

Abstract
Purpose
This open-label, randomized phase II trial assessed efficacy and tolerability of two low-dose regimens of subcutaneous (SC) decitabine in patients with low- or intermediate-1–risk myelodysplastic syndrome (MDS).
Patients and Methods
Patients received decitabine 20 mg/m2 SC per day for 3 consecutive days on days 1, 2, and 3 every 28 days (schedule A) or 20 mg/m2 SC per day once every 7 days on days 1, 8, and 15 every 28 days (schedule B) for up to 1 year. Primary efficacy end point was overall improvement rate (OIR: complete remission [CR], partial remission [PR], marrow CR [mCR], or hematologic improvement [HI]). Secondary end points were HI, transfusion independence, cytogenetic response, overall survival (OS), and time to acute myeloid leukemia or death.
Results
Efficacy and safety populations were identical: schedule A, n = 43; schedule B, n = 22. Median time from MDS diagnosis to treatment was 3.6 months; 89% had de novo MDS. The trial was terminated early on achievement of protocol-defined OIR superiority of schedule A over schedule B; OIR was 23% for schedule A (seven CRs, three HIs) and 23% for schedule B (one mCR, one PR, three HIs). No differences were observed in secondary end points. Median OS was not reached; approximately 70% of patients were alive at 500 days. Patients in schedule A (67%) and schedule B (59%) were RBC/platelet independent on study. The most frequent drug-related adverse events overall were neutropenia (28% v 36%), anemia (23% v 18%), and thrombocytopenia (16% v 32%).
Conclusion
In this phase II study, low-dose decitabine showed promising results in patients with low- or intermediate-1–risk MDS.
INTRODUCTION
Myelodysplastic syndromes (MDSs) are a heterogeneous group of myeloid malignancies. Patients are generally divided into risk groups on the basis of International Prognostic Scoring System (IPSS) criteria. Lower-risk groups are those with IPSS low and intermediate-1 scores; higher-risk groups are those with IPSS intermediate-2 and high scores.1 Recently, it was proposed that the prognosis for patients with lower-risk disease is heterogeneous, with a substantial fraction of these patients having poor survival.2 Furthermore, the cause of death in patients with lower-risk disease is more commonly related to complications intrinsic to MDSs (particularly infection) rather than transformation to acute myeloid leukemia (AML).3 Recent data also indicate that poor prognosis mutational events are common in patients with poor-risk or lower-risk MDSs.4,5 These data suggest the need for the development of treatment strategies for specific subsets of patients with lower-risk and poor-risk MDSs. Treatment options for patients with low- or intermediate-1–risk MDSs include lenalidomide, hypomethylating agents, and immunosuppression.6,7 In the United States, lenalidomide is indicated for treatment of patients with transfusion-dependent anemia due to low- or intermediate-1–risk MDS associated with a del(5q) abnormality.8 Two hypomethylating agents, decitabine and azacitidine, are approved for treatment of MDSs.9,10 Azacitidine is indicated for treatment of patients with MDSs of all French-American-British (FAB) categories.10 Decitabine is indicated for treatment of patients with MDS in all FAB categories and intermediate-1–risk, intermediate-2–risk, and higher-risk IPSS categories.9 However, decitabine has not been evaluated in patients with lower-risk MDS, so the optimal dose and schedule of decitabine in this setting is not known. A recent phase I study of oral azacitidine suggested that lower drug exposure has activity in MDSs, potentially with a better toxicity profile.11 A study of three low-dose decitabine regimens, including a 5-day schedule at 20 mg/m2 intravenously (IV), a 5-day schedule at 20 mg/m2 subcutaneously (SC), and a 10-day schedule at 10 mg/m2 IV, suggested that low-dose regimens may be more effective and less toxic than higher-dose regimens.12 The SC schedule, studied in 14 patients, was well tolerated and active.12
On the basis of these data, with the hypothesis that lower doses of decitabine may be active and well tolerated in patients with MDSs, this study aimed to determine efficacy, safety, and tolerability of two low-dose SC regimens of decitabine in patients with low- or intermediate-1–risk MDS.
PATIENTS AND METHODS
Patients
Patients age ≥ 18 years with de novo or secondary IPSS low- or intermediate-1–risk MDS were eligible if they had Eastern Cooperative Oncology Group (ECOG) performance status (PS) of 0 to 2, adequate renal and hepatic function (creatinine < 2× upper limit of normal [ULN], total bilirubin < 2× ULN, and AST and ALT ≤ 2× ULN), unless proven to be related to disease infiltration. Transfusion-independent patients were eligible for the study. Patients were excluded if they were pregnant or nursing; had received prior therapy with decitabine or azacitidine; had received growth factor support, lenalidomide, or an investigational agent within 30 days before first decitabine dose; had an active uncontrolled systemic infection; or had concurrent disease that made the patient inappropriate for inclusion. Participants with child-bearing potential were required to use contraception throughout the study.
This study was conducted following the guidelines of each institution. The institutional review board of each center approved the protocol, and all patients provided written informed consent.
Treatment
This multicenter, open-label, randomized phase II study registered patients through an interactive voice response system. Patients were randomly assigned by using a central procedure to receive decitabine 20 mg/m2 SC per day for 3 consecutive days on days 1, 2, and 3 every 28 days (schedule A) or decitabine 20 mg/m2 SC per day once every 7 days on days 1, 8, and 15 (schedule B) every 28 days. Injections were administered at suitable anatomic sites (eg, abdomen, thigh, upper arm) on a rotating basis. Multiple injections during the same administration session were permitted for patients requiring more than 2 mL of drug.
Patients received their second course of therapy without interruption, regardless of their degree of myelosuppression. After the first course, intervals between subsequent cycles could be spaced out ± 3 days at the investigator's discretion. Treatment duration was up to 1 year or until progressive disease (PD), allogeneic bone marrow transplantation, intercurrent illness, patient request, or changes in condition rendering further treatment unacceptable in the judgment of the investigator. However, patients could continue treatment off protocol at that time. If prolonged myelosuppression (≥ 42 days of absolute neutrophil count [ANC] < 1 × 109/L and platelet count < 30 × 109/L) was reported after cycle 1, subsequent cycles of decitabine were given at the next lower dose (15 mg/m2 per day, then 10 mg/m2 per day, then 5 mg/m2 per day) after recovery (ANC ≥ 1 × 109/L and platelet count > 50 × 109/L). If a patient had a grade 3 or 4 nonhematologic toxicity and a subsequent course of therapy was appropriate (as judged by the investigator), the patient received the subsequent course at a reduced dose (15 mg/m2 per day). Patients could receive granulocyte colony-stimulating factor for fever of unknown origin, infection, and/or ANC less than 0.75 × 109/L, if indicated. Use of other growth factors, lenalidomide, azacitidine, or investigational agents was not permitted during the study.
Statistical Analyses
By using an adaptive, blocked randomization procedure of Bayesian design, the first 40 patients enrolled were randomly assigned 1:1 to schedule A or B, and subsequent patients were randomly assigned to schedule A or B on the basis of response data from the previous patients until a preferred schedule was identified or a maximum of 80 patients was enrolled. There was 80% statistical power to detect a superior dosing schedule. Superiority was defined as a posterior probability of more than 95% that the objective response (complete response [CR], marrow CR [mCR], partial response, or hematologic improvement [HI]) in one arm was superior to that of the other (see Data Supplement).
Categorical variables were compared by using Fisher's exact test, and continuous variables were compared by using one-way analysis of variance; survival was analyzed with Kaplan-Meier, Cox regression, and log-rank methods. All statistical assessments were two-sided with a significance level of less than .05. The efficacy (modified intent-to-treat [mITT]) and safety populations both comprised all randomly assigned patients who received at least one dose of study treatment.
Efficacy Assessments
The primary end point was overall improvement rate (OIR), defined as CR, partial response, mCR, or HI, measured at the end of each cycle by using each patient's best response. Response was assessed by using modified International Working Group 2006 criteria.13 Secondary end points included HI, cytogenetic response, overall survival (OS), and time to AML transformation or death. Transfusion independence, also a secondary end point, was defined as a patient being transfusion-free for ≥ 8 consecutive weeks between first dose of study drug and treatment discontinuation. Patients were considered independent at baseline if they did not receive any transfusions in the 8 weeks before the first dose; transfusions occurring on the date of the first dose were considered to be on study. An exploratory end point was assessment of the molecular effects of decitabine by using DNA methylation and gene expression assays on patients with available samples (assessments provided in Data Supplement).
Safety Evaluations
Toxicity was a secondary end point, assessed on the basis of adverse events (AEs), exposure to study drug, medical history, physical examinations, vital signs, concomitant medications, and laboratory assessments. All AEs were coded by using the Medical Dictionary for Regulatory Activities (MedDRA), v13.0. Toxicities were graded according to the National Cancer Institute Common Terminology Criteria for Adverse Events v3.0.
RESULTS
Patients
In total, 67 patients were randomly assigned from May 2008 to October 2009 at five US sites. The mITT and safety populations each comprised 65 patients (schedule A, n = 43; schedule B, n = 22). Baseline demographics and clinical characteristics are summarized in Table 1. Both groups were balanced, although there was a larger percentage of men in schedule B versus schedule A (P = .01). Patient disposition is shown in Figure 1.
Table 1.
Patient Demographics and Baseline Characteristics
| Characteristic | Schedule A (n = 43) |
Schedule B (n = 22) |
Overall (N = 65) |
|||
|---|---|---|---|---|---|---|
| No. | % | No. | % | No. | % | |
| Age, years | ||||||
| Mean | 67 | 71 | 68 | |||
| SD | 14 | 10 | 13 | |||
| Male sex* | 25 | 58 | 20 | 91 | 45 | 69 |
| Time from diagnosis to treatment, months | ||||||
| Median | 3.7 | 3.2 | 3.6 | |||
| Range | 0.2-118 | 0-51 | 0-118 | |||
| De novo MDS | 39 | 91 | 19 | 86 | 58 | 89 |
| IPSS intermediate-1 | 31 | 72 | 15 | 68 | 46 | 71 |
| ECOG PS 0-1 | 41 | 95 | 20 | 91 | 61 | 94 |
| Baseline cytogenetics | ||||||
| Diploid | 29 | 67 | 16 | 73 | 45 | 69 |
| −Y | 2 | 5 | 0 | 0 | 2 | 3 |
| del (5q) | 2 | 5 | 1 | 5 | 3 | 5 |
| del (20q) | 3 | 7 | 1 | 5 | 4 | 6 |
| +8 | 3 | 7 | 1 | 5 | 4 | 6 |
| Other | 5 | 12 | 2 | 9 | 7 | 11 |
| Complex (≥ three abnormalities) | 1 | 2 | 1 | 5 | 2 | 3 |
| < 5% blasts in bone marrow | 27 | 63 | 16 | 73 | 43 | 66 |
| Prior nonradiology oncologic therapy | 13 | 30 | 8 | 36 | 21 | 32 |
| Prognostic model score2† | ||||||
| Category 1 (score 0-2) | 7 | 18 | 2 | 9 | 9 | 15 |
| Category 2 (score 3-4) | 21 | 53 | 11 | 50 | 32 | 52 |
| Category 3 (score 5-7) | 12 | 30 | 9 | 41 | 21 | 34 |
Abbreviations: ECOG PS, Eastern Cooperative Oncology Group performance status; IPSS, International Prognostic Scoring System; MDS, myelodysplastic syndrome; SD, standard deviation.
P = .001 between groups.
N = 62 (schedule A, n = 40; schedule B, n = 22). Three patients did not have baseline bone marrow blasts reported.
Fig 1.

Patient disposition.
Overall, 15 patients (23%) completed 12 cycles of study treatment (schedule A, n = 11 [26%]; schedule B, n = 4 [18%]). Of 52 patients who discontinued, reasons included investigator decision (29% overall), PD (23%), and patient withdrawal of consent (23%). Patients on schedule A received a median of 7.0 treatment cycles (range, 1 to 13 treatment cycles), and patients on schedule B received a median of 5.5 treatment cycles (range, 2 to 16 treatment cycles). In total, 32% of patients had prior nonradiologic oncologic therapy. Of these, six patients (46%) in schedule A and three patients (38%) in schedule B had prior chemotherapy; one patient in schedule A had prior immunologic therapy, and one patient in schedule B had prior biologic therapy. No patient had received hormone therapy. In schedules A and B, respectively, four (9%) of 43 patients and one (5%) of 22 patients had prior radiation therapy.
Efficacy
The trial was terminated early following achievement of protocol-defined superiority, when the posterior probability of more than 95% was met that OIR of schedule A was superior to that of schedule B during the adaptive design phase. Posterior probability that schedule A was superior to schedule B was 95.5% when eight successes (OIR) and two failures (PD) with schedule A were compared with two successes (OIR) and three failures (PD) with schedule B. Thus, randomization to schedule B was terminated in October 2009. Enrollment onto schedule A was terminated in December 2009, based on sponsor review and confirmation of achievement of protocol- defined superiority.
Although protocol-defined superiority was reached, no significant difference in OIR was detected between groups (Table 2). The OIR was 23% for both schedules A (10 of 43) and B (five of 22; 95% CI, −21.1 to 22.1 for difference). No relevant between-group differences were detected in OIR when patients were classified by subgroups of age, IPSS risk assessment, time from MDS diagnosis, type of MDS, receipt or not of prior MDS therapy, baseline cytogenetic abnormalities, or ECOG PS.
Table 2.
Response to Treatment
| Variable | Schedule A (n = 43) |
Schedule B (n = 22) |
||
|---|---|---|---|---|
| No. | % | No. | % | |
| Response | ||||
| Overall improvement rate | 10 | 23 | 5 | 23 |
| Complete response | 7 | 16 | 0 | 0 |
| Marrow complete response | 0 | 0 | 1 | 5 |
| Partial response | 0 | 0 | 1 | 5 |
| Hematologic improvement | 3 | 7 | 3 | 14 |
| Transfusion status | ||||
| RBC baseline dependent/on-study independent | 6 of 17 | 35 | 4 of 8 | 50 |
| PLT baseline dependent/on-study independent | 3 of 4 | 75 | 1 of 4 | 25 |
| RBC/PLT baseline dependent/on-study independent | 7 of 18 | 39 | 4 of 10 | 40 |
| RBC baseline and on-study independent | 24 of 26 | 92 | 11 of 14 | 79 |
| PLT baseline and on-study independent | 34 of 39 | 87 | 17 of 18 | 94 |
| RBC/PLT baseline and on-study independent | 22 of 25 | 88 | 9 of 12 | 75 |
Abbreviation: PLT, platelet.
No significant differences in HI were seen between schedules A and B (schedule A, n = 3 [7%]; schedule B, n = 3 [14%]; P = not significant). Table 2 shows the proportion of patients who were independent from transfusion of RBCs, platelets, or both at baseline and on study. Overall, 29 patients (67%) on schedule A and 13 (59%) on schedule B were both RBC and platelet independent on study. There was no difference between groups in duration of transfusion independence of RBCs, platelets, or both. No relevant between-group differences in duration of transfusion independence were detected when patients were classified by subgroup. No patient in either group had a complete or partial cytogenetic response.
Survival
Median duration of follow-up was 14.6 months (range, 0.8 to 22.2 months) for schedule A and 15.5 months (range, 4.6 to 24.0 months) for schedule B. At time of analysis, median OS had not been reached; approximately 70% of all patients were alive at 500 days (Fig 2A), and no significant difference was noted between groups (hazard ratio, 1.5; 95% CI, 0.5 to 4.5). In addition, the groups did not differ significantly in time to AML transformation or death, and median time had not been reached in either group at the time of analysis (Fig 2B).
Fig 2.
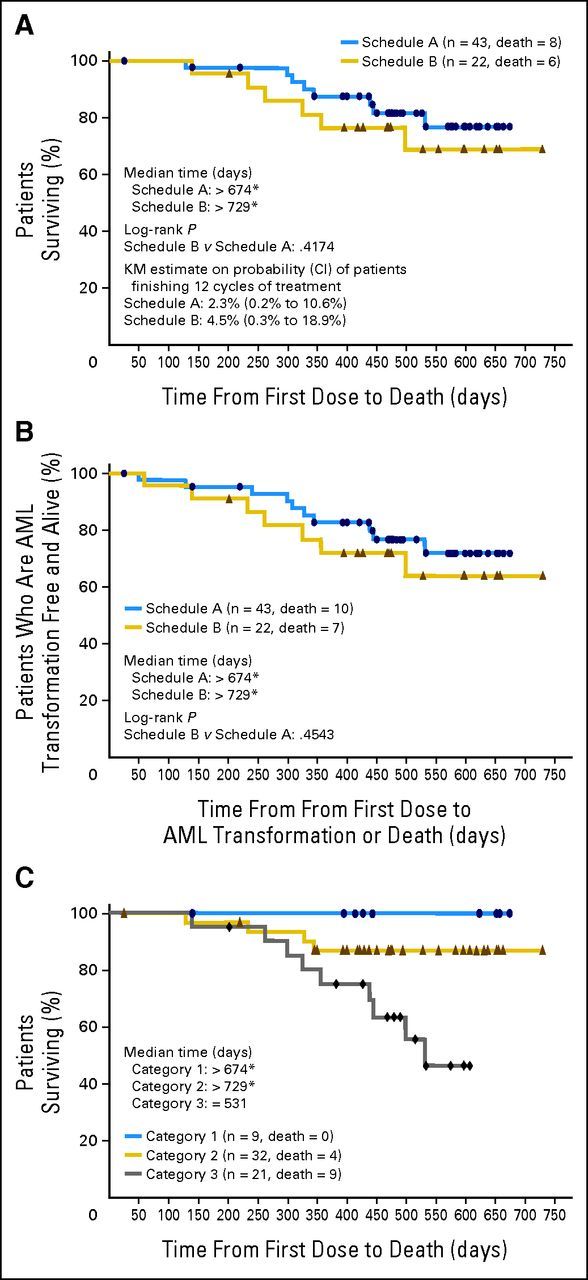
(A) Overall survival. Median was not reached. (B) Time to acute myeloid leukemia (AML) transformation or death. Median was not reached. (C) Median overall survival by myelodysplastic syndrome prognostic score category (n = 62). Note that three patients in the modified intent-to-treat population (n = 65) were excluded because they had missing bone marrow blast results and so could not be categorized. Median was not reached. KM, Kaplan-Meier. (*) Showing last data point.
Safety
All patients experienced at least one treatment-emergent AE. Drug-related AEs were reported in 26 patients (61%) receiving schedule A and 15 patients (68%) receiving schedule B; most were hematologic. The most frequently reported drug-related AEs for schedules A and B are summarized in Table 3. Drug-related AEs of grade 3 or higher (Table 3) were experienced by 40% of patients who received schedule A and 46% of patients who received schedule B; most were hematologic.
Table 3.
Drug-Related Adverse Events Occurring in ≥ 5% of Patients in Either Group
| Adverse Event | Schedule A (n = 43) |
Schedule B (n = 22) |
Overall (N = 65) |
|||
|---|---|---|---|---|---|---|
| No. | % | No. | % | No. | % | |
| Any grade | ||||||
| Neutropenia | 12 | 28 | 8 | 36 | 20 | 31 |
| Anemia | 10 | 23 | 4 | 18 | 14 | 22 |
| Thrombocytopenia | 7 | 16 | 7 | 32 | 14 | 22 |
| Fatigue | 8 | 19 | 2 | 9 | 10 | 15 |
| Leukopenia | 4 | 9 | 6 | 27 | 10 | 15 |
| Injection site pain | 5 | 12 | 3 | 14 | 8 | 12 |
| Nausea | 5 | 12 | 1 | 5 | 6 | 9 |
| Peripheral edema | 3 | 7 | 0 | 0 | 3 | 5 |
| Grade ≥ 3 | ||||||
| Anemia | 6 | 14 | 4 | 18 | 10 | 15 |
| Leukopenia | 3 | 7 | 3 | 14 | 6 | 9 |
| Neutropenia | 12 | 28 | 7 | 32 | 19 | 29 |
| Pancytopenia | 0 | 0 | 1 | 5 | 1 | 2 |
| Thrombocytopenia | 5 | 12 | 5 | 23 | 10 | 15 |
By study termination, eight (19%) of 43 patients who received schedule A and six (27%) of 22 patients who received schedule B had died. Reasons for deaths were PD (schedule A, n = 5; schedule B, n = 3), AE (n = 1 in each schedule), multiorgan failure (schedule B, n = 1), and unknown (schedule A, n = 2; schedule B, n = 1). No AEs led to death in either treatment group within the first 8 weeks on study. Two patients (5%) receiving schedule A had a drug-related serious AE leading to study discontinuation. One patient experienced angina and one patient had pseudomonal sepsis. No drug-related deaths occurred in either group.
DNA Methylation
Results of exploratory and gene-specific promoter DNA methylation analyses are provided in the Data Supplement.
Expected Survival Based on Lower-Risk Prognostic Model
In a post hoc analysis, we applied a lower-risk MDS scoring system2 to 62 of the 65 patients in the mITT population (three patients were excluded because bone marrow blasts were not reported). Of the three possible score categories (1, 2, or 3), approximately half the patients in each group (schedule A, 53%; schedule B, 50%) had a prognostic score category of 2, based on baseline cytogenetics, age, hemoglobin, platelets, and bone marrow blast percentage (Table 1). Analyzing OS by MDS prognostic score category, median OS was more than 674 days (median not reached) for patients in category 1, more than 729 days (median not reached) for patients in category 2, and 531 days for patients in category 3 (Fig 2C).
DISCUSSION
Decitabine is currently indicated as an IV injection for the treatment of patients with intermediate-1, intermediate-2, and high-risk MDS.9 We hypothesized that a low-dose SC formulation might be active in patients with lower-risk MDS. In our comparison of two low-dose regimens of SC decitabine in patients with low- or intermediate-1–risk MDS, protocol-defined superiority was achieved for schedule A over schedule B. That said, no between-group differences were found in OIR, HI, transfusion independence, or cytogenetic response. On-study RBC and platelet independence was achieved by 67% of patients on schedule A and 59% on schedule B. Median OS and median time to AML transformation or death had not been reached at the time of this analysis, but results appeared similar between groups. Both groups had an OIR of 23%, and there were no differences between groups when analyzed by various demographic and baseline characteristics. In addition, the AE profiles of both SC regimens of decitabine investigated in this study were consistent with the known safety profile of the IV formulation.9 Given these results, we recommend decitabine 20 mg/m2 per day SC for 3 consecutive days on days 1, 2, and 3 every 28 days (schedule A) for this patient population.
Results from exploratory analyses of DNA methylation suggested induction of long interspersed nucleotide elements hypomethylation on day 8 for schedule A but not for schedule B. However, the sample size was small (n = 17), so no conclusions can be drawn, and no correlation with clinical outcome was feasible. Levels of promoter methylation were analyzed in five genes reported to be methylated in MDS,14,15 but only a decrease in promoter methylation of PGRB was found. Finally, miR29b levels were generally low in these patients, and no association could be made with response.
Treatment options for patients with lower-risk MDS are not optimal. Stem-cell transplantation is usually not recommended as first-line therapy. Lenalidomide is the only therapy specifically indicated in the United States for patients with transfusion-dependent anemia due to low- or intermediate-1–risk MDS associated with a del(5q) abnormality, with or without additional cytogenetic abnormalities.6,8 Other treatment options for patients with lower-risk MDS include supportive care with transfusions and/or growth factor support.6 Decitabine is effective for treatment of patients with intermediate- and higher-risk MDS6,10 but has not been investigated extensively in patients with lower-risk disease. Results from this study demonstrate that low-dose SC decitabine warrants further evaluation in these patients.
A considerable proportion of patients with MDS have poor prognoses not identified by their IPSS classification. In a recent analysis of 856 patients with low- to intermediate-1–risk MDS, 80% of patients had a poor prognosis if they were untreated.2 Prognostic factors for survival in patients with low- to intermediate-1–risk MDS have been identified, and a scoring system has been developed to predict outcomes in these patients.2 Characteristics predictive of poor survival included platelets less than 50 × 109/L or 50 to 200 × 109/L, hemoglobin less than 10 g/dL, age 60 years or older, ≥ 4% bone marrow blasts, and unfavorable cytogenetics.2 In this study, patients with the highest prognostic score category (ie, 3) had the lowest median OS, as would be predicted by this model. However, medians were not reached for patients in categories 1 and 2.
This study is limited by its open-label design and by the small number of patients enrolled. These factors may limit the interpretation and extrapolation of the results to the larger population of adult patients with low- or intermediate-1–risk MDS. Consequently, larger studies are needed to confirm these data and to further understand the molecular effects of the intervention. Furthermore, we used a subcutaneous route that is not currently approved to administer decitabine but was chosen for patient convenience on the basis of prior experience. In addition, one of the basic assumptions for Bayesian adaptive randomization design, specifically the construction of predictive probability, is the continuity of the trend in data observed up to the decision point. Small sample size studies for low-probability events may result in volatile sample event rates over a certain period of time as the study is ongoing. Such high variance of an estimator, especially for studies of Bayesian adaptive randomization design in which the criteria defined a priori will be evaluated repeatedly against continuously accumulated data, may increase the chance of early false rejection of the null hypothesis and may result in a higher type I error. Furthermore, inherent in the analysis of OIR or any standard event rate analysis, is that the duration for the event of interest under observation is not addressed in the analysis. There was no significant difference in time to best OIR between the two schedules, although there was a high censoring rate for patients, and the long exposure time resulted in the median time to best response being not estimable for schedule A. Finally, for open-label phase II trials such as this, it is possible that recruitment patterns may change over the course of the trial because recruiters know that randomization is favoring the investigational treatment arm.16
In summary, in this study, lower dose schedules of decitabine administered subcutaneously appeared to be active and have an acceptable safety profile in patients with lower-risk MDS. Future studies targeting poor prognosis lower-risk patients with MDS should be considered.
Supplementary Material
Acknowledgment
We thank Yvonne E. Yarker, PhD, CMPP, of Peloton Advantage for providing drafts during the development of this article.
Footnotes
Supported by Eisai Pharmaceuticals. Medical writing, editing, and graphics assistance were provided by Peloton Advantage and funded by Eisai Pharmaceuticals.
Authors' disclosures of potential conflicts of interest and author contributions are found at the end of this article.
Clinical trial information: NCT00619099.
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
Although all authors completed the disclosure declaration, the following author(s) and/or an author's immediate family member(s) indicated a financial or other interest that is relevant to the subject matter under consideration in this article. Certain relationships marked with a “U” are those for which no compensation was received; those relationships marked with a “C” were compensated. For a detailed description of the disclosure categories, or for more information about ASCO's conflict of interest policy, please refer to the Author Disclosure Declaration and the Disclosures of Potential Conflicts of Interest section in Information for Contributors
Employment or Leadership Position: None Consultant or Advisory Role: Laurent Kassalow, Eisai Pharmaceuticals (C) Stock Ownership: None Honoraria: Hagop Kantarjian, Eisai Pharmaceuticals Research Funding: Guillermo Garcia-Manero, Eisai Pharmaceuticals; Gautam Borthakur, Eisai Pharmaceuticals; Nashat Gabrail, Amgen, Celgene, sanofi-aventis Expert Testimony: None Patents: None Other Remuneration: None
AUTHOR CONTRIBUTIONS
Conception and design: Guillermo Garcia-Manero, Laurent Kassalow
Provision of study materials or patients: Elias Jabbour, Lucy A. Godley, Nashat Gabrail
Collection and assembly of data: Guillermo Garcia-Manero, Gautam Borthakur, Stefan Faderl, Hui Yang, Sirisha Maddipoti, Lucy A. Godley, Jesus G. Berdeja, Ahmed Nadeem, Laurent Kassalow
Data analysis and interpretation: Guillermo Garcia-Manero, Elias Jabbour, Zeev Estrov, Lucy A. Godley, Nashat Gabrail, Jesus G. Berdeja, Laurent Kassalow, Hagop Kantarjian
Manuscript writing: All authors
Final approval of manuscript: All authors
REFERENCES
- 1.Greenberg P, Cox C, LeBeau MM, et al. International scoring system for evaluating prognosis in myelodysplastic syndromes. Blood. 1997;89:2079–2088. [PubMed] [Google Scholar]
- 2.Garcia-Manero G, Shan J, Faderl S, et al. A prognostic score for patients with lower risk myelodysplastic syndrome. Leukemia. 2008;22:538–543. doi: 10.1038/sj.leu.2405070. [DOI] [PubMed] [Google Scholar]
- 3.Dayyani F, Conley AP, Strom SS, et al. Cause of death in patients with lower-risk myelodysplastic syndrome. Cancer. 2010;116:2174–2179. doi: 10.1002/cncr.24984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bejar R, Stevenson K, Caughey B, et al. Validation of a prognostic model and the impact of SF3B1, DNMT3A, and other mutations in 289 genetically characterized lower risk MDS patient samples. Blood. 2011:118. (abstr 969) [Google Scholar]
- 5.Kantarjian HM, Larson RA, Guilhot F, et al. Efficacy of imatinib dose escalation in patients with chronic myeloid leukemia in chronic phase. Cancer. 2009;115:551–560. doi: 10.1002/cncr.24066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.National Comprehensive Cancer Network. Fort Washington, PA: NCCN; NCCN Clinical Practice Guidelines in Oncology: Myelodysplastic Syndromes, v2.2011. www.nccn.org. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Garcia-Manero G. Integrating care for patients with lower risk myelodysplastic syndrome. Semin Oncol. 2011;38:658–666. doi: 10.1053/j.seminoncol.2011.06.002. [DOI] [PubMed] [Google Scholar]
- 8.Celgene. Summit, NJ: Celgene; 2010. Revlimid (lenalidomide) package insert. [Google Scholar]
- 9.Esai. Woodcliff Lake, NJ: Eisai; 2010. Dacogen (decitabine) package insert. [Google Scholar]
- 10.Celgene. Summit, NJ: Celgene; 2011. Vidaza (azacitidine) package insert. [Google Scholar]
- 11.Garcia-Manero G, Gore SD, Cogle C, et al. Phase I study of oral azacitidine in myelodysplastic syndromes, chronic myelomonocytic leukemia, and acute myeloid leukemia. J Clin Oncol. 2011;29:2521–2527. doi: 10.1200/JCO.2010.34.4226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kantarjian H, Oki Y, Garcia-Manero G, et al. Results of a randomized study of 3 schedules of low-dose decitabine in higher-risk myelodysplastic syndrome and chronic myelomonocytic leukemia. Blood. 2007;109:52–57. doi: 10.1182/blood-2006-05-021162. [DOI] [PubMed] [Google Scholar]
- 13.Cheson BD, Greenberg PL, Bennett JM, et al. Clinical application and proposal for modification of the International Working Group (IWG) response criteria in myelodysplasia. Blood. 2006;108:419–425. doi: 10.1182/blood-2005-10-4149. [DOI] [PubMed] [Google Scholar]
- 14.Figueroa ME, Lugthart S, Li Y, et al. DNA methylation signatures identify biologically distinct subtypes in acute myeloid leukemia. Cancer Cell. 2010;17:13–27. doi: 10.1016/j.ccr.2009.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shen L, Kantarjian H, Guo Y, et al. DNA methylation predicts survival and response to therapy in patients with myelodysplastic syndromes. J Clin Oncol. 2010;28:605–613. doi: 10.1200/JCO.2009.23.4781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Korn EL, Freidlin B. Outcome: Adaptive randomization—Is it useful? J Clin Oncol. 2011;29:771–776. doi: 10.1200/JCO.2010.31.1423. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.