

Significance
Some inherited autosomal dominant disorders are caused by dominant negative mutations whose gene product adversely affects the normal gene product expressed from the other allele. Gene editing with engineered site-specific endonucleases leads to a double-strand break, which promotes nonhomologous end-joining (NHEJ). NHEJ is error-prone and conducive to the generation of frameshift mutations, leading to intentional knockout of a gene. Therefore, gene editing with engineered site-specific endonucleases is applicable to dominant negative disorders, to target only the mutant allele and leave the normal allele intact. This technique could provide a significant benefit for patients with dominant negative disorders in combination with induced pluripotent stem cell technology that acquires multipotential differentiation and unlimited self-renewal capacity.
Keywords: CRISPR/Cas, TALENs, epidermolysis bullosa, gene editing, dominant negative effect
Abstract
Genome editing with engineered site-specific endonucleases involves nonhomologous end-joining, leading to reading frame disruption. The approach is applicable to dominant negative disorders, which can be treated simply by knocking out the mutant allele, while leaving the normal allele intact. We applied this strategy to dominant dystrophic epidermolysis bullosa (DDEB), which is caused by a dominant negative mutation in the COL7A1 gene encoding type VII collagen (COL7). We performed genome editing with TALENs and CRISPR/Cas9 targeting the mutation, c.8068_8084delinsGA. We then cotransfected Cas9 and guide RNA expression vectors expressed with GFP and DsRed, respectively, into induced pluripotent stem cells (iPSCs) generated from DDEB fibroblasts. After sorting, 90% of the iPSCs were edited, and we selected four gene-edited iPSC lines for further study. These iPSCs were differentiated into keratinocytes and fibroblasts secreting COL7. RT-PCR and Western blot analyses revealed gene-edited COL7 with frameshift mutations degraded at the protein level. In addition, we confirmed that the gene-edited truncated COL7 could neither associate with normal COL7 nor undergo triple helix formation. Our data establish the feasibility of mutation site-specific genome editing in dominant negative disorders.
Genome editing with engineered site-specific endonucleases is an approach being used to correct genetic mutations, in contrast to conventional gene therapy methods of gene replacement, such as viral or nonviral transfection of cDNA (1). The technique leads to double-strand breaks (DSBs), which stimulate cellular DNA repair through either the homology-directed repair (HDR) pathway or the nonhomologous end-joining (NHEJ) pathway (2). The HDR pathway uses a donor DNA template to guide repair and can be used to create specific sequence changes to the genome, including the targeted addition of whole genes (3). In contrast, the NHEJ pathway is error-prone and thus conducive to generating frameshift mutations, leading to intentional knockout of a gene or correction of a disrupted reading frame (4).
Based on the DNA recognition motif, four distinct platforms of engineered nucleases have been developed: meganucleases (MNs), zinc-finger nucleases (ZFNs), transcription activator-like effector nucleases (TALENs), and the clustered regularly interspaced short palindromic repeat (CRISPR)/CRISPR-associated protein (Cas) system (3, 5–7). Compared with MNs and ZFNs, TALENs and CRISPR/Cas offer more flexibility in target site design, which enables the targeting of mutation-specific sites in patients with genetic diseases.
Dominant dystrophic epidermolysis bullosa (DDEB) is a rare genetic blistering skin disorder with no known cure (8, 9). DDEB is caused by dominant negative mutations in the COL7A1 gene encoding type VII collagen (COL7). Homotrimeric COL7 is secreted from both keratinocytes and fibroblasts, and is the main protein component of anchoring fibrils, which attach the dermis and epidermis (10). Glycine substitution or in-frame small insertion/deletion (indel) mutations in one allele of COL7A1 result in DDEB, in which one-eighth of all trimers are normal and seven-eighths of all trimers are disrupted by the abnormal protein (11).
Here we show that mutation site-specific NHEJ using CRISPR/Cas9 and TALENs can be applied to DDEB. We postulate that the disease can be treated simply by knocking out the mutant allele, while leaving the wild-type allele unchanged. The potential for generating patient-specific keratinocytes and fibroblasts treated by NHEJ could provide a significant benefit for patients with DDEB in combination with induced pluripotent stem cell (iPSC) technologies.
Results
Patient Information.
The patient with DDEB was a 34-y-old Asian male. Multiple erosions, scarring pruriginous papules, and lichenoid plaques were observed on his trunk and extremities. Direct DNA sequencing of genomic DNA obtained from blood detected a heterozygous complex indel COL7A1 mutation (c.8068_8084delinsGA) in exon 109 (Fig. 1A) (12).
Fig. 1.

Mutation analysis of the genomic DNA from the DDEB patient and schematic design and validation of CRISPR/Cas9 and TALENs. (A) Normal and mutant COL7A1 sequences. The DDEB patient has a heterozygous indel COL7A1 mutation (c.8068_8084delinsGA) in exon 109 (12). The 17-nucleotide deletion (framed rectangle in the normal COL7A1 allele) from c.8068–8084 with a GA insertion (framed rectangle in the mutant COL7A1 allele) resulted in a 15-nucleotide deletion within the collagenous domain. (B) Design of CRISPR/Cas9 and TALENs targeted to c.8068_8084delinsGA in COL7A1. Red characters indicate CRISPR and TAL binding sites, and blue characters indicate the protospacer adjacent motif. The mutation sites are highlighted by the framed rectangles. Two uppercase letters within the rectangles represent TALE repeat variable diresidues. (C) Schematic design of partial COL7A1 gene with the mutation in HEK293 cells. To evaluate CRISPR/Cas9- and TALEN-mediated genetic disruption by NHEJ, we generated HEK293 cells transfected with the partial COL7A1 gene (red arrows) containing the 15-indel mutation (asterisk). We amplified only the mutant sequence using the vector sequence-specific primer (green arrow). (D) Evaluating CRISPR/Cas9 and TALENs using Surveyor nuclease assays. gDNA extracted from HEK293 cells was assessed for modification by the Surveyor nuclease assay, which evaluated their NHEJ activity. Arrows denote expected cleavage band sizes indicative of NHEJ activity. n.d., not detected. (E) Schematic representation of normal (intrinsic) sequences. Normal sequence was amplified with primer (black arrow) located 3′ downstream of the DNA fragment transfected into HEK293 cells. (F) Surveyor nuclease analysis for the normal COL7A1 sequence. The CRISPR/Cas9 and TALENs did not exhibit NHEJ activity against the normal COL7A1 sequence. gDNA extracted from the DDEB patient’s fibroblasts served as a positive control.
The 17-nucleotide deletion with a GA insertion results in a 15-nucleotide deletion within the collagenous domain, which does not disrupt the downstream COL7A1 ORF (12). Consequently, the deletion of 15 nucleotides (five amino acids) interferes with the collagen triple helix (Gly–X–Y repeat) and causes the DDEB phenotype, likely in a dominant negative fashion. The indel mutation, c.8068_8084delinsGA, is extremely suitable for this approach, because CRISPR/Cas9 and TALENs can target the unique mutation site with high specificity.
Design and Validation of CRISPR/Cas9 and TALENs Targeting the COL7A1 Gene.
One CRISPR/Cas9 and three pairs of TALENs were designed to target the mutation site of the COL7A1 gene using in silico software (Fig. 1B) (13, 14). To evaluate CRISPR/Cas9- and TALEN-mediated gene editing via NHEJ, we transfected HEK293 cells with a partial COL7A1 genomic construct containing the mutation c.8068_8084delinsGA (Fig. 1C). CRISPR/Cas9 or each of the three TALEN pairs was transfected into the HEK293 cells. To analyze the mutant (transfected) and normal (intrinsic) COL7A1 sequences separately, we designed two primer pairs that could amplify them independently (Fig. 1 C and E). The amplified DNA was assessed for modification by the Surveyor nuclease assay, which can detect the frequency of allelic modifications (15). All engineered site-specific endonucleases showed NHEJ activity at the mutant sequence, with CRISPR/Cas9 exhibiting a substantially higher mutation frequency (6.5%) than the TALEN pairs (Fig. 1D). Importantly, the CRISPR/Cas9 and TALENs did not introduce NHEJ in the normal sequence (Fig. 1F). These results indicate that the CRISPR/Cas9 and TALENs specifically targeted only the mutant sequence of COL7A1. Based on these experiments, we used CRISPR/Cas9 for further mutation site-specific gene editing experiments in our DDEB iPSCs.
CRISPR/Cas9-Mediated Gene Editing in DDEB iPSCs.
We generated iPSCs from primary fibroblasts isolated from a healthy individual and the DDEB patient by transfection of integration-free episomal vectors (Figs. S1 and S2) (16, 17). To select iPSCs transfected with both Cas9 and guide RNA, we transfected pCas9_GFP and pgRNA_DsRed, which coexpress Cas9 and GFP and guide RNA and DsRed, respectively, into DDEB-iPSCs (Fig. S3A). At 48 h after electroporation of pCas9_GFP and pgRNA_DsRed into DDEB iPSCs, the transfected iPSCs were enzymatically dissociated to single cells, and the GFP and DsRed double-positive single iPSCs were sorted using flow cytometry methods (Fig. S3B).
Fig. S1.

Phase-contrast and immunofluorescence studies of normal and DDEB iPSCs. The morphology of normal and DDEB iPSCs and immunocytochemical analysis of stem cells markers are shown, including alkaline phosphatase, OCT4, Sox2, SSEA3, TRA1–60, and TRA1–81. The differentiation capabilities of iPSC lines into all three germ layers were determined by embryoid body formation in vitro. β-III tubulin (TUBIII), smooth muscle actin (SMA), and α-fetoprotein (AFP) indicate ectodermal, mesoderm, and endoderm differentiation, respectively. AP, alkaline phosphatase; EB, embryoid body. (Scale bars: 200 μm for phase-contrast, alkaline phosphatase stain, and embryoid body formation; 100 μm for immunofluorescence.)
Fig. S2.

Characteristics of iPSCs. (A) RT-PCR analysis of stem cell markers in normal, DDEB, and gene-edited DDEB iPSCs. The four reprogramming factors were endogenously expressed in these iPSC lines. RT-PCR analysis showed that additional pluripotent markers were expressed in normal, DDEB, and gene-edited DDEB iPSCs. Normal fibroblasts served as a negative control. FB, fibroblasts. (B) Methylation analysis of the NANOG promoter region in iPSCs. Bisulfite sequencing revealed that the NANOG promoter region in DDEB iPSCs and gene-edited iPSCs were demethylated in a pattern similar to that seen in normal iPSCs, demonstrating that our iPSCs were epigenetically reprogrammed and that the NANOG promoter was reactivated in these cells. ○, unmethylated CpGs; ●, methylated CpGs. (C) Karyotype analysis of iPSCs in the healthy control and DDEB patient. iPSCs of both individuals had a normal male karyotypes (46,XY[20] and 46,XY[20], respectively), consistent with the starting fibroblasts. (D) Teratoma formation analysis of iPSCs of the healthy control and the DDEB patient. The in vivo differentiation capabilities of iPSCs are shown by teratoma formation after intradermal injection of iPSCs into nude mice. No differences were detected in the characteristics of normal and DDEB-iPSCs, indicating that the iPSCs had the capacity to differentiate into all three germ layers. (Scale bars: 200 μm.)
Fig. S3.

Schematic design for generating gene-edited iPSCs and cell sorting of iPSCs cotransfected with pCas9_GFP and pgRNA_DsRed. (A) Schematic representation of CRISPR/Cas9 transfection into DDEB-iPSCs. The guide RNA was subcloned into DsRed-expression vector (pgRNA_DsRed). At the time of gene correction with CRISPR/Cas9, both the pgRNA_DsRed and pCas9_GFP, which coexpress Cas9 nuclease and GFP, were transfected. After electroporation of the two plasmids into iPSCs, the GFP- and DsRed-positive single iPSCs were sorted. (B) Flow cytometry analysis and cell sorting of GFP- and DsRed (PE)-double-positive cells. At 48 h after electroporation of pCas9_GFP and pgRNA_DsRed, the transfected iPSCs were enzymatically dissociated to single cells, and the GFP- and DsRed-double-positive single iPSCs were sorted using an BD Influx Cell Sorter. (Upper) Dot plots in arbitrary units on a linear scale showing forward scatter and side scatter (Left) and forward scatter and trigger pulse width (Right). FSC, forward scatter; SSC, side scatter. Each gate is used to exclude cell debris and select cells for dual fluorescence analysis. (Lower) Dot plot showing fluorescence intensity of iPSCs double-positive with GRP/FITC (x-axis) and DsRed (PE; y-axis), measured in arbitrary units on a log scale.
Once the single iPSCs formed colonies, we selected and expanded 50 colonies and performed direct sequence analysis. Gene editing was successfully induced in the mutant allele in 45 of the 50 clones (90%; 31 deletions, 12 insertions, and 2 indel mutations) (Table 1). Notably, gene editing of the normal COL7A1 allele was not detected, indicating high specificity for the mutant allele.
Table 1.
Sequence analysis of gene-edited DDEB-iPSCs induced by CRISPR/Cas9
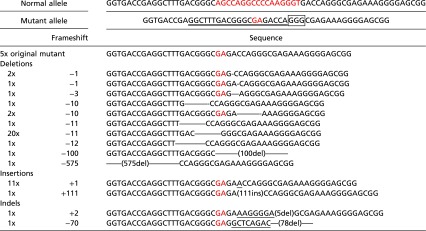 |
We selected 50 gene-edited DDEB-iPSC colonies and performed direct sequence analysis. Double underscores indicates CRISPR binding sites. The rectangle indicates the protospacer adjacent motif. Underscores indicate insertion sequences. Mutation sites are highlighted in red font.
We selected four gene-edited iPSC lines for further study (frameshift mutations: c.8073^8074insA, c.8073delC, and c.8072delA; in-frame mutation: c.8072_8074delACC). To further determine the frequency of mutagenesis at potential off-target sites, we analyzed the four clones at five distinct genomic regions with the greatest homology to the target site predicted by ZiFiT Targeter (zifit.partners.org/ZiFiT/) (13, 18) and CRISPR RGEN Tools (www.rgenome.net) (19). We found that none of these sites had undergone nonspecific cleavage (Table S1).
Table S1.
Summary of the potential off-target site
 |
Mutation sites are in blue type.
Characterization of Keratinocytes and Fibroblasts Derived from Gene-Edited DDEB iPSC Lines.
Before the differentiation of iPSCs into keratinocytes and fibroblasts, we first analyzed and evaluated the expression of stem cell markers and differentiation capacity in embryoid body formation assays in vitro to confirm the pluripotency of the four iPSC lines (Fig. S4). iPSC-derived keratinocytes and fibroblasts were generated as described previously (20, 21). No differences were detected among normal, untreated DDEB, and gene-edited DDEB iPSC lines in the process of differentiation into keratinocytes and fibroblasts. Immunofluorescence studies showed the expression of cell type-specific proteins in iPSC-derived keratinocytes and fibroblasts (Fig. 2 and Fig. S5). Furthermore, there were no differences between corrected and wild-type keratinocytes and fibroblasts, including in COL7 expression pattern (Fig. 2). There also were no notable differences in growth and survival rates among normal, untreated DDEB, and gene-edited DDEB iPSC-derived keratinocytes and fibroblasts.
Fig. S4.

Phase-contrast and immunofluorescence studies of iPSC clones. The morphology of gene-edited DDEB iPSC clones and immunocytochemical analysis of stem cell markers are shown. The iPSC colonies were identified by positive alkaline phosphatase staining and the expression of several stem cell markers, including OCT4, SOX2, SSEA3, TRA1–60, and TRA1–81. We confirmed the differentiation capabilities of iPSC lines into all three germ layers by embryoid body formation in vitro. β-III tubulin, smooth muscle actin, and α-fetoprotein indicate ectodermal, mesoderm, and endoderm differentiation, respectively. (Scale bars: 200 μm for phase-contrast, alkaline phosphatase stain, and embryoid body formation; 100 μm for immunofluorescence.)
Fig. 2.

Phase-contrast and immunofluorescence study of keratinocytes (KC) and fibroblasts (FB) derived from iPSCs. (A) Keratinocytes derived from normal, DDEB, and gene-edited iPSCs expressed keratinocyte markers, including keratin 14, p63, and COL7, similar to normal primary keratinocytes. (Scale bars: 100 μm for phase-contrast; 50 μm for immunofluorescence.) (B) Fibroblasts derived from normal iPSCs, DDEB iPSCs, and gene-edited DDEB iPSCs expressed fibroblast markers, including type I collagen (COL1), type III collagen (COL3), and COL7, similar to normal fibroblasts. (Scale bars: 200 μm for phase-contrast; 100 μm for immunofluorescence.)
Fig. S5.

Immunofluorescence study of fibroblasts derived from iPSCs. Fibroblasts derived from normal iPSCs, DDEB iPSCs, and gene-edited DDEB iPSCs expressed fibroblast markers, including COL4, PH4B, and vimentin, similar to normal fibroblasts. FB, fibroblast. (Scale bars: 100 μm.)
Analysis of COL7A1 mRNA Expression in Gene-Edited Fibroblasts.
To investigate the expression of COL7A1 transcripts in the gene-edited cell lines, we performed RT-PCR using total RNA extracted from iPSC-derived fibroblasts. The COL7A1 cDNA was amplified using primers designated in exons 105, 113, and 114. Direct sequencing of the RT-PCR product showed a heterozygous RNA sequence and the same expression level of gene-edited or untreated mutant cells as that of normal cells (Fig. S6A).
Fig. S6.

RT-PCR and TA cloning analyses of iPSC-derived fibroblasts. (A) RT-PCR of iPSC-derived fibroblasts. To investigate the expression of COL7A1 transcripts in the gene-edited cell lines, RT-PCR was performed using total RNA extracted from iPSC-derived fibroblasts. The gene-edited or untreated mutant RNA expression level was the same as that of normal RNA. FB, fibroblasts. (B) TA cloning analysis of cDNA derived from iPSC-derived fibroblasts. TA cloning of the amplified cDNA was performed. TA cloning analysis showed that the splicing event of gene-edited COL7A1 was the same as that of the normal and untreated mutant COL7A1. The transcripts of gene-edited COL7A1 resulting in a frameshift led to premature termination codon (PTC) mutations (c.8073^8074insA: p.Asp2691Glufs*22; c.8073delC: p.Gln2692Argfs*88; c.8072delA: p.Asp2691Alafs*89). These findings indicate that c.8073^8074insA, c.8073delC, and c.8072delA result in PTC mutations, but do not lead to NMD.
To further characterize the gene-edited COL7A1 mRNA, we performed TA cloning of the amplified cDNA (Fig. S6B). Our TA cloning analysis showed that the splicing event of gene-edited COL7A1 was the same as that of the normal and untreated mutant COL7A1. The resultant transcripts of gene-edited COL7A1 resulting in a frameshift led to premature termination codon (PTC) mutations (c.8073^8074insA: p.Asp2691Glufs*22, c.8073delC: p.Gln2692Argfs*88 and c.8072delA: p.Asp2691Alafs*89). These findings indicate that c.8073^8074insA, c.8073delA, and c.8072delA result in PTC mutations, but do not lead to nonsense-mediated mRNA decay (NMD).
Analysis of COL7 Protein Expression in Gene-Edited Fibroblasts.
To analyze COL7 protein expressed by gene-edited fibroblasts, we performed Western blot analysis of cell lysate. Cytoplasmic truncated COL7 protein was slightly detected in the c.8073^8074insA, c.8073delC, and c.8072delA fibroblasts (Fig. 3A).
Fig. 3.

Western blot analysis of COL7 produced by iPSC-derived fibroblasts. (A) Western blot analysis using cell lysate. Cytoplasmic truncated COL7 protein was detected at low levels in the c.8073^8074insA, c.8073delC, and c.8072delA fibroblasts. In contrast, the truncated COL7 was absent in normal human fibroblasts or uncorrected patient and in-frame c.8072_8074delACC. Antibody to vimentin served as the loading control for cell lysates. (B) Western blot analysis using fibroblast culture medium was performed to determine whether the truncated COL7 proteins were intracellularly degraded. The secreted COL7 band pattern showed no difference between gene-edited fibroblasts and the cells from a normal individual and the DDEP patient. These findings suggest that truncated COL7 proteins expressed from frameshift gene-edited fibroblasts are degraded intracellularly.
We next performed Western blot analysis using fibroblast culture medium. On immunoblot analysis of supernatant, the secreted COL7 band pattern showed no differences among gene-edited fibroblasts and the cells from a normal individual and the DDEB patients (Fig. 3B). These results suggest that truncated COL7 proteins expressed from frameshift gene-edited fibroblasts may be degraded in the cytoplasm.
Triple-Helix Formation Analysis of the Truncated Recombinant COL7 Protein.
It is known that three pro-COL7 polypeptides associate through their carboxyl-terminal ends within the intracellular space of keratinocytes and fibroblasts, and, subsequently, COL7 trimer is secreted into the extracellular space (22). To determine whether the gene-edited COL7 protein can be secreted appropriately, we generated a COL7 expression construct with the same mutation by site-directed mutagenesis and assessed the ability of the recombinant COL7 to form trimers. Transfection of the normal sequence, the original mutant, and c.8072_8074delACC COL7A1 cDNA into HEK293 cells resulted in the secretion of a 290-kDa COL7 monomer and a 900-kDa COL7 trimer. In contrast, the COL7 trimer was not detected in cells expressing the truncated COL7 with the frameshift mutation (Fig. 4A).
Fig. 4.

Trimer formation analysis and co-IP assay of recombinant COL7. (A) Trimer formation analysis of recombinant COL7 protein. To assess the ability of the COL7 isoforms to form trimers, the COL7 expression construct with the same mutation was generated by site-directed mutagenesis, and Western blot analysis was performed under nonreducing conditions. Transfection of the normal, original mutant and c.8072_8074delACC COL7A1 cDNA into HEK293 cells resulted in secretion of a 290-kDa monomer and a 900-kDa trimer of COL7. In contrast, the COL7 trimer was not detected in the truncated COL7 with frameshift mutation. (B) Co-IP of normal and gene-edited mutant COL7. HEK293 cells were transfected with FLAG-normal COL7 and HA-normal or mutant COL7, and cell media were subjected to IP using anti–FLAG-tagged mAb magnetic beads. IP materials were analyzed with anti-HA antibody.
Interaction Analysis Between the Gene-Edited Mutant and Normal COL7.
To evaluate the potential interactions between gene-edited mutant and normal COL7, we generated FLAG-tagged normal COL7 and HA-tagged normal and mutant COL7 expression vectors and cotransfected the two different tagged vectors into HEK293 cells. Coimmunoprecipitation (co-IP) assays showed the association of HA-tagged normal, original mutant, and gene-edited in-frame mutant (c.8072_8074delACC) COL7 with FLAG-tagged normal COL7. In contrast, the HA-tagged truncated COL7 failed to associate with FLAG-tagged normal COL7 (Fig. 4B).
Discussion
Eliminating the mutant protein while sparing the normal protein is a desirable approach to developing therapies for dominant negative disorders. In principle, this can be achieved through degradation of the mutant mRNA or protein or through disruption or substitution of the mutant gene. To degrade the mutant mRNA, RNA interference has been used as a potential therapy for dominant negative disorders (23); however, this method requires repeated injection and improvements in delivery techniques. It was recently reported that engineered site-specific endonucleases together with a DNA repair template can replace a mutant allele with a wild-type sequence via HDR (24).
In this study, we succeeded in knocking out the COL7A1 mutant allele in DDEB via mutation site-specific mutagenesis NHEJ using CRISPR/Cas9 and TALENs. The NHEJ pathway offers several potential advantages over the HDR pathway. NHEJ does not require a donor template, which may cause nonspecific insertional mutagenesis. In addition, Cre/loxP or Flp/FRT recombination systems are used to remove the unwanted selection cassettes for isolating clones that have undergone HDR, which can leave behind single loxP or FRT sites (25, 26). These small ectopic sequences have the potential to interfere with transcriptional regulatory elements of surrounding genes (27).
Although the use of CRISPR/Cas9 in iPSCs has been reported in the literature, the efficiency of targeting rate was relatively low (7). Ding et al. (28) generated pCas9 nuclease coexpressed with GFP (pCas9_GFP), resulting in much more efficient gene editing after cell selection using flow cytometry (51–79%). We used that strategy in this study and generated guide RNA coexpressed with DsRed, and transfected both pCas9_GFP and pgRNA_DsRed into DDEB iPSCs. Unexpectedly, the efficiency of gene editing was ∼90% after sorting of double-positive cells. The high efficiency of our strategy allows for decreases in Cas9 and guide RNA concentrations for transfection into iPSCs. The targeting specificity of Cas9 nucleases is of particular concern, especially for clinical applications and gene therapy.
Enzymatic concentration is an important factor in determining Cas9 off-target mutagenesis (29, 30). Therefore, this technique could be manipulated to decrease Cas9 and guide RNA concentration and improve the on-target to off-target ratio at the expense of the efficiency of on-target cleavage. In addition, the flow cytometry step in the gene editing strategy enables selection of iPSCs transfected with appropriates amount of Cas9 and guide RNA by setting the ranges of GFP and DsRed intensities. Every cell has a different dose of plasmid or linear DNA after electroporation or lipofection, even though the cells are of a monoclonal cell line. In our strategy, we used flow cytometry to select iPSCs expressed with GFP and DsRed by fluorescence intensity, which reflects the expression level of Cas9 and guide RNA, resulting in exclusion of iPSCs overexpressed with Cas9 or guide RNA at the single-cell level.
iPSCs constitute a unique cell type that can be generated directly from patients and have the capacity for multipotential differentiation and unlimited self-renewal (31). There were two benefits of using iPSCs in this study. The first is the ability to study multipotential differentiation. Within the intracellular space of keratinocytes and fibroblasts, three pro-COL7 polypeptides associate through their carboxyl-terminal ends, and their collagenous domains fold into a characteristic triple-helical conformation. After being secreted into the extracellular space, triple-helical COL7 molecules form antiparallel dimers. Subsequently, a large number of dimer molecules assemble laterally in register to form anchoring fibrils below the basement membrane zone (22). Given that secreted COL7 trimers are expressed from both keratinocytes and fibroblasts and could interfere with each other, it would be beneficial to gene-correct both keratinocytes and fibroblasts in a therapeutic strategy for DDEB. The gene-corrected iPSCs could provide a robust source of patient-specific keratinocytes and fibroblasts with which to perform cultured epidermal autograft and systemic or local injections.
The second advantage of using iPSCs is their unlimited self-renewal capacity, given the need to expand single cell clones after gene editing. A critical consideration for the clinical use of gene editing reagents is the potential for off-target effects. Engineered nucleases can cause target site-dependent and -independent cleavages, resulting in small indels. Such indels may damage tumor-suppressor genes, and possibly ultimately lead to tumorigenicity. To decrease the possibility of off-target mutagenesis, it is better to expand and analyze single cell clones than polyclonal cell populations. Gene editing could provide a significant benefit for DDEB patients in combination with iPSC technology, which affords multipotential cellular differentiation and potentially unlimited self-renewal capacity.
We generated four gene-edited iPSC lines (frame shift mutations: c.8073^8074insA, c.8073delC, and c.8072delA; in-fame mutation: c.8072_8074delACC). The frameshift gene editing led to a PTC mutation, which would be expected to degrade at the mRNA level by the NMD pathway (32); however, in our RT-PCR analysis, the expression level of mRNA with the PTC mutations was similar to that of normal COL7A1 mRNA (Fig. S6A). Although Western blot analysis using cell lysate detected truncated COL7 isoforms in the frameshift gene-edited fibroblasts (Fig. 3A), the secreted COL7 band pattern showed no differences among gene-edited fibroblasts and the counterparts from a normal control and the DDEB patient (Fig. 3B), indicating that the proteins translated from the COL7A1 allele with the frameshift mutations might be degraded at the protein level.
To confirm this hypothesis, we generated a COL7 expression construct with the same mutation, and found that the truncated recombinant protein produced from HEK293 cells transfected with the frameshift mutation failed to form a COL7 trimer (Fig. 4A). In fact, it has been reported that DDEB patients with the E2857X mutation in COL7A1 express truncated COL7 protein at the dermal–epidermal junction, although the nonsense mutation is located more than 50 nucleotides upstream of the most 3′ exon–exon junction (33). Interestingly, in a previous immunohistological study, two DDEB patients with c.8074delG and c.8091delG, resulting in PTC mutations at p.2785 and p.2799, respectively, did not express COL7 at the dermal–epidermal junction (34, 35). Taken together, our findings indicate that truncated COL7 protein with a PTC mutation at p.2857 might not be degraded in cytoplasm, whereas shorter COL7 proteins with PTC mutations at p.2785 and p.2779 likely may be degraded in cytoplasm.
We have developed successful genome editing approaches with both CRISPR/Cas9 and TALENs targeting the DDEB c.8068_8084delinsGA site-specific sequence. Gene editing was applied to DDEB-iPSCs, which we reprogrammed in this study. We tested the differentiation capacity of gene-corrected iPSCs into keratinocytes and fibroblasts that expressed COL7. The potential for generating functional keratinocytes and fibroblasts from gene-edited iPSCs could provide a significant benefit for DDEB patients.
Our data establish the feasibility of site-specific genome editing to generate null alleles using CRISPR/Cas9 and TALENs for selective targeting of the mutation site in dominant negative disorders, which can be coupled with directed differentiation of iPSCs into skin cell types in a therapeutic approach for DDEB.
Materials and Methods
Ethics Statement.
Informed consent was obtained from all subjects, and approval for this study was provided by the Institutional Review Board of Columbia University in accordance with the Declaration of Helsinki.
Generation of HEK293 Cells Transfected with Partial COL7A1 Gene Including c.8068_8084delinsGA.
To evaluate CRISPR/Cas9 and TALENs-mediated genetic disruption by NHEJ, we generated HEK293 cells transfected with partial COL7A1 gene with c.8068_8084delinsGA (Fig. 1C). The Surveyor nuclease assay detecting mismatch of heteroduplex DNA was used to evaluate the efficiency of NHEJ (15). The gene-targeted DNA fragment covering exon 109 and 110 was amplified with the primers of intron 108 F and intron 110–1 R (Table S2). The DNA fragment with the mutation was subcloned into the EcoRI site of pIRES-puro-GFP vector (Addgene plasmid 16616) (36). HEK293 cells were transfected with the plasmid using FuGENE HD transfection reagent (Promega) and selected with 2 μg/mL puromycin.
Table S2.
Primers used in this study
| Gene | Primer sequence | Aim |
| cMYC | Forward: ttcgggtagtggaaaaccag | RT-PCR |
| Reverse: cagcagctcgaatttcttcc | ||
| SOX2 | Forward: acaccaatcccatccacact | RT-PCR |
| Reverse: gcaaacttcctgcaaagctc | ||
| OCT4 | Forward: gtactcctcggtccctttcc | RT-PCR |
| Reverse: caaaaaccctggcacaaact | ||
| KLF4 | Forward: cccacacaggtgagaaacct | RT-PCR |
| Reverse: ttctggcagtgtgggtcata | ||
| DPPA2 | Forward: tcagccaggaattcaagacc | RT-PCR |
| Reverse: acgcttggttccatttgttc | ||
| DPPA4 | Forward: gggttcacctgcagtttcat | RT-PCR |
| Reverse: tctccagcttttctgccatt | ||
| ESG1 | Forward: ccgaagacctgaaagatcca | RT-PCR |
| Reverse: gactggagcatccacttggt | ||
| LIN28 | Forward: tgaggcagtggagttcacct | RT-PCR |
| Reverse: cttgcattccttggcatgat | ||
| NANOG | Forward: gatttgtgggcctgaagaaa | RT-PCR |
| Reverse: aagtgggttgtttgcctttg | ||
| TDGF1 | Forward: acagaacctgctgcctgaat | RT-PCR |
| Reverse: atcacagccgggtagaaatg | ||
| TERT | Forward: gcgtttggtggatgatttct | RT-PCR |
| Reverse: cagggcctcgtcttctacag | ||
| ZIC3 | Forward: cccacacaggtgagaaacct | RT-PCR |
| Reverse: tatagcgggtggagtggaag | ||
| GAPDH | Forward: tcatctctgccccctctgct | RT-PCR |
| Reverse: cgacgcctgcttcaccacct | ||
| NANOG promoter | Forward: tggttaggttggttttaaatttttg | Methylation analysis |
| Reverse: aacccacccttataaattctcaatta | ||
| COL7A1 exon 105 F | Forward: aggagatgttggcttcatgg | RT-PCR |
| COL7A1 exon 113–114 R | Reverse: caggcacagtgctgactcat | RT-PCR |
| COL7A1 intron 108 F | Forward: agggtctgtggacagtggac | Surveyor nuclease assay |
| COL7A1 intron 110–1 R | Reverse: atgaggttggaagggtaggg | Surveyor nuclease assay |
| COL7A1 intron 110–2 R | Reverse: atcagagtcaggcaggttgg | Surveyor nuclease assay |
| GFP R | Reverse: agaacggcatcaaggtgaac | Surveyor nuclease assay |
To analyze the mutant (transfected) and normal (intrinsic) sequences separately, we used two primer pairs (Table S2). To amplify only the mutant sequence, we used primers placed in intron 108-F and GFP-R placed in the vector sequence (Fig. 1C). To amplify only the normal (intrinsic) sequence, we used primers placed in intron 108-F and intron 110–2-R located 3′ downstream of the DNA fragment subcloned into the vector (Fig. 1E).
Gene Correction of iPSCs Derived from the DDEB Patient.
Because the CRISPR/Cas9 vector was the most efficient editing system, we used the CRISPR/Cas9 vector in this study. iPSCs derived from the DDEB patient were cultured using feeder-free adherent culture conditions in chemically defined mTeSR1 media (Stem Cell Technologies) on dishes precoated with Matrigel (BD Biosciences). For 3 h before electroporation, iPSCs were pretreated with 10 μM ROCK inhibitor (Y-27632; EMD Millipore). After dissociation with Accutase (Invitrogen), 7.5 μg of pgRNA-DsRed and 7.5 μg of pCas9_GFP vectors were electroporated into 4 × 106 iPSCs with the Amaxa Nucleofector 2 Device (Lonza; program A-023).
After electroporation, the transfected iPSCs were cultured in mTeSR1 with 10 μM ROCK inhibitor. At 48 h following electroporation, the transfected iPSCs were dissociated to single cells with Accutase, and the GFP and DsRed double-positive single iPSCs were sorted with an Influx Cell Sorter (BD Biosciences) and then reseeded onto mitomycin C (MMC)-treated mouse embryo fibroblasts (MEFs) (Fig. S3 A and B) (28, 37). Once the GFP and DsRed double-positive iPSCs formed colonies, the cells were split into two wells, and direct sequence analysis was performed.
Differentiation into Keratinocytes and Fibroblasts.
iPSC-derived keratinocytes and fibroblasts were generated as described previously (20, 21). These iPSC-derived keratinocytes and fibroblasts were characterized by immunofluorescence studies as described previously (Table S3) (20, 21).
Table S3.
Antibodies used in this study
| Antibody | Dilution | Company |
| Primary antibodies | ||
| Anti-human/mouse OCT-3/4 | 1:25 | R&D Systems |
| Rabbit anti-SOX2 | 1:100 | Invitrogen |
| Rat anti-SSEA3 | 1:100 | Invitrogen |
| Anti–TRA-1–60 | 1:200 | Millipore |
| Anti–TRA-1–81 | 1:200 | Millipore |
| Rabbit polyclonal to neuron-specific beta-III tubulin | 1:300 | Abcam |
| Anti-smooth muscle actin | 1:100 | Abcam |
| Anti–α-fetoprotein | 1:200 | Abcam |
| Anti-keratin 14 | 1:1,000 | Covance |
| Anti-p63 (4A4) | 1:100 | Santa Cruz Biotechnology |
| Anti-type VII collagen | 1:100 | Calbiochem |
| Anti-type I collagen | 1:300 | Abcam |
| Anti-type III collagen | 1:100 | Fitzgerald Industries |
| Anti-type IV collagen | 1:300 | Chemicon |
| Anti-PH4B | 1:300 | Novus Biologicals |
| Anti-vimentin | 1:100 | Santa Cruz Biotechnology |
| Secondary antibodies | ||
| Alexa Fluor 488 donkey anti-rat IgG (H+L) | 1:1,000 | Invitrogen |
| Alexa Fluor 488 goat anti-rat IgM (μ chain) | 1:1,000 | Invitrogen |
| Alexa Fluor 488 goat anti-rabbit IgG (H+L) | 1:1,000 | Invitrogen |
| Alexa Fluor 488 goat anti-mouse IgG (H+L) | 1:1,000 | Invitrogen |
| Alexa Fluor 488 goat anti-mouse IgM (μ chain) | 1:1,000 | Invitrogen |
Generation of Recombinant COL7 DNA Constructs.
Site-directed mutagenesis was performed on the COL7A1 cDNA in the modified pCMVβ by replacing LacZ with human full-length COL7A1 cDNA using the KOD Plus Mutagenesis Kit (Toyobo) (38). For co-IP assays, a FLAG tag and an HA tag were introduced into the normal COL7A1 cDNA and the normal and mutant COL7A1 cDNA, respectively, after amino acid 23 (39).
Characterization of Mutant COL7.
For evaluation of the function of triple-helix formation, the COL7 expression construct was transfected into HEK293 cells using FuGENE HD. After 48 h of transfection, the media were collected, and Western blot analysis was performed under nonreducing conditions.
To investigate the association of gene-edited COL7 protein with the normal counterpart, we performed co-IP assays. The FLAG-tagged normal COL7 and the HA-tagged normal or mutant COL7 expression vectors were cotransfected into HEK293 cells. After 48 h, the cell medium was immunoprecipitated with anti–FLAG-tagged mAb magnetic beads (MBL) and then eluted with Laemmli sample buffer at 95 °C for 5 min. The proteins were separated on SDS/PAGE and immunoblotted with anti-HA tag antibody.
SI Materials and Methods
Construction of Guide RNA and TALEN Expression Vectors Targeting the Mutant Site-Specific Sequence.
The CRISPR/Cas9 candidate molecules targeted to the mutant site were designed using ZiFiT Targeter (Fig. 1B) (13). A 469-bp fragment flanked by the HindIII restriction site contains all of the components necessary for guide RNA expression, including the U6 promoter, target sequence, guide RNA scaffold, and termination signal. The fragment was synthesized as gBlocks Gene Fragments (Integrated DNA Technologies) (7). The DNA fragment was subcloned into the HindIII site of pCAG-DsRed (Addgene plasmid 11151) (pgRNA_DsRed) (40).
Using the TAL Effector Nucleotide Targeter (https://tale-nt.cac.cornell.edu/) and ZiFiT Targeter, three TALEN pairs were designed to target the mutation site (Fig. 1B) (13, 14). Expression vectors of TALENs were constructed via the Golden Gate Assembly method (Addgene; catalog no. 1000000024) (41). The TALE repeat arrays were inserted into a homodimeric form of a truncated CAG promoter-driven FokI endonuclease (pC-GoldyTALEN; Addgene plasmid 38143) (42).
Generation and Evaluation of iPSCs.
Fibroblasts were isolated from the skin of a healthy individual and of the DDEB patient and expanded in DMEM supplemented with 10% (vol/vol) FBS and 1% penicillin-streptomycin (Invitrogen). Integration-free iPSCs from fibroblasts were generated using transfection of integration-free episomal plasmids. To generate iPSCs from fibroblasts, on day 0 four reprogramming vectors (pCXLE-hOCT3/4-shp53-F: OCT3/4 and p53 shRNA, pCXLE-hSK: SOX2 and KLF4, pCXLE-hUL: l-Myc and LIN28, pCXWB-EBNA1: EBNA1; Addgene plasmids 27077, 27078, 27080, and 37624) were electroporated into fibroblasts with the Amaxa Nucleofector 2 Device (Lonza) with a NHDF Nucleofector Kit (16, 17). Transfected fibroblasts were trypsinized on day 7 and reseeded onto an MMC-treated MEF feeder layer. These cells were maintained in iPSC medium [KO DMEM (Invitrogen) supplemented with 20% (vol/vol) KO serum replacement (Invitrogen), 1% GlutaMAX-I (Invitrogen), 1% nonessential amino acid (Invitrogen), 1% penicillin-streptomycin, and 4 ng/mL basic FGF (R&D Systems)] (20). After 25 days posttransfection, colonies with a flat human embryonic stem cell-like morphology were mechanically picked up and replated on MMC-treated MEFs for expansion and characterization. To confirm their pluripotency, the iPSCs were analyzed for expression of stem cell markers and differentiation capacity from embryoid body formation in vitro (Tables S2 and S3) (20).
Evaluating CRISPR/Cas9 and TALENs Using the Surveyor Nuclease Assay.
The pgRNA_DsRed and pCas9_GFP (Addgene plasmid 44719) and the left and right TALEN pairs were cotransfected into HEK293 cells transfected with a partial COL7A1 gene, including the mutation site, using Fugene HD transfection reagent. After transfection, cells were cultured for 2 days at 32 °C and 37 °C, and whole gDNA was extracted using the Qiagen DNeasy Blood and Tissue Kit. The targeted mutant sequence was amplified using intron 108-F and GFP-R primers, whereas the normal sequence was amplified using intron 108-F and intron 110-2-R primers. The amplified PCR products were subjected to Surveyor nuclease treatment as described previously (15). The digestion products were loaded onto a 1.5% agarose gel stained with ethidium bromide, and images were captured with a ChemiDoc MP Imaging System (Bio-Rad). Densitometry was performed using ImageJ as described previously (15).
RNA Isolation and RT-PCR.
Total RNA was isolated from iPSC-derived fibroblasts using the Qiagen RNeasy Mini Kit and treated with DNase (Invitrogen) to remove gDNA contaminations. cDNA was synthesized using 3 μg of RNA by SuperScript III reverse transcriptase (Invitrogen) and Oligo-dT primer (Invitrogen) according to the manufacturer’s instructions. Using the first-strand cDNA as a template, the COL7A1 cDNA was amplified using primers designed in exons 105, 113, and 114 of the COL7A1 cDNA (Table S2). The PCR products were TA-cloned into the pCRII-TOPO vector (Invitrogen), after which each allele was sequenced separately.
Western Blot Analysis of iPSC-Derived Fibroblasts.
iPSC-derived fibroblasts were maintained for 48 h after the medium was changed to serum-free DMEM supplemented with 50 μg/mL ascorbic acid (Sigma-Aldrich). After 48 h of incubation, the medium was collected and equilibrated to protease inhibitor mixture (Sigma-Aldrich) and 5 mM EDTA, and then concentrated 5- to 10-fold (Centricon-30; Amicon). The cell lysate was extracted using RIPA buffer. Proteins were separated on 4% and 4–20% gradient SDS/PAGE gels. The samples were mixed with an equal volume of sample buffer supplemented with β-mercaptoethanol. All samples were boiled for 5 min before electrophoresis. Blots were transferred to a PVDF membrane that was blocked with PBS containing 2% (wt/vol) skim milk and 0.05% (vol/vol) Tween 20, incubated with rabbit anti-COL7 polyclonal antibody (Calbiochem) for 1 h at room temperature, and then treated with HRP-conjugated anti-rabbit IgG antibody for 45 min at room temperature. The blots were detected using SuperSignal West Pico Chemiluminescent Substrate (Thermo Scientific). The images were obtained with a ChemiDoc MP Imaging System (Bio-Rad).
Acknowledgments
We thank Ming Zhang, Emily Chang, and Yui Shinkuma for their expert assistance with the immunofluorescence studies, animal experiments, and gene analyses, respectively. We also thank Drs. Munenari Itoh, Noriko Arao-Umegaki, and Wataru Nishie for stimulating discussions and Dr. Siu-hong Ho, Yanan Ding, and Dr. Murty Vundavalli for expert assistance with cell sorter and karyotype analysis. Funding for this work was provided by the Skin Disease Research Center in the Department of Dermatology, Columbia University (National Institutes of Health/National Institute of Arthritis and Musculoskeletal and Skin Diseases Grant P30AR44535), the Japan Society for the Promotion of Science (Grants-in-Aid for Research Activity Start-Up Grant 15H05999 and a Postdoctoral Fellowship for Research Abroad), the New York State Stem Cell Science program (Grant SDH C024321), and the Helmsley Trust.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1512028113/-/DCSupplemental.
References
- 1.Perez-Pinera P, Ousterout DG, Gersbach CA. Advances in targeted genome editing. Curr Opin Chem Biol. 2012;16(3-4):268–277. doi: 10.1016/j.cbpa.2012.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Joung JK, Sander JD. TALENs: A widely applicable technology for targeted genome editing. Nat Rev Mol Cell Biol. 2013;14(1):49–55. doi: 10.1038/nrm3486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Urnov FD, et al. Highly efficient endogenous human gene correction using designed zinc-finger nucleases. Nature. 2005;435(7042):646–651. doi: 10.1038/nature03556. [DOI] [PubMed] [Google Scholar]
- 4.Porteus MH, Baltimore D. Chimeric nucleases stimulate gene targeting in human cells. Science. 2003;300(5620):763. doi: 10.1126/science.1078395. [DOI] [PubMed] [Google Scholar]
- 5.Arnould S, et al. The I-CreI meganuclease and its engineered derivatives: Applications from cell modification to gene therapy. Protein Eng Des Sel. 2011;24(1-2):27–31. doi: 10.1093/protein/gzq083. [DOI] [PubMed] [Google Scholar]
- 6.Bogdanove AJ, Voytas DF. TAL effectors: Customizable proteins for DNA targeting. Science. 2011;333(6051):1843–1846. doi: 10.1126/science.1204094. [DOI] [PubMed] [Google Scholar]
- 7.Mali P, et al. RNA-guided human genome engineering via Cas9. Science. 2013;339(6121):823–826. doi: 10.1126/science.1232033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fine JD, et al. The classification of inherited epidermolysis bullosa (EB): Report of the Third International Consensus Meeting on Diagnosis and Classification of EB. J Am Acad Dermatol. 2008;58(6):931–950. doi: 10.1016/j.jaad.2008.02.004. [DOI] [PubMed] [Google Scholar]
- 9.Shinkuma S. Dystrophic epidermolysis bullosa: A review. Clin Cosmet Investig Dermatol. 2015;8:275–284. doi: 10.2147/CCID.S54681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shinkuma S, McMillan JR, Shimizu H. Ultrastructure and molecular pathogenesis of epidermolysis bullosa. Clin Dermatol. 2011;29(4):412–419. doi: 10.1016/j.clindermatol.2011.01.010. [DOI] [PubMed] [Google Scholar]
- 11.Dang N, Murrell DF. Mutation analysis and characterization of COL7A1 mutations in dystrophic epidermolysis bullosa. Exp Dermatol. 2008;17(7):553–568. doi: 10.1111/j.1600-0625.2008.00723.x. [DOI] [PubMed] [Google Scholar]
- 12.Sawamura D, Nizeki H, Miyagawa S, Shinkuma S, Shimizu H. A novel indel COL7A1 mutation 8068del17insGA causes dominant dystrophic epidermolysis bullosa. Br J Dermatol. 2006;154(5):995–997. doi: 10.1111/j.1365-2133.2006.07148.x. [DOI] [PubMed] [Google Scholar]
- 13.Sander JD, et al. ZiFiT (Zinc Finger Targeter): An updated zinc finger engineering tool. Nucleic Acids Res. 2010;38(Web Server issue):W462–W468. doi: 10.1093/nar/gkq319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Doyle EL, et al. TAL Effector-Nucleotide Targeter (TALE-NT) 2.0: Tools for TAL effector design and target prediction. Nucleic Acids Res. 2012;40(Web Server issue):W117–W122. doi: 10.1093/nar/gks608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Guschin DY, et al. A rapid and general assay for monitoring endogenous gene modification. Methods Mol Biol. 2010;649:247–256. doi: 10.1007/978-1-60761-753-2_15. [DOI] [PubMed] [Google Scholar]
- 16.Okita K, Nakagawa M, Hyenjong H, Ichisaka T, Yamanaka S. Generation of mouse induced pluripotent stem cells without viral vectors. Science. 2008;322(5903):949–953. doi: 10.1126/science.1164270. [DOI] [PubMed] [Google Scholar]
- 17.Okita K, et al. An efficient nonviral method to generate integration-free human-induced pluripotent stem cells from cord blood and peripheral blood cells. Stem Cells. 2013;31(3):458–466. doi: 10.1002/stem.1293. [DOI] [PubMed] [Google Scholar]
- 18.Sander JD, et al. In silico abstraction of zinc finger nuclease cleavage profiles reveals an expanded landscape of off-target sites. Nucleic Acids Res. 2013;41(19):e181. doi: 10.1093/nar/gkt716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bae S, Kweon J, Kim HS, Kim JS. Microhomology-based choice of Cas9 nuclease target sites. Nat Methods. 2014;11(7):705–706. doi: 10.1038/nmeth.3015. [DOI] [PubMed] [Google Scholar]
- 20.Itoh M, Kiuru M, Cairo MS, Christiano AM. Generation of keratinocytes from normal and recessive dystrophic epidermolysis bullosa-induced pluripotent stem cells. Proc Natl Acad Sci USA. 2011;108(21):8797–8802. doi: 10.1073/pnas.1100332108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Itoh M, et al. Generation of 3D skin equivalents fully reconstituted from human induced pluripotent stem cells (iPSCs) PLoS One. 2013;8(10):e77673. doi: 10.1371/journal.pone.0077673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chung HJ, Uitto J. Type VII collagen: The anchoring fibril protein at fault in dystrophic epidermolysis bullosa. Dermatol Clin. 2010;28(1):93–105. doi: 10.1016/j.det.2009.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pendaries V, et al. siRNA-mediated allele-specific inhibition of mutant type VII collagen in dominant dystrophic epidermolysis bullosa. J Invest Dermatol. 2012;132(6):1741–1743. doi: 10.1038/jid.2012.11. [DOI] [PubMed] [Google Scholar]
- 24.Sun N, Abil Z, Zhao H. Recent advances in targeted genome engineering in mammalian systems. Biotechnol J. 2012;7(9):1074–1087. doi: 10.1002/biot.201200038. [DOI] [PubMed] [Google Scholar]
- 25.Hockemeyer D, et al. Efficient targeting of expressed and silent genes in human ESCs and iPSCs using zinc-finger nucleases. Nat Biotechnol. 2009;27(9):851–857. doi: 10.1038/nbt.1562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sebastiano V, et al. In situ genetic correction of the sickle cell anemia mutation in human induced pluripotent stem cells using engineered zinc finger nucleases. Stem Cells. 2011;29(11):1717–1726. doi: 10.1002/stem.718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Meier ID, et al. Short DNA sequences inserted for gene targeting can accidentally interfere with off-target gene expression. FASEB J. 2010;24(6):1714–1724. doi: 10.1096/fj.09-140749. [DOI] [PubMed] [Google Scholar]
- 28.Ding Q, et al. Enhanced efficiency of human pluripotent stem cell genome editing through replacing TALENs with CRISPRs. Cell Stem Cell. 2013;12(4):393–394. doi: 10.1016/j.stem.2013.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hsu PD, et al. DNA targeting specificity of RNA-guided Cas9 nucleases. Nat Biotechnol. 2013;31(9):827–832. doi: 10.1038/nbt.2647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hsu PD, Lander ES, Zhang F. Development and applications of CRISPR-Cas9 for genome engineering. Cell. 2014;157(6):1262–1278. doi: 10.1016/j.cell.2014.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126(4):663–676. doi: 10.1016/j.cell.2006.07.024. [DOI] [PubMed] [Google Scholar]
- 32.Couttet P, Grange T. Premature termination codons enhance mRNA decapping in human cells. Nucleic Acids Res. 2004;32(2):488–494. doi: 10.1093/nar/gkh218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Saito M, Masunaga T, Teraki Y, Takamori K, Ishiko A. Genotype-phenotype correlations in six Japanese patients with recessive dystrophic epidermolysis bullosa with the recurrent p.Glu2857X mutation. J Dermatol Sci. 2008;52(1):13–20. doi: 10.1016/j.jdermsci.2008.03.005. [DOI] [PubMed] [Google Scholar]
- 34.Gardella R, et al. Genotype-phenotype correlation in Italian patients with dystrophic epidermolysis bullosa. J Invest Dermatol. 2002;119(6):1456–1462. doi: 10.1046/j.1523-1747.2002.19606.x. [DOI] [PubMed] [Google Scholar]
- 35.Kon A, Pulkkinen L, Ishida-Yamamoto A, Hashimoto I, Uitto J. Novel COL7A1 mutations in dystrophic forms of epidermolysis bullosa. J Invest Dermatol. 1998;111(3):534–537. doi: 10.1046/j.1523-1747.1998.00326.x. [DOI] [PubMed] [Google Scholar]
- 36.Torrance CJ, Agrawal V, Vogelstein B, Kinzler KW. Use of isogenic human cancer cells for high-throughput screening and drug discovery. Nat Biotechnol. 2001;19(10):940–945. doi: 10.1038/nbt1001-940. [DOI] [PubMed] [Google Scholar]
- 37.Ding Q, et al. A TALEN genome-editing system for generating human stem cell-based disease models. Cell Stem Cell. 2013;12(2):238–251. doi: 10.1016/j.stem.2012.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ito K, et al. Keratinocyte-/fibroblast-targeted rescue of Col7a1-disrupted mice and generation of an exact dystrophic epidermolysis bullosa model using a human COL7A1 mutation. Am J Pathol. 2009;175(6):2508–2517. doi: 10.2353/ajpath.2009.090347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Villone D, et al. Supramolecular interactions in the dermo-epidermal junction zone: Anchoring fibril-collagen VII tightly binds to banded collagen fibrils. J Biol Chem. 2008;283(36):24506–24513. doi: 10.1074/jbc.M802415200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Matsuda T, Cepko CL. Electroporation and RNA interference in the rodent retina in vivo and in vitro. Proc Natl Acad Sci USA. 2004;101(1):16–22. doi: 10.1073/pnas.2235688100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cermak T, et al. Efficient design and assembly of custom TALEN and other TAL effector-based constructs for DNA targeting. Nucleic Acids Res. 2011;39(12):e82. doi: 10.1093/nar/gkr218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Carlson DF, et al. Efficient TALEN-mediated gene knockout in livestock. Proc Natl Acad Sci USA. 2012;109(43):17382–17387. doi: 10.1073/pnas.1211446109. [DOI] [PMC free article] [PubMed] [Google Scholar]