

Abstract
The endoplasmic reticulum (ER) is an organelle in which proteins form their appropriate structures. However, several of these proteins become unfolded or misfolded when exposed to stimuli, including hyperglycemia, oxidative stress, ischemia, disturbance of calcium homeostasis and overexpression of abnormal proteins, which activates ER stress and the unfolded protein response (UPR). To date, investigations have demonstrated that ER stress is important in diabetic myocardial fibrosis by inducing cardiac cell apoptosis. Therefore, in the present study, the polymerase chain reaction, western blotting analysis and tissue staining were performed to identify the changes in UPR signaling proteins and apoptotic proteins in diabetic rats at different time points, and to determine whether the myocardial fibrosis is associated with ER-stress-mediated apoptosis using a diabetes mellitus (DM) rat model. It was found that the upregulation of ER stress markers and apoptotic molecules developed over time. It was also demonstrated that anti-apoptotic markers and proapoptotic markers were activated early following model establishment, and then decreased in months 4 and 5. The changes in myocardial fibrosis were found to accelerate in a time-dependent manner with apoptosis in the DM rats.
Keywords: apoptosis, unfolded protein response, endoplasmic reticulum stress, myocardial fibrosis
Introduction
Diabetes mellitus (DM) is a disease characterized by hyperglycemia resulting from defects in insulin secretion, insulin action, or the two together, and is often accompanied by hyperlipidemia. The endoplasmic reticulum (ER) is a complex, intracellular membranous network, which regulates protein synthesis. If the cell suffers from adverse conditions, including overabundance of glucose, infection, hypoxia, ischemia or angiotensin-II stimulation, homeostasis in the ER is disrupted, leading to the accumulation of multiple unfolded or misfolded proteins in the ER. This results in ER stress and subsequent UPR (1). There are two functions of the UPR. Initially, it induces the inhibition of protein synthesis and subsequently, when ER stress is prolonged in the presence of hyperglycemia, an apoptotic pathway is activated (2) through the activation of the C/EBP homologous protein (CHOP/GADD153) (3), the Jun N-terminal kinase (JNK)4 pathway (4) and caspase-12 (5). In mammalian cells, ER stress initially activates intracellular signaling pathways mediated by three ER-resident proteins: Protein kinase R-like ER kinase (PERK) (6), activating transcription factor 6 (ATF6) and inositol-requiring kinase-1 (IRE-1) (7). Under normal conditions, these molecules straddle the ER membrane, positioning their N-terminus in the lumen of the ER membranes and their C-terminus in the cytosol. The N-terminus is often associated with the chaperone, glucose regulating protein 78 (GRP78) (8). When unfolded proteins aggregate in the ER, PERK dislocates from GRP78 to dimerize and autophosphorylate, and is then activated to phosphorylate eukaryotic initiation factor-α (9). Initiation factor-dependent protein synthesis can be inhibited to reduce the protein load in the ER (10). However, rather than the transcription of ER stress response (ERSE) genes being reduced, it increases due to their specific structures and selective advantages (11). IRE1 is central to the ER stress response (12). Following its activation, it functions as a kinase and an endoribonuclease, which can cleave specific exon-intron sites in the mRNA that encode the transcription factor, X-box-binding protein 1. This cleavage initiates an unconventional splicing reaction, which leads to the production of an active transcription factor and the induction of various adaptive genes, particularly ERSE (13). The mechanism of activation of ATF6 differs from those of PERK and IRE1. GRP78 dissociation from ATF6 promotes its translocation to the Golgi, where it undergoes cleavage by site-1 and site-2 proteases (14). The cleaved and activated N-ATF6 migrates to the nucleus and increases the expression of ERSE (15).
ER stress is a pathogenic mechanism associated with type 1 and type 2 diabetes. In type 2 diabetes, insulin resistance is the underlying problem, and is often associated with obesity. It is now recognized that a high-fat diet causes ER stress. If hyperglycemia is persistent, ER stress is prolonged and, when cells are not sufficiently relieved from stress, they undergo apoptosis and self-destruction (16). The apoptotic course is complicated as it involves three pathways, which interact with each other to induce the apoptosis of cardiomyocytes. IRE-1 recruits tumor necrosis factor receptor associated factor 2 (TRAF2) and apoptotic signal-regulation kinase 1 (ASK-1) to form an IRE-1-TRAF2-ASK1 complex, which then activates JNK and other downstream molecules (17). Additionally, PERK upregulates the translation of ATF4 and induces the transcription of CHOP/GADD153, a member of the C/EBP family of basic leucine zipper transcription factors, which potentiates apoptosis (18). The caspase pathway is also commonly activated in ER stress, particularly caspase-12, although, the precise mechanism remains to be elucidated. The important mechanisms underlying ER stress in heart disease have been reported previously. For example, ischemia/reperfusion injury in the heart has been shown to induce ATF6 markers (19–21). Studies have also shown that ER stress is involved in the pathogenesis of various cardiovascular diseases, including as coronary heart disease, cardiac ischemia-reperfusion injury, cardiomyopathy and heart failure (19,22,23). The PERK and ATF6 pathways are the major determining pathways involved in ER stress-mediated metabolic dysfunction. These pathways eventually lead to diabetic cardiomyopathy in non-obese type 2 DM rats when sacrificed at 32 weeks (24). Limited investigations have been performed in animal models. In the present study, the expression levels of the predominant markers of UPR signaling proteins were examined in DM rats at different time points, to determine whether myocardial fibrosis is accompanied by ER stress-mediated apoptosis.
Materials and methods
Animals
Male Sprague-Dawley rats (n=55; age, 8 weeks; Beijing Vital River Laboratory Animal Technology Co., Ltd., SPF, Beijing, China), weighing 210–250 g were used in the present study. The animals were maintained under conditions at a temperature of 18–26°C and humidity of 40–70%, in a 12-h light cycle. The present study was approved by the ethics committee of Capital Medical University (Beijing, China).
Reagents
Streptozocin was purchased from Sigma-Aldrich (St. Louis, MO, USA). Normal fodder (crude protein, 18%; crude fat, 4%; and crude fiber, 5%) and high-glucose fodder (crude protein, 18%; crude fat, 20%; crude fiber, 5%; and sucrose, 20%) were acquired from Beijing Keao Xieli Fodder Co., Ltd. (Beijing, China)
Experimental protocol
All the rats were adapted to the housing conditions and fed the same diet of normal fodder for 1 week, following which they were randomly divided into an experimental group (n=30) and a control group (n=25) with 6 rats/month. To create the type 2 DM model, the experimental rats were fed with high-glucose and high-fat fodder for 28 days, whereas the control rats were fed with normal fodder.
All rats were fasted overnight with free access to water to calculate the low dose of streptozocin. Solution A (2.1 g citric acid + 100 ml distilled water) and solution B (2.94 g + 100 ml distilled water) were mixed and buffered to pH 4.2–4.5 to produce solution C. Streptozocin (1 g) was added to 100 ml solution C to produce 1% streptozocin solution. The rats were administered with a single peritoneal injection of streptozocin (30 mg/kg body weight), and the control rats received the samedose of sodium citrate/citric acid buffer. The post-injection dietary structure in the respective groups remained the same as that pre-injection. After 1 week, blood was collected via the tail vein to measure glucose levels, and a successful type 2DM model was established in 24 of the 30 rats. The blood glucose levels were between 16.8 and 29.7 mmol/l in 10 rats, and were above the upper limit of 30.5 mmol/l in 14 rats. If the blood glucose was >16.7 mmol/l and this state was sustained for 4 weeks, construction of the model was considered to be successful. The body weights and blood glucose levels were assessed periodically following establishment of the model, and six animals were sacrificed at 1, 4, 5 and 7 months.
Ultrasonography
Following fasting overnight, the 6 rats in each group were anesthetized with chloral hydrate (30 mg/kg; Sigma-Aldrich), and placed on a current-conducting plate (VisualSonics, Inc., Toronto, Canada). The precordial region was shaved, and ventricular ejection fraction was measured by an experienced laboratory technician using an RMV-716 probe, Vevo 770™ microscopy and high-resolution ultrasound (VisualSonics, Inc.). Following measurement, the rats were sacrificed by administration of 2 ml gas to induce air emboli.
Measurement of heart and left ventricle weights
Following sacrifice of the animals, the hearts were rapidly removed and washed in cold 0.9% saline, with removal of connective tissue and contaminating blood, and placed on ice. The right ventricle and left ventricle were then dissected. The heart and left ventricle were weighed using a precision electronic balance (XPE105; Mettler-Toledo, Columbus, OH, USA) and the transverse diameters were measured using the same ruler. Subsequently, the heart weight index (HWI) was calculated (HWI = weight of the heart / the weight of the body) and left ventricular weight index (LVWI) were calculated (LVWI = the weight of the left ventricle / the weight of the body). The tissues were then stored at -80°C until analyzed using reverse transcription-quantitative polymerase chain reaction (RT-qPCR analysis).
Tissue analysis
All the tissues were stained using hematoxylin and eosin (Sigma-Aldrich) to evaluate the inflammatory reaction and the deformation of myocardial cells. Masson's trichrome staining (Sigma-Aldrich) was used to more clearly visualize fibrosis. Apoptotic cells were detected using terminal deoxynucleotidyl transferase-mediated dUTP-biotin nick end labeling (TUNEL) staining with the DeadEnd™ Fluorometric TUNEL system (Promega Corporation, Madison, WI, USA). TUNEL staining was performed according to the manufacturer's protocol. Briefly, the cells were fixed with 4% paraformaldehyde (Sigma-Aldrich) in phosphate-buffered saline (PBS), rinsed with PBS, treated with 0.1% Triton X-100 (Sigma-Aldrich) for 2 min on ice, and incubated with TUNEL for 1 h at 37°C. The number of apoptotic cells was observed using XSZ-A08 microscope (Wuzhou Optical Instrument Factory, Wuzhou, China), expressed as the percentage of apoptotic cells of the normal cells.
RT-qPCR
RT-qPCR was performed using the BIOER Linegene 9600 system (BIOER, Hangzhou, China) to monitor the mRNA expression levels of the ER-associated molecules. β-actin was selected as the endogenous control for relative quantification in the RT-qPCR analysis. mRNA was extracted using chloroform, Spin Column RM (Beijing Kang Century Biotechnology Co., Ltd., Beijing, China) and RLT buffer (Sangon Biotech Co., Ltd., Shanghai, China) and 80 μl DNase I was administered. The RT reaction mixture containing 2 μl RNA, 2 μl primers and 11 μl RNase was incubated for 5 min at 65°C. The RT mix (4 μl; 5X) and 1 μl HiFi-MMLV enzyme mix (Beijing Kang Century Biotechmology Co., Ltd.) were added and incubated for 40 min at 37°C and subsequently at 70°C for 10 min. The PCR reaction was conducted using 10 μl UltraSYBR Mixture (Beijing Kang Century Biotechnology Co., Ltd.), 1 μl forward primer, 1 μl reverse primer, 2 μl cDNA template and 6 μl RNase-free water. The reaction conditions were as follows: Initial incubation step of 5 min at 65°C, reverse transcription of 40 min at 37°C and 70°C for 10 min, followed by 45 cycles of 15 sec at 95°C for denaturation, and 60 sec at 58°C for annealing and extension. The following primers (Beijing Central Yatai Ke Biomedical Technology Co., Ltd., Beijing, China) were used: GRP78, forward 5′-CCATGCATTGTTTGT GCC GA-3′ and reverse 3′-CTGAAGCTGTTGGGAGGGAG-5′ (139 bp); PERK, forward 5′-GTGAAGGTCGAGAGGCGTCG-3′ and reverse 3′-AATGCCGTATCCGATGTGGG-5′ (248 bp); ATF6, forward 5′-GAGGTGTCTGTTTCGGGGAA-3′ and reverse 3′-AGAACAACTGGAGGAAGCCG-5′ (309 bp); CHOP, forward 5′-GCCAGTGCCAAGCATGTA TC-3′ and reverse 3′-GTAAAGCCCCTCCTTTGG-5′ (78 bp); caspase-12, forward 5′-GGCCCATGTGGAGACAGATT-3′ and reverse 3′-AGCCACTCTTGCCTACCTTC-5′ (80 bp); JNK, forward 5′-GCTGCTTCCTGAGTCACAGT-3′ and reverse 3′-AGAATTGCTGGGGTCATGGGC-5′ (238 bp); collagen I, forward 5′-TGGTGAGACGTGGAAACCTG-3′ and reverse 3′-CTTGGGTCCCTCGACTCCTA-5′ (193 bp); collagen III, forward 5′-TTGGCACAGCAGTCCAATGTA -3′ and reverse 3′-GGGCAGTCTAGTGGCTCATC-5′ (164 bp); and β-actin, forward 5′-CCCATCTATGAGGGTTACGC-3′ and reverse, 3′-TTTAATGTCACGCACGATTTC-5′ (150 bp). The 2−∆∆Cq method was used to analyze the results (25).
Western blotting
The cells were lysed and protein was extracted using radioimmunoprecipitation assay lysis buffer (Youkezhuoye Biotech Ltd., Beijing, China), containing 50 mM Tris (pH 7.4), 1% NP-40, 0.25% Na-deoxycholate, 150 mM NaCl, 1 mM EDTA). The protein was quantified using a BCA Protein assay kit (Beijing Kang Century Biotechnology Co., Ltd.) and 20 μg samples were resolved on 5% SDS-PAGE, wet transferred onto nitrocellulose membrane (Sigma-Aldrich) at 300 mA for 90 min and immunoblotted. The membrane was blocked with 5% bovine serum albumin in Tris-buffered saline-Tween 20 (TBST; Beijing Solarbio Science & Technology Co., Ltd., Beijing, China). The monoclonal rabbit anti-PERK (cat. no. 3192; dilution, 1:1,000) primary antibodies were obtained from Cell Signaling Technology, Inc. (Danvers, MA, USA) and polyclonal rabbit anti-caspase-12 (cat. no. ab62484; dilution, 1:1,000) primary antibodies were obtained from Abcam (Cambridge, MA, USA) and incubated with the membrane at 4°C for 12 h. The membranes were then washed three times with TBST for 10 min and incubated with horseradish peroxidase-conjugated goat anti-rabbit (dilution, 1:10,000; Jackson ImmunoResearch Laboratories, Inc., West Grove, PA, USA; cat. no. 111-035-003) or goat anti-mouse IgG (dilution, 1:10,000; Jackson ImmunoResearch Laboratories, Inc.; cat. no. 115-035-033) secondary antibodies at 25°C for 40 min. The membranes were washed again three times for 10 min. Image analysis was performed using Gelpro 3.0 software (Media Cybernetics, Inc., Rockville, MD, USA).
Statistical analysis
All the data are expressed as the mean ± standard deviation. The differences in each group were evaluated using an independent-samples t-test. The differences between groups and between different time points were evaluated by two-way analysis of variance using IBM SPSS 19 statistical software (IBM SPSS, Armonk, NY, USA. Multiple comparison between the groups was performed using the least significant difference or Bonferroni method. P<0.05 was considered to indicate a statistically significant difference.
Results
Ratios of the heart and left ventricle weights to whole weight
After 1 month, the weights of the heart and left ventricle were increased in the DM rats, compared with the normal rats, however, the difference was not statistically significant. The same trend was noted at months 4 and 5. After 7 months, the increases in the weight of the heart and left ventricle in the DM rats were significantly higher, compared with the control rats (Fig. 1A and B).
Figure 1.
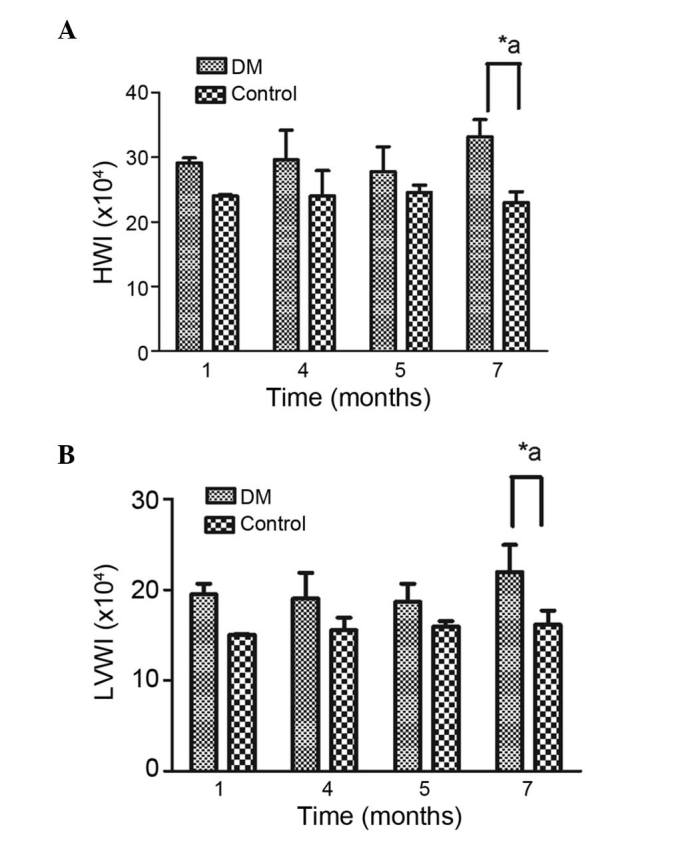
Ratios of (A) heart weight and (B) left ventricle weight to whole body weight. Data are expressed as the mean ± standard deviation. *P<0.05 vs. the DM group at the same time point; aP<0.05, DM group vs. the control group between each month. HWI, heart weight index; LVWI, left ventricle weight index; DM, diabetes mellitus.
Change in ventricular EF in UCG
It is known that structural changes are generally followed by functional changes. This was apparent in the present study, which showed a significant decrease in EF in the DM rats, compared with the normal rats at 5 and 7 months. Despite the unexplained marginal increase in EF at 7 months, the data suggested that, overall, EF significantly decreased over time (Fig. 2).
Figure 2.

Left ventricular EF. The left ventricular EF was compared between the control and DM groups. Data are expressed as the mean ± standard deviation. *P<0.05 vs. the DM group at the same time point; aP<0.05, DM group vs. the control group between each month. EF, ejection fraction; DM, diabetes mellitus.
Myocardial fibrosis in DM and normal rats
After 1 month, clear areas of tissue damage were noted, with loss and degeneration of cardiomyocytes, a pattern that persisted during the following months. It is well known that myocardial tissue is ultimately replaced by granulation tissue due to a deficient process of regeneration. Granulation tissue is characterized by early cellular infiltration, an increase in nuclear density and subsequently by extracellular matrix deposition. The present study used Masson's trichrome staining to enable clearer evaluation of granulation, and it was observed that the sizes of the blue-stained regions expanded significantly over time, (Fig. 3A and B).
Figure 3.

(A) H&E and Masson's trichome staining for tissue analysis. H&E staining of myocardial tissue was performed in the (a) control group and after (b) 1, (c) 4, (d) 5 and (e) 7 months in the DM group (magnification, ×200). Left ventricle endomyocardial histological samples were obtained from control and DM rats. Pink staining indicates cardiac myocytes; blue staining indicates nuclei. Tissues were stained using Masson's trichrome staining in the (f) control group and after (g) 1, (h) 4, (i) 5 and (j) 7 months in the DM group to visualize collagen deposition (blue staining). (B) Ratios of the area of collagen to myocardial tissue. The expression levels of markers of myocardial fibrosis, (C) collagen I and (D) collagen III, are shown. Data are expressed as the mean ± standard deviation. *P<0.05 vs. the DM group at the same time point;**P<0.01, DM group vs. the control at 1, 4, 5 and 7 months; aP<0.05, DM group vs. the control group between each month; bP<0.05, 1 month vs. the DM group at 4, 5 and 7 months. H&E, hematoxylin and eosin; DM, diabetes mellitus.
The present study also found that the expression of collagen I remained higher in the DM rats, compared with the normal rats. This pattern of expression was different for collagen III, which was increased significantly early in the experiment, and then decreased prior to increasing again, however, this was not to the level observed at month 1 (Fig. 3C and D).
Changes in the mRNA levels of GRP78, ATF6 and PERK in DM rats and normal rats
The ER chaperone, GRP78, possesses antiapoptotic properties and is a central regulator of ER homeostasis. Its upregulation is widely used as an essential marker for ER stress under pathological conditions (26). In the present study, a significant increase in GRP78 was observed in the DM rats at 1 month, followed by normalization in months 4 and 5, and subsequent reactivation, which indicated that GRP78 was activated early in the process of ER stress (Fig. 4A).
Figure 4.

mRNA and protein expression levels of ER stress markers. The expression levels of (A) GRP78, (B) ATF6 and (C) PERK, and the protein expression levels of (D and E) PERK are shown. Data are expressed as the mean ± standard deviation. *P<0.05 vs. the DM group at the same time point;**P<0.01, DM group vs. the control at 1, 4, 5 and 7 months; aP<0.05, DM group vs. the control group between each month; bP<0.05, 1 month vs. the DM group at 4, 5 and 7 months. DM, diabetes mellitus; ER, endoplasmic reticulum; GRP78, glucose regulating protein 78; ATF6, activating transcription factor 6; PERK, protein kinase R-like ER kinase.
The activation of ATF6 is an important branch of the three pathways of ER stress (Fig. 4B). The response of ATF6 to ER stress was similar to that of GRP78.
PERK is an important pathway in ER stress and is associated with cellular apoptosis (27). PERK increased significantly at 1 month, following which the mRNA levels reached a plateau (Fig. 4C). By contrast, the present study found that the increase in protein levels was sustained, suggesting that this may reflect more rapid degradation of RNA, compared with protein. It may be that this unstable RNA does not accurately reflect the changes, or that other unknown factors may be stimulating protein expression. However, taken together, the data obtained in the present study indicated that the expression of PERK increased over time (Fig. 4D and E).
TUNEL staining and the mRNA expression levels of caspase-12, CHOP and JNK in DM rats and normal rats
In the present study, TUNEL staining was used to determine the number of apoptotic nuclei, which revealed that there was a significant increase in the number of apoptotic nuclei over time (Fig. 5A and B). The mRNA expression levels of JNK, CHOP and caspase-12 were also assessed (Fig. 5C–E) and were found to increase significantly in the first month. This activation occurred simultaneously with the UPR.
Figure 5.

TUNEL staining of apoptotic nuclei. (A) Nuclei of TUNEL-positive cells were stained brown-yellow (solid arrow) and those of normal cells were stained blue. (B) Ratios of apoptotic nuclei to myocardial tissue. mRNA expression levels of (C) CHOP, (D) JNK and (E) caspase-12 ER stress markers at different time points is shown. (F and G) Protein expression levels of caspase-12. Data are expressed as the mean ± standard deviation. *P<0.05 vs. the DM group at the same time point;**P<0.01, DM group vs. the control at 1, 4, 5 and 7 months; aP<0.05, DM group vs. the control group between each month; bP<0.05, 1 month vs. the DM group at 4, 5 and 7 months. DM, diabetes mellitus; CHOP, C/EBP homologous protein; JNK, Jun N-terminal kinase; TUNEL, terminal deoxynucleotidyl transferase-mediated dUTP-biotin nick end labeling.
With respect to caspase-12, the results of the mRNA and protein analyses in month 1 were consistent, with higher expression levels in the DM group, compared with the control group. However, this increase was reduced in months 5 and 7, which was followed by an increase (Fig. 5F).
Discussion
In the present study, it was found that ER stress and ER stress-mediated apoptosis comprised the two phases of activation of ER stress and apoptosis, followed by myocardial fibrosis, in rats. When the rats were induced to exhibit high blood glucose for 1 month, the expression levels of the antiapoptotic markers, GRP78, PERK and ATF6, and the proapoptotic markers, JNK, CHOP and caspase-12 were all significantly higher. ER stress induced cellular apoptosis, which resulted in fibrosis and tissue remodeling. Although the present study performed extensive measurements of changes in the mRNA and protein expression levels of various molecules involved in the ER stress reaction, these are purely observational data. No evidence was presented to determine whether the changes in the ER pathway are simply associated with the progression of DM fibrosis, or whether they are involved in mediating the pathogenesis, and to what extent. The mechanisms underlying the external pathway and the mitochondrial pathway of apoptosis in the same model remain to be elucidated, and evidence of causation requires the use of specific inhibitors and/or knockout or knockin experiments of specific genes.
The present study also found that the functional alterations began to appear at 7 months, rather than early in the process. The early and significant responses of ER stress and apoptosis indicated that the activation of apoptosis occurred simultaneously with anti-apoptosis, which is an early protective response to attenuate damage to the heart. Of note, under constant glucose stimulation, the expression levels of antiapototic and proapoptotic markers decreased, suggesting that the cells were developing mechanisms of adaptation. Whether this adaptation persists over time with minimal effect on function remains to be elucidated. An important pathway by which CHOP induces apoptosis is by regulating the balance between proapoptotic and antiapoptotic proteins from the B cell lymphoma-2 (Bcl-2) family. CHOP mediates the transcriptional repression and activation of proteins from the antiapoptotic Bcl-2 family (28). CHOP also mediates the direct transcriptional induction of Bcl-2-interacting mediator of cell death, which is a proapoptotic protein from the Bcl-2 family (29). Therefore, the Bcl-2 family may be an important factor, which determines cell survival or apoptosis. The present study did not monitor the changes in Bcl-2 family molecules. A more detailed comprehension of the signals determining the outcome of myocytes may offer potential in predicting the prognosis of patients and improve treatment programs at earlier stages. A previous study demonstrated that ER luminal proteins are released into the cytosol during ER stress-induced apoptosis in a Bcl-2 homologous antagonist killer/Bcl-2-associated X protein-dependent manner (30). This provides an approach to locate apoptosis-associated molecules, rather than purely monitoring their levels of expression.
The rapid increase in the protein level of PERK observed in the present study was not mirrored by the changes in mRNA expression. It is known that the PERK signaling arm is uniquely important for promoting adaptation and survival, as downstream eukaryotic initiation factor-2α can regulate transcription, and PERK is also regulated by cytokines (31). This may be the reason that PERK is a central marker, as it regulates, but is also regulated, although the present study did not monitor functional phosphorylated-PERK.
After 7 months, all proapoptotic markers were expressed at high levels again. This late response was associated with structural remodeling and heart failure, however, there is no evidence for this two-phase response in other models of heart disease. The present study also observed that the mRNA expression of collagen III increased significantly, and that the differences in HWI and LVWI between the DM rats, compared with the control rats, became evident at 7 months. A previous study demonstrated that, when diabetes-induced cardiac cell death is prevented by metallothionein at the early stage, a significant prevention in cardiac remodeling and dysfunction is observed at 5 or 6 months (32). This finding indicates that the differences in remodeling and dysfunction begin to appear at ~6 months, and not earlier. The present study hypothesized that, if a stressful stimuli is maintained for 7 months, as in the DM rats used in the present study, the left ventricle is damaged causing irreversible changes to the structure and function of the heart. Therefore, identifying and interrupting the activation of proapoptotic markers during the early stages is important for inhibiting myocardial fibrosis.
In the DM rats examined in the present study, sequential changes were described in ER stress and the ER stress-mediated apoptosis in myocardial fibrosis in rats. It was found that sustained DM induced myocardial fibrosis. The early activation of ER stress and apoptosis were observed after 1 month, and a turning point was observed after 7 months with ER stress and the presence of irreversible functional abnormality. However, the value of these findings depend, in part, on the identification of a protein, which can portend the direction of the outcome.
Acknowledgments
The present study was supported by the National Natural Science Foundation of China (grant no. 81570220).
References
- 1.Malhotra JD, Kaufman RJ. Endoplasmic reticulum stress and oxidative stress: A vicious cycle or a double-edged sword. Antioxid Redox Signal. 2007;9:2277–2293. doi: 10.1089/ars.2007.1782. [DOI] [PubMed] [Google Scholar]
- 2.Ma Y, Hendershot LM. The unfolding tale of the unfolded protein response. Cell. 2001;107:827–830. doi: 10.1016/S0092-8674(01)00623-7. [DOI] [PubMed] [Google Scholar]
- 3.Wang XZ, Lawson B, Brewer JW, Zinszner H, Sanjay A, Mi LJ, Boorstein R, Kreibich G, Hendershot LM, Ron D. Signals from the stressed endoplasmic reticulum induce C/EBP-homologous protein (CHOP/GADD153) Mol Cell Biol. 1996;16:4273–4280. doi: 10.1128/MCB.16.8.4273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hotamisligil GS. Role of endoplasmic reticulum stress and c-Jun NH2-terminal kinase pathways in inflammation and origin of obesity and diabetes. Diabetes. 2005;54(Suppl 2):S73–S78. doi: 10.2337/diabetes.54.suppl_2.S73. [DOI] [PubMed] [Google Scholar]
- 5.Nakagawa T, Zhu H, Morishima N, Li E, Xu J, Yankner BA, Yuan J. Caspase-12 mediates endoplasmic-reticulum-specific apoptosis and cytotoxicity by amyloid-beta. Nature. 2000;403:98–103. doi: 10.1038/47513. [DOI] [PubMed] [Google Scholar]
- 6.Haze K, Yoshida H, Yanagi H, Yura T, Mori K. Mammalian transcription factor ATF6 is synthesized as a transmembrane protein and activated by proteolysis in response to endoplasmic reticulum stress. Mol Biol Cell. 1999;10:3787–3799. doi: 10.1091/mbc.10.11.3787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tirasophon W, Welihinda AA, Kaufman RJ. A stress response pathway from the endoplasmic reticulum to the nucleus requires a novel bifunctional protein kinase/endoribonuclease (Ire1p) in mammalian cells. Genes Dev. 1998;12:1812–1824. doi: 10.1101/gad.12.12.1812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mannan MA, Shadrick WR, Biener G, Shin BS, Anshu A, Raicuet V, Frick DN, Dey M. An ire1-phk1 chimera reveals a dispensable role of autokinase activity in endoplasmic reticulum stress response. J Mol Biol. 2013;425:2083–2099. doi: 10.1016/j.jmb.2013.02.036. [DOI] [PubMed] [Google Scholar]
- 9.Bertolotti A, Zhang Y, Hendershot LM, Harding HP, Ron D. Dynamic interaction of BiP and ER stress transducers in the unfolded-protein response. Nat Cell Biol. 2000;2:326–332. doi: 10.1038/35014014. [DOI] [PubMed] [Google Scholar]
- 10.Shi Y, Vattem KM, Sood R, An J, Liang J, Stramm L, Wek RC. Identification and characterization of pancreatic eukaryotic initiation factor 2 alpha-subunit kinase, PEK, involved in translational control. Mol Cell Biol. 1998;18:7499–7509. doi: 10.1128/MCB.18.12.7499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ma Y, Hendershot LM. Delineation of a negative feedback regulatory loop that controls protein translation during endoplasmic reticulum stress. J Biol Chem. 2003;278:34864–34873. doi: 10.1074/jbc.M301107200. [DOI] [PubMed] [Google Scholar]
- 12.Korennykh AV, Egea PF, Korostelev AA, Finer-Moore J, Zhang C, Shokat KM, Stroud RM, Walter P. The unfolded protein response signals through high-order assembly of Ire1. Nature. 2009;457:687–693. doi: 10.1038/nature07661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yoshida H, Matsui T, Yamamoto A, Okada T, Mori K. XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell. 2001;107:881–891. doi: 10.1016/S0092-8674(01)00611-0. [DOI] [PubMed] [Google Scholar]
- 14.Shen J, Chen X, Hendershot L, Prywes R. ER stress regulation of ATF6 localization by dissociation of BiP/GRP78 binding and unmasking of Golgi localization signals. Dev Cell. 2002;3:99–111. doi: 10.1016/S1534-5807(02)00203-4. [DOI] [PubMed] [Google Scholar]
- 15.Glembotski CC. Endoplasmic reticulum stress in the heart. Circ Res. 2007;101:975–984. doi: 10.1161/CIRCRESAHA.107.161273. [DOI] [PubMed] [Google Scholar]
- 16.Sundar Rajan S, Srinivasan V, Balasubramanyam M, Tatu U. Endoplasmic reticulum (ER) stress and diabetes. Indian J Med Res. 2007;125:411–424. [PubMed] [Google Scholar]
- 17.Nishitoh H, Matsuzawa A, Tobiume K, Saegusa K, Takeda K, Inoue K, Hori S, Kakizuka A, Ichijo H. ASK1 is essential for endoplasmic reticulum stress-induced neuronal cell death triggered by expanded polyglutamine repeats. Genes Dev. 2002;16:1345–1355. doi: 10.1101/gad.992302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ma Y, Brewer JW, Diehl JA, Hendershot LM. Two distinct stress signaling pathways converge upon the CHOP promoter during the mammalian unfolded protein response. J Mol Biol. 2002;318:1351–1365. doi: 10.1016/S0022-2836(02)00234-6. [DOI] [PubMed] [Google Scholar]
- 19.Martindale JJ, Fernandez R, Thuerauf D, Whittaker R, Gude N, Sussman MA, Glembotski CC. Endoplasmic reticulum stress gene induction and protection from ischemia/reperfusion injury in the hearts of transgenic mice with a tamoxifen-regulated form of ATF6. Circ Res. 2006;98:1186–1193. doi: 10.1161/01.RES.0000220643.65941.8d. [DOI] [PubMed] [Google Scholar]
- 20.Okada K, Minamino T, Tsukamoto Y, Liao Y, Tsukamoto O, Takashima S, Hirata A, Fujita M, Nagamachi Y, Nakatani T, et al. Prolonged endoplasmic reticulum stress in hypertrophic and failing heart after aortic constriction: Possible contribution of endoplasmic reticulum stress to cardiac myocyte apoptosis. Circulation. 2004;110:705–712. doi: 10.1161/01.CIR.0000137836.95625.D4. [DOI] [PubMed] [Google Scholar]
- 21.Dally S, Monceau V, Corvazier E, Bredoux R, Raies A, Bobe R, del Monte F, Enouf J. Compartmentalized expression of three novel sarco/endoplasmic reticulum Ca2+ATPase 3 isoforms including the switch to ER stress, SERCA3f, in non-failing and failing human heart. Cell Calcium. 2009;45:144–156. doi: 10.1016/j.ceca.2008.08.002. [DOI] [PubMed] [Google Scholar]
- 22.Mao W, Fukuoka S, Iwai C, Liu J, Sharma VK, Sheu SS, Fu M, Liang CS. Cardiomyocyte apoptosis in autoimmune cardiomyopathy: Mediated via endoplasmic reticulum stress and exaggerated by norepinephrine. Am J Physiol Heart Circ Physiol. 2007;293:H1636–1645. doi: 10.1152/ajpheart.01377.2006. [DOI] [PubMed] [Google Scholar]
- 23.Severino A, Campioni M, Straino S, Salloum FN, Schmidt N, Herbrand U, Freude S, Toietta G, Di Rocco, Bussani R, et al. Identification of protein disulfide isomerase as a cardiomyocyte survival factor in ischemic cardiomyopathy. J Am Coll Cardiol. 2007;50:1029–1037. doi: 10.1016/j.jacc.2007.06.006. [DOI] [PubMed] [Google Scholar]
- 24.Lakshmanan AP, Harima M, Suzuki K, Soetikno V, Nagata M, Nakamura T, Takahashi T, Sone H, Kawachi H, Watanabe K. The hyperglycemia stimulated myocardial endoplasmic reticulum (ER) stress contributes to diabetic cardiomyopathy in the transgenic non-obese type 2 diabetic rats: A differential role of unfolded protein response (UPR) signaling proteins. Int J Biochem Cell Biol. 2013;45:438–447. doi: 10.1016/j.biocel.2012.09.017. [DOI] [PubMed] [Google Scholar]
- 25.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−delta delta C(T)) method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 26.Dong D, Ni M, Li J, Xiong S, Ye W, Virrey JJ, Mao C, Ye R, Wang M, Pen L, et al. Critical role of the stress chaperone GRP78/BiP in tumor proliferation, survival and tumor angio-genesis in transgene-induced mammary tumor development. Cancer Res. 2008;68:498–505. doi: 10.1158/0008-5472.CAN-07-2950. [DOI] [PubMed] [Google Scholar]
- 27.Chen YJ, Su JH, Tsao CY, Huang CT, Chao HH, Lin JJ, Liao MH, Yang ZY, Huang HH, Tsai FJ, et al. Sinulariolide induced hepatocellular carcinoma apoptosis through activation of mitochondrial-related apoptotic and PERK/eIF2α/ATF4/CHOP pathway. Molecules. 2013;18:10146–10161. doi: 10.3390/molecules180910146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.McCullough KD, Martindale JL, Klotz LO, Aw TY, Holbrook NJ. Gadd153 sensitizes cells to endoplasmic reticulum stress by down-regulating Bcl2 and perturbing the cellular redox state. Mol Cell Biol. 2001;21:1249–1259. doi: 10.1128/MCB.21.4.1249-1259.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Puthalakath H, O'Reilly LA, Gunn P, Lee L, Kelly PN, Huntington ND, Hughes PD, Michalak EM, McKimm-Breschkin J, Motoyama N, et al. ER stress triggers apoptosis by activating BH3-only protein Bim. Cell. 2007;129:1337–1349. doi: 10.1016/j.cell.2007.04.027. [DOI] [PubMed] [Google Scholar]
- 30.Wang X, Olberding KE, White C, Li C. Bcl-2 proteins regulate ER membrane permeability to luminal proteins during ER stress-induced apoptosis. Cell Death Differ. 2011;18:38–47. doi: 10.1038/cdd.2010.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cojocari D, Vellanki RN, Sit B, Uehling D, Koritzinsky M, Wouters BG. New small molecule inhibitors of UPR activation demonstrate that PERK, but not IRE1α signaling is essential for promoting adaptation and survival to hypoxia. Radiother Oncol. 2013;108:541–547. doi: 10.1016/j.radonc.2013.06.005. [DOI] [PubMed] [Google Scholar]
- 32.Xu J, Wang G, Wang Y, Liu Q, Xu W, Tan Y, Cai L. Diabetes- and angiotensin II-induced cardiac endoplasmic reticulum stress and cell death: Metallothionein protection. J Cell Mol Med. 2009;13:1499–1512. doi: 10.1111/j.1582-4934.2009.00833.x. [DOI] [PMC free article] [PubMed] [Google Scholar]