

Abstract
M13 bacteriophages act as versatile scaffolds capable of organizing single-walled carbon nanotubes and fabricating three-dimensional conducting nanocomposites. The morphological, electrical, and electrochemical properties of the nanocomposites are presented, as well as its ability to disperse and utilize single-walled carbon nanotubes effectively.
Keywords: conducting polymer, M13 bacteriophage, polyaniline, single-walled carbon nanotubes, three-dimensional network
Conductive polymers have received tremendous interests due to their widespread use in both scientific and engineering applications.[1–3] Polyaniline (PANI) is one of the most interesting conductive polymers due to its low cost, good processability, environmental stability, high conductivity and redox activity,[4] and various morphologies.[5–7] Incorporating carbon nanomaterials into PANI-based structures is a promising approach to improving physicochemical properties of nanocomposite films.[8] Among various carbon nanomaterials, pristine single-walled carbon nanotubes (SWNTs) have long been considered as ideal components due to their excellent electrical conductivity and high aspect ratio. Nanocomposites of SWNT and PANI have been extensively studied using different assembly techniques, including assembly from colloidal mixtures,[9] electrostatic deposition,[10] electropolymerization,[11–13] radical polymerization in solution,[14] and direct polymerization on SWNT supports.[15–18]
However, these approaches are limited by non-homogeneous suspensions and have a propensity for aggregation of both the PANI nanostructures and SWNTs, resulting in nanocomposites with unwanted physiochemical properties. While aggregation can be addressed by chemical modification of SWNTs,[14,16] or by adding surfactants[15,18] and binders,[11,12] these methods often lead to further deterioration of the electrical properties or non-uniform distribution of these materials in the nanocomposites. The thin films fabricated with these techniques require a high loading of SWNTs in the nanocomposites to enhance the electrochemical performance, so the scale-up potential of such composites is severely limited by the high material cost.[11,12,15] Therefore, an efficient and economical approach is highly desirable to incorporate SWNTs into the PANI-based nanocomposites effectively.
Biological materials as templates enable the environmentally friendly synthesis and organization of hybrid materials at the nanoscale. One particularly versatile bio-template is the M13 bacteriophage, a filamentous virus that is 880 nm in length and 6.5 nm in diameter. 2700 copies of its pVIII coat protein can be engineered to display affinity for specific materials, making these phage excellent templates for nanoscale manipulation of metals,[19] semiconductors,[20] metal oxides,[21] conducting polymers, and SWNTs.[22] Through genetically engineering approaches, the M13 virus can generate versatile scaffolds to organize nanoscale materials into three-dimensional (3D) nanostructures for various device applications, including piezoelectric generators,[23] batteries,[20] catalysts, bio-sensing,[24] photovoltaics,[25] and electrochemical capacitors.
Herein, we utilize M13 viruses that are genetically programmed to bind SWNTs in a controlled fashion by aligning the nanotubes along the length of the phage, and enabling clustering of SWNTs without aggregation. The SWNTs-phage complexes are used as the basis for the formation of crosslinked hydrogel scaffolds for the fabrication of continuous 3D porous PANI nanostructures which integrate SWNTs into the films homogeneously. The templated PANI nanowire (NW)-based network provides high surface area and directional pathways for electron transport. The resulting PANI-coated SWNT nanocomposites further improve the electrical conductivity of thin films. The virus-templated approach allows an efficient utilization of the high market price SWNTs in nanocomposites and improves the specific capacity of the electrochemical capacitors (from 379 to 664 F g−1) with a relatively low loading of SWNTs (less than 4.3 wt.%). In addition, an effective cost analysis demonstrates that this work enables the fabrication of nanocomposites with the lowest effective cost in USD F−1 among all SWNT/PANI-based nanocomposites in the literature. The assembly of viral hydrogels as templates promises the fabrication of 3D conducting nanocomposites and demonstrates great potential in sensing, energy conversion, and energy storage applications.
As a first step, M13 virus are genetically programmed to bind pristine SWNTs and disperse them homogeneously without any surfactant stabilization or chemical functionalization.[22] To bind SWNTs non-covalently along the virus surface proteins, a library of surface proteins is constructed, and the virus with the highest binding affinity towards SWNTs is identified using a bio-panning method.[22] Among several identified clones, a specific virus with the pVIII insert sequence DSPHTELP is selected for SWNT-binding. During dialysis, the virus (transmission electron microscopy (TEM) image shown in Figure 1A) and HipCo SWNTs are complexed to create SWNT@virus assemblies. High-resolution TEM imaging shown in Figure 1B reveals the SWNT bound along the major surface proteins without creating bundled aggregates.
Figure 1.

A. TEM image of three strings of M13 viruses. B. High-resolution TEM image of SWNT@virus complex, demonstrating SWNT bound along the major surface proteins without creating aggregates. C. Schematic illustration of the fabrication process of the virus-templated PANI and SWNT/PANI thin films, including (1) SWNT binding, (2) virus crosslinking, and (3) PANI coating. The photos in the bottom (from left to right) are the solutions of M13 virus and SWNT@virus, thin-film hydrogel coatings and free-standing hydrogel, and virus-templated PANI film coated on flexible substrate, respectively. D. SEM image of the lyophilized virus hydrogel.
The virus-enabled fabrication of 3D PANI thin films is shown in Figure 1C. To generate 3D scaffolds for porous PANI thin films, a virus solution (M13 virus or SWNT-binding virus) is drop-casted onto various substrates (e.g., glass, plastic); the substrates are then placed upside-down on the surface of an aqueous crosslinking agent containing 50 wt.% glutaraldehyde solution. The M13 virus and SWNT-binding virus scaffolds become hydrogels in this step. At a near-neutral pH value, the highly reactive aldehyde groups instantaneously react with the free amine groups in the viral surface proteins. The crosslinking reaction occurs as the glutaraldehyde diffuses upward across the fluid interface into the virus solution. The first reaction that takes place is the formation of a Schiff base bond between the virus and crosslinking agent (Equation 1), and a large variety of subsequent reactions may be involved in the crosslinking process.[26]
| (1) |
These covalent bonds allow the formation of free-standing films or thin-film hydrogel coatings depending on the concentration and volume of the virus solution (shown in Figure 1C).[25] Figure 1D shows a scanning electron microscopy (SEM) image of the lyophilized viral hydrogel where the diameter of the crosslinked viral NW is around 20–50 nm. This observation suggests that each crosslinked NW is a bundle of viruses based on a comparison to the size of individual viral particles, which are ~7 nm in diameter (Figure 1A). It should be mentioned that the porosity of the lyophilized hydrogel is much larger than that in the hydrated state due to the expansion of water that takes place during the freeze-drying process.
After the hydrogel scaffold is attained, a PANI thin film is generated directly on the scaffold by the polymerization of aniline using ammonium persulfate ((NH4)2S2O8) as an initiator in 2.0 M hydrochloric acid (HCl) solution (Equation 2).[27,28]
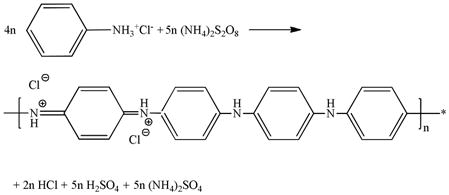 |
(2) |
During the polymerization, formation of PANI in the emeraldine state is indicated by the appearance of a dark greenish color. The formation of PANI is confirmed by Fourier transform infrared (FTIR) measurements (Figure 2A), and the spectrum shows three peaks at 1310 cm−1, 1490 cm−1 and 1585 cm−1, which are attributed to C-N stretching, C=C stretching in the benzenoid ring and C=C stretching in the quinoid ring, respectively. Furthermore, the X-ray photoelectron spectroscopy (XPS) survey scan shown in Figure 2B indicates the presence of nitrogen and chloride from PANI in the emeraldine state doped by HCl. In addition, the different carbon bonds composing the PANI backbone can be identified in the C1s scan (Figure 2C). These include C-N, C-C, and =CH- moieties. Carbonyl groups and other carbon chemistries at higher binding energies belong to the viral scaffold. The aspect ratio of the PANI NWs and the quality of thin films depend mainly on the shape of the crosslinked viral scaffold and the thickness of the PANI coating. Accordingly, we have systematically investigated three related parameters to fine tune the synthesis, namely (1) reaction time of the crosslinking process, (2) reaction time of the PANI coating, and (3) concentration of the aniline solution. With longer crosslinking process time, we observe higher film coverage and longer, denser crosslinked viral templates, leading to PANI NWs with higher aspect ratios in the network (SEM images shown in Figure S1A–S1C). Also, longer polymerization time results in thicker PANI coatings on the scaffolds. Thicker PANI coating on the scaffold can incorporate more active materials (i.e., PANI), enhancing the electrochemical performance of the PANI-based thin films. However, when the polymerization period exceeds ten hours, excessive coating leads to pore filling and random aggregation on the viral template (SEM images shown in Figure S1G–S1I), causing a drastic decrease in surface area and an increase in the resistance to electrolyte diffusion during electrochemical testing.
Figure 2.

A. FTIR spectra of the virus hydrogel and virus-templated PANI thin film. B. XPS survey scan of the virus-templated PANI thin film. C. XPS C1s spectrum and deconvolution of peaks for the virus-templated PANI thin film. D.–F. SEM images of the top-down view of virus-templated PANI thin films at different magnification. G. SEM images of the cross-section view of virus-templated PANI thin films. H. Pore size distribution calculated by the top-down SEM images. I. Thickness of the PANI films templated by the virus hydrogels crosslinked in different concentrations of the virus solution.
In addition, concentration of the aniline solution affects the morphology of thin films. As shown in Figure S2A and S2B, lower concentrations of the aniline solutions (0.04 M and 0.1 M) give rise to non-uniform and low coverage of the NW-based films. As shown in Figure S2C, an optimal concentration (0.8 M) of the aniline solution can produce uniform virus-templated networks. However, when the concentration of the aniline solution is increased to 2.0 M, larger aggregates are nucleated on the viral template, leading to lower aspect ratio wires and reduced surface-to-volume ratio structures (Figure S2D).
When the virus-templated PANI-based thin films are fabricated under the optimized conditions (two-hour crosslinking, two-hour polymerization using 0.8 M aniline solution with 0.2 M (NH4)2S2O8 solution), an uniform network composed of interconnected PANI NWs can be obtained, as imaged by SEM in Figure 2D–2F. The dimensions of the virus-templated PANI NWs within the network are approximately 300–400 nm in diameter and 3–4 μm in length (Figure 2E), which are much larger than the diameters of the previously observed virus NWs found in the lyophilized templates prior to polymerization (~20–50 nm). The increase in diameter indicates that the overall quantity of the deposited active material (i.e. PANI) is much greater than that of the viral scaffold itself. In addition, the templated synthesis enables a continuous coating along the joints of the network (Figure 2F), which reduces the contact resistance between the NWs and thus improves the overall electrical conductivity of thin films. The virus-templated PANI thin film shows higher conductivity (5.3 S cm−1) than the electrostatic multilayered PANI NW-based thin films (0.8 S cm−1).[29] The nanostructure also exhibits numerous macro-sized pores (Figure 2F), resulting in a high surface area; the porous and interconnected NW-based network persists across the entire film (thickness ~3 μm), providing direct pathways for efficient electron transport in the thin films (Figure 2G). The porosity and pore size distribution of the film are examined by analyzing the SEM image (Figure 2E) with ImageJ, shown in Figure 2H. The equivalent pore diameter ranges from 200 nm (or less) to 2 μm with an average pore diameter of ~350 nm, and the overall porosity of the film is 48.4%. Such a hierarchical nanostructure with interconnected pores maximizes the interfacial contact with the electrolyte, thereby increasing the mobility of the redox couples and minimizing electron recombination. The concentration of the virus solution mainly impacts the thickness of PANI-based films (Figure 2I), while the morphology of nanostructures remains unchanged. Using similar fabrication approaches, the virus-templated SWNT/PANI nanocomposites can be obtained (SEM image in Figure S3), that demonstrate similar morphologies to that of the virus-templated PANI films.
To further investigate the inner structure of the virus-templated SWNT/PANI nanocomposites, an ultra-sonication was performed to disassemble the SWNT/PANI NW complexes. After disassembly, some of the rigid SWNT@virus complexes stay intact (TEM images in Figure 3A and 3B), which could not be observed in the virus-templated PANI samples. The well-dispersed SWNT@virus complexes are able to maximize the contact area between the bound SWNTs and the coated PANI material. In addition, the presence of SWNTs in the nanocomposites is confirmed by Raman spectroscopy, Figure 3C, and the G-band peaks from SWNTs at ~1600 cm−1 and ~2600 cm−1 are observed both in the dried SWNT@virus hydrogels and the SWNT/PANI films. The loading of SWNTs in the nanocomposite can be controlled systematically by adjusting the ratio between the virus solution and SWNT solution during the complexation. To quantify the composition of the nanocomposites, thermogravimetric analyses (TGA) have been performed for the virus-templated PANI and SWNT/PANI samples under nitrogen environment. In Figure 3D, the weight of the purified SWNTs remains nearly identical even at high temperature ~900 °C in nitrogen environment. Loading of SWNTs in the nanocomposite (virus:SWNT = 1:8) is estimated to be 4.3 wt.% by comparing the difference between the two thermograms at 900 °C. The virus-enabled incorporation of highly conductive SWNTs enhances the electrical conductivity of thin films, and the improvement correlates strongly with the loading of SWNTs in the nanocomposites (Figure 3E). A remarkable improvement in electrical conductivity by nearly 340% (from 5.3 to 23.5 S cm−1) is observed as the loading of SWNTs is increased from 0 to 4.3 wt.%.
Figure 3.

A, B. TEM images of the virus-templated SWNT/PANI complex. The SWNT@virus are present and well-dispersed inside the PANI NW. C. Raman spectra of the virus hydrogel, SWNT@virus hydrogel, virus-templated PANI and SWNT/PANI films. D. TGA measurements of the purified SWNTs, virus-templated PANI and SWNT/PANI films (virus:SWNT = 1:8). Loading of SWNTs in the SWNT/PANI films (virus:SWNT = 1:8) is estimated ~4.3 wt.%. E. Electrical conductivity measurement on the virus-templated PANI and SWNT/PANI thin films. F. Impedance measurements on the virus-templated PANI and SWNT/PANI (with the loading of SWNTs ~ 4.3 wt.%) thin films. G. CV curves of the virus-templated PANI and SWNT/PANI (with the loading of SWNTs ~ 4.3 wt.%) thin films at the scan rate of 10 mV s−1. H. Galvanostatic charge-discharge curves of the virus-templated PANI and SWNT/PANI (with the loading of SWNTs ~ 4.3 wt.%) thin films, and the inset is the specific capacitances as a function of charge-discharge current densities. I. Analysis on the effective cost in USD F−1 of nanocomposites in this work and in the literature. The red, grey, green, and orange bars are the effective cost of the virus-templated SWNT/PANI thin films, SWNT/PANI composite electrodes, MWNT/PANI composite electrode, and rGO/PANI composite electrode, respectively.
Electrochemical impedance spectroscopy is used to analyze the interfacial resistances and provides additional insight into the effects of the incorporation of SWNTs on the electrochemical performance of nanocomposites (Figure 3F). In the PANI-based thin films, two major resistances limit the overall electrochemical performance: the charge-transfer resistance at the electrode-electrolyte interface, and the diffusion resistance of redox couples into the network. In Figure 3F, the semicircle at high frequency region is attributed to the resistance of charge-transfer process, and the diameter indicates the magnitude of charge-transfer resistance (RCT). In addition, the 45°-sloped portion of the curves in the lower frequency region indicates the Warburg resistance of ion transport. From the fitting results, RCT of the virus-templated SWNT/PANI thin film (RCT = 0.8 Ω) is smaller than that of the virus-templated PANI thin film (RCT = 2.0 Ω). This indicates that the SWNT/PANI matrix facilitates electron transport and reduces RCT, as a result of the enhanced electrical conductivity. In the lower frequency region in Figure 3F, both of the PANI-based thin films exhibit negligible Warburg resistances and the responses yield near-vertical lines, which are indicative of the ideal capacitive behavior. This ideal behavior results from the multi-scale morphology of the virus-templated structures, which allows electrolyte to maximize the contact area at the electrolyte/electrode interface and thereby minimize the Warburg resistance.
To evaluate the electrochemical behavior of both virus-templated PANI and SWNT/PANI thin films, cyclic voltammetry (CV) measurements are performed in a potential range of −0.2 V to 0.8 V (versus Ag/AgCl) in 1.0 M H2SO4 electrolyte with Pt as the counter electrode. In Figure S4, the virus-enabled nanostructures remain stable under a wide range of scan rates from 1 to 100 mV s−1. In Figure 3G, three pairs of the redox peaks are evident for all of the PANI-based films, and are labeled as A/A′, B/B′ and C/C′. The A/A′ and C/C′ peaks are associated with the redox nature of PANI molecules (leucoemeraldine and pernigraniline species); the weak B/B′ peaks are attributed to the double electron redox transition between benzoquinone and hydroquinone through the hydrolysis reaction of PANI.[30] In addition, the virus-templated SWNT/PANI thin film exhibits higher current density than PANI alone. Furthermore, the galvanostatic charging and discharging behaviors of both thin films are measured from 0 to 0.8 V at a discharging current of 5 mA cm−2 in 1.0 M H2SO4 electrolyte (shown in Figure 3H). The specific capacitance value, CS [F g−1], of the nanocomposites are estimated from the discharging data according to Equation 3 and the results are presented in the inset of Figure 3H.
| (3) |
where I is the charge-discharge current [A], m is the mass of the active materials (including the virus scaffold) [g], and ΔV/Δt is the rate of change [V s−1] in the discharging potential obtained from the charge-discharge trace. The highest CS of the virus-templated PANI thin film is ~379 F g−1, and ~ 664 F g−1 for the SWNT/PANI thin film with the loading of SWNTs around 4.3 wt.% in the nanocomposite. The high specific capacitance originates from two charge storage mechanism in SWNT/PANI nanocomposites, e.g., the electric double layer charge (EDLC) storage in SWNTs, additional contribution comes from the PANI nanowires due to its surface redox-chemistry (pseudocapacitance). The improvement on the electrochemical performance originates from the highly percolated electron conduction pathway and increased contact surface area between the well-dispersed SWNTs and coated PANI matrix. The cycle stability from the charge-discharge behavior is calculated and presented in Figure S5 at a current rate 1 mA cm−2 for over 2000 cycles. The SWNT/PANI electrode shows cycling stability with a drop (~ 17.7%) in specific capacitance after 2000 cycles. Moreover, to highlight the effectiveness of this approach, an effective cost analysis on this fabrication process is performed and the result is compared to the literature in Figure S6 and Figure 3I (see Supporting Information for the details).[9–18,29,31] To the best of our knowledge, the virus-templated approach reported here enables the fabrication of SWNT/PANI nanocomposites with the lowest effective cost among all SWNT/PANI-based nanocomposites reported in the literature. Furthermore, the virus-templated SWNT/PANI nanocomposites exhibits comparable or better effective cost compared to the multi-walled carbon nanotube (MWNT)/PANI and reduced graphene oxide (rGO)/PANI electrodes.
In conclusion, we have demonstrated the successful bio-templating with M13 bacteriophages to create 3D virus-templated SWNT/PANI nanocomposites with enhanced electrical conductivity and electrochemical performance. The macroporous virus-templated PANI films provide directional pathways for electron transport. The affinity of the viral surface proteins to SWNTs enables the formation of SWNT@virus complexes to further improve the electrical conductivity of thin films. A maximal specific capacity of 668 F g−1 is attained with the loading of SWNTs around 4.3 wt.% in the nanocomposite that achieves the lowest effective cost in USD F−1 among other SWNT/PANI nanocomposites reported in the literature. The bio-templated approach based on an aqueous-based synthetic process at a relatively low cost is expected to be translatable to large-scale production. The facile assembly of 3D NW-based conducting network and their nanocomposites are expected to be useful in many other applications, including, but not limited to solar cells, pseudocapacitors, batteries, and sensors.
Supplementary Material
Acknowledgments
This work was supported by Eni, S.p.A (Italy) through the MIT Energy Initiative Program, by NSF for the Center for Chemical Innovation under the NSF Center CHE-1305124, and by Institute for Collaborative Biotechnologies from the U.S. Army Research Office. P.-Y.C. and N.-M.D.C. acknowledge support from the MIT Energy Initiative Eni-MIT Energy Fellowship. M.N.H acknowledges a postdoctoral fellowship and N.-M.D.C. also acknowledges a postgraduate Scholarship, both from the Natural Sciences and Engineering Research Council of Canada.
Footnotes
The authors wish to dedicate this paper to the memory of Officer Sean Collier, for his caring service to the MIT community and for his sacrifice.
Supporting Information, including Experimental Section, Figure S1–S6, and Table S1, is available from the Wiley Online Library or from the author.
Contributor Information
Po-Yen Chen, Department of Chemical Engineering, The David H. Koch Institute for Integrative Cancer Research, Massachusetts Institute of Technology, 77 Massachusetts Avenue Cambridge, MA, 02139, USA.
Dr. Md Nasim Hyder, Department of Chemical Engineering, The David H. Koch Institute for Integrative Cancer Research, Massachusetts Institute of Technology, 77 Massachusetts Avenue Cambridge, MA, 02139, USA
David Mackanic, Department of Mechanical Engineering, Virginia Polytechnic Institute and State University, Blacksburg, VA 24061, USA.
Noémie-Manuelle Dorval Courchesne, Department of Chemical Engineering, The David H. Koch Institute for Integrative Cancer Research, Massachusetts Institute of Technology, 77 Massachusetts Avenue Cambridge, MA, 02139, USA.
Dr. Jifa Qi, Department of Biological Engineering, Department of Materials Science and Engineering, The David H. Koch Institute for Integrative Cancer Research, Massachusetts Institute of Technology, 77 Massachusetts Avenue Cambridge, MA, 02139, USA
Prof. Angela M. Belcher, Email: belcher@mit.edu, Department of Biological Engineering, Department of Materials Science and Engineering, The David H. Koch Institute for Integrative Cancer Research, Massachusetts Institute of Technology, 77 Massachusetts Avenue Cambridge, MA, 02139, USA
Prof. Paula T. Hammond, Email: hammond@mit.edu, Department of Chemical Engineering, The David H. Koch Institute for Integrative Cancer Research, Massachusetts Institute of Technology, 77 Massachusetts Avenue Cambridge, MA, 02139, USA
References
- 1.Lange U, Roznyatouskaya NV, Mirsky VM. Anal Chim Acta. 2008;614:1. doi: 10.1016/j.aca.2008.02.068. [DOI] [PubMed] [Google Scholar]
- 2.Green RA, Lovell NH, Wallace GG, Poole-Warren LA. Biomaterials. 2008;29:3393. doi: 10.1016/j.biomaterials.2008.04.047. [DOI] [PubMed] [Google Scholar]
- 3.Snook GA, Kao P, Best AS. J Power Sources. 2011;196:1. [Google Scholar]
- 4.Snook GA, Kao P, Best AS. Journal of Power Sources. 2011;196:1. [Google Scholar]
- 5.Park HW, Kim T, Huh J, Kang M, Lee JE, Yoon H. ACS Nano. 2012;6:7624. doi: 10.1021/nn3033425. [DOI] [PubMed] [Google Scholar]
- 6.Wang ZL, Guo R, Li GR, Lu HL, Liu ZQ, Xiao FM, Zhang M, Tong YX. J Mater Chem. 2012;22:2401. [Google Scholar]
- 7.Wang K, Huang J, Wei Z. J Phys Chem C. 2010;114:8062. [Google Scholar]
- 8.Frackowiak E, Béguin F. Carbon. 2001;39:937. [Google Scholar]
- 9.Liu Q, Nayfeh MH, Yau ST. J Power Sources. 2010;195:7480. [Google Scholar]
- 10.Hur J, Im K, Kim SW, Kim UJ, Lee J, Hwang S, Song J, Kim S, Hwang S, Park N. J Mater Chem A. 2013;1:14460. [Google Scholar]
- 11.Gupta V, Miura N. Electrochim Acta. 2006;52:1721. [Google Scholar]
- 12.Gupta V, Miura N. J Power Sources. 2006;157:616. [Google Scholar]
- 13.Zhou Y-k, He B-l, Zhou W-j, Li H-l. J Electrochem Soc. 2004;151:A1052. [Google Scholar]
- 14.Abdiryim T, Ubul A, Jamal R, Rahman A. Materials. 2012;5:1219. [Google Scholar]
- 15.Ge J, Cheng G, Chen L. Nanoscale. 2011;3:3084. doi: 10.1039/c1nr10424a. [DOI] [PubMed] [Google Scholar]
- 16.Liu J, Sun J, Gao L. J Phys Chem C. 2010;114:19614. [Google Scholar]
- 17.Niu Z, Luan P, Shao Q, Dong H, Li J, Chen J, Zhao D, Cai L, Zhou W, Chen X, Xie S. Energy Environ Sci. 2012;5:8726. [Google Scholar]
- 18.Wang K, Zhao P, Zhou X, Wu H, Wei Z. J Mater Chem. 2011;21:16373. [Google Scholar]
- 19.Lee YJ, Lee Y, Oh D, Chen T, Ceder G, Belcher AM. Nano Lett. 2010;10:2433. doi: 10.1021/nl1005993. [DOI] [PubMed] [Google Scholar]
- 20.Chen X, Gerasopoulos K, Guo J, Brown A, Wang C, Ghodssi R, Culver JN. ACS Nano. 2010;4:5366. doi: 10.1021/nn100963j. [DOI] [PubMed] [Google Scholar]
- 21.Chen PY, Ladewski R, Miller R, Dang X, Qi J, Liau F, Belcher AM, Hammond PT. J Mater Chem A. 2013;1:2217. [Google Scholar]
- 22.Lee YJ, Yi H, Kim WJ, Kang K, Yun DS, Strano MS, Ceder G, Belcher AM. Science. 2009;324:1051. doi: 10.1126/science.1171541. [DOI] [PubMed] [Google Scholar]
- 23.Lee BY, Zhang J, Zueger C, Chung WJ, Yoo SY, Wang E, Meyer J, Ramesh R, Lee SW. Nat Nanotechnol. 2012;7:351. doi: 10.1038/nnano.2012.69. [DOI] [PubMed] [Google Scholar]
- 24.Arter JA, Taggart DK, McIntire TM, Penner RM, Weiss GA. Nano Lett. 2010;10:4858. doi: 10.1021/nl1025826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chen P-Y, Dang X, Klug MT, Qi J, Dorval Courchesne N-M, Burpo FJ, Fang N, Hammond PT, Belcher AM. ACS Nano. 2013;7:6563. doi: 10.1021/nn4014164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Olde Damink LHH, Dijkstra PJ, Luyn MJA, Wachem PB, Nieuwenhuis P, Feijen J. J Mater Sci - Mater Med. 1995;6:460. [Google Scholar]
- 27.Chiang JC, MacDiarmid AG. Synt Met. 1986;13:193. [Google Scholar]
- 28.Sapurina I, Stejskal J. Polym Int. 2008;57:1295. [Google Scholar]
- 29.Hyder MN, Lee SW, Cebeci F, Schmidt DJ, Shao-Horn Y, Hammond PT. ACS Nano. 2011;5:8552. doi: 10.1021/nn2029617. [DOI] [PubMed] [Google Scholar]
- 30.Chang CM, Weng CJ, Chien CM, Chuang TL, Lee TY, Yeh JM, Wei Y. J Mater Chem A. 2013;1:14719. [Google Scholar]
- 31.Wang DW, Li F, Zhao J, Ren W, Chen ZG, Tan J, Wu ZS, Gentle I, Lu GQ, Cheng HM. ACS Nano. 2009;3:1745. doi: 10.1021/nn900297m. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.