

Abstract
The primary effect of the endoplasmic reticulum (ER) stress response or unfolded protein response (UPR) is to reduce the load of unfolded protein and promote survival. However, prolonged and severe ER stress leads to tissue injury and serious diseases. Thus, it is important to identify drugs that can attenuate ER stress for the treatment of diseases. Natural products continue to provide lead compounds for drug discovery and front-line pharmacotherapy for people worldwide. Previous studies have indicated that selenoprotein S (SelS) is a sensitive and ideal maker of ER stress. In the present study, a firefly luciferase reporter driven by the SelS gene promoter was used to screen for natural compounds capable of attenuating ER stress. From this, paclitaxel (PTX) was identified to efficiently inhibit the promoter activity of the SelS gene, and further results revealed that PTX significantly inhibited the tunicamycin-induced upregulation of SelS at the mRNA and protein levels in HepG2 and HEK293T cells. In addition, PTX was able to efficiently inhibit the expression of the ER stress marker, glucose-regulated protein 78, in ER stress, indicating that PTX may reverse ER stress. Taken together, these results suggest that PTX is able to inhibit SelS expression during ER stress and attenuate ER stress.
Keywords: paclitaxel, endoplasmic reticulum stress, selenoprotein S, natural product, compound screening
Introduction
The endoplasmic reticulum (ER) is the main organelle involved in protein modification and folding, lipid biogenesis, and is an intracellular Ca2+ store, and is important for the maintenance of homeostasis. Perturbations in ER homeostasis, for example hypoxia, nutrient deprivation, acidosis and a reduction in luminal Ca2+ concentration, can lead to an accumulation of unfolded proteins, which results in the ER stress response, also known as the unfolded protein response (UPR) (1,2). The UPR aims to re-establish homeostasis by the inhibition of new protein synthesis, the induction of ER chaperones and the activation of the ER-associated degradation (ERAD) system, which translocates and removes misfolded proteins by proteasomal degradation (3–5). In doing so, UPR signaling acts to promote survival and adaptation to ER stress. However, persistent and severe ER stress activates apoptotic pathways and therefore contributes to organ damage. ER stress has been implicated in the pathogenesis of diseases including cardiovascular diseases, neurodegenerative diseases, diabetes and liver disease (6–9). This suggests that screening drugs that can reverse ER stress may be an effective strategy in the treatment of diseases such as Parkinson's disease, Alzheimer's disease and diabetes.
Natural products are recognized as rich resources for drug discovery in disease treatment. Natural compounds can offer powerful leads with favorable absorption, distribution, metabolism, excretion and toxicity characteristics (10). Additionally, they are frequently more cost-effective, easily preserved and standardized. Furthermore, their effects on inhibiting and activating the function of specific proteins are often reversible and may be finely tuned by varying the concentration (11,12).
Selenoprotein S (SelS; also known as SEPS1, VIMP, Tanis and SELENOS) was identified by Walder et al (13) in 2002. Previous studies have reported the role of SelS in ER stress. In the ER membrane, SelS forms a complex with Derlin-1 and the p97 ATPase. The complex mediates the retrotranslocation of misfolded proteins out of the ER towards cytosolic degradation, a process also known as ERAD, and thereby reduces ER stress (14). SelS expression was immediately and markedly increased by ER stress agents such as tunicamycin (TM), thapsigargin, dithiothreitol (DTT), cycloheximide, staurosporine, β-mercaptoethanol and sodium selenite during ER stress (15,16). Conversely, SelS expression was observed to be markedly downregulated following a reduction in ER stress (15). Taken together, this indicates SelS to be a sensitive and ideal marker of ER stress for the screening of natural compounds that are able to attenuate ER stress.
In the current study, a firefly luciferase reporter screening system driven by SelS promoter was established, and greater than 300 purified natural compounds were screened, from which paclitaxel (PTX) was identified to effectively inhibit TM-induced upregulation of SelS at the mRNA and protein levels in HepG2 and HEK293T cells. Furthermore, PTX was able to efficiently inhibit the expression levels of a marker of ER stress, glucose-regulated protein 78 (GRP78), in ER stress. These results suggest that PTX is a novel small molecule able to reduce ER stress, and is a potential drug for the treatment of diseases associated with ER stress.
Materials and methods
Cell lines and cell culture
HepG2 human hepatocellular carcinoma cells and HEK293T human embryonic kidney cells were obtained from the Chinese Academy of Sciences Shanghai Institute for Biological Sciences Cell Resource Center (Shanghai, China). Cells were cultured in Dulbecco's modified Eagle's medium (DMEM; Invitrogen; Thermo Fisher Scientific, Inc., Waltham, MA, USA), which was supplemented with 10% fetal bovine serum (FBS; Invitrogen; Thermo Fisher Scientific, Inc.), 100 U penicillin and 100 µg/ml streptomycin (Ameresco, LLC Solon, OH, USA) at 37°C with 5% CO2.
Reagents and natural compounds
DTT, 3-(4,5-dimethyl-thiazol-2-yl)-2,5-diphenyl-2H-tetrazolium bromide (MTT) and TM were obtained from Sigma-Aldrich (St. Louis, MO, USA). PTX was purchased from the National Institutes for Food and Drug Control (lot number 100382-201102; Beijing, China), and the purity of PTX was 99.6%. The other natural compounds used in the study were extracted from plants and animals in our laboratory and the purity was greater than 95%. All compounds were dissolved in 100% dimethyl sulfoxide (DMSO; Sigma-Aldrich) as a 10 mg/ml stock.
Screening of potential inhibitors of SelS expression
A pSelS-luc reporter plasmid was constructed as described previously (17). In the primary screening assay, HEK293T cells were plated at 6×105 cells/well in a 6-well plate. After 24 h, cells were transfected with 3–4 µg of the pSelS-luc reporter plasmid using the Calcium Phosphate Cell Transfection kit (Beyotime Institute of Biotechnology, Shanghai, China), and were maintained in DMEM. After 4 h, the transfected cells were replated in 96-well plates. At 24 h later, cells were treated with the compounds at final concentration of 5 µg/ml in DMEM containing 3% FBS (v/v) (to reduce the complex interference caused by the composition of serum) for 24 h. Luciferase activity was measured as described previously (18).
In the secondary screening assay, HEK293T cells were plated at a concentration of 1×105 cells/well in a 24-well plate. After 24 h, cells were transfected with 1 µg of pSelS-luc plasmids or 1–1.5 µg of pGL3-basic vector plasmids per well plus 0.1 µg of pCMV-β-galactosidase plasmids using Calcium Phosphate Cell Transfection kit according to the manufacturer's instructions. The cells were incubated for 24 h and then treated with the compounds at a final concentration of 5 µg/ml or 0.05% DMSO for 24 h. Subsequently, luciferase activity was measured and normalized to the β-galactosidase activity using a FLUOstar OPTIMA system (BMG Labtech, Offenburg, Germany).
RNA extraction and reverse transcription-polymerase chain reaction (RT-PCR)
HepG2 cells were plated at a concentration of 5×105 cells/well in a 6-well plate. Following culture for 24 h, cells were treated with TM (5 µg/ml) and PTX (the identified concentrations) in 2 ml DMEM containing 3% FBS (v/v) for 12 h. Total RNA was prepared from the cells using Trizol reagent (Invitrogen; Thermo Fisher Scientific, Inc.) following the manufacturer's instructions. RNA was quantified by measuring the absorbance at 260/280 nm (E-SPECT, Malcom Co., Ltd., Tokyo, Japan). A total of 1 µg of total RNA was reverse transcribed by oligo (dT) primers using the Reverse Transcription System (Takara Biotechnology, Co., Ltd., Dalian, China). The RT-PCR kit was obtained from Beijing TransGen Biotech Co., Ltd. (Beijing, China). SelS mRNA was amplified from cDNA templates using the following primer sequences: Sense, 5′-GTTGCGTTGAATGATGTCTTCCT-3′ and antisense, 5′-AGAAACAAACCCCATCAACTGT-3′. The sequences of the primers of the β-actin internal control were: Sense, 5′-TCGTGCGTGACATTAAGGAG-3′ and antisense, 5′-ATGCCAGGGTACATGGTGGT-3′. PCR was performed for 25 cycles, with each cycle consisting of 94°C for 30 sec, 55°C for 40 sec, and 72°C for 30 sec. The PCR products were analyzed by electrophoresis on a 1% agarose gel (Sigma-Aldrich) stained with cyanine (Nanjing KeyGEN Biotech Co., Ltd., Nanjing, China) and visualized using UV light (DNR Bio-imaging Systems Ltd., Jerusalem, Israel).
Protein preparation and western blot analysis
To determine the expression of associated proteins, the whole-cell lysates were prepared and western blotting was performed. Briefly, cells were harvested, resuspended in cell lysis buffer (1% Triton X-100, 0.015 M NaCl, 10 mM Tris-HCl, 1 mM ethylenediaminetetraacetic acid, 1 mM phenylmethylsulfonyl fluoride, 10 µg/ml leupeptin and 10 µg/ml pepstain A) and then incubated on ice for 30 min. The cell lysates were centrifuged at 12,000 x g for 10 min at 4°C, and the supernatants were mixed with one quarter volume of 4x sodium dodecyl sulfate (SDS) sample buffer, boiled for 10 min, and then separated by 12% SDS-polyacrylamide gel electrophoresis. Following electrophoresis, proteins were transferred to polyvinylidene fluoride membranes (Beyotime Institute of Biotechnology) and blocked with 5% non-fat dry milk in Tris-buffered saline-Tween (TBST) buffer (20 mM Tris-HCl pH 7.6, 150 mM NaCl and 0.05% Tween-20) for 2 h at room temperature. Subsequently, the membranes were probed with the following primary antibodies in TBST buffer overnight at 4°C: Mouse monoclonal anti-SelS (1:1,000; sc-365498; Santa Cruz Biotechnology, Inc., Dallas, TX, USA); rabbit polyclonal anti-GRP78 (1:1,000; sc-13968; Santa Cruz Biotechnology, Inc.); mouse monoclonal anti-glyceraldehyde-3-phosphate dehydrogenase (GAPDH; 1:5,000; KC-5G4; Kangcheng Biotech, Shanghai, China). The membranes were then washed three times with TBST, and incubated with horseradish peroxidase-conjugated goat anti-mouse (1:2,000; A0216; Beyotime Institute of Biotechnology) and goat anti-rabbit (1:2,000; A0208; Beyotime Institute of Biotechnology) secondary antibodies for 30 min at room temperature, and washed extensively with TBST. Finally, protein bands were identified using enhanced chemiluminescence detection (ECL-Plus kit, Beyotime Institute of Biotechnology) and were digitally captured (MicroChemi, DNR Bio-imaging Systems, Ltd.). Differences in protein loading were controlled by blotting for GAPDH.
Immunoreactive bands were quantified by densitometry of unsaturated images with background density subtracted (ImageJ; National Institutes of Health, Bethesda, MD, USA). GRP78 and SelS immunoblotting intensities were normalized by dividing by the corresponding GAPDH immunoblotting intensities from the same sample labeled on the same gel. Each experiment was repeated a minmum of three times to obtain the mean values and standard deviations. The normalized intensity values were statistically analyzed with SPSS software, version 17.0 (SPSS, Inc., Chicago, IL, USA) using one-way analysis of variance followed by Dunnett's post-hoc tests.
MTT assay
HepG2 or HEK293T cells were seeded into 96-well plates at a concentration of 1×104 cells/well for 24 h at 37°C and then treated with PTX (0.0625, 0.125, 0.25, 0.5, 1, 2 and 4 µg/ml) in the presence of 3% FBS (v/v) medium for 44 h at 37°C. Subsequently, 20 µl MTT solution (5 µg/ml in phosphate-buffered saline) was added to each well and incubated for 4 h at 37°C. The culture medium was removed and 100 µl DMSO was added and agitated for 10 min at room temperature. Cell viability was determined by measuring the absorbance at 570 nm by plate reader (Bio-Rad Laboratories, Inc., Hercules, CA, USA).
Statistical analysis
All experiments were repeated a minimum of three times. SPSS software, version 17.0 (SPSS, Inc.) was used for statistical analysis. Student's t-test and one-way analysis of variance followed by Dunnett's multiple comparisons tests were used to compare the results from each group. P<0.05 was considered to indicate a statistically significant difference. All data are presented as the mean ± standard deviation.
Results
TM upregulates SelS expression and induces ER stress
Prior to the screening of ER stress inhibitors, an ER stress model was generated using TM. TM, which is an inhibitor of protein glycosylation, has been widely used as an inducer of ER stress (19). As presented in Fig. 1A and B, TM was able to significantly promote SelS expression at different concentrations (5, 10 and 15 µg/ml) and at varying durations of treatment (12, 24 and 36 h).
Figure 1.

TM upregulates SelS expression and induces endoplasmic reticulum stress. (A) The effects of treatment with 5, 10 and 15 µg/ml TM on SelS expression. (B) The effects of 5 µg/ml TM on SelS expression following treatment for 12, 24 and 36 h. (C) TM induced upregulation of GRP78 following 12 h treatment with 5, 10 and 15 µg/ml TM. The data are presented as the mean ± standard deviation (*P<0.05 and **P<0.01 vs. the control group). TM, tunicamycin; SelS, selenoprotein S; GRP78, glucose-regulated protein 78; GAPDH, glyceraldehyde 3-phosphate dehydrogenase.
GRP78 (also known as BiP or heat shock 70kDa protein 5) is a major ER chaperone protein, which is able to increase the protein folding capacity of the ER and regulate the activation of ER transmembrane signaling molecules (20). GRP78 has an anti-apoptotic function to prevent ER stress-induced cell death and is widely used as a marker of ER stress (21). As presented in Fig. 1C, TM markedly increased GRP78 expression at different concentrations (5, 10 and 15 µg/ml), suggesting that TM results in significant ER stress.
Screening of SelS expression inhibitors
In order to directly test SelS expression, a firefly luciferase reporter screening system driven by SelS promoter (pSelS-luc) was generated, and greater than 300 natural compounds were screened in HEK239T cells. Fig. 2A presents the fold induction and P-value of each compound. With the thresholds of fold induction <1.00 and P<0.05, 54 of the tested compounds were selected for further analysis.
Figure 2.

Screening of SelS expression inhibitors. (A) Primary screening of HEK293T cells transfected with a pSelS-luc reporter plasmid and treated with the compounds (5 µg/ml). (B) Secondary screening of HEK293T cells co-transfected with a pSelS-luc reporter plasmid or pGL3-basic vector and a pCMV-β-galactosidase plasmid, and treated with the identified compounds (5 µg/ml). Results are expressed as the fold induction (over the activity of the negative control) and the P-value (compared with negative control). The black data point represents paclitaxel. SelS, selenoprotein S.
In the secondary screening assay, HEK239T cells were plated in 24-well plates. After 24 h, cells were transfected with pSelS-luc plasmids or pGL3-basic vector plasmids plus pCMV-β-galactosidase. The cells were incubated for 24 h prior to treatment with the identified compounds at final concentration of 5 µg/ml or 0.05% DMSO for 24 h. The fold induction and P-value of each compound are presented in Fig. 2B. PTX (Fig. 3A) exhibited a fold induction of 0.368 (P<0.05) and the most marked inhibitory effect on SelS promoter activity. Therefore, the subsequent investigations focused on PTX.
Figure 3.
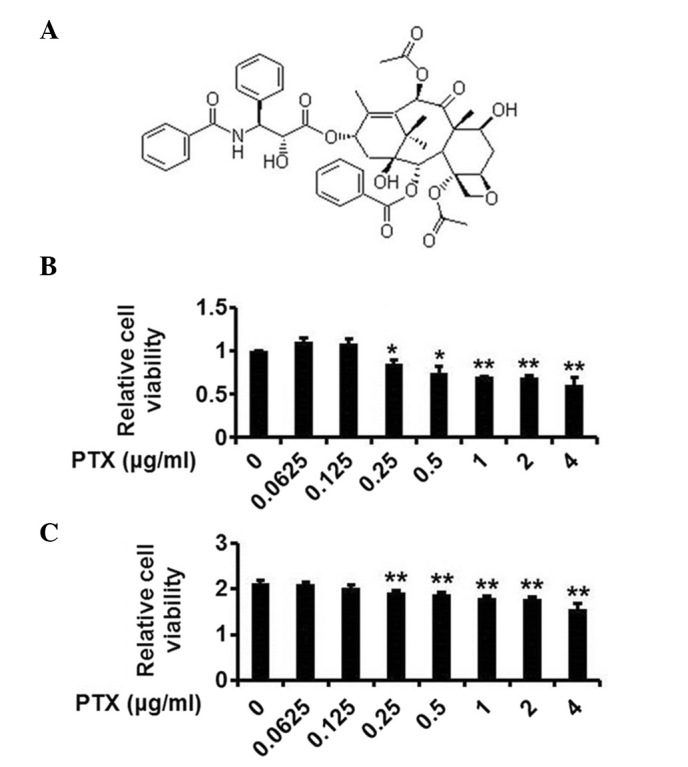
The structure and cytotoxicity of PTX. (A) The structure of PTX. (B) The cytotoxicity of PTX to HepG2 cells. HepG2 cells were treated with 0.0625, 0.125, 0.25, 0.5, 1, 2 and 4 µg/ml PTX and cell viability was assessed. (C) The cytotoxicity of PTX to HEK293T cells. HEK293T cells were treated with 0.0625, 0.125, 0.25, 0.5, 1, 2 and 4 µg/ml PTX and cell viability was assessed. The data are presented as the mean ± standard deviation (n=3; *P<0.05, **P<0.01 vs. the control group). PTX, paclitaxel.
The cytotoxicity of PTX was subsequently investigated using an MTT assay in HepG2 and HEK293T cells. As presented in Fig. 3B and C, PTX at the concentrations of 0.0625 and 0.125 µg/ml was not toxic to HepG2 and HEK293T cells.
PTX inhibits TM-induced upregulation of SelS in HepG2 cells
To confirm whether PTX is able to reverse the TM-induced upregulation of SelS expression, the expression levels of SelS were measured by RT-PCR in HepG2 cells. As presented in Fig. 4A–C, TM increased SelS expression, and treatment with PTX reversed the upregulation of SelS induced by TM at different concentrations (0.03125, 0.0625 and 0.125 µg/ml). Western blotting also indicated that PTX markedly reversed the upregulation of SelS at different concentrations (0.0625 and 0.125 µg/ml) in HepG2 cells (Fig. 4D and E). Taken together, these results indicate that PTX may reverse the TM-induced upregulation of SelS, and that PTX may be an ER stress inhibitor.
Figure 4.

PTX inhibits the upregulation of SelS in ER stress and attenuates ER stress in HepG2 cells. PTX inhibits TM-induced upregulation of SelS mRNA expression levels in HepG2 cells treated with (A) 0.03125, (B) 0.0625 and (C) 0.125 µg/ml PTX and 5 µg/ml TM. (D–E) PTX inhibits the TM-induced upregulation of SelS and GRP78 protein expression in HepG2 cells treated with 5 µg/ml TM and (D) 0.0625 and (E) 0.125 µg/ml PTX. (F) PTX inhibits the DTT-induced upregulation of SelS and GRP78. HepG2 cells were treated with 0.09258 µg/ml DTT and 0.03125 µg/ml PTX. The data are presented as the mean ± standard deviation (*P<0.05 compared with the TM-induced group). PTX, paclitaxel; SelS, selenoprotein S; ER, endoplasmic reticulum; TM, tunicamycin; GRP78, glucose-regulated protein 78; DTT, dithiothreitol; GAPDH, glyceraldehyde 3-phosphate dehydrogenase.
PTX attenuates ER stress in HepG2 cells
Considering that PTX is able to inhibit SelS expression, it was subsequently investigated whether PTX was able to reverse ER stress. As presented in Fig. 4D and E, TM induced ER stress as indicated by the upregulation of GRP78 levels. Following treatment with PTX (0.0625 and 0.125 µg/ml), the expression levels of GRP78 were significantly reduced, demonstrating that PTX is able to reverse TM-induced ER stress. As presented in Fig. 4F, PTX was additionally able to inhibit DTT-induced upregulation of GRP78, suggesting that PTX may attenuate DTT-induced ER stress.
PTX inhibits TM-induced upregulation of SelS and attenuates ER stress in HEK293T cells
In agreement with the observation in HepG2 cells, PTX markedly attenuated TM-induced upregulation of SelS expression at different concentrations (0.03125 and 0.0625 µg/ml) in HEK293T cells (Fig. 5A and B). Additionally, PTX was able to inhibit the TM-induced upregulation of GRP78 (Fig. 5C), suggesting that PTX may reverse ER stress in HEK293T cells.
Figure 5.

PTX inhibits the TM-induced upregulation of SelS and endoplasmic reticulum stress in HEK293T cells. PTX inhibits the TM-induced upregulation of SelS protein expression in HEK293T cells treated with 5 µg/ml TM and (A) 0.03125 and (B) 0.0625 µg/ml PTX. (C) PTX inhibits the TM-induced upregulation of GRP78 protein expression in HEK293T cells treated with 5 µg/ml TM and 0.03125 µg/ml PTX. The data are presented as the mean ± standard deviation (*P<0.05 vs. the TM-induced group). PTX, paclitaxel; TM, tunicamycin; SelS, selenoprotein S; GRP78, glucose-regulated protein 78; GAPDH, glyceraldehyde 3-phosphate dehydrogenase.
Discussion
ER stress is involved in the pathogenesis of a number of diseases including neurodegenerative diseases, liver diseases, cardiovascular diseases and diabetes (22–25). The identification of compounds that can attenuate ER stress may be beneficial to patients. Natural products, characterized by high chemical diversity and biochemical specificity, have been widely explored as potential therapeutics for a variety of diseases (10). As SelS is a sensitive marker of ER stress (26,27) it was selected as a marker for the screening of compounds that are able to reverse ER stress.
The liver and the kidneys are important organs in the human body, and perform numerous essential functions. ER stress in hepatocytes and nephrocytes is induced in a number of serious diseases, including viral hepatitis, hepatocellular carcinoma, glomerular injury and renal ischemia reperfusion injury (28,29). The identification of drugs that can attenuate ER stress in liver and kidney cells will be advantageous, therefore, liver and kidney cell lines (HepG2 and HEK293T cells) were selected for the screening compounds able to reduce ER stress.
The pSelS-luci reporter plasmid was constructed by subcloning the SelS promoter region (−1073 to +39 bp) into the pGL3-basic plasmid. This region contains an ER stress response element that is activated by ER stress (17). Previous studies have indicated that a number of transcription factor binding sites are located in the SelS promoter region and that the promoter is important for regulating the basal transcription of SelS (26). Therefore, a firefly luciferase reporter driven by the SelS gene promoter was used in the present study to screen the compounds that were able to inhibit SelS gene promoter activity. Following screening of >300 compounds, PTX was observed to markedly inhibit the activity of the SelS promoter (Fig. 2A and B). Further results revealed that PTX was able to inhibit the TM-induced upregulation of SelS mRNA and protein at different concentrations (Fig. 4). However, the possibility that PTX may influence the activity of other genes in the screening assay cannot be excluded.
The current study demonstrated that PTX at concentrations <0.125 µg/ml were able to attenuate ER stress in HepG2 and HEK293T cells (Figs. 4 and 5), and that these concentrations of PTX did not exhibit cytotoxicity, as presented in Fig. 3B and C. These results suggest that PTX may attenuate ER stress at sub-toxic concentrations in HepG2 and HEK293T cells. However, whether PTX can attenuate ER stress at higher (toxic) concentrations in these cells requires further investigation.
The ER stress response, also known as the UPR, is an adaptive response used to protect ER function. The mechanisms of the UPR reduce the load of unfolded protein and restore ER homeostasis by reducing protein translation and inducing the transcription of components of the ER machinery involved in folding, N-glycosylation, ERAD, quality control, redox and lipid biogenesis (30). The mechanisms by which PTX inhibits ER stress is unknown and requires further study to be fully elucidated.
In conclusion, the current study demonstrates the role of PTX in inhibiting SelS expression during ER stress and attenuating ER stress. The present study identifies a novel natural small molecule able to attenuate ER stress, and provides new insight into the function and therapeutic application of PTX.
Acknowledgments
The present study was supported by the Fundamental Research Funds for the Central Universities (grant no. 11QNJJ021), and the Research Foundation of Jilin Provincial Science & Technology Development (grant no. 20110938).
References
- 1.Gentile CL, Frye M, Pagliassotti MJ. Endoplasmic reticulum stress and the unfolded protein response in nonalcoholic fatty liver disease. Antioxid Redox Signal. 2011;15:505–521. doi: 10.1089/ars.2010.3790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Marciniak SJ, Ron D. Endoplasmic reticulum stress signaling in disease. Physiol Rev. 2006;86:1133–1149. doi: 10.1152/physrev.00015.2006. [DOI] [PubMed] [Google Scholar]
- 3.Meusser B, Hirsch C, Jarosch E, Sommer T. ERAD: The long road to destruction. Nat Cell Biol. 2005;7:766–772. doi: 10.1038/ncb0805-766. [DOI] [PubMed] [Google Scholar]
- 4.Sozen E, Karademir B, Ozer NK. Basic mechanisms in endoplasmic reticulum stress and relation to cardiovascular diseases. Free Radic Biol Med. 2015;78:30–41. doi: 10.1016/j.freeradbiomed.2014.09.031. [DOI] [PubMed] [Google Scholar]
- 5.Zhao L, Ackerman SL. Endoplasmic reticulum stress in health and disease. Curr Opin Cell Biol. 2006;18:444–452. doi: 10.1016/j.ceb.2006.06.005. [DOI] [PubMed] [Google Scholar]
- 6.Halliday M, Mallucci GR. Targeting the unfolded protein response in neurodegeneration: A new approach to therapy. Neuropharmacology. 2014;76:169–174. doi: 10.1016/j.neuropharm.2013.08.034. [DOI] [PubMed] [Google Scholar]
- 7.Imai Y, Soda M, Inoue H, Hattori N, Mizuno Y, Takahashi R. An unfolded putative transmembrane polypeptide, which can lead to endoplasmic reticulum stress, is a substrate of Parkin. Cell. 2001;105:891–902. doi: 10.1016/S0092-8674(01)00407-X. [DOI] [PubMed] [Google Scholar]
- 8.Kaufman RJ. Orchestrating the unfolded protein response in health and disease. J Clin Invest. 2002;110:1389–1398. doi: 10.1172/JCI0216886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Minamino T, Kitakaze M. ER stress in cardiovascular disease. J Mol Cell Cardiol. 2010;48:1105–1110. doi: 10.1016/j.yjmcc.2009.10.026. [DOI] [PubMed] [Google Scholar]
- 10.Sulaiman RS, Basavarajappa HD, Corson TW. Natural product inhibitors of ocular angiogenesis. Exp Eye Res. 2014;129:161–171. doi: 10.1016/j.exer.2014.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Corson TW, Crews CM. Molecular understanding and modern application of traditional medicines: Triumphs and trials. Cell. 2007;130:769–774. doi: 10.1016/j.cell.2007.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hou P, Li Y, Zhang X, Liu C, Guan J, Li H, Zhao T, Ye J, Yang W, Liu K, et al. Pluripotent stem cells induced from mouse somatic cells by small-molecule compounds. Science. 2013;341:651–654. doi: 10.1126/science.1239278. [DOI] [PubMed] [Google Scholar]
- 13.Walder K, Kantham L, McMillan JS, Trevaskis J, Kerr L, De Silva A, Sunderland T, Godde N, Gao Y, Bishara N, et al. Tanis: A link between type 2 diabetes and inflammation? Diabetes. 2002;51:1859–1866. doi: 10.2337/diabetes.51.6.1859. [DOI] [PubMed] [Google Scholar]
- 14.Ye Y, Shibata Y, Yun C, Ron D, Rapoport TA. A membrane protein complex mediates retro-translocation from the ER lumen into the cytosol. Nature. 2004;429:841–847. doi: 10.1038/nature02656. [DOI] [PubMed] [Google Scholar]
- 15.Du S, Liu H, Huang K. Influence of SelS gene silence on beta-Mercaptoethanol-mediated endoplasmic reticulum stress and cell apoptosis in HepG2 cells. Biochim Biophys Acta. 2010;1800:511–517. doi: 10.1016/j.bbagen.2010.01.005. [DOI] [PubMed] [Google Scholar]
- 16.Kim KH, Gao Y, Walder K, Collier GR, Skelton J, Kissebah AH. SEPS1 protects RAW264.7 cells from pharmacological ER stress agent-induced apoptosis. Biochem Biophys Res Commun. 2007;354:127–132. doi: 10.1016/j.bbrc.2006.12.183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gao Y, Feng HC, Walder K, Bolton K, Sunderland T, Bishara N, Quick M, Kantham L, Collier GR. Regulation of the seleno-protein SelS by glucose deprivation and endoplasmic reticulum stress-SelS is a novel glucose-regulated protein. FEBS Lett. 2004;563:185–190. doi: 10.1016/S0014-5793(04)00296-0. [DOI] [PubMed] [Google Scholar]
- 18.Zhang Y, Bao YL, Wu Y, Yu CL, Sun Y, Li YX. Identification and characterization of the human SLC5A8 gene promoter. Cancer Genet Cytogenet. 2010;196:124–132. doi: 10.1016/j.cancergencyto.2009.09.004. [DOI] [PubMed] [Google Scholar]
- 19.Lee AS. The glucose-regulated proteins: Stress induction and clinical applications. Trends Biochem Sci. 2001;26:504–510. doi: 10.1016/S0968-0004(01)01908-9. [DOI] [PubMed] [Google Scholar]
- 20.Iwasa K, Nambu Y, Motozaki Y, Furukawa Y, Yoshikawa H, Yamada M. Increased skeletal muscle expression of the endoplasmic reticulum chaperone GRP78 in patients with myasthenia gravis. J Neuroimmunol. 2014;273:72–76. doi: 10.1016/j.jneuroim.2014.05.006. [DOI] [PubMed] [Google Scholar]
- 21.Ma KX, Chen GW, Shi CY, Cheng FF, Dou H, Feng CC, Liu DZ. Molecular characterization of the glucose-regulated protein 78 (GRP78) gene in planarian Dugesia japonica. Comp Biochem Physiol B Biochem Mol Biol. 2014;171:12–17. doi: 10.1016/j.cbpb.2014.03.001. [DOI] [PubMed] [Google Scholar]
- 22.Dara L, Ji C, Kaplowitz N. The contribution of endoplasmic reticulum stress to liver diseases. Hepatology. 2011;53:1752–1763. doi: 10.1002/hep.24279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liu H, Cao MM, Wang Y, Li LC, Zhu LB, Xie GY, Li YB. Endoplasmic reticulum stress is involved in the connection between inflammation and autophagy in type 2 diabetes. Gen Comp Endocrinol. 2015;210:124–129. doi: 10.1016/j.ygcen.2014.09.006. [DOI] [PubMed] [Google Scholar]
- 24.Luo T, Kim JK, Chen B, Abdel-Latif A, Kitakaze M, Yan L. Attenuation of ER stress prevents post-infarction-induced cardiac rupture and remodeling by modulating both cardiac apoptosis and fibrosis. Chem Biol Interact. 2015;225:90–98. doi: 10.1016/j.cbi.2014.10.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Torres M, Matamala JM, Duran-Aniotz C, Cornejo VH, Foley A, Hetz C. ER stress signaling and neurodegeneration: At the intersection between Alzheimer's disease and Prion-related disorders. Virus Res. 2015;207:69–75. doi: 10.1016/j.virusres.2014.12.018. [DOI] [PubMed] [Google Scholar]
- 26.Gao Y, Hannan NR, Wanyonyi S, Konstantopolous N, Pagnon J, Feng HC, Jowett JB, Kim KH, Walder K, Collier GR. Activation of the selenoprotein SEPS1 gene expression by pro-inflammatory cytokines in HepG2 cells. Cytokine. 2006;33:246–251. doi: 10.1016/j.cyto.2006.02.005. [DOI] [PubMed] [Google Scholar]
- 27.Speckmann B, Gerloff K, Simms L, Oancea I, Shi W, McGuckin MA, Radford-Smith G, Khanna KK. Selenoprotein S is a marker but not a regulator of endoplasmic reticulum stress in intestinal epithelial cells. Free Radic Biol Med. 2014;67:265–277. doi: 10.1016/j.freeradbiomed.2013.11.001. [DOI] [PubMed] [Google Scholar]
- 28.Malhi H, Kaufman RJ. Endoplasmic reticulum stress in liver disease. J Hepatol. 2011;54:795–809. doi: 10.1016/j.jhep.2010.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Taniguchi M, Yoshida H. Endoplasmic reticulum stress in kidney function and disease. Curr Opin Nephrol Hypertens. 2015;24:345–350. doi: 10.1097/MNH.0000000000000141. [DOI] [PubMed] [Google Scholar]
- 30.Cnop M, Foufelle F, Velloso LA. Endoplasmic reticulum stress, obesity and diabetes. Trends Mol Med. 2012;18:59–68. doi: 10.1016/j.molmed.2011.07.010. [DOI] [PubMed] [Google Scholar]