

Abstract
Hepatic steatosis is common in obesity and insulin resistance and results from a net retention of lipids in the liver. A key mechanism to prevent steatosis is to increase secretion of triglycerides (TG) packaged as VLDLs. Insulin controls nutrient partitioning via signaling through its cognate receptor in peripheral target organs such as liver, muscle, and adipose tissue and via signaling in the central nervous system (CNS) to orchestrate organ cross talk. While hepatic insulin signaling is known to suppress VLDL production from the liver, it is unknown whether brain insulin signaling independently regulates hepatic VLDL secretion. Here, we show that in conscious, unrestrained male Sprague Dawley rats the infusion of insulin into the third ventricle acutely increased hepatic TG secretion. Chronic infusion of insulin into the CNS via osmotic minipumps reduced the hepatic lipid content as assessed by noninvasive 1H-MRS and lipid profiling independent of changes in hepatic de novo lipogenesis and food intake. In mice that lack the insulin receptor in the brain, hepatic TG secretion was reduced compared with wild-type littermate controls. These studies identify brain insulin as an important permissive factor in hepatic VLDL secretion that protects against hepatic steatosis.
Introduction
Nonalcoholic fatty liver disease (NAFLD) is a major health burden in both developed and developing countries due to its potential progression to cirrhosis and end-stage liver failure. NAFLD is common in obesity and represents the hepatic manifestation of the metabolic syndrome (1). Increased hepatic triglyceride (TG) synthesis can be the result of 1) increased de novo lipogenesis in the liver (2), 2) increased delivery of fatty acid (FA) to the liver due to excessive dietary lipid intake and/or unrestrained lipolysis in white adipose tissue (WAT) (3), and 3) decreased hepatic lipid oxidation (4). In the insulin resistant state, hepatic de novo lipogenesis and lipolysis in WAT are increased, while hepatic lipid oxidation declines, which is reflected in decreased hepatic ATP levels and turnover (5–7). While mitochondrial dysfunction in the liver can result in steatohepatitis (8), humans with simple NAFLD initially exhibit increased oxidative activity in hepatic mitochondria, which likely represents a compensatory adaptation to the increased lipid flux to the liver (9,10). However, when NAFLD progresses to steatohepatitis, mitochondrial function declines owing to excessive oxidative stress (10).
An important contributor to hepatic fat accumulation is the insufficient hepatic export of lipids in the form of VLDL particles. This is demonstrated in genetic diseases of impaired VLDL particle secretion such as abetalipoproteinemia or hypobetalipoproteinemia (11) and in mice that lack hepatic microsomal TG transfer protein (MTP), the rate-limiting enzyme in VLDL assembly (12). Obese subjects with moderate NAFLD partially compensate for the excess supply of lipids to the liver by increased TG export (13), but with disease progression, hepatic VLDL secretion reaches a plateau and this failure of the liver to further increase TG export results in greater net retention of lipids (14).
The principal mechanism through which the liver mobilizes and disposes of lipids is assembly of TGs with apolipoprotein (apo)B to VLDL particles that are secreted into circulation. In insulin-sensitive individuals, insulin acutely reduces VLDL appearance in the blood stream (15), which facilitates chylomicron clearance, but also causes transient postprandial steatosis that likely resolves during fasting (16). In agreement with this notion, hyperinsulinemia leads to transient mild hepatic steatosis, suggesting that the acute decrease in VLDL secretion translates into measureable lipid accumulation (17,18). Thus, it is conceivable that in insulin-resistant individuals, relative hyperinsulinemia in the fasting state contributes to steatosis.
A prevailing paradigm is that hyperinsulinemia decreases hepatic VLDL secretion by inhibiting apoB synthesis (19), promoting apoB degradation (20), and reducing forkhead box O1–mediated MTP expression (21) through induction of insulin signaling within the hepatocyte. Failure of insulin to inactivate forkhead box O1 (22) or insulin resistance impairs the suppression of glucose-induced VLDL secretion by insulin (23), which could also be interpreted as a compensatory mechanism to promote TG export in order to prevent steatosis. However, the moderate increase in hepatic VLDL secretion is insufficient to prevent hepatic steatosis induced by weight gain in obese individuals (13). This insufficient VLDL secretion could be explained by an increase of the direct effects of insulin on the liver in the setting of obesity and hyperinsulinemia, while counteracting factors that promote hepatic VLDL secretion seem to decline, resulting in hepatic lipid accumulation and ultimately in steatosis. Such counterbalancing signals that augment hepatic VLDL secretion can be conveyed via the brain and the autonomic nervous system (24–26). Interestingly, glucose sensing in the central nervous system (CNS) can attenuate VLDL secretion, leading to a consecutive increase in hepatic TGs (27). Here, we hypothesize that brain insulin signaling augments hepatic TG mobilization, thereby counterbalancing the effects of brain glucose and, vice versa, that brain insulin resistance results in impaired hepatic TG secretion leading to hepatic steatosis.
Research Design and Methods
Animals
All infusion experiments were performed either in 10-week-old male Sprague Dawley (SD) rats fed with standard diet (Rodent Diet 5001, LabDiet, or ssniff R/M-H Alleinfutter, ssniff Spezialdiaeten GmbH, Soest, Germany) or high-fat diet (cat. no. D12492; Research Diets, Brogaarden, Denmark) (60% of calories from fat) or in mice. Rats were fitted with stereotaxic cannulae targeting the third ventricle (intracerebroventricular) or the mediobasal hypothalamus (MBH) and jugular venous and carotid artery (clamped rats only) catheters as described elsewhere (28). All animal protocols were approved by the International Animal Care and Use Committee of Mount Sinai School of Medicine, New York, NY, and the Austrian Federal Ministry of Science, Research and Economy (BMWFW-66.009/0246-WF/V/3b/2015). Experiments presented in Figs. 1–3 were performed in the U.S., whereas the experiments shown in Fig. 4 were performed in Europe.
Figure 1.

Intracerebroventricular (ICV) insulin infusion increases TG secretion. A: Protocol. B: Plasma insulin concentration at baseline (−60 min), at the end (180 min) of the intracerebroventricular insulin infusion, and after intravenous (IV) insulin infusion (n ≥ 5/group). C: Plasma TG accumulation in intracerebroventricular or intravenous insulin– and tyloxapol–injected rats (n ≥ 5/group, except intravenous insulin group: n = 4). D: The calculated VLDL secretion rate combining two independent experimental cohorts (n ≥ 11/group, except intravenous insulin group: n = 5). E: Plasma FA profiles as assessed by gas chromatography; depicted are the absolute changes from time point 0–180 min (n ≥ 6/group). All error bars are SEM. *P < 0.05, **P < 0.01, ***P < 0.001 vs. intracerebroventricular vehicle group; §P < 0.05, §§P < 0.01, §§§P < 0.001 vs. intracerebroventricular insulin group.
Figure 3.

Deletion of neuronal insulin receptor signaling reduces TG secretion, while the targeted knockout of insulin receptors restricted to the periphery increases TG secretion rates. A: Experimental protocol. B: Body weights of Nirko mice and littermate controls (n ≥ 9/group). C: Blood glucose levels of Nirko and littermate controls at baseline of the tyloxapol infusion experiment (n ≥ 9/group). D: Plasma TG levels at baseline (0) and 1 and 2 h after a tyloxapol injection in Nirko vs. littermate control mice (n ≥ 9/group). E–G: VLDL secretion rates as assessed after tyloxapol infusion experiments in Nirko, IR∆PER (n = 5/group), and IR∆WB (n ≥ 6/group) mice vs. their respective controls. All error bars are SEM. *P < 0.05 vs. controls. hrs, hours; N.S., not significant.
Figure 4.

Chronic brain insulin infusion reduces liver TG content independent of changes in body weight and food intake. A: Study protocol. MRT I–III indicate the time points where 1H-MRS measurements were performed. B: Liver fat content measured using 1H-MRS in anesthetized rats on days −3, 8, and 28. C: FA profiles from liver tissue harvested on day 30. Data are depicted as % change vs. vehicle-infused animals. D: Body weight. E: Food intake. F: Comparison of mRNA copy numbers of DNL genes in liver tissue samples from intracerebroventricular (ICV) insulin– or vehicle–treated rats. All error bars are SEM. *P < 0.05, **P < 0.01, ***P < 0.001 vs. respective control group. n = 8/group.
Nirko mice were generated as previously described (28) and subjected to tyloxapol infusion studies at the age of 30 weeks after implanting of jugular venous catheters.IRΔPER and IRΔWB mice, two mouse models of inducible insulin receptor deletion, were generated pharmacologically, induced using tamoxifen and doxycycline, respectively, and genotyped as previously described (29) and implanted with jugular venous catheters.
Signaling studies were performed with a 10-µL bolus of either vehicle (artificial cerebrospinal fluid [CSF]) or insulin (30 µU). Fifteen minutes after the intracerebroventricular (3rd ventricle) bolus, the arcuate nucleus (ARC) was dissected using N.I.H. style neuro punches (Fine Science Tools) and snap frozen in liquid nitrogen. Correct cannula placement was verified by food dye infusion.
Rat tyloxapol experiments were conducted as outlined in Fig. 1A and previously described (27). The tyloxapol (Triton WR-1339; Sigma-Aldrich) injection solution was prepared at a concentration of 80 mg/mL by dissolution in PBS, shaking overnight, and titration to a pH of 7.4 with HCl. Rats fit with indwelling cannulae targeting the brain region of interest and a jugular venous catheter for blood sampling were placed into individual cages. Animals were fasted for 5–6 h to exclude that chylomicron secretion from the gastrointestinal tract contributes to TG appearance in plasma and causes overestimation of hepatic VLDL flux. After fasting animals were connected to the catheters and a continuous infusion (time point −60 min; 0.083 µL/min i.c.v.; MBH 0.003 µL/min/side) was started and maintained for the entire study (Fig. 1A). Rats received insulin (5 µU/h i.c.v.; MBH 0.18 µU/h/side), vehicle (artificial CSF, Harvard Apparatus: 5 μL/h i.c.v.; MBH 0.18 µL/h), glucose (2 mmol/L; 0.18 µL/h/side), or insulin-like growth factor I (IGF-I) (0.77 µg over 4 h). We allowed animals to recover from the potential stress experienced during the connecting of catheter and stereotactic cannula for 1 h before we injected a tyloxapol bolus (600 mg/kg body wt i.v.) at time point 0 min. Blood samples were collected at time points −60, 0, 30, 60, 90, 120, 150, and 180 min for TG determination, after which rats were killed by intravenous injection of ketamine followed by decapitation.
Hyperinsulinemic-clamp studies were performed with an insulin dose of 3 mU/kg/min, which induces moderate hyperinsulinemia, as previously described (28). We simultaneously initiated the clamp and the tyloxapol study as illustrated in Fig. 1A. To ensure that the potential stress caused by the tyloxapol injection did not introduce a bias, and to have a time course comparable with that of the intracerebroventricular experiments, we extended the clamp for a total of 3 h. We adjusted the glucose infusion rate every 10 min based on arterial blood glucose measurements (Supplementary Fig. 2A and B).
VLDL secretion rate was calculated from the slope (micrograms per microliter per minute) of the plasma TG rise over time by linear regression analysis (24–27), under the assumption that the average molecular weight (MW) of TGs is 884 g/mol (MW oleic acid ∼282 g/mol * 3; MW glycerol-backbone ∼38 g/mol) and the average volume of distribution of VLDLs is 0.042 mL/g body wt (i.e., plasma volume 4.2%) using the following formula:
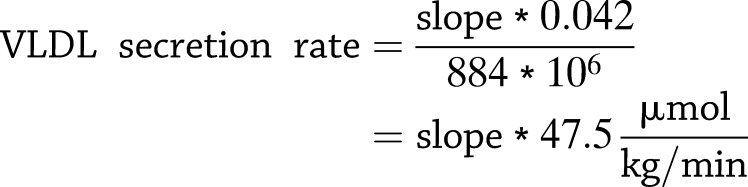 |
Plasma samples for FA profiles of SD rats, which did not receive tyloxapol (Fig. 2C), were derived from previously described studies (28).
Figure 2.

MBH infusion of insulin or glucose did not increase plasma TG accumulation in tyloxapol-injected rats. A: Plasma TG accumulation in MBH insulin– or MBH glucose–infused, tyloxapol-injected rats (n ≥ 5/group). B: Plasma FA profiles as assessed by gas chromatography; depicted are the absolute changes from time point 0–180 min (n = 5/group). C: Changes from baseline (−120 min) of the indicated FA species measured in plasma collected from rats that were infused with either intracerebroventricular (ICV) or MBH insulin for 6 h while rats were subjected to a basal insulin or hyperinsulinemic-euglycemic clamp to control for circulating insulin and glucose levels. The sum of fatty acids represents the sum of all measured lipid species (n ≥ 4/group). All error bars are SEM. *P < 0.05 vs. intracerebroventricular vehicle group; §P < 0.05, §§P < 0.01 vs. intracerebroventricular insulin group. IV, intravenous.
Mouse Tyloxapol Studies
Mice implanted with a jugular venous catheter were fasted for 5–6 h. At time point 0 min, a baseline blood sample was collected by tail vein sampling. Tyloxapol (500 mg/kg body wt i.v.) was then injected. Mice were restrained during the tyloxapol injection (2–3 mins) but were able to move freely thereafter, except for during rapid collection of additional blood samples (time points 1 and 2 h). After the final blood draw, mice were anesthetized with isoflurane and killed by cervical dislocation.
Chronic intracerebroventricular insulin infusion was conducted as outlined in Fig. 4A. Rats were surgically equipped with MRI-compatible brain infusion kits targeting the third ventricle (PlasticsOne), which were connected to subcutaneously implanted osmotic minipumps (pump rate ∼0.25 µL/h, model 2004; Durect) infusing insulin (5 µU/h) or vehicle (artificial CSF with 5% BSA). Postsurgery rats were single-housed, and food intake and body weight were monitored. Hepatic lipid content was measured noninvasively under isoflurane anesthesia using 1H-MRS as described below (method validation in Supplementary Fig. 3A and B). Thirty days after pump implantation, rats were killed and livers were harvested for FA profiling.
Liver 1H-MRS
We measured hepatic lipid content using 1H-MRS triggered to breath motion of the animals using a Medspec 3T MR system (Bruker, Karlsruhe, Germany) equipped with a BGA-12 microgradient insert (200 mT/m) and using a 72-mm 1H tuned resonator. Animals were anesthetized with isoflurane. Measurements of intrahepatic lipids were performed by a short-echo time single-voxel MRS sequence (stimulated echo acquisition mode, echo time 20 ms, repetition time 5 s, number of scans = 64) without water signal suppression. The volume of interest (8 × 10 × 10 mm3) was selected within the liver parenchyma, and data acquisition was synchronized with breath movements of the animal. MRS data were later processed off-line, and signal intensities of water (W) and methylene groups of lipids (L) (CH2: 1.25 parts per million) were used to quantify lipid content in the liver as intrahepatic lipids = [L/(L+W)] and given as % of the total MRS signal.
Gene Expression Analysis
Total RNA was isolated from liver samples using QIAzol (Qiagen) according to the manufacturer’s instructions. cDNA was reverse transcribed with Superscript II from 0.5 µg RNA. Expression of de novo lipogenesis (DNL) genes was determined by TaqMan real-time PCR (assays by Applied Biosystems: Fasn, Rn00569117_m1; Scd1, Rn00594894_g1; and Srebp1, Rn01495759_m1) and normalization to TATA-binding protein mRNA (Tbp, forward primer 5′-ACTTCGTGCCAGAAATGCTGAATAT-3′, reverse primer 5′-CGTGGCTCTCTTATTCTCATGATGA-3′, probe context sequence 5′-TCCCAAGCGGTTTGC-3′).
Hepatic and plasma FA profiles were performed using gas chromatography as previously described (30). Body composition was measured in 20-week-old Nirko and littermate control mice using EchoMRI-100 (EchoMRI, LLC).
Analytic Procedures
Plasma TGs and free glycerol levels were measured with a colorimetric assay (Sigma-Aldrich). Plasma free FA levels were measured with a colorimetric assay (Wako Chemicals, Neuss, Germany). Plasma insulin was measured by an ELISA kit (Mercodia, Uppsala, Sweden). Blood glucose levels were measured with a strip-based glucose meter.
Western blots were performed as previously described (28). Primary antibodies against Acc, phospho-Acc Ser79, Atpcl, phospho-Atpcl Ser455, phospho-Akt Ser473, phospho-Hsl Ser563, and phospho–insulin receptor β (all Cell Signaling Technology); GAPDH and β-actin (both Abcam); insulin receptor β (Santa Cruz Biotechnology); and FA synthase (BD Bioscience) were used.
Statistics
Data are represented as mean ± SEM. Comparisons among groups were made with one-way ANOVA followed by, as appropriate, one-sample or two-sample two-tailed Student t tests. Differences were considered statistically significant at P < 0.05.
Results
For assessment of the role of brain insulin signaling on hepatic TG secretion, fasted and unrestrained male SD rats received a stereotaxic infusion of insulin into the third ventricle of the brain according to the protocol depicted in Fig. 1A. Given that the volume of CSF in a 300-g rat is ∼0.6 mL (31), the average CSF insulin concentration is 2.8 µU/mL in SD rats, (32) and the half-life of insulin is 26 min (33), an insulin infusion rate of 5 µU/h i.c.v. is estimated to approximately double CSF insulin, which is comparable with the physiological increment of insulin in CSF postprandially (34). Intracerebroventricular insulin administration activated the insulin signaling cascade in the brain (Supplementary Fig. 1A), while plasma insulin levels during a 4-h intracerebroventricular infusion of insulin or vehicle were similar, indicating that the direct effects of systemic insulin on the liver and other insulin sensitive organs were comparable (Fig. 1B). This was further substantiated with signaling studies, where no changes in liver Akt phosphorylation were noted (Supplementary Fig. 1B).
To next compare the effects of selectively induced brain insulin signaling with those of systemic hyperinsulinemia, we subjected a cohort of rats to hyperinsulinemic-euglycemic clamps, during which circulating insulin was raised by 60%. The increase in systemic insulin levels induces insulin signaling in the whole body including brain and liver (Fig. 1B and Supplementary Fig. 2B).
Hepatic VLDL secretion was studied after administration of a tyloxapol bolus. Tyloxapol is an inhibitor of VLDL hydrolysis and prevents clearance of VLDL particles, leading to accumulation of VLDL in plasma. This VLDL accumulation in plasma allows for an estimation of the hepatic VLDL secretion rate in the fasting state (35), since chylomicrons originating from the gut are not a significant source of plasma TGs (25). Compared with vehicle infusions, intracerebroventricular insulin increased hepatic VLDL secretion rate by 30%, indicating that brain insulin mobilizes hepatic TGs, while systemic hyperinsulinemia suppresses TG secretion (Fig. 1C and D), consistent with prior reports (4).
The MBH has been identified as a region critical for CNS-mediated insulin action on hepatic glucose production and adipose tissue metabolism (28,36). Hence, we examined whether insulin infused directly into the MBH would replicate the effects of the intracerebroventricular infusion experiments. To our surprise, MBH insulin infusion did not alter VLDL secretion rates (Fig. 2A). Likewise, the infusion of glucose into the MBH failed to affect hepatic VLDL secretion (Fig. 2B), although glucose infusion into the third ventricle has been shown to lower hepatic TG secretion and promote hepatic lipid accumulation (27). These results argue against a critical role of the MBH in CNS control of hepatic VLDL secretion. Consistent with our previous observation that MBH insulin reduces the activation state of hormone-sensitive lipase in adipose tissue (28), we found that phosphorylation of hormone-sensitive lipase at Ser563 was reduced in epigonadal fat after hypothalamic insulin infusion (Supplementary Fig. 1C).
Plasma FA profiles from these tyloxapol studies revealed that intracerebroventricular insulin infusion increased major TG-associated FAs such as palmitic, stearic, and oleic acid, while hyperinsulinemia reduced these same FA species (Fig. 1E). MBH insulin infusion did not alter FA composition (Fig. 2B). In the absence of tyloxapol, intracerebroventricular insulin infusion increased, whereas systemic hyperinsulinemia decreased major TG-associated plasma FA species, while MBH insulin infusions did not change FA profiles (Fig. 2C). The results suggest that the major effect of brain insulin in altering hepatic lipid handling was due to the regulation of VLDL secretion, not through increased DNL, which was further supported by our finding that the protein expression and activation state of major DNL enzymes such as FA synthase, ATP citrate lyase, and acetyl-CoA carboxylase was not altered in the liver after intracerebroventricular insulin administration (Supplementary Fig. 1D).
Since insulin can also signal through the IGF-I receptor and intracerebroventricular administration of IGF-I has been shown to affect hepatic glucose production (37), we tested whether intracerebroventricular infusion of IGF-I would similarly increase hepatic TG secretion using the protocol in Fig. 1A. Intracerebroventricular IGF-I infusion at a dose of 0.77 µg over 4 h did not alter hepatic TG secretion (Supplementary Fig. 1E), suggesting that the effects of brain insulin are mediated through the insulin and not the IGF receptor.
We next used genetic approaches to assess whether brain insulin resistance alters hepatic TG secretion. To this end, mice that lack the insulin receptor in neurons (Nirko) (38) received a tyloxapol bolus via a preimplanted jugular catheter. We sampled blood after 1 and 2 h via the tail vein to measure TG concentration (Fig. 3A). Nirko mice exhibited a reduced rate of hepatic TG secretion (Fig. 3D and E), which was independent of changes in body weight, blood glucose levels, and body composition (Fig. 3B and C and Supplementary Fig. 2C and D) and consistent with a role of brain insulin signaling as a permissive factor in hepatic VLDL secretion.
Since the observed metabolic phenotype in the Nirko mice may be due to developmental impairments and/or the activation of compensatory pathways in this lifelong insulin receptor deletion model, we studied two mouse models of inducible insulin receptor deletion (29) that lack the insulin receptor either in the whole body (IR∆WB) or in peripheral tissues only but not in the brain (IR∆PER). Upon doxycycline administration, IR∆WB mice express a short hairpin RNA that inhibits translation of the insulin receptor mRNA in peripheral and central tissues. In contrast, IR∆PER mice express an inducible Cre deleter in peripheral tissues, but only at low levels in the brain, that is activated by tamoxifen. Due to the low blood-brain barrier penetration of tamoxifen and the low brain Cre expression, deletion of the insulin receptor is minimal or absent in the CNS but is complete in peripheral tissues such as liver, adipose tissue, and muscle (29). Both mouse models developed comparable hyperglycemia and hyperinsulinemia (60-fold increased insulin levels) upon induction of the insulin receptor deletions (data not shown).
During tyloxapol studies, the IR∆WB mice demonstrated no change in hepatic TG secretion, whereas IR∆PER mice exhibited a markedly increased TG secretion rate, which is likely due to the enhanced brain insulin action resulting from hyperinsulinemia in the presence of functional CNS insulin receptors (Fig. 3F and G).
Having demonstrated that intracerebroventricular insulin acutely enhanced hepatic TG secretion, we next examined whether prolonged low-dose insulin infusion into the third ventricle of SD rats would reduce hepatic TG levels (Fig. 4A). Indeed, after 4 weeks of intracerebroventricular insulin infusion via osmotic pumps, rats exhibited a 40% reduction of hepatic TG content as assessed by noninvasive liver 1H-MRS (Fig. 4B and Supplementary Fig. 3A). This noninvasive method allows the longitudinal assessment of liver lipids in a single rat and compares well with conventional liver TG measurements using Folch extraction (Supplementary Fig. 3B). FA profiling of hepatic TGs revealed reductions in almost every FA species indicating that chronic induction of brain insulin signaling reduces steatosis (Fig. 4C). Importantly, the reduction in hepatic lipid content was not accompanied by any changes in food intake, body weight (Fig. 4D and E), or circulating insulin levels (Supplementary Fig. 3C). Plasma TG, free FA, and glycerol levels were also unchanged in intracerebroventricular insulin–infused rats (Supplementary Fig. 3D). Similarly to the short-term intracerebroventricular insulin infusions, the mRNA expression of key DNL enzymes and of sterol regulatory element–binding protein 1 (Srebp1) was not altered by brain insulin (Fig. 4F).
Discussion
Here, we demonstrate that CNS insulin signaling augments TG export from the liver and reduces liver lipid content independent of peripheral and, in particular, hepatic insulin signaling. These findings suggest that brain insulin is an important permissive regulator of hepatic VLDL secretion that balances the effect of direct hepatic insulin signaling, which enhances hepatic lipogenesis and acutely reduces VLDL secretion. Conversely, the experimental induction of brain insulin resistance resulted in lower hepatic VLDL secretion. Since overnutrition rapidly induces brain insulin resistance in humans and rodents (39), and obesity and type 2 diabetes can be associated with brain insulin resistance (40,41), these findings provide a potential new mechanism that might in part explain the high incidence of steatohepatosis in obesity and diabetes. Our study also demonstrates that brain insulin balances the effects of brain glucose sensing that suppresses VLDL secretion and increases hepatic TG content (27).
Of note, we did not observe hypertriglyceridemia during intracerebroventricular insulin administration (Supplementary Fig. 3D), despite increased hepatic VLDL secretion rates. This is likely explained by the finding that brain insulin signaling also increases FA uptake into WAT, at least in C57Bl/6J mice (42). An additional mechanism by which brain insulin protects from NAFLD is the suppression of lipolysis in adipose tissue, which restricts lipid flux to the liver (28,43).
Genetic mice models of either CNS, whole-body, or peripherally restricted insulin receptor deletions further support the novel paradigm of brain insulin being a permissive factor of VLDL secretion. Lifelong deficiency of the neuronal insulin receptor in the Nirko mice resulted in a decrease in hepatic TG secretion (Fig. 3D and E), while enhancing neuronal insulin signaling in the setting of a peripheral loss of insulin receptors (IR∆PER) promoted hepatic TG export (Fig. 3F). At first sight, these results seem to be in conflict with reports from studies of liver-specific insulin receptor knockout mice (LIRKO). LIRKO mice are also hyperinsulinemic (10-fold) compared with control subjects but show a decrease in TG accumulation after tyloxapol infusion; yet, surprisingly, hepatic TG content is unchanged. An explanation for this apparent discrepancy may lie in the observation that serum apoB levels are markedly increased in LIRKO mice, suggesting that, actually, more VLDL particles with lower TG content are secreted (44). The latter may be due to the pronounced reduction of hepatic DNL in the LIRKO mice. Conversely, the marked hyperinsulinemia that the LIRKO mice develop in the setting of preserved brain insulin signaling may lead to an enhancement of brain insulin action. The latter may reduce hepatic steatosis by enhancing hepatic VLDL secretion. Hence, we believe that the phenotype of the LIRKO mice does not contradict the concept of brain insulin signaling enhancing VLDL secretion when liver insulin receptors are lost.
A surprising finding of our studies is that insulin infusion directly into the MBH did not alter VLDL secretion, suggesting that insulin signaling in another, as of yet unidentified brain region regulates VLDL secretion (Fig. 2A). In most studies that examined a role of neuropeptides, hormones, or nutrients within the CNS in regulating hepatic TG flux, brain delivery was accomplished by intracerebroventricular injections (24–27). Bruinstroop et al. (24), on the other hand, have argued that an intact ARC and a functioning sympathetic nervous system are necessary for brain neuropeptide Y to promote liver VLDL secretion in rats. However, the ARC was not anatomically targeted in these studies; instead, rats had been treated with monosodium glutamate as a means to pharmacologically lesion the ARC. This approach has limitations, since it is unspecific and the lesion is not confined to the ARC, as it has been demonstrated that monosodium glutamate also damages the paraventricular nucleus (45) and affects cortical excitability (46). Recent data suggest that the dorsal vagal complex of the brainstem via the parasympathetic nervous system might affect hepatic VLDL flux (47), and future studies will have to identify such regions.
Our findings provide a possible explanation for the recent observations in humans where intranasal insulin administration (a delivery method that preferentially delivers insulin to the CNS) reduced hepatic lipid content assessed by 1H-MRS, while intravenous insulin increased hepatic lipid content (18). Hence, the opposing role of insulin signaling in brain versus the liver in regulating hepatic lipid flux is likely due to the differential regulation of hepatic VLDL secretion. Hence, the physiology that we have studied in rodents also seems to be applicable to humans.
Insulin transport across the blood-brain barrier is saturable in humans (48). As insulin levels rise with aggravating obesity and concomitant insulin resistance, the brain is exposed to relative hypoinsulinemia. Simultaneously, the liver is exposed to high insulin levels disrupting the balance between CNS and liver insulin signaling, promoting hepatic TG accumulation. Thus, enhancing brain insulin signaling in selective neurons that remain insulin sensitive may be effective in ameliorating NAFLD.
This study provides an explanation for why insulin analogs that preferentially target the liver cause hepatic steatosis and elevated liver function tests, as has been seen with LY2605541 (49,50). The hope is that such liver-specific insulins that appear to have lower hypoglycemia rates also cause less weight gain. However, this seems to come at the price of increased hepatic steatosis in the setting of brain insulin resistance. Future studies should carefully assess in humans whether indeed steatosis risk is increased by these insulin analogs through noninvasive assessments of hepatic lipid content, as was done in our rodent studies.
In conclusion, insulin signaling in the brain protects from ectopic lipid accumulation in the liver by stimulating hepatic TG secretion without the induction of hypertriglyceridemia. Conversely, restoration of brain insulin signaling should ameliorate the susceptibility to hepatic steatosis in conditions such as obesity and diabetes.
Article Information
Acknowledgments. The authors thank the team of the Core Unit for Biomedical Research, Medical University of Vienna, for professional support and for excellent animal care.
Funding. This study was supported by Austrian Science Fund grant P26766 to T.S.; German Research Foundation (DFG) grant SFB 841: liver inflammation: infection, immune regulation, and consequences to J.H.; and NIH grants DK-074873, DK-083568, and DK-082724, and American Diabetes Association award 7-11-CD-02 to C.B.
Duality of Interest. This study was also supported by an unrestricted grant from Novo Nordisk Ges.m.b.H Austria to the Clinical Division of Endocrinology and Metabolism, Department of Medicine III, Medical University of Vienna. No other potential conflicts of interest relevant to this article were reported.
Author Contributions. T.S. designed and performed experiments, supervised experimentation, analyzed data, coordinated the project, and wrote the manuscript. C.L., J.O., and E.Z. assisted with the tyloxapol infusion experiments. M.H. performed part of the chronic intracerebroventricular infusion studies and lipid analyses. A.F., S.B.-P., K.T., J.H., and L.S. performed lipid species analysis and lipogenic gene expression analysis. M.K. performed the liver 1H-MRS measurements. C.F. helped with the chronic intracerebroventricular infusion studies and wrote the manuscript. C.B. designed experiments, supervised experimentation, analyzed data, coordinated the project, and wrote the manuscript. T.S. and C.B. are the guarantors of this work and, as such, had full access to all the data in the study and take responsibility for the integrity of the data and the accuracy of the data analysis.
Prior Presentation. Preliminary results were presented orally at the 94th Annual Meeting and Expo of the Endocrine Society, Houston, TX, 23–26 June 2012 and the 15th Congress of the European Neuroendocrine Association, Vienna, Austria, 12–15 September 2012 and presented as a poster at the Keystone Symposium on Neuronal Control of Appetite, Metabolism and Weight, Keystone, CO, 24–29 January 2010.
Footnotes
This article contains Supplementary Data online at http://diabetes.diabetesjournals.org/lookup/suppl/doi:10.2337/db15-1552/-/DC1.
See accompanying article, p. 1481.
References
- 1.Kwon YM, Oh SW, Hwang SS, Lee C, Kwon H, Chung GE. Association of nonalcoholic fatty liver disease with components of metabolic syndrome according to body mass index in Korean adults. Am J Gastroenterol 2012;107:1852–1858 [DOI] [PubMed] [Google Scholar]
- 2.Lambert JE, Ramos-Roman MA, Browning JD, Parks EJ. Increased de novo lipogenesis is a distinct characteristic of individuals with nonalcoholic fatty liver disease. Gastroenterology 2014;146:726–735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Donnelly KL, Smith CI, Schwarzenberg SJ, Jessurun J, Boldt MD, Parks EJ. Sources of fatty acids stored in liver and secreted via lipoproteins in patients with nonalcoholic fatty liver disease. J Clin Invest 2005;115:1343–1351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Grefhorst A, Hoekstra J, Derks TG, et al. Acute hepatic steatosis in mice by blocking beta-oxidation does not reduce insulin sensitivity of very-low-density lipoprotein production. Am J Physiol Gastrointest Liver Physiol 2005;289:G592–G598 [DOI] [PubMed] [Google Scholar]
- 5.Schmid AI, Szendroedi J, Chmelik M, Krssák M, Moser E, Roden M. Liver ATP synthesis is lower and relates to insulin sensitivity in patients with type 2 diabetes. Diabetes Care 2011;34:448–453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Szendroedi J, Chmelik M, Schmid AI, et al. Abnormal hepatic energy homeostasis in type 2 diabetes. Hepatology 2009;50:1079–1086 [DOI] [PubMed] [Google Scholar]
- 7.Valkovič L, Gajdošík M, Traussnigg S, et al. Application of localized ³¹P MRS saturation transfer at 7 T for measurement of ATP metabolism in the liver: reproducibility and initial clinical application in patients with non-alcoholic fatty liver disease. Eur Radiol 2014;24:1602–1609 [DOI] [PubMed] [Google Scholar]
- 8.Cortez-Pinto H, Chatham J, Chacko VP, Arnold C, Rashid A, Diehl AM. Alterations in liver ATP homeostasis in human nonalcoholic steatohepatitis: a pilot study. JAMA 1999;282:1659–1664 [DOI] [PubMed] [Google Scholar]
- 9.Sunny NE, Parks EJ, Browning JD, Burgess SC. Excessive hepatic mitochondrial TCA cycle and gluconeogenesis in humans with nonalcoholic fatty liver disease. Cell Metab 2011;14:804–810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Koliaki C, Szendroedi J, Kaul K, et al. Adaptation of hepatic mitochondrial function in humans with non-alcoholic fatty liver is lost in steatohepatitis. Cell Metab 2015;21:739–746 [DOI] [PubMed] [Google Scholar]
- 11.Kneeman JM, Misdraji J, Corey KE. Secondary causes of nonalcoholic fatty liver disease. Therap Adv Gastroenterol 2012;5:199–207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Raabe M, Véniant MM, Sullivan MA, et al. Analysis of the role of microsomal triglyceride transfer protein in the liver of tissue-specific knockout mice. J Clin Invest 1999;103:1287–1298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fabbrini E, Tiemann Luecking C, Love-Gregory L, et al.. Physiological mechanisms of weight gain-induced steatosis in people with obesity. Gastroenterology 2016;150:79–81.e2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fabbrini E, Mohammed BS, Magkos F, Korenblat KM, Patterson BW, Klein S. Alterations in adipose tissue and hepatic lipid kinetics in obese men and women with nonalcoholic fatty liver disease. Gastroenterology 2008;134:424–431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Malmström R, Packard CJ, Watson TD, et al. Metabolic basis of hypotriglyceridemic effects of insulin in normal men. Arterioscler Thromb Vasc Biol 1997;17:1454–1464 [DOI] [PubMed] [Google Scholar]
- 16.Petersen KF, Dufour S, Savage DB, et al. The role of skeletal muscle insulin resistance in the pathogenesis of the metabolic syndrome. Proc Natl Acad Sci U S A 2007;104:12587–12594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Anderwald C, Bernroider E, Krssak M, et al. Effects of insulin treatment in type 2 diabetic patients on intracellular lipid content in liver and skeletal muscle. Diabetes 2002;51:3025–3032 [DOI] [PubMed] [Google Scholar]
- 18.Gancheva S, Koliaki C, Bierwagen A, et al. Effects of intranasal insulin on hepatic fat accumulation and energy metabolism in humans. Diabetes 2015;64:1966–1975 [DOI] [PubMed] [Google Scholar]
- 19.Sparks JD, Sparks CE. Insulin modulation of hepatic synthesis and secretion of apolipoprotein B by rat hepatocytes. J Biol Chem 1990;265:8854–8862 [PubMed] [Google Scholar]
- 20.Sparks JD, Phung TL, Bolognino M, Sparks CE. Insulin-mediated inhibition of apolipoprotein B secretion requires an intracellular trafficking event and phosphatidylinositol 3-kinase activation: studies with brefeldin A and wortmannin in primary cultures of rat hepatocytes. Biochem J 1996;313:567–574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kamagate A, Qu S, Perdomo G, et al. FoxO1 mediates insulin-dependent regulation of hepatic VLDL production in mice. J Clin Invest 2008;118:2347–2364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wu K, Cappel D, Martinez M, Stafford JM. Impaired-inactivation of FoxO1 contributes to glucose-mediated increases in serum very low-density lipoprotein. Endocrinology 2010;151:3566–3576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Malmström R, Packard CJ, Caslake M, et al. Defective regulation of triglyceride metabolism by insulin in the liver in NIDDM. Diabetologia 1997;40:454–462 [DOI] [PubMed] [Google Scholar]
- 24.Bruinstroop E, Pei L, Ackermans MT, et al. Hypothalamic neuropeptide Y (NPY) controls hepatic VLDL-triglyceride secretion in rats via the sympathetic nervous system. Diabetes 2012;61:1043–1050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Stafford JM, Yu F, Printz R, Hasty AH, Swift LL, Niswender KD. Central nervous system neuropeptide Y signaling modulates VLDL triglyceride secretion. Diabetes 2008;57:1482–1490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.van den Hoek AM, Voshol PJ, Karnekamp BN, et al. Intracerebroventricular neuropeptide Y infusion precludes inhibition of glucose and VLDL production by insulin. Diabetes 2004;53:2529–2534 [DOI] [PubMed] [Google Scholar]
- 27.Lam TK, Gutierrez-Juarez R, Pocai A, et al. Brain glucose metabolism controls the hepatic secretion of triglyceride-rich lipoproteins. Nat Med 2007;13:171–180 [DOI] [PubMed] [Google Scholar]
- 28.Scherer T, O’Hare J, Diggs-Andrews K, et al. Brain insulin controls adipose tissue lipolysis and lipogenesis. Cell Metab 2011;13:183–194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Koch L, Wunderlich FT, Seibler J, et al. Central insulin action regulates peripheral glucose and fat metabolism in mice. J Clin Invest 2008;118:2132–2147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Scheja L, Toedter K, Mohr R, et al. Liver TAG transiently decreases while PL n-3 and n-6 fatty acids are persistently elevated in insulin resistant mice. Lipids 2008;43:1039–1051 [DOI] [PubMed] [Google Scholar]
- 31.Lai YL, Smith PM, Lamm WJ, Hildebrandt J. Sampling and analysis of cerebrospinal fluid for chronic studies in awake rats. J Appl Physiol 1983;54:1754–1757 [DOI] [PubMed] [Google Scholar]
- 32.Hu SH, Jiang T, Yang SS, Yang Y. Pioglitazone ameliorates intracerebral insulin resistance and tau-protein hyperphosphorylation in rats with type 2 diabetes. Exp Clin Endocrinol Diabetes 2013;121:220–224 [DOI] [PubMed] [Google Scholar]
- 33.Cashion MF, Banks WA, Kastin AJ. Sequestration of centrally administered insulin by the brain: effects of starvation, aluminum, and TNF-alpha. Horm Behav 1996;30:280–286 [DOI] [PubMed] [Google Scholar]
- 34.Stein LJ, Dorsa DM, Baskin DG, et al. Immunoreactive insulin levels are elevated in the cerebrospinal fluid of genetically obese Zucker rats. Endocrinology 1983;113:2299–2301 [DOI] [PubMed] [Google Scholar]
- 35.Schotz MC, Scanu A, Page IH. Effect of triton on lipoprotein lipase of rat plasma. Am J Physiol 1957;188:399–402 [DOI] [PubMed] [Google Scholar]
- 36.Obici S, Zhang BB, Karkanias G, Rossetti L. Hypothalamic insulin signaling is required for inhibition of glucose production. Nat Med 2002;8:1376–1382 [DOI] [PubMed] [Google Scholar]
- 37.Muzumdar RH, Ma X, Fishman S, et al. Central and opposing effects of IGF-I and IGF-binding protein-3 on systemic insulin action. Diabetes 2006;55:2788–2796 [DOI] [PubMed] [Google Scholar]
- 38.Brüning JC, Gautam D, Burks DJ, et al. Role of brain insulin receptor in control of body weight and reproduction. Science 2000;289:2122–2125 [DOI] [PubMed] [Google Scholar]
- 39.Tschritter O, Preissl H, Hennige AM, et al. The cerebrocortical response to hyperinsulinemia is reduced in overweight humans: a magnetoencephalographic study. Proc Natl Acad Sci U S A 2006;103:12103–12108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Scherer T, Lindtner C, Zielinski E, O’Hare J, Filatova N, Buettner C. Short term voluntary overfeeding disrupts brain insulin control of adipose tissue lipolysis. J Biol Chem 2012;287:33061–33069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hallschmid M, Benedict C, Schultes B, Born J, Kern W. Obese men respond to cognitive but not to catabolic brain insulin signaling. Int J Obes 2008;32:275–282 [DOI] [PubMed] [Google Scholar]
- 42.Coomans CP, Geerling JJ, Guigas B, et al. Circulating insulin stimulates fatty acid retention in white adipose tissue via KATP channel activation in the central nervous system only in insulin-sensitive mice. J Lipid Res 2011;52:1712–1722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Iwen KA, Scherer T, Heni M, et al. Intranasal insulin suppresses systemic but not subcutaneous lipolysis in healthy humans. J Clin Endocrinol Metab 2014;99:E246–E251 [DOI] [PMC free article] [PubMed]
- 44.Biddinger SB, Hernandez-Ono A, Rask-Madsen C, et al. Hepatic insulin resistance is sufficient to produce dyslipidemia and susceptibility to atherosclerosis. Cell Metab 2008;7:125–134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Abe M, Saito M, Shimazu T. Neuropeptide Y in the specific hypothalamic nuclei of rats treated neonatally with monosodium glutamate. Brain Res Bull 1990;24:289–291 [DOI] [PubMed] [Google Scholar]
- 46.Lima CB, Soares GdeS, Vitor SM, Castellano B, Andrade da Costa BL, Guedes RC. Neonatal treatment with monosodium glutamate lastingly facilitates spreading depression in the rat cortex. Life Sci 2013;93:388–392 [DOI] [PubMed] [Google Scholar]
- 47.Yue JT, Abraham MA, LaPierre MP, et al. A fatty acid-dependent hypothalamic-DVC neurocircuitry that regulates hepatic secretion of triglyceride-rich lipoproteins. Nat Commun 2015;6:5970. [DOI] [PubMed] [Google Scholar]
- 48.Banks WA, Jaspan JB, Kastin AJ. Selective, physiological transport of insulin across the blood-brain barrier: novel demonstration by species-specific radioimmunoassays. Peptides 1997;18:1257–1262 [DOI] [PubMed] [Google Scholar]
- 49.Rosenstock J, Bergenstal RM, Blevins TC, et al. Better glycemic control and weight loss with the novel long-acting basal insulin LY2605541 compared with insulin glargine in type 1 diabetes: a randomized, crossover study. Diabetes Care 2013;36:522–528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bergenstal RM, Rosenstock J, Arakaki RF, et al. A randomized, controlled study of once-daily LY2605541, a novel long-acting basal insulin, versus insulin glargine in basal insulin-treated patients with type 2 diabetes. Diabetes Care 2012;35:2140–2147 [DOI] [PMC free article] [PubMed] [Google Scholar]