

Abstract
Restoring functional β-cell mass is an important therapeutic goal for both type 1 and type 2 diabetes (1). While proliferation of existing β-cells is the primary means of β-cell replacement in rodents (2), it is unclear whether a similar principle applies to humans, as human β-cells are remarkably resistant to stimulation of division (3,4). Here, we show that 5-iodotubercidin (5-IT), an annotated adenosine kinase inhibitor previously reported to increase proliferation in rodent and porcine islets (5), strongly and selectively increases human β-cell proliferation in vitro and in vivo. Remarkably, 5-IT also increased glucose-dependent insulin secretion after prolonged treatment. Kinome profiling revealed 5-IT to be a potent and selective inhibitor of the dual-specificity tyrosine phosphorylation–regulated kinase (DYRK) and cell division cycle–like kinase families. Induction of β-cell proliferation by either 5-IT or harmine, another natural product DYRK1A inhibitor, was suppressed by coincubation with the calcineurin inhibitor FK506, suggesting involvement of DYRK1A and nuclear factor of activated T cells signaling. Gene expression profiling in whole islets treated with 5-IT revealed induction of proliferation- and cell cycle–related genes, suggesting that true proliferation is induced by 5-IT. Furthermore, 5-IT promotes β-cell proliferation in human islets grafted under the kidney capsule of NOD-scid IL2Rgnull mice. These results point to inhibition of DYRK1A as a therapeutic strategy to increase human β-cell proliferation.
Introduction
The loss of β-cell mass is a central feature of both type 1 and type 2 diabetes. Thus, understanding the mechanisms involved in increasing β-cell mass is an area of major research interest in diabetes. Although concerted efforts to differentiate β-like cells from embryonic stem cells or induced pluripotent (adult) stem cells are in progress, low conversion efficiency continues to be a challenge for developing cell-based therapies (6). Other approaches to enhance mammalian β-cell mass include the identification of small molecules or secreted factors that have the ability to replicate existing β-cells (1,4,7–11).
The replication of preexisting β-cells in rodents has been extensively studied at the molecular level, and several signaling pathways that promote β-cell regeneration have been proposed (2,12,13). In contrast, adult human β-cell replication has been reported to be virtually absent, suggesting that the capacity to replicate plateaus at ∼10 years of age (14,15). Nonetheless, reports from several independent laboratories studying humans with long-standing type 1 diabetes demonstrate their ability to increase circulating C-peptide levels in response to a mixed meal, as well as the presence of islet cells positive for Ki67 and insulin (16–18). These observations suggest that adult human β-cells in type 1 diabetes are functional and retain their ability to replicate, albeit at very low levels. These reports provide confidence that efforts to identify small molecules that safely and specifically enhance β-cell numbers in a controlled manner would be an attractive therapeutic approach to correct insulin deficiency in diabetes.
In order to discover small molecules capable of inducing β-cell proliferation, we developed a high-throughput system to culture dissociated human islet cells and measure proliferation in response to various conditions (19,20). In an untreated state, we measured a small but nonzero level of β-cell proliferation, as measured by incorporation of the thymidine analog 5-ethynyl-2’-deoxyuridine (EdU) (Supplementary Fig. 1) (21). As a positive control, we also observed a large increase in EdU-positive β-cells after adenoviral infection with cyclin-dependent kinase 6 (CDK6) and cyclin D1 (Supplementary Fig. 1) (22,23).
Recently, the adenosine kinase inhibitor 5-iodotubercidin (5-IT) was shown to increase rodent and porcine β-cell proliferation (5). Here, we show that 5-IT also potently promotes human β-cell proliferation both in vitro and in vivo, but mechanism-of-action studies suggest that 5-IT acts by inhibiting the dual-specificity tyrosine phosphorylation–regulated kinase 1A (DYRK1A). These results are consistent with recent reports that DYRK1A inhibition induces human β-cell proliferation (24,25). Our study provides proof of concept that small molecule–induced human β-cell proliferation is achievable, and lends considerable promise to the goals of regenerative medicine for diabetes treatment.
Research Design and Methods
Human Islets
Human islets were obtained through the Integrated Islet Distribution Program and the National Disease Research Interchange and cultured, stained, and imaged as described previously (19). Islets were washed with PBS and incubated in CMRL medium (Cellgro) supplemented with 10% FBS, 2 mmol/L glutamine, 100 units/mL penicillin, and 100 μg/mL streptomycin. Intact islets were stored in 60-mm Petri dishes in a 37°C incubator at ∼5,000 islet equivalents (IEQ) per 10 mL media. Donor information for each figure is provided in Supplementary Fig. 2.
Cell Lines
HTB-9 cells were obtained from American Type Culture Collection. Rat INS-1E cells (provided by Claes Wollheim and Pierre Maechler, University of Geneva, Geneva, Switzerland) (26) were maintained in RPMI 1640, containing 11 mmol/L glucose, 10% FBS, 10 mmol/L HEPES, 50 μmol/L 2-mercaptoethanol, and 1 mmol/L sodium pyruvate, and cultivated at 37°C with 5% CO2 in a humidified atmosphere.
Human Islet Dissociation
To dissociate tissue, islets were pelleted, washed in PBS, and centrifuged at 1,000 rpm for 5 min at room temperature. Pelleted islets were incubated at 5,000 IEQ/mL in Accutase (Life Technologies) at 37°C for 20 min. The pellet was resuspended in CMRL complete media, and an aliquot was removed for cell counting with a hemacytometer. Trypan blue was used to determine viability. We seeded ∼30,000 cells per well in 96-well plates or ∼15,000 cells per well in 384-well plates with a Multidrop Combi automated liquid dispenser (Thermo Scientific).
Proliferation Assay
Cells were incubated with a modified thymidine analog, EdU (Click-iT EdU; Invitrogen), throughout the treatment period. EdU incorporation and detection were performed as described by the manufacturer’s protocol.
Immunofluorescence
Cultures were fixed for 15 min at room temperature using freshly prepared 3% paraformaldehyde and washed twice with PBS. Cells were permeabilized for 20 min at room temperature in PBS containing 0.2% Triton-X 100 and blocked overnight at 4°C with 2% BSA in PBS. The C-peptide antibody GN-ID4 was obtained from the Developmental Studies Hybridoma Bank developed under the auspices of the Eunice Kennedy Shriver National Institute of Child Health and Human Development and maintained by the University of Iowa, Department of Biology (Iowa City, IA). Primary antibodies were diluted in antibody dilution buffer (ADB) (1% BSA in PBS) and incubated overnight at 4°C, followed by three washes in ADB. Cultures were incubated in secondary antibody (Alexa Fluor 594 goat anti-rat; Life Technologies) diluted in ADB for 1 h at room temperature, followed by five washes with PBS. Plates were stored foil sealed at 4°C with 100 μL/well PBS. Plates were imaged with an ImageXpress Micro automated microscope (Molecular Devices), and automated image analysis was performed using the Multiwavelength Cell Scoring Application Module in MetaXpress software. This method identifies nuclei using the DAPI channel and calculates fluorescein isothiocyanate (EdU) and tetramethylrhodamine (C-peptide) fluorescence based on this nuclear mask. β-Cells positive for EdU incorporation were also counted manually. High-magnification images were taken using a Zeiss Cell Observer.
Glucose-Stimulated Insulin Secretion
Dissociated human islet cells were incubated for 2 h in KRBH buffer (135 mmol/L NaCl, 3.6 mmol/L KCl, 5 mmol/L NaHCO3, 0.5 mmol/L NaH2PO4, 0.5 mmol/L MgCl2, 1.5 mmol/L CaCl2, 10 mmol/L HEPES, pH 7.4, 0.1% BSA) lacking glucose. Cells were subsequently incubated with KRBH buffer containing 1.67 mmol/L or 16.7 mmol/L glucose for 1 h. The supernatant was collected for measurement of secreted insulin by ELISA (ALPCO).
Kinase Profiling
Human kinome profiling was performed on 5-IT and ABT-702 by Millipore and Life Technologies according to the manufacturer’s protocols. ATP concentrations were within 15 μmol/L of the apparent KM for each enzyme.
Gene Expression Analysis
RNA was extracted using an RNeasy Plus kit (Qiagen), and gene expression profiling was performed with GeneChip Human Genome U133 Plus 2.0 Array (Affymetrix). For gene expression assays, cDNA was synthesized with random hexamers using a high-capacity cDNA reverse transcription kit (Life Technologies), followed by real-time PCR quantification using TaqMan primers. To estimate cell mass changes based on gene expression, we used previously published data (27) to determine which cells were most affected by 5-IT treatment. Let ai = Ai/Si (bi = Bi/Si, ci = Ci/Si, di = Di/Si, and ei = Ei/Si) be the fold change of expression of gene i in α (β, acinar, large duct, and small duct) cells, where Ai (Bi, Ci, Di, Ei, and Fi) are the corresponding intensity levels, the sum Si = Ai + Bi + Ci + Di + Ei + Fi, and Fi is expression of gene i in uncharacterized islet cells. Let α (β, γ, δ, ε, and φ) be the change of α (β, acinar, large duct, small duct, and uncharacterized) cell mass after treatment by 5-IT and let yi be the fold change of expression of gene i after islet treatment by 5-IT. Our goal is to determine α, β, γ, δ, ε, and φ using only fold-change values of FACS islet cells ai, bi, ci, di, and ei and fold-change values of 5-IT–treated islet cells yi. Since yi are fold-change values of 5-IT–treated versus untreated islet cells, we have
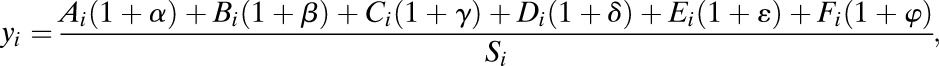 |
which can be reformulated as
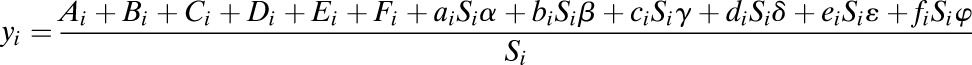 |
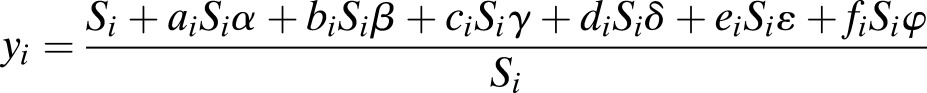 |
assuming that average gene expression of uncharacterized islet cells does not change with 5-IT treatment. The last equation means that the relationship can be seen as five-dimensional linear regression and the least-squares method (lm function in R or regress function in MATLAB) can be used to determine values for α, β, γ, δ, ε, and φ. To assess significance, we randomized the assignment of sorted replicate samples to individual cell type subpopulations.
Genetic Knockdown
Adenoviral-based knockdown of DYRK1A by short hairpin RNA (shRNA) was performed as previously described (22).
Western Blotting
INS-1E cells were seeded at the appropriate density, and after overnight incubation, cells were treated with compounds for 1 h, followed by lysis in RIPA buffer. Total protein was separated by 4–12% SDS-PAGE, transferred to a polyvinylidene fluoride membrane, and probed with the appropriate antibody. Blots were developed using the chemiluminescence detection system SuperSignal, and light emission was captured using an Imaging Station 4000MM Pro (CareStream).
Animal Studies
Eight- to ten-week-old male NOD.Cg-PrkdcscidIl2rgtm1Wjl/SzJ (NOD-scid IL2Rgnull [NSG]) mice (of n = 4) were used for experiments and grafted with human islets (1,000 IEQ) under the kidney capsule as described previously (28,29). One week posttransplantation, when the islets developed vascular anastomosis, mice were injected intraperitoneally with vehicle or 5-IT (0.25 mg/kg/body weight [BW], 50% [volume for volume] DMSO) twice a week for 3 weeks. Four weeks posttransplantation, mice were intraperitoneally injected with bromodeoxyuridine (BrdU; Sigma-Aldrich, St. Louis, MO) (100 mg/kg/BW) for 3 days and killed 6 h after receiving the final BrdU injection. Blood was collected before and 2 and 4 weeks after islets were grafted for the measurement of human insulin (Human Insulin ELISA Kit; Mercodia) and human C-peptide (C-Peptide ELISA Kit; Mercodia). In vivo glucose tolerance test (3 g/kg/BW, 20% dextrose) was performed at 4 weeks posttransplantation after overnight fasting. The graft-containing kidneys and pancreata were paraffin embedded and prepared for immunohistological analysis (30). Sections were blocked and incubated with primary antibodies (e.g., anti-BrdU, rabbit anti-Ki67 [BD Bioscience], anti–phosphohistone H3 [Millipore], or guinea pig anti-insulin [Linco]). The bound antibodies were detected with appropriate immunofluorescent secondary antibodies and counterstained with DAPI. At least 1,000–2,000 β-cell nuclei were counted per animal and data were expressed as percentage of BrdU+, Ki67+, or pHH3+ β-cells. All experiments were performed after approval from the Institutional Animal Care and Use Committee protocols at Joslin Diabetes Center and in accordance with National Institutes of Health guidelines.
Statistical Analysis
All data are expressed as the mean ± SD, unless indicated otherwise. Results were tested for significance using an unpaired two-tailed Student t test.
Results
5-IT Promotes Human β-Cell Proliferation
We tested the effects of 5-IT, an adenosine kinase inhibitor that increases proliferation of mouse, rat, and porcine β-cells (5), on EdU incorporation in human islet cells. After 6 days of treatment, we observed a striking increase in EdU incorporation in β-cells (Fig. 1A and B) that led to a 10- to 12-fold increase in β-cell proliferation, with a maximal effect at 1 μmol/L 5-IT. The findings were consistent across eight human islet donors, irrespective of age, sex, BMI, or cause of death (Fig. 1C and Supplementary Fig. 3). Other compounds, such as Bay K8644 (31) or WS-6 and analogs (32), have been reported in the literature to induce human β-cell proliferation; we had previously found that Bay K8644 induces nonspecific cellular proliferation (19). To assess the novelty of our findings, we compared the effects of WS-3, a commercially available analog of WS-6, with those of 5-IT. After a 6-day treatment, there was no significant difference between WS-3– and vehicle-treated islets (Supplementary Fig. 3). Further, WS-3 induced significant toxicity at 1 μmol/L. On the other hand, 5-IT induced a 20-fold increase in pair-matched islets (Supplementary Fig. 4), indicating that the cells were responding as expected. These results demonstrate that 5-IT reproducibly induces human β-cell proliferation in cell culture.
Figure 1.

Induction of human β-cell proliferation by 5-IT. A: Images showing insulin immunofluorescence and EdU incorporation in dissociated islets treated for 6 days with 1 μmol/L 5-IT. Representative EdU-positive β-cells are indicated with a white arrow. Scale bar, 100 μm. B: Higher-magnification image of EdU-positive β-cells. C: Heat map of EdU-positive β-cells in eight individual islet donors. Donor information is provided in Supplementary Fig. 2. D: GSIS after treatment of dissociated islets for 12 days with the indicated concentration of 5-IT. Cells were challenged with 1.67 mmol/L (black bars) or 16.7 mmol/L glucose (gray bars) for 1 h. Data represent the mean ± SD of four independent wells. *P < 0.01 and **P < 0.001, compared with DMSO treatment (high glucose), two-tailed Student t test. #P < 0.001, compared with DMSO treatment; ##P < 10−4, compared with DMSO treatment (low glucose), two-tailed Student t test.
Importantly, 5-IT did not induce proliferation of all cell types in the heterogeneous culture system (Supplementary Fig. 5A), suggesting a cell type–restricted effect. However, we did observe a few cells that were positive for EdU incorporation that appeared to weakly express glucagon (Supplementary Fig. 5B), indicating that 5-IT may exert its effects on other endocrine cells in cell culture. To determine whether other quiescent cell types could be stimulated by 5-IT, we tested 5-IT on primary rat neurons in culture, but saw no effect (Supplementary Fig. 5C and D). When we compared the results of EdU incorporation with Ki67 immunofluorescence in islet cells from the same donor treated with 5-IT, we observed a stronger dose-dependent increase in EdU incorporation upon 5-IT treatment than Ki67 positivity (Supplementary Fig. 6), suggesting that the cumulative readout provided by thymidine analog incorporation, coupled with prolonged (6-day) incubation in the presence of EdU, was required to observe a response to compound treatment.
In order to evaluate whether the proliferation induced in human β-cells caused the loss of β-cell function (33,34), we measured glucose-stimulated insulin secretion (GSIS) on cells treated for 12 days with 5-IT. In the DMSO-treated state, glucose stimulated an approximately twofold increase in insulin secretion in dissociated islet cells (Fig. 1D). After treatment with 5-IT, we observed an increase in GSIS upon 5-IT treatment that did not appear to rely upon the compound concentration (Fig. 1D). These results demonstrate that treated human islet cells are not only alive and normal in appearance but also function appropriately after prolonged compound treatment.
Promotion of Human β-Cell Proliferation Is Not Dependent Upon Adenosine Kinase Inhibition
Since 5-IT is primarily annotated as an adenosine kinase inhibitor, we tested the effects of the structurally unrelated adenosine kinase inhibitor ABT-702, which has an IC50 of 1.7 nmol/L (35,36). We found that ABT-702, tested up to 20 μmol/L, had no effect on EdU incorporation (Fig. 2A), suggesting that the mechanism of action of 5-IT in human β-cells may be different. Because 5-IT is a close analog of adenosine, we reasoned that it may act competitively in the ATP-binding pocket of many kinases, and it was likely to inhibit multiple kinases. Thus, we compared the effects of 100 nmol/L 5-IT with 500 nmol/L ABT-702 on the activity of 253 human protein kinases (37). Surprisingly, both compounds were quite selective, with ABT-702 inhibiting the tyrosine kinase TXK only moderately (60% remaining activity), whereas 5-IT strongly inhibited a related class of the CMGC family (which includes CDKs, mitogen-activated protein [MAP] kinases, glycogen synthase kinases [GSKs], and cell division cycle [CDC]–like kinases [CLKs]) kinases: DYRK1A, DYRK1B, DYRK2, DYRK3, and DYRK4 and CLK1, CLK2, CLK3, and CLK4, each inhibited to <10% remaining activity at 100 nmol/L (Fig. 2B). Closer examination of the effect of 5-IT on these kinases revealed IC50 values of 13, 10, 6, 3, 129, 36, 10, 195, and 14 nmol/L, respectively (Fig. 2C and Supplementary Fig. 7). As adenosine kinase is not a protein kinase, it was not included in the screening panel. These results suggest that the CMGC kinase family may be involved in the activity of 5-IT on β-cells.
Figure 2.

Inhibition of DYRK and CLK kinase families by 5-IT. A: Comparison of the effects of 6-day treatment with 5-IT and the structurally unrelated adenosine kinase inhibitor ABT-702. The percent of β-cells positive for EdU was measured and calculated. Data represent the mean ± SD of four independent wells and nine fields of view per well. **P < 0.01, compared with DMSO treatment, two-tailed Student t test. B: Kinome profiling of 253 human kinases. Tested kinases are indicated by closed circles, kinases inhibited by 5-IT are indicated in red, and the kinase inhibited by ABT-702 is indicated in green. C: Dose-dependent inhibition of indicated DYRK and CLK by 5-IT.
These results led us to hypothesize that the stimulation of human β-cell proliferation by 5-IT is due to inhibition of this subset of the CMGC kinase family. To test this hypothesis, we measured the effects of harmine, a natural product and established DYRK1A inhibitor (38), on EdU incorporation in human islets. Subsequent to our experiments, harmine was also reported to induce human β-cell proliferation (25). Consistent with those results, harmine induced a dose-dependent increase in EdU-positive cells at 5 and 10 μmol/L after 6-day treatment (Fig. 3A), suggesting that DYRK1A itself was the relevant target for inducing proliferation in these cells. We reasoned that the superior potency of 5-IT in stimulating β-cell proliferation could be explained by its greater potency for DYRK1A inhibition. To test this hypothesis, we developed a biochemical assay for DYRK1A activity, using the purified catalytic domain, and measured the activity of harmine and closely related structural analogs. Harmine was the most potent compound in our hands, with an IC50 of ∼300 nmol/L (Supplementary Fig. 8). We then compared the effects on β-cell proliferation of harmine and harmaline, which has no activity toward DYRK1A. Harmaline had no effect on EdU incorporation (Supplementary Fig. 8), lending further evidence to the important role for DYRK1A inhibition in stimulating β-cell proliferation.
Figure 3.

DYRK1A inhibitor harmine also induces β-cell proliferation. A: Effect of 6-day treatment with indicated concentrations of 5-IT or harmine. Data represent the mean ± SD of four independent wells and nine fields of view per well. *P < 0.05, **P < 0.01, and ***P < 0.001, compared with DMSO treatment, two-tailed Student t test. C-pep, C-peptide. B: Fluorescent image of dissociated islets treated for 6 days with 10 μmol/L harmine. Scale bar, 100 μm. EdU-positive β-cells are circled in white. C: Fluorescent image of dissociated islets treated with 10 μmol/L harmine and 1 μmol/L FK506. D: Fluorescent image of dissociated islets treated for 6 days with 0.5 μmol/L 5-IT. E: Fluorescent image of dissociated islets treated with 0.5 μmol/L 5-IT and 1 μmol/L FK506. F: Western blot of p27KIP phosphorylation (S10) in INS-1E cells in response to 24- or 48-h treatment with 10 μmol/L harmine or 0.5 μmol/L 5-IT. Actin was included as a loading control. G: Fluorescent image of dissociated islets untreated (left) or infected with adenovirus expressing sh-DYRK1A (right). H: Quantification of EdU-positive β-cells in untreated islets or islets in which DYRK1A has been knocked down by two different constructs. Data represent 12 independent wells per condition, imaged at nine sites per well. Error bars represent the 95% CI of the mean. NT, untreated.
DYRK1A Inhibition Plays a Key Role in Small-Molecule Activity
We reasoned that inhibition of DYRK1A may act to stimulate nuclear factor of activated T cells (NFAT) signaling to promote proliferation in β-cells. We would thus expect that FK506, an inhibitor of calcineurin and NFAT activation, should suppress the effects of 5-IT or harmine on β-cells. Indeed, cotreatment with 1 μmol/L FK506 completely blocked the effects of 1 μmol/L 5-IT or 10 μmol/L harmine, resulting in the absence of EdU-positive β-cells in the treated cultures (Fig. 3B–E). Recent evidence showed that DYRK1A phosphorylates the cell cycle inhibitor p27KIP, and that DYRK1A overexpression blocked cell proliferation (39); therefore, we tested the effects of 5-IT and harmine on rat INS-1E cells. We observed a decrease in p27KIP phosphorylation after 24 or 48 h (Fig. 3F), consistent with β-cell entry into the cell cycle. Finally, knockdown of DYRK1A by shRNA (22) resulted in a strong increase in EdU-positive β-cells (Fig. 3G and H), providing further evidence for the role of DYRK1A in β-cell proliferation.
Gene Expression Analysis of Compound-Treated Islets Reveals a Proliferative Response
These results were obtained in dissociated islet cells, which one could argue may alter or remove important cell contacts that more faithfully mimic the environment in vivo. Therefore, we treated intact human islets from three individual donors with 1 μmol/L 5-IT or DMSO for 6 days and performed gene expression profiling (Fig. 4A). 5-IT most strongly upregulated the genes NEFM, MMP3, and SLC2A2 while downregulating REG1B, REG3A, and OLFM4; these effects were confirmed by quantitative PCR (Supplementary Fig. 9). Interestingly, SLC2A2 encodes GLUT2, a major glucose transporter for the β-cell; its upregulation may explain, in part, the increase in glucose-induced insulin secretion seen after 12-day treatment. Gene set enrichment analysis (GSEA) (40) revealed that 10 of the top 30 enriched gene sets increased by 5-IT treatment were involved in proliferation (Fig. 4B and Supplementary Fig. 10). The top leading-edge genes contributing to GSEA results (41) include many involved in cell cycle machinery, members of the mini-chromosome maintenance complex, and topoisomerases (Fig. 4C). These data indicate that 5-IT induces changes in gene expression consistent with cell proliferation.
Figure 4.

Gene expression profiling of 5-IT–treated islets consistent with increased β-cell proliferation. A: Scatter plot of gene expression results of intact islets treated for 6 days with DMSO or 1 μmol/L 5-IT, expressed as the geometric average across three donors. Gray lines show the threshold for genes with threefold increase or decrease in gene expression. B: GSEA results reveal a number of sets associated with cell division. Representative gene set analysis shown. Statistical analysis performed as previously described (23,24). C: Top leading-edge genes contributing to GSEA. D: Estimated changes in cell mass (α, β, acinar, large duct, small duct, and uncharacterized) after treatment with 5-IT, determined by five-dimensional linear regression.
Because gene expression profiling was performed on intact islets without sorting cells, we sought to quantify the contribution of each cell population to the response to 5-IT treatment. We therefore compared our expression dataset to published data generated in fluorescently sorted human endocrine, acinar, and ductal cells (27), aiming to dissect the overall gene expression changes in terms of how much of that change is due to an increase in individual cell types (e.g., β-cells). Using five-dimensional linear regression and the least-squares method, we were able to determine the proportional changes in each cell population required to be responsible for the overall changes in gene expression induced by 5-IT. Our results indicate that a β-cell mass increase of 7.5% (95% CI 5.8–9.2) could account for these gene expression changes (Fig. 4D). Similarly, a 6.1% increase in the α-cell mass (95% CI 4.6–7.5) is predicted by the gene expression results, with relatively little change in the other populations (Fig. 4D). These results suggest that an increase in β-cell mass could account for these changes in gene expression. Despite these trends, permutation testing revealed that our analysis is likely underpowered (P = 0.1282), possibly due to the use of only three individual islet donors in the gene expression study. Future efforts to accumulate data from more islet donors may reveal more significant trends. Nonetheless, our results indicate that the expression of proliferation-associated genes is elevated in response to 5-IT. Additional studies to measure β-cell numbers after long-term treatment or to perform single-cell analysis will be necessary to dissect the cell selectivity of small-molecule response to DYRK1A inhibition.
5-IT Promotes Human β-Cell Proliferation Transplanted Into NSG Mice
Finally, we used an animal model to further evaluate the stimulatory effect of 5-IT on human β-cell replication in vivo. We transplanted 1,000 human IEQ from two different donors under the kidney capsule of 8-week-old male NSG mice (Fig. 5A). One week posttransplantation, mice receiving grafts were treated with 0.25 mg/kg/BW 5-IT (based on dose optimization studies) or vehicle (DMSO), whereas sham-operated mice did not receive human islets or 5-IT treatment. Graft survival and functionality were examined by insulin secretory response to glucose in mice that received human islets compared with sham controls (Fig. 5B). The detection of circulating human insulin and C-peptide levels in the posttransplantation periods clearly demonstrated functional human islets (Fig. 5C and D). Immunohistochemical analysis of kidney sections bearing human islet grafts showed an approximately fivefold (P < 0.01) increase in BrdU incorporation in the 5-IT–treated group, indicating a transition from the G1 to the S phase of the cell cycle (Fig. 6A and B). The enhanced mitosis with an approximately sixfold increase in the treated group was confirmed using Ki67 staining (P < 0.05) (Fig. 6C), and a dramatic increase (∼10-fold) in pHH3 immunostaining confirmed a progression of the β-cells into the G2 or M phases (P < 0.05) (Fig. 6D). We also confirmed proliferation by using another marker, PDX1. Whereas the PDX1/BrdU double-positive cells showed an approximately fourfold increase in human islets treated with 5-IT (P < 0.05) (Fig. 6E and F), the PDX1/Ki67 double-positive cells were approximately sixfold increased in the treated group (P < 0.05) (Fig. 6E and G). Furthermore, consistent with our in vitro analysis, endogenous NSG pancreatic islets showed an approximate 2.5-fold increase in BrdU+ β-cells in the 5-IT–treated group compared with nontreated or sham-operated mice (Fig. 7A and B).
Figure 5.

Study design and functionality of human islets grafted under the kidney capsule of NSG mice. A: Experimental strategy showing human islet transplantation under the kidney capsule of male NSG mice followed by either vehicle or intraperitoneal 5-IT (0.25 mg/kg/BW) injection twice a week. BrdU (100 mg/kg/BW) was injected 3 weeks posttransfer for 3 days, and 6 h after the last injection, pancreata were harvested for immunohistochemical analyses. Image shows human islets under the kidney at the end of the follow-up. B: Glucose tolerance test performed 4 weeks after human islet transplantation on sham-operated, vehicle (HI Tx), or 5-IT–treated (HI Tx + 5-IT) mice grafted with human islets (n = 3–4). Glucose was measured at the indicated time (minutes) of the tolerance test. Human insulin (C) and human C-peptide (D) ELISA results assayed from three different groups 2 and 4 weeks post–human islet transplantation. Representative experiment from n = 4. Error bars indicate ±SEM. P values were determined by unpaired Student t test. *P < 0.05; **P < 0.01. §Sham vs. HI Tx; *sham vs. HI Tx + 5-IT. ND, not detectable.
Figure 6.

5-IT treatment promotes human β-cell proliferation in vivo. A: Immunohistochemistry of vehicle or 5-IT–treated kidney sections grafted with human islets (10×) showing BrdU (green), insulin (red), and DAPI (blue). Yellow arrows define the kidney and human islet grafts. White arrows mark proliferating β-cells. Magnified images (×40) show each immunofluorescent staining separately (indicating nuclear gap for replicating β-cell surrounded by insulin) for the proliferating β-cell. Representative experiment from n = 3–4. Quantification of proliferating β-cells for BrdU/insulin (B), Ki67/insulin (C), and pHH3/insulin (D) double-positive cells from human islet grafts from A. Between 1,000 and 2,000 β-cells were counted in each section. E: Immunohistochemistry for another β-cell marker (PDX-1) in human islet–grafted kidney sections treated with or without 5-IT, using PDX-1 (red), Ki67/BrdU (green), and DAPI (blue). Representative experiment from n = 4. Quantification of proliferating β-cells for BrdU/PDX-1 (F) and Ki67/BrdU (G) double-positive cells. Error bars indicate ±SEM. P values were determined by unpaired Student t test. *P < 0.05; **P < 0.01. Absolute numbers of β-cells counted are provided in Supplementary Fig. 13. HI, human islet; HI Tx, vehicle treated; HI Tx + 5IT, 5-IT treated.
Figure 7.

5-IT effects on endogenous mouse pancreas. A: Immunohistochemistry for proliferating β-cells in pancreatic sections of human islet graft–bearing mice showing insulin (red), BrdU (green), and DAPI (blue). Magnified images (×40) mark proliferating β-cells from 10× merged samples. Representative experiment from n = 4. B: Quantification of proliferating β-cells for BrdU/insulin. Error bars indicate ±SEM. P values were determined by unpaired Student t test. *P < 0.05. HI, human islet; HI Tx, vehicle treated; HI Tx + 5IT, 5-IT treated.
Although our in vitro findings demonstrated a few glucagon-expressing cells that were positive for EdU incorporation, careful analyses of α-cell proliferation in both human islets grafted under the mouse kidney capsule and in endogenous mouse pancreas showed no significant alterations in proliferation between the treated and nontreated groups, suggesting a β-cell–specific effect of 5-IT (Supplementary Fig. 11). The difference between in vitro and in vivo results in terms of α-cell proliferation might be due to the single cell state and warrants additional research. Indeed, the lack of vascularization in cultured islets may possibly account for these differences. To further investigate whether 5-IT leads to morphological changes and/or stimulates replication in other cell types in vivo, we examined liver, adipose, muscle and kidney tissue harvested from 3-week 5-IT–treated mice. We observed no significant differences between control and 5-IT–treated mice in the proliferation of any of these tissues (Fig. 8 and Supplementary Fig. 12). Thus, treatment with 5-IT can be effectively used to selectively promote human β-cell proliferation.
Figure 8.

5-IT treatment did not increase cell proliferation in other tissues. Representative images (A) and quantification (B) of nuclei BrdU+ in indicated tissues. Insets show BrdU+ proliferating cells. For each mouse, a minimum of 3,000 cells were counted in sections from liver and muscle and 1,000–2,000 cells were counted in sections from visceral (Visc.) and subcutaneous (Sc.) adipose tissue. Representative experiment from n = 4. Error bars indicate ±SEM. HI, human islet; HI Tx, vehicle treated; HI Tx + 5IT, 5-IT treated.
Discussion
These results demonstrate successful small-molecule induction of β-cell proliferation in human islets. Using kinome profiling, chemical epistasis analysis, and structure-activity relationship analysis, we have shown that inhibition of DYRK1A kinase by 5-IT is an attractive approach to enhance β-cell proliferation, likely by causing NFAT activation in β-cells. We also observed enhanced glucose-dependent insulin secretion after 5-IT treatment, which appears to emerge too early to be the result of an increase in β-cell number. Further studies are required to understand the mechanism of this effect. Our results are consistent with a parallel study demonstrating that harmine induces β-cell proliferation through inhibition of DYRK1A and activation of Myc signaling in β-cells (25). Interestingly, harmine was discovered in that study after high-throughput screening for moderate induction of Myc. However, 5-IT displays superior potency against DYRK1A (IC50 = 14 nmol/L, as opposed to 80–300 nmol/L, based on our results and literature reports, for harmine). This difference in potency correlates well with cellular efficacy in stimulating β-cell proliferation; 5-IT shows optimal activity at 0.5–1 μmol/L, whereas harmine is active at 5–10 μmol/L. Together, these independent studies strongly suggest that small-molecule inhibition of DYRK1A is an effective strategy to induce human β-cell regeneration through proliferation.
The DYRK kinases play key roles in the control of cell proliferation and differentiation. DYRK1A and DYRK1B are negative regulators of the cell cycle that promote the switch to a quiescent cellular state (42,43). Further, overexpression of human DYRK1A suppresses cell proliferation in multiple cell lines (42). The DYRK1A gene is located on chromosome 21, and increased gene dosage is associated with cognitive defects of Down syndrome, primarily due to increased activity toward its substrate, NFAT (44). The role of NFATc1 in β-cell development, proliferation, and insulin secretion was studied in greater detail, revealing that calcineurin and NFAT signaling were important for each of these functions, and was conserved across rodent and human β-cells, consistent with a negative role for DYRK1A (45).
Recent reports have shown that, in mice, DYRK1A is positively correlated with β-cell proliferation (46,47). However, the large differences between rodent and human β-cell replication (3), the importance of DYRK1A in development (44), and the divergence of genetic and acute small-molecule perturbation, especially for kinases (48), all make further study of selective and acute DYRK1A inhibition of paramount importance to β-cell regeneration. As is the case when promoting cell proliferation, cellular selectivity will be desirable to avoid oncogenesis. According to T1DBase (http://www.t1dbase.org), DYRK1A is highly expressed in human islets but moderately expressed in mouse and rat islets, as measured by Affymetrix profiling. Although ubiquitously expressed, DYRK1A is the most highly expressed of these kinases in primary human β-cells, suggesting that the potency of 5-IT against DYRK1A may explain why human cells are reproducibly responsive to this compound. For example, a higher concentration of 5-IT may be required to observe proliferation of other cell types. Second, although DYRK1A is expressed ubiquitously, its kinase targets (such as NFAT) may not be, leading to substrate-level regulation of β-cell specificity. Third, there may be polypharmacological effects in which the inhibition of multiple kinases by 5-IT or harmine is required for β-cell selectivity. To bypass this concern, future development of β-cell–specific probes to deliver DYRK1A inhibitors may help impart unique selectivity. In addition to cell selectivity, the high research interest in, yet difficulty in separating small-molecule activity of, the DYRK and CLK families (49,50) make identifying selective small-molecule inhibitors of DYRK1A the next challenge for studying β-cell proliferation and inducing therapeutically relevant β-cell regeneration.
Article Information
Acknowledgments. The authors are grateful to Dr. A. Stewart and Dr. P. Wang (Mt. Sinai School of Medicine) for generously providing adenoviral shRNA constructs (supported by Einstein-Mount Sinai Diabetes Research Center NIH/National Institute of Diabetes and Digestive and Kidney Diseases [NIDDK] P-30 DK 0205241); to Dr. P. Kilian (JDRF), Dr. O. Plettenburgh (Sanofi), and Dr. P. Strop (Sanofi) for helpful suggestions and support; to Dr. S. Schreiber and Dr. A. Shamji (Broad Institute) for helpful comments and advice; and to R. Ng (Joslin Diabetes Center) for assistance with analyses of pancreas sections.
Funding. This work was supported by a JDRF Strategic Research Agreement (17-2011-642). E.D. is supported by a Senior JDRF Fellowship (JDRF-3-APF-2014-220-A-N), and R.N.K. acknowledges support from the National Institute of Diabetes and Digestive and Kidney Diseases (R01 DK67536, R01 DK103215, and P30 DK036836) and JDRF (2-SRA-2015-17).
Duality of Interest. No potential conflicts of interest relevant to this article were reported.
Author Contributions. E.D. researched data, contributed to discussion, and reviewed and edited the manuscript. D.W., A.V., and S.M.B. researched data and contributed to discussion. B.C.M., S.K., J.H., T.J.G., and D.E.O. researched data. V.D. and P.A.C. analyzed data. R.N.K. and B.K.W. designed the study, contributed to discussion, and wrote the manuscript. R.N.K. and B.K.W. are the guarantors of this work and, as such, had full access to all the data in the study and take responsibility for the integrity of the data and the accuracy of the data analysis.
Footnotes
This article contains Supplementary Data online at http://diabetes.diabetesjournals.org/lookup/suppl/doi:10.2337/db15-1127/-/DC1.
S.M.B. is currently affiliated with Intellia Therapeutics, Inc., Cambridge, MA.
See accompanying article, p. 1496.
References
- 1.Vetere A, Choudhary A, Burns SM, Wagner BK. Targeting the pancreatic β-cell to treat diabetes. Nat Rev Drug Discov 2014;13:278–289 [DOI] [PubMed] [Google Scholar]
- 2.Dor Y, Brown J, Martinez OI, Melton DA. Adult pancreatic beta-cells are formed by self-duplication rather than stem-cell differentiation. Nature 2004;429:41–46 [DOI] [PubMed] [Google Scholar]
- 3.Bernal-Mizrachi E, Kulkarni RN, Scott DK, Mauvais-Jarvis F, Stewart AF, Garcia-Ocaña A. Human β-cell proliferation and intracellular signaling part 2: still driving in the dark without a road map. Diabetes 2014;63:819–831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kulkarni RN, Mizrachi EB, Ocana AG, Stewart AF. Human β-cell proliferation and intracellular signaling: driving in the dark without a road map. Diabetes 2012;61:2205–2213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Annes JP, Ryu JH, Lam K, et al. Adenosine kinase inhibition selectively promotes rodent and porcine islet β-cell replication. Proc Natl Acad Sci U S A 2012;109:3915–3920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Maehr R, Chen S, Snitow M, et al. Generation of pluripotent stem cells from patients with type 1 diabetes. Proc Natl Acad Sci U S A 2009;106:15768–15773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vetere A, Wagner BK. Chemical methods to induce beta-cell proliferation. Int J Endocrinol 2012;2012:925143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dirice E, Kahraman S, Jiang W, et al. Soluble factors secreted by T cells promote β-cell proliferation. Diabetes 2014;63:188–202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.El Ouaamari A, Kawamori D, Dirice E, et al. Liver-derived systemic factors drive β cell hyperplasia in insulin-resistant states. Cell Reports 2013;3:401–410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.El Ouaamari A, Dirice E, Gedeon N, et al. SerpinB1 promotes pancreatic beta cell proliferation. Cell Metab 2016;23:194–205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dirice E, Kulkarni RN. Harnessing immune cells to enhance β-cell mass in type 1 diabetes. J Investig Med 2016;64:14–20 [DOI] [PubMed] [Google Scholar]
- 12.Georgia S, Hinault C, Kawamori D, et al. Cyclin D2 is essential for the compensatory beta-cell hyperplastic response to insulin resistance in rodents. Diabetes 2010;59:987–996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhou JX, Dhawan S, Fu H, et al. Combined modulation of polycomb and trithorax genes rejuvenates β cell replication. J Clin Invest 2013;123:4849–4858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gregg BE, Moore PC, Demozay D, et al. Formation of a human β-cell population within pancreatic islets is set early in life. J Clin Endocrinol Metab 2012;97:3197–3206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Meier JJ, Butler AE, Saisho Y, et al. Beta-cell replication is the primary mechanism subserving the postnatal expansion of beta-cell mass in humans. Diabetes 2008;57:1584–1594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Keenan HA, Sun JK, Levine J, et al. Residual insulin production and pancreatic ß-cell turnover after 50 years of diabetes: Joslin Medalist Study. Diabetes 2010;59:2846–2853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Oram RA, Jones AG, Besser RE, et al. The majority of patients with long-duration type 1 diabetes are insulin microsecretors and have functioning beta cells. Diabetologia 2014;57:187–191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sherr JL, Ghazi T, Wurtz A, Rink L, Herold KC. Characterization of residual β cell function in long-standing type 1 diabetes. Diabetes Metab Res Rev 2014;30:154–162 [DOI] [PubMed] [Google Scholar]
- 19.Walpita D, Hasaka T, Spoonamore J, et al. A human islet cell culture system for high-throughput screening. J Biomol Screen 2012;17:509–518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Walpita D, Wagner BK. Evaluation of compounds in primary human islet cell culture. Curr Protoc Chem Biol 2014;6:157–168 [DOI] [PubMed] [Google Scholar]
- 21.Salic A, Mitchison TJ. A chemical method for fast and sensitive detection of DNA synthesis in vivo. Proc Natl Acad Sci U S A 2008;105:2415–2420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fiaschi-Taesch N, Bigatel TA, Sicari B, et al. Survey of the human pancreatic beta-cell G1/S proteome reveals a potential therapeutic role for cdk-6 and cyclin D1 in enhancing human beta-cell replication and function in vivo. Diabetes 2009;58:882–893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fiaschi-Taesch NM, Salim F, Kleinberger J, et al. Induction of human beta-cell proliferation and engraftment using a single G1/S regulatory molecule, cdk6. Diabetes 2010;59:1926–1936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shen W, Taylor B, Jin Q, et al. Inhibition of DYRK1A and GSK3B induces human β-cell proliferation. Nat Commun 2015;6:8372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang P, Alvarez-Perez JC, Felsenfeld DP, et al. A high-throughput chemical screen reveals that harmine-mediated inhibition of DYRK1A increases human pancreatic beta cell replication. Nat Med 2015;21:383–388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Merglen A, Theander S, Rubi B, Chaffard G, Wollheim CB, Maechler P. Glucose sensitivity and metabolism-secretion coupling studied during two-year continuous culture in INS-1E insulinoma cells. Endocrinology 2004;145:667–678 [DOI] [PubMed] [Google Scholar]
- 27.Dorrell C, Schug J, Lin CF, et al. Transcriptomes of the major human pancreatic cell types. Diabetologia 2011;54:2832–2844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Diiorio P, Jurczyk A, Yang C, et al. Hyperglycemia-induced proliferation of adult human beta cells engrafted into spontaneously diabetic immunodeficient NOD-Rag1null IL2rγnull Ins2Akita mice. Pancreas 2011;40:1147–1149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Levitt HE, Cyphert TJ, Pascoe JL, et al. Glucose stimulates human beta cell replication in vivo in islets transplanted into NOD-severe combined immunodeficiency (SCID) mice. Diabetologia 2011;54:572–582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Flier SN, Kulkarni RN, Kahn CR. Evidence for a circulating islet cell growth factor in insulin-resistant states. Proc Natl Acad Sci U S A 2001;98:7475–7480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang W, Walker JR, Wang X, et al. Identification of small-molecule inducers of pancreatic beta-cell expansion. Proc Natl Acad Sci U S A 2009;106:1427–1432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shen W, Tremblay MS, Deshmukh VA, et al. Small-molecule inducer of β cell proliferation identified by high-throughput screening. J Am Chem Soc 2013;135:1669–1672 [DOI] [PubMed] [Google Scholar]
- 33.Ouziel-Yahalom L, Zalzman M, Anker-Kitai L, et al. Expansion and redifferentiation of adult human pancreatic islet cells. Biochem Biophys Res Commun 2006;341:291–298 [DOI] [PubMed] [Google Scholar]
- 34.Russ HA, Bar Y, Ravassard P, Efrat S. In vitro proliferation of cells derived from adult human beta-cells revealed by cell-lineage tracing. Diabetes 2008;57:1575–1583 [DOI] [PubMed] [Google Scholar]
- 35.Jarvis MF, Yu H, Kohlhaas K, et al. ABT-702 (4-amino-5-(3-bromophenyl)-7-(6-morpholinopyridin-3-yl)pyrido[2, 3-d]pyrimidine), a novel orally effective adenosine kinase inhibitor with analgesic and anti-inflammatory properties: I. In vitro characterization and acute antinociceptive effects in the mouse. J Pharmacol Exp Ther 2000;295:1156–1164 [PubMed] [Google Scholar]
- 36.Zheng GZ, Lee C, Pratt JK, et al. Pyridopyrimidine analogues as novel adenosine kinase inhibitors. Bioorg Med Chem Lett 2001;11:2071–2074 [DOI] [PubMed] [Google Scholar]
- 37.Gao Y, Davies SP, Augustin M, et al. A broad activity screen in support of a chemogenomic map for kinase signalling research and drug discovery. Biochem J 2013;451:313–328 [DOI] [PubMed] [Google Scholar]
- 38.Bain J, Plater L, Elliott M, et al. The selectivity of protein kinase inhibitors: a further update. Biochem J 2007;408:297–315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Soppa U, Schumacher J, Florencio Ortiz V, Pasqualon T, Tejedor FJ, Becker W. The Down syndrome-related protein kinase DYRK1A phosphorylates p27(Kip1) and cyclin D1 and induces cell cycle exit and neuronal differentiation. Cell Cycle 2014;13:2084–2100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Subramanian A, Tamayo P, Mootha VK, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 2005;102:15545–15550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Subramanian A, Kuehn H, Gould J, Tamayo P, Mesirov JP. GSEA-P: a desktop application for gene set enrichment analysis. Bioinformatics 2007;23:3251–3253 [DOI] [PubMed] [Google Scholar]
- 42.Litovchick L, Florens LA, Swanson SK, Washburn MP, DeCaprio JA. DYRK1A protein kinase promotes quiescence and senescence through DREAM complex assembly. Genes Dev 2011;25:801–813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mercer SE, Friedman E. Mirk/Dyrk1B: a multifunctional dual-specificity kinase involved in growth arrest, differentiation, and cell survival. Cell Biochem Biophys 2006;45:303–315 [DOI] [PubMed] [Google Scholar]
- 44.Arron JR, Winslow MM, Polleri A, et al. NFAT dysregulation by increased dosage of DSCR1 and DYRK1A on chromosome 21. Nature 2006;441:595–600 [DOI] [PubMed] [Google Scholar]
- 45.Goodyer WR, Gu X, Liu Y, Bottino R, Crabtree GR, Kim SK. Neonatal β cell development in mice and humans is regulated by calcineurin/NFAT. Dev Cell 2012;23:21–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rachdi L, Kariyawasam D, Aïello V, et al. Dyrk1A induces pancreatic β cell mass expansion and improves glucose tolerance. Cell Cycle 2014;13:2221–2229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rachdi L, Kariyawasam D, Guez F, et al. Dyrk1a haploinsufficiency induces diabetes in mice through decreased pancreatic beta cell mass. Diabetologia 2014;57:960–969 [DOI] [PubMed] [Google Scholar]
- 48.Knight ZA, Shokat KM. Chemical genetics: where genetics and pharmacology meet. Cell 2007;128:425–430 [DOI] [PubMed] [Google Scholar]
- 49.Rosenthal AS, Tanega C, Shen M, et al. Potent and selective small molecule inhibitors of specific isoforms of Cdc2-like kinases (Clk) and dual specificity tyrosine-phosphorylation-regulated kinases (Dyrk). Bioorg Med Chem Lett 2011;21:3152–3158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Coombs TC, Tanega C, Shen M, et al. Small-molecule pyrimidine inhibitors of the cdc2-like (Clk) and dual specificity tyrosine phosphorylation-regulated (Dyrk) kinases: development of chemical probe ML315. Bioorg Med Chem Lett 2013;23:3654–3661 [DOI] [PMC free article] [PubMed] [Google Scholar]