99. doi: 10.1113/jphysiol.2002.031161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furie B, Furie BC. Mechanisms of thrombus formation. N Engl J Med. 2008;359:938–49. doi: 10.1056/NEJMra0801082. [DOI] [PubMed] [Google Scholar]
- Galko MJ, Krasnow MA. Cellular and genetic analysis of wound healing in Drosophila larvae. PLoS Biol. 2004;2:e239. doi: 10.1371/journal.pbio.0020239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geuskens M, Tencer R. An ultrastructural study of the effects of wheat germ agglutinin (WGA) on cell cortex organization during the first cleavage of Xenopus laevis eggs. II Cortical wound healing. J Cell Sci. 1979;37:59–67. doi: 10.1242/jcs.37.1.59. [DOI] [PubMed] [Google Scholar]
- Gingell D. Contractile responses at the surface of an amphibian egg. J Embryol Exp Morphol. 1970;23:583–609. [PubMed] [Google Scholar]
- Godell CM, Smyers ME, Eddleman CS, Ballinger ML, Fishman HM, et al. Calpain activity promotes the sealing of severed giant axons. Proc Natl Acad Sci USA. 1997;94:4751–56. doi: 10.1073/pnas.94.9.4751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godin LM, Vergen J, Prakash YS, Pagano RE, Hubmayr RD. Spatiotemporal dynamics of actin remodeling and endomembrane trafficking in alveolar epithelial type I cell wound healing. Am J Physiol Lung Cell Mol Physiol. 2011;300:L615–23. doi: 10.1152/ajplung.00265.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grembowicz KP, Sprague D, McNeil PL. Temporary disruption of the plasma membrane is required for c-fos expression in response to mechanical stress. Mol Biol Cell. 1999;10:1247–57. doi: 10.1091/mbc.10.4.1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu Y, Forostyan T, Sabbadini R, Rosenblatt J. Epithelial cell extrusion requires the sphingosine-1-phosphate receptor 2 pathway. J Cell Biol. 2011;193:667–76. doi: 10.1083/jcb.201010075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurtner GC, Werner S, Barrandon Y, Longaker MT. Wound repair and regeneration. Nature. 2008;453:314–21. doi: 10.1038/nature07039. [DOI] [PubMed] [Google Scholar]
- Hambleton P. Repair of wall damage in Escherichia coli recovered from an aerosol. J Gen Microbiol. 1971;69:81–88. doi: 10.1099/00221287-69-1-81. [DOI] [PubMed] [Google Scholar]
- Han R, Bansal D, Miyake K, Muniz VP, Weiss RM, et al. Dysferlin-mediated membrane repair protects the heart from stress-induced left ventricular injury. J Clin Investig. 2007;117:1805–13. doi: 10.1172/JCI30848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heasman SJ, Ridley AJ. Mammalian Rho GTPases: new insights into their functions from in vivo studies. Nat Rev Mol Cell Biol. 2008;9:690–701. doi: 10.1038/nrm2476. [DOI] [PubMed] [Google Scholar]
- Ho M, Post CM, Donahue LR, Lidov HG, Bronson RT, et al. Disruption of muscle membrane and phenotype divergence in two novel mouse models of dysferlin deficiency. Hum Mol Genet. 2004;13:1999–2010. doi: 10.1093/hmg/ddh212. [DOI] [PubMed] [Google Scholar]
- Hopkinson-Woolley J, Hughes D, Gordon S, Martin P. Macrophage recruitment during limb development and wound healing in the embryonic and foetal mouse. J Cell Sci. 1994;107(Pt. 5):1159–67. doi: 10.1242/jcs.107.5.1159. [DOI] [PubMed] [Google Scholar]
- Huh CG, Factor VM, Sanchez A, Uchida K, Conner EA, et al. Hepatocyte growth factor/c-met signaling pathway is required for efficient liver regeneration and repair. Proc Natl Acad Sci USA. 2004;101:4477–82. doi: 10.1073/pnas.0306068101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humphreys BD, Lin SL, Kobayashi A, Hudson TE, Nowlin BT, et al. Fate tracing reveals the pericyte and not epithelial origin of myofibroblasts in kidney fibrosis. Am J Pathol. 2010;176:85–97. doi: 10.2353/ajpath.2010.090517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Idone V, Tam C, Goss JW, Toomre D, Pypaert M, et al. Repair of injured plasma membrane by rapid Ca2+-dependent endocytosis. J Cell Biol. 2008;180:905–14. doi: 10.1083/jcb.200708010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Illa I, Serrano-Munuera C, Gallardo E, Lasa A, Rojas-Garcia R, et al. Distal anterior compartment myopathy: a dysferlin mutation causing a new muscular dystrophy phenotype. Ann Neurol. 2001;49:130–34. [PubMed] [Google Scholar]
- Jacinto A, Wood W, Balayo T, Turmaine M, Martinez-Arias A, et al. Dynamic actin-based epithelial adhesion and cell matching during Drosophila dorsal closure. Curr Biol. 2000;10:1420–26. doi: 10.1016/s0960-9822(00)00796-x. [DOI] [PubMed] [Google Scholar]
- Jacinto A, Wood W, Woolner S, Hiley C, Turner L, et al. Dynamic analysis of actin cable function during Drosophila dorsal closure. Curr Biol. 2002;12:1245–50. doi: 10.1016/s0960-9822(02)00955-7. [DOI] [PubMed] [Google Scholar]
- Jaiswal JK, Andrews NW, Simon SM. Membrane proximal lysosomes are the major vesicles responsible for calcium-dependent exocytosis in nonsecretory cells. J Cell Biol. 2002;159:625–35. doi: 10.1083/jcb.200208154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaiswal JK, Rivera VM, Simon SM. Exocytosis of post-Golgi vesicles is regulated by components of the endocytic machinery. Cell. 2009;137:1308–19. doi: 10.1016/j.cell.2009.04.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janis JE, Kwon RK, Lalonde DH. A practical guide to wound healing. Plast Reconstr Surg. 2010;125:230e–44e. doi: 10.1097/PRS.0b013e3181d9a0d1. [DOI] [PubMed] [Google Scholar]
- Jeon KW, Jeon MS. Cytoplasmic filaments and cellular wound healing in Amoeba proteus. J Cell Biol. 1975;67:243–49. doi: 10.1083/jcb.67.1.243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshi SD, Von Dassow M, Davidson LA. Experimental control of excitable embryonic tissues: three stimuli induce rapid epithelial contraction. Exp Cell Res. 2010;316:103–14. doi: 10.1016/j.yexcr.2009.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiehart DP, Galbraith CG, Edwards KA, Rickoll WL, Montague RA. Multiple forces contribute to cell sheet morphogenesis for dorsal closure in Drosophila. J Cell Biol. 2000;149:471–90. doi: 10.1083/jcb.149.2.471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kite GL. The relative permeability of the surface and interior portions of the cytoplasm of animal and plant cells. Biol Bull. 1913;25:1–7. [Google Scholar]
- Klinge L, Laval S, Keers S, Haldane F, Straub V, et al. From T-tubule to sarcolemma: damage-induced dysferlin translocation in early myogenesis. FASEB J. 2007;21:1768–76. doi: 10.1096/fj.06-7659com. [DOI] [PubMed] [Google Scholar]
- Kwon YC, Baek SH, Lee H, Choe KM. Nonmuscle myosin II localization is regulated by JNK during Drosophila larval wound healing. Biochem Biophys Res Commun. 2010;393:656–61. doi: 10.1016/j.bbrc.2010.02.047. [DOI] [PubMed] [Google Scholar]
- La Claire JW., Jr Inducement of wound motility in intact giant algal cells. Exp Cell Res. 1983;145:63–69. doi: 10.1016/s0014-4827(83)80008-1. [DOI] [PubMed] [Google Scholar]
- Larson BJ, Longaker MT, Lorenz HP. Scarless fetal wound healing: a basic science review. Plast Reconstr Surg. 2010;126:1172–80. doi: 10.1097/PRS.0b013e3181eae781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lennon NJ, Kho A, Bacskai BJ, Perlmutter SL, Hyman BT, et al. Dysferlin interacts with annexins A1 and A2 and mediates sarcolemmal wound-healing. J Biol Chem. 2003;278:50466–73. doi: 10.1074/jbc.M307247200. [DOI] [PubMed] [Google Scholar]
- Lesch C, Jo J, Wu Y, Fish GS, Galko MJ. A targeted UAS-RNAi screen in Drosophila larvae identifies wound closure genes regulating distinct cellular processes. Genetics. 2010;186:943–57. doi: 10.1534/genetics.110.121822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levin DE. Cell wall integrity signaling in Saccharomyces cerevisiae. Microbiol Mol Biol Rev. 2005;69:262–91. doi: 10.1128/MMBR.69.2.262-291.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liechty KW, Crombleholme TM, Cass DL, Martin B, Adzick NS. Diminished interleukin-8 (IL-8) production in the fetal wound healing response. J Surg Res. 1998;77:80–84. doi: 10.1006/jsre.1998.5345. [DOI] [PubMed] [Google Scholar]
- Liu J, Aoki M, Illa I, Wu C, Fardeau M, et al. Dysferlin, a novel skeletal muscle gene, is mutated in Miyoshi myopathy and limb girdle muscular dystrophy. Nat Genet. 1998;20:31–36. doi: 10.1038/1682. [DOI] [PubMed] [Google Scholar]
- Lotz MM, Rabinovitz I, Mercurio AM. Intestinal restitution: progression of actin cytoskeleton rearrangements and integrin function in a model of epithelial wound healing. Am J Pathol. 2000;156:985–96. doi: 10.1016/S0002-9440(10)64966-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mace KA, Pearson JC, Mcginnis W. An epidermal barrier wound repair pathway in Drosophila is mediated by grainy head. Science. 2005;308:381–85. doi: 10.1126/science.1107573. [DOI] [PubMed] [Google Scholar]
- Madara JL. Maintenance of the macromolecular barrier at cell extrusion sites in intestinal epithelium: physiological rearrangement of tight junctions. J Membr Biol. 1990;116:177–84. doi: 10.1007/BF01868675. [DOI] [PubMed] [Google Scholar]
- Mandato CA, Bement WM. Contraction and polymerization cooperate to assemble and close actomyosin rings around Xenopus oocyte wounds. J Cell Biol. 2001;154:785–97. doi: 10.1083/jcb.200103105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandato CA, Bement WM. Actomyosin transports microtubules and microtubules control actomyosin recruitment during Xenopus oocyte wound healing. Curr Biol. 2003;13:1096–105. doi: 10.1016/s0960-9822(03)00420-2. [DOI] [PubMed] [Google Scholar]
- Marchiando AM, Shen L, Graham WV, Edelblum KL, Duckworth CA, et al. The epithelial barrier is maintained by in vivo tight junction expansion during pathologic intestinal epithelial shedding. Gastroenterology. 2011;140:1208–18. doi: 10.1053/j.gastro.2011.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin P. Wound healing—aiming for perfect skin regeneration. Science. 1997;276:75–81. doi: 10.1126/science.276.5309.75. [DOI] [PubMed] [Google Scholar]
- Martin P, D’Souza D, Martin J, Grose R, Cooper L, et al. Wound healing in the PU. 1 null mouse—tissue repair is not dependent on inflammatory cells. Curr Biol. 2003;13:1122–28. doi: 10.1016/s0960-9822(03)00396-8. [DOI] [PubMed] [Google Scholar]
- Martin P, Leibovich SJ. Inflammatory cells during wound repair: the good, the bad and the ugly. Trends Cell Biol. 2005;15:599–607. doi: 10.1016/j.tcb.2005.09.002. [DOI] [PubMed] [Google Scholar]
- Martin P, Lewis J. Actin cables and epidermal movement in embryonic wound healing. Nature. 1992;360:179–83. doi: 10.1038/360179a0. [DOI] [PubMed] [Google Scholar]
- Martin P, Nobes CD. An early molecular component of the wound healing response in rat embryos—induction of c-fos protein in cells at the epidermal wound margin. Mech Dev. 1992;38:209–15. doi: 10.1016/0925-4773(92)90054-n. [DOI] [PubMed] [Google Scholar]
- Mathias JR, Perrin BJ, Liu TX, Kanki J, Look AT, et al. Resolution of inflammation by retrograde chemotaxis of neutrophils in transgenic zebrafish. J Leukoc Biol. 2006;80:1281–88. doi: 10.1189/jlb.0506346. [DOI] [PubMed] [Google Scholar]
- McCluskey J, Martin P. Analysis of the tissue movements of embryonic wound healing—DiI studies in the limb bud stage mouse embryo. Dev Biol. 1995;170:102–14. doi: 10.1006/dbio.1995.1199. [DOI] [PubMed] [Google Scholar]
- McNeil AK, Rescher U, Gerke V, McNeil PL. Requirement for annexin A1 in plasma membrane repair. J Biol Chem. 2006;281:35202–7. doi: 10.1074/jbc.M606406200. [DOI] [PubMed] [Google Scholar]
- McNeil PL, Ito S. Gastrointestinal cell plasma membrane wounding and resealing in vivo. Gastroenterology. 1989;96:1238–48. doi: 10.1016/s0016-5085(89)80010-1. [DOI] [PubMed] [Google Scholar]
- McNeil PL, Ito S. Molecular traffic through plasma membrane disruptions of cells in vivo. J Cell Sci. 1990;96(Pt. 3):549–56. doi: 10.1242/jcs.96.3.549. [DOI] [PubMed] [Google Scholar]
- McNeil PL, Khakee R. Disruptions of muscle fiber plasma membranes. Role in exercise-induced damage. Am J Pathol. 1992;140:1097–109. [PMC free article] [PubMed] [Google Scholar]
- McNeil PL, Miyake K, Vogel SS. The endomembrane requirement for cell surface repair. Proc Natl Acad Sci USA. 2003;100:4592–97. doi: 10.1073/pnas.0736739100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNeil PL, Muthukrishnan L, Warder E, D’Amore PA. Growth factors are released by mechanically wounded endothelial cells. J Cell Biol. 1989;109:811–22. doi: 10.1083/jcb.109.2.811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNeil PL, Steinhardt RA. Plasma membrane disruption: repair, prevention, adaptation. Annu Rev Cell Dev Biol. 2003;19:697–731. doi: 10.1146/annurev.cellbio.19.111301.140101. [DOI] [PubMed] [Google Scholar]
- McNeil PL, Vogel SS, Miyake K, Terasaki M. Patching plasma membrane disruptions with cytoplasmic membrane. J Cell Sci. 2000;113(Pt. 11):1891–902. doi: 10.1242/jcs.113.11.1891. [DOI] [PubMed] [Google Scholar]
- Mellgren RL. A plasma membrane wound proteome: reversible externalization of intracellular proteins following reparable mechanical damage. J Biol Chem. 2010;285:36597–607. doi: 10.1074/jbc.M110.110015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyake K, McNeil PL, Suzuki K, Tsunoda R, Sugai N. An actin barrier to resealing. J Cell Sci. 2001;114:3487–94. doi: 10.1242/jcs.114.19.3487. [DOI] [PubMed] [Google Scholar]
- Montagnac G, Echard A, Chavrier P. Endocytic traffic in animal cell cytokinesis. Curr Opin Cell Biol. 2008;20:454–61. doi: 10.1016/j.ceb.2008.03.011. [DOI] [PubMed] [Google Scholar]
- Moreira S, Stramer B, Evans I, Wood W, Martin P. Prioritization of competing damage and developmental signals by migrating macrophages in the Drosophila embryo. Curr Biol. 2010;20:464–70. doi: 10.1016/j.cub.2010.01.047. [DOI] [PubMed] [Google Scholar]
- Morgan BP, Dankert JR, Esser AF. Recovery of human neutrophils from complement attack: removal of the membrane attack complex by endocytosis and exocytosis. J Immunol. 1987;138:246–53. [PubMed] [Google Scholar]
- Mori R, Power KT, Wang CM, Martin P, Becker DL. Acute downregulation of connexin43 at wound sites leads to a reduced inflammatory response, enhanced keratinocyte proliferation and wound fibroblast migration. J Cell Sci. 2006;119:5193–203. doi: 10.1242/jcs.03320. [DOI] [PubMed] [Google Scholar]
- Mori R, Shaw TJ, Martin P. Molecular mechanisms linking wound inflammation and fibrosis: knockdown of osteopontin leads to rapid repair and reduced scarring. J Exp Med. 2008;205:43–51. doi: 10.1084/jem.20071412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naik-Mathuria B, Gay AN, Zhu X, Yu L, Cass DL, et al. Age-dependent recruitment of neutrophils by fetal endothelial cells: implications in scarless wound healing. J Pediatr Surg. 2007;42:166–71. doi: 10.1016/j.jpedsurg.2006.09.058. [DOI] [PubMed] [Google Scholar]
- Niethammer P, Grabher C, Look AT, Mitchison TJ. A tissue-scale gradient of hydrogen peroxide mediates rapid wound detection in zebrafish. Nature. 2009;459:996–99. doi: 10.1038/nature08119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novo E, Parola M. Redox mechanisms in hepatic chronic wound healing and fibrogenesis. Fibrogenes Tissue Repair. 2008;1:5. doi: 10.1186/1755-1536-1-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oeckler RA, Hubmayr RD. Cell wounding and repair in ventilator injured lungs. Respir Physiol Neurobiol. 2008;163:44–53. doi: 10.1016/j.resp.2008.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olutoye OO, Yager DR, Cohen IK, Diegelmann RF. Lower cytokine release by fetal porcine platelets: a possible explanation for reduced inflammation after fetal wounding. J Pediatr Surg. 1996;31:91–95. doi: 10.1016/s0022-3468(96)90326-7. [DOI] [PubMed] [Google Scholar]
- Pearson JC, Juarez MT, Kim M, Drivenes O, McGinnis W. Multiple transcription factor codes activate epidermal wound-response genes in Drosophila. Proc Natl Acad Sci USA. 2009;106:2224–29. doi: 10.1073/pnas.0810219106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollard TD. Mechanics of cytokinesis in eukaryotes. Curr Opin Cell Biol. 2010;22:50–56. doi: 10.1016/j.ceb.2009.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pujol N, Cypowyj S, Ziegler K, Millet A, Astrain A, et al. Distinct innate immune responses to infection and wounding in the C. elegans epidermis. Curr Biol. 2008;18:481–89. doi: 10.1016/j.cub.2008.02.079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramet M, Lanot R, Zachary D, Manfruelli P. JNK signaling pathway is required for efficient wound healing in Drosophila. Dev Biol. 2002;241:145–56. doi: 10.1006/dbio.2001.0502. [DOI] [PubMed] [Google Scholar]
- Redd MJ, Kelly G, Dunn G, Way M, Martin P. Imaging macrophage chemotaxis in vivo: studies of microtubule function in zebrafish wound inflammation. Cell Motil Cytoskelet. 2006;63:415–22. doi: 10.1002/cm.20133. [DOI] [PubMed] [Google Scholar]
- Reddy A, Caler EV, Andrews NW. Plasma membrane repair is mediated by Ca2+-regulated exocytosis of lysosomes. Cell. 2001;106:157–69. doi: 10.1016/s0092-8674(01)00421-4. [DOI] [PubMed] [Google Scholar]
- Repertinger SK, Campagnaro E, Fuhrman J, El-Abaseri T, Yuspa SH, et al. EGFR enhances early healing after cutaneous incisional wounding. J Investig Dermatol. 2004;123:982–89. doi: 10.1111/j.0022-202X.2004.23478.x. [DOI] [PubMed] [Google Scholar]
- Riesgo-Escovar JR, Jenni M, Fritz A, Hafen E. The Drosophila Jun-N-terminal kinase is required for cell morphogenesis but not for DJun-dependent cell fate specification in the eye. Genes Dev. 1996;10:2759–68. doi: 10.1101/gad.10.21.2759. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Diaz A, Toyama Y, Abravanel DL, Wiemann JM, Wells AR, et al. Actomyosin purse strings: renewable resources that make morphogenesis robust and resilient. HFSP J. 2008;2:220–37. doi: 10.2976/1.2955565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenblatt J, Raff MC, Cramer LP. An epithelial cell destined for apoptosis signals its neighbors to extrude it by an actin- and myosin-dependent mechanism. Curr Biol. 2001;11:1847–57. doi: 10.1016/s0960-9822(01)00587-5. [DOI] [PubMed] [Google Scholar]
- Russo JM, Florian P, Shen L, Graham WV, Tretiakova MS, et al. Distinct temporal-spatial roles for rho kinase and myosin light chain kinase in epithelial purse-string wound closure. Gastroenterology. 2005;128:987–1001. doi: 10.1053/j.gastro.2005.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schafer M, Werner S. Transcriptional control of wound repair. Annu Rev Cell Dev Biol. 2007;23:69–92. doi: 10.1146/annurev.cellbio.23.090506.123609. [DOI] [PubMed] [Google Scholar]
- Schapire AL, Valpuesta V, Botella MA. Plasma membrane repair in plants. Trends Plant Sci. 2009;14:645–52. doi: 10.1016/j.tplants.2009.09.004. [DOI] [PubMed] [Google Scholar]
- Schapire AL, Voigt B, Jasik J, Rosado A, Lopez-Cobollo R, et al. Arabidopsis synaptotagmin 1 is required for the maintenance of plasma membrane integrity and cell viability. Plant Cell. 2008;20:3374–88. doi: 10.1105/tpc.108.063859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuh AC, Keating SJ, Monteclaro FS, Vogt PK, Breitman ML. Obligatory wounding requirement for tumorigenesis in v-jun transgenic mice. Nature. 1990;346:756–60. doi: 10.1038/346756a0. [DOI] [PubMed] [Google Scholar]
- Shaw TJ, Martin P. Wound repair at a glance. J Cell Sci. 2009;122:3209–13. doi: 10.1242/jcs.031187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen SS, Tucker WC, Chapman ER, Steinhardt RA. Molecular regulation of membrane resealing in 3T3 fibroblasts. J Biol Chem. 2005;280:1652–60. doi: 10.1074/jbc.M410136200. [DOI] [PubMed] [Google Scholar]
- Singer AJ, Clark RA. Cutaneous wound healing. N Engl J Med. 1999;341:738–46. doi: 10.1056/NEJM199909023411006. [DOI] [PubMed] [Google Scholar]
- Sipos P, Gyory H, Hagymasi K, Ondrejka P, Blazovics A. Special wound healing methods used in ancient Egypt and the mythological background. World J Surg. 2004;28:211–16. doi: 10.1007/s00268-003-7073-x. [DOI] [PubMed] [Google Scholar]
- Slattum G, McGee KM, Rosenblatt J. P115 RhoGEF and microtubules decide the direction apoptotic cells extrude from an epithelium. J Cell Biol. 2009;186:693–702. doi: 10.1083/jcb.200903079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spaeth CS, Boydston EA, Figard LR, Zuzek A, Bittner GD. A model for sealing plasmalemmal damage in neurons and other eukaryotic cells. J Neurosci. 2010;30:15790–800. doi: 10.1523/JNEUROSCI.4155-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinhardt RA, Bi G, Alderton JM. Cell membrane resealing by a vesicular mechanism similar to neurotransmitter release. Science. 1994;263:390–93. doi: 10.1126/science.7904084. [DOI] [PubMed] [Google Scholar]
- Stramer B, Wood W, Galko MJ, Redd MJ, Jacinto A, et al. Live imaging of wound inflammation in Drosophila embryos reveals key roles for small GTPases during in vivo cell migration. J Cell Biol. 2005;168:567–73. doi: 10.1083/jcb.200405120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szubinska B. “New membrane” formation in Amoeba proteus upon injury of individual cells. Electron microscope observations. J Cell Biol. 1971;49:747–72. doi: 10.1083/jcb.49.3.747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szubinska B. Closure of the plasma membrane around microneedle in Amoeba proteus. An ultrastructural study. Exp Cell Res. 1978;111:105–15. doi: 10.1016/0014-4827(78)90241-0. [DOI] [PubMed] [Google Scholar]
- Takada H, Nishimura M, Asayama Y, Mannse Y, Ishiwata S, et al. Atf1 is a target of the mitogen-activated protein kinase Pmk1 and regulates cell integrity in fission yeast. Mol Biol Cell. 2007;18:4794–802. doi: 10.1091/mbc.E07-03-0282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamada M, Perez TD, Nelson WJ, Sheetz MP. Two distinct modes of myosin assembly and dynamics during epithelial wound closure. J Cell Biol. 2007;176:27–33. doi: 10.1083/jcb.200609116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taura K, Miura K, Iwaisako K, Osterreicher CH, Kodama Y, et al. Hepatocytes do not undergo epithelial-mesenchymal transition in liver fibrosis in mice. Hepatology. 2010;51:1027–36. doi: 10.1002/hep.23368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor DL, Wang YL, Heiple JM. Contractile basis of ameboid movement. VII The distribution of fluorescently labeled actin in living amebas. J Cell Biol. 1980;86:590–98. doi: 10.1083/jcb.86.2.590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terasaki M, Miyake K, McNeil PL. Large plasma membrane disruptions are rapidly resealed by Ca2+-dependent vesicle-vesicle fusion events. J Cell Biol. 1997;139:63–74. doi: 10.1083/jcb.139.1.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Therrien C, Di Fulvio S, Pickles S, Sinnreich M. Characterization of lipid binding specificities of dysferlin C2 domains reveals novel interactions with phosphoinositides. Biochemistry. 2009;48:2377–84. doi: 10.1021/bi802242r. [DOI] [PubMed] [Google Scholar]
- Ting SB, Caddy J, Hislop N, Wilanowski T, Auden A, et al. A homolog of Drosophila grainy head is essential for epidermal integrity in mice. Science. 2005;308:411–3. doi: 10.1126/science.1107511. [DOI] [PubMed] [Google Scholar]
- Togo T. Long-term potentiation of wound-induced exocytosis and plasma membrane repair is dependent on cAMP-response element-mediated transcription via a protein kinase C- and p38 MAPK-dependent pathway. J Biol Chem. 2004;279:44996–5003. doi: 10.1074/jbc.M406327200. [DOI] [PubMed] [Google Scholar]
- Togo T. Disruption of the plasma membrane stimulates rearrangement of microtubules and lipid traffic toward the wound site. J Cell Sci. 2006;119:2780–86. doi: 10.1242/jcs.03006. [DOI] [PubMed] [Google Scholar]
- Togo T, Krasieva TB, Steinhardt RA. A decrease in membrane tension precedes successful cell-membrane repair. Mol Biol Cell. 2000;11:4339–46. doi: 10.1091/mbc.11.12.4339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tucker PK, Evans IR, Wood W. Ena drives invasive macrophage migration in Drosophila embryos. Dis Model Mech. 2011;4:126–34. doi: 10.1242/dmm.005694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vacquier VD. The connection of blastomeres of sea urchin embryos by filopodia. Exp Cell Res. 1968;52:571–81. doi: 10.1016/0014-4827(68)90497-7. [DOI] [PubMed] [Google Scholar]
- Vaughan EM, Miller AL, Yu HY, Bement WM. Control of local Rho GTPase crosstalk by Abr. Curr Biol. 2011;21:270–77. doi: 10.1016/j.cub.2011.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velnar T, Bailey T, Smrkolj V. The wound healing process: an overview of the cellular and molecular mechanisms. J Int Med Res. 2009;37:1528–42. doi: 10.1177/147323000903700531. [DOI] [PubMed] [Google Scholar]
- Volkmann D, Baluska F. Actin cytoskeleton in plants: from transport networks to signaling networks. Microsc Res Tech. 1999;47:135–54. doi: 10.1002/(SICI)1097-0029(19991015)47:2<135::AID-JEMT6>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- Walter ND, Rice PL, Redente EF, Kauvar EF, Lemond L, et al. Wound healing after trauma may predispose to lung cancer metastasis: review of potential mechanisms. Am J Respir Cell Mol Biol. 2011;44:591–96. doi: 10.1165/rcmb.2010-0187RT. [DOI] [PubMed] [Google Scholar]
- Wang S, Tsarouhas V, Xylourgidis N, Sabri N, Tiklova K, et al. The tyrosine kinase Stitcher activates Grainy head and epidermal wound healing in Drosophila. Nat Cell Biol. 2009;11:890–95. doi: 10.1038/ncb1898. [DOI] [PubMed] [Google Scholar]
- Wang X, Xie W, Zhang Y, Lin P, Han L, et al. Cardioprotection of ischemia/reperfusion injury by cholesterol-dependent MG53-mediated membrane repair. Circ Res. 2010;107:76–83. doi: 10.1161/CIRCRESAHA.109.215822. [DOI] [PubMed] [Google Scholar]
- Weber KS, Klickstein LB, Weber PC, Weber C. Chemokine-induced monocyte transmigration requires cdc42-mediated cytoskeletal changes. Eur J Immunol. 1998;28:2245–51. doi: 10.1002/(SICI)1521-4141(199807)28:07<2245::AID-IMMU2245>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]
- Werner S, Smola H, Liao X, Longaker MT, Krieg T, et al. The function of KGF in morphogenesis of epithelium and reepithelialization of wounds. Science. 1994;266:819–22. doi: 10.1126/science.7973639. [DOI] [PubMed] [Google Scholar]
- Whitby DJ, Longaker MT, Harrison MR, Adzick NS, Ferguson MW. Rapid epithelialisation of fetal wounds is associated with the early deposition of tenascin. J Cell Sci. 1991;99(Pt. 3):583–86. doi: 10.1242/jcs.99.3.583. [DOI] [PubMed] [Google Scholar]
- Wood W, Faria C, Jacinto A. Distinct mechanisms regulate hemocyte chemotaxis during development and wound healing in Drosophila melanogaster. J Cell Biol. 2006;173:405–16. doi: 10.1083/jcb.200508161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood W, Jacinto A, Grose R, Woolner S, Gale J, et al. Wound healing recapitulates morphogenesis in Drosophila embryos. Nat Cell Biol. 2002;4:907–12. doi: 10.1038/ncb875. [DOI] [PubMed] [Google Scholar]
- Woolner S, Jacinto A, Martin P. The small GTPase Rac plays multiple roles in epithelial sheet fusion—dynamic studies of Drosophila dorsal closure. Dev Biol. 2005;282:163–73. doi: 10.1016/j.ydbio.2005.03.005. [DOI] [PubMed] [Google Scholar]
- Wu JQ, Ye Y, Wang N, Pollard TD, Pringle JR. Cooperation between the septins and the actomyosin ring and role of a cell-integrity pathway during cell division in fission yeast. Genetics. 2010;186:897–915. doi: 10.1534/genetics.110.119842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y, Brock AR, Wang Y, Fujitani K, Ueda R, et al. A blood-borne PDGF/VEGF-like ligand initiates wound-induced epidermal cell migration in Drosophila larvae. Curr Biol. 2009;19:1473–77. doi: 10.1016/j.cub.2009.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoo SK, Deng Q, Cavnar PJ, Wu YI, Hahn KM, et al. Differential regulation of protrusion and polarity by PI3K during neutrophil motility in live zebrafish. Dev Cell. 2010;18:226–36. doi: 10.1016/j.devcel.2009.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoo SK, Huttenlocher A. Spatiotemporal photolabeling of neutrophil trafficking during inflammation in live zebrafish. J Leukoc Biol. 2011;89:661–67. doi: 10.1189/jlb.1010567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young PE, Pesacreta TC, Kiehart DP. Dynamic changes in the distribution of cytoplasmic myosin during Drosophila embryogenesis. Development. 1991;111:1–14. doi: 10.1242/dev.111.1.1. [DOI] [PubMed] [Google Scholar]
- Young PE, Richman AM, Ketchum AS, Kiehart DP. Morphogenesis in Drosophila requires nonmuscle myosin heavy chain function. Genes Dev. 1993;7:29–41. doi: 10.1101/gad.7.1.29. [DOI] [PubMed] [Google Scholar]
- Zallen JA. Planar polarity and tissue morphogenesis. Cell. 2007;129:1051–63. doi: 10.1016/j.cell.2007.05.050. [DOI] [PubMed] [Google Scholar]