

Abstract
Alternative polyadenylation (APA) and alternative splicing (AS) provide mRNAs with the means to avoid microRNA repression through selective shortening or differential usage of 3′UTRs. The two glutaminase (GLS) mRNA isoforms, termed KGA and GAC, contain distinct 3′UTRs with the KGA isoform subject to repression by miR-23. We show that depletion of the APA regulator CFIm25 causes a strong shift to the usage of a proximal poly(A) site within the KGA 3′UTR and also alters splicing to favor exclusion of the GAC 3′UTR. Surprisingly, we observe that while miR-23 is capable of down-regulating the shortened KGA 3′UTR, it has only minor impact on the full-length KGA 3′UTR, demonstrating that additional potent negative regulation of GLS expression exists beyond this single microRNA targeting site. Finally, we show that the apoptosis induced upon down-regulation of the GAC isoform can be alleviated through concurrent reduction in CFIm25 expression, revealing the sensitivity of glutaminase expression to the levels of RNA processing factors. These results exemplify the complex interplay between RNA processing and microRNA repression in controlling glutamine metabolism in cancer cells.
Keywords: alternative polyadenylation, glutaminase, glutamine addiction, c-Myc, cell growth, miRNA
INTRODUCTION
Cleavage and polyadenylation was initially viewed as a constitutive event, but numerous bioinformatics analyses have shown that more than half of human genes contain multiple polyadenylation signals, leading to the generation of different mRNA isoforms (Beaudoing et al. 2000; Beaudoing and Gautheret 2001; Tian et al. 2005). This process, termed alternative polyadenylation (APA), can therefore create mRNAs with different lengths and regulatory features. The most common form of APA involves the use of multiple polyadenylation sites within the same terminal exon (termed UTR-APA). This results in the formation of mature mRNA transcripts that code for the same protein but possess different 3′UTR lengths and sequence information (Yan and Marr 2005). Transcripts with truncated 3′UTRs are potentially more stable due to the loss of negative regulation by microRNA and/or AU-rich RNA destabilizing element (ARE) (Chen and Shyu 1995; Friedman et al. 2009; Mayr and Bartel 2009; Masamha et al. 2014). The less common type of APA involves the use of distinct 3′UTRs and allows for the expression of unique protein isoforms containing different carboxyl terminal sequences. The most characterized form of this regulation, termed “splicing-APA,” is in the Immunoglobulin heavy chain pre-mRNA where an alternative PAS is used upon B-cell activation (Takagaki et al. 1996). Aside from this example, only limited mechanistic investigation of splicing-APA has been done.
Recent studies have suggested that genome-wide changes in APA may occur in rapidly proliferating cells and cancer (Sandberg et al. 2008; Mayr and Bartel 2009; Elkon et al. 2012). The implication of these studies is that controlling APA may represent a novel means by which actively dividing cells reprogram their transcriptome to meet the needs associated with cell growth. In order to adapt to the demands of rapid proliferation, cancer cells also undergo metabolic reprogramming, which includes the enhanced utilization of glucose through aerobic glycolysis (the Warburg effect) and up-regulated glutamine catabolism through glutaminolysis (for review, see DeBerardinis and Cheng 2010; Oermann et al. 2012; Ward and Thompson 2012). Among other functions, glutamine is essential for the replenishment of TCA cycle intermediates (anaplerosis) for ATP synthesis and acts as a nitrogen donor in critical biosynthetic pathways that support rapidly dividing cancer cells (for review, see Wise and Thompson 2010; Oermann et al. 2012).
The first step in glutaminolysis is mediated by the enzyme glutaminase (GLS), which converts glutamine to glutamate. Oncogenic levels of c-Myc are often associated with increased levels of GLS and glutaminolysis, resulting in glutamine-dependent survival in cancer, a phenomenon termed “glutamine addiction” (Yuneva et al. 2007; Wise et al. 2008; Gao et al. 2009; Cassago et al. 2012). Regulation of human GLS by c-Myc occurs at the posttranscriptional level (Gao et al. 2009). Overexpression of c-Myc leads to a direct transcriptional repression of several microRNAs (Chang et al. 2008) including miR-23a/b, which binds to a single site within the 3′UTR of GLS to silence its expression (Gao et al. 2009). Confounding this observation, however, is the complicated gene structure of GLS. It encodes two different isoforms (called GAC and KGA) possessing unique 3′UTRs, which are a result of alternative splicing (AS). The KGA isoform houses the target site for miR-23a/b as well as a potent ARE that has been shown to function in rat cells (Fig. 1A; Laterza et al. 1997; Laterza and Curthoys 2000b). Moreover, while c-Myc transcriptional repression of miR-23a/b causes the de-repression of the KGA isoform, large-scale analyses of clinical samples from TCGA data sets identified that tumors often switch use of 3′UTRs from the KGA to the GAC isoform, thus circumventing the need to repress miR-23a/b (Xia et al. 2014). More investigation of GLS RNA processing is therefore warranted, given how little is understood about both the regulation of splicing-APA events and its importance to tumor biology.
FIGURE 1.
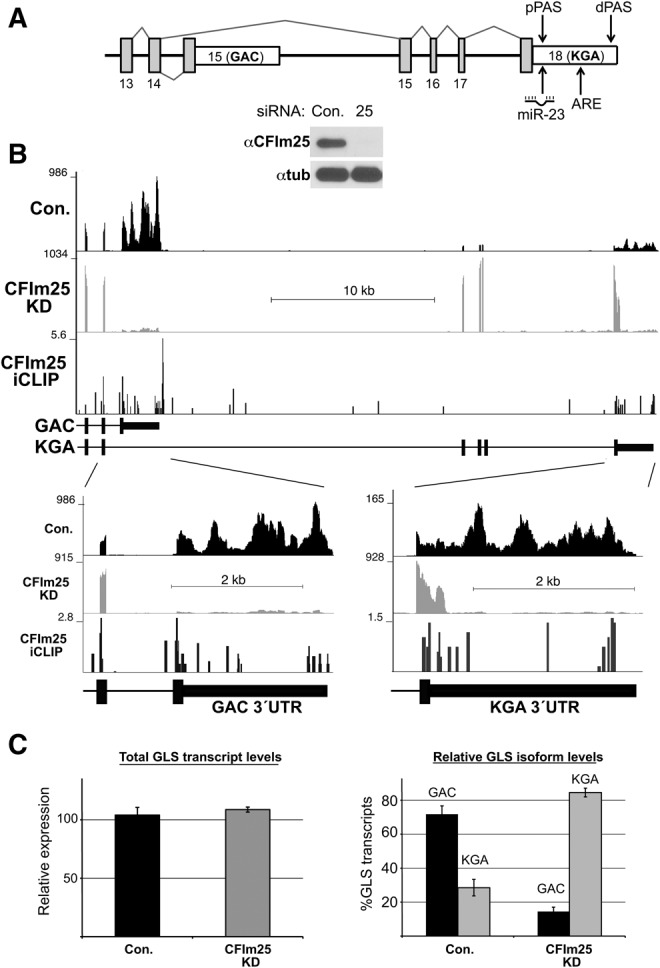
Changes in the levels of GLS1 isoform transcripts upon CFIm25 knockdown in HeLa cells. (A) Genomic structure of the 3′ end of the GLS1 gene. The final exons are labeled and the two distinct 3′UTRs for the two GLS1 mRNA isoforms (GAC and KGA) are shown. The upward pointing arrow indicates the approximate location of the targeting site for miR-23; the other arrowheads indicate the approximate locations of the pPAS and dPAS in the 3′UTR of KGA isoform. Open rectangles depict regions of 3′UTR for both isoforms. (B) Read density of RNA-seq data from the corresponding region of GLS1 isoforms after treating HeLa cells with control siRNA (con) or CFIm25 siRNA (CFIm25 KD). The number of sequence reads is represented on the y-axis and zoom-in views of the two 3′UTRs are shown below. The bottom track depicts the mapped CFIm25 iCLIP tags identified in 293T cells as described in Martin et al. (2012). The figure inset is a Western blot of CFIm25 knockdown in HeLa cells. (C) Bar graphs for qRT-PCR results showing the levels of total GLS transcripts and the changes in the percentage of the GAC and KGA isoform transcripts after CFIm25 depletion (CFIm25KD) compared to control (Con.). Results were normalized to 7SK and data represented the average ± SD (n = 3).
We and others have identified CFIm25 as a global regulator of APA (Gruber et al. 2012; Martin et al. 2012; Masamha et al. 2014), whose knockdown not only induces a global switch to the usage of the poly(A) signal most proximal to the stop codon (pPAS) sites, but also enhances cell proliferation (Masamha et al. 2014). Here, we show that one of the major gene targets of CFIm25 is GLS, which undergoes altered splicing-APA after CFIm25 down-regulation. Upon knockdown of CFIm25, we observe a significant 3′UTR shortening of the KGA isoform. Intriguingly, CFIm25 knockdown also impacts alternative splicing such that GAC exon inclusion is repressed in cells that normally use GAC isoform. Further, we observe that the GAC 3′UTR stimulates glutaminase expression more efficiently than the KGA 3′UTR and that the KGA 3′UTR is under multiple forms of negative regulation in addition to miR-23. Finally, we found that specific depletion of the GAC isoform by RNAi induces apoptosis but this can be alleviated if CFIm25 is also knocked down simultaneously to induce splicing-APA that favors shortened KGA isoform expression. These results reveal the complex posttranscriptional gene control of GLS where RNA processing events, including AS and APA, play an equally important regulatory role as c-Myc and miR-23.
RESULTS AND DISCUSSION
Knockdown of CFIm25 induces GLS isoform switching and 3′UTR shortening of the newly switched isoform
Within the human genome there are two genes that encode glutaminase enzymes. The first is GLS/GLS1 (or kidney-type), located on chromosome 2 and the other is GLS2 (or liver type) located on chromosome 12. Based upon Ensembl gene annotation, GLS/GLS1 will be referred to as “GLS” herein. GLS has two major splice variants termed KGA and GAC, which are generated by AS near the 3′ end with both isoforms having slightly different C termini and distinct 3′UTRs (Fig. 1A; Elgadi et al. 1999; Aledo et al. 2000; de la Rosa et al. 2009). Intriguingly, a single miR-23a/b targeting site within the 2.6 kb 3′UTR of the KGA isoform appears capable of silencing expression from this transcript (Gao et al. 2009). Further, the rat KGA 3′UTR has been shown to contain a highly potent AU-rich RNA destabilizing element (ARE) that is bound by the ARE-binding protein AUF1, which mediates its instability (Porter et al. 2002; Schroeder et al. 2006). An ARE with six AUUUA signature motifs embedded in an AU-rich region spanning ∼650 nt is also found at the 3′ end portion of human KGA 3′UTR.
To investigate the possibility that glutaminase is subject to APA regulation by CFIm25, we depleted HeLa cells of CFIm25 using siRNA-mediated knockdown. The efficiency of knockdown was confirmed by Western blot analysis of lysates from transfected cells (Fig. 1B, inset). RNA from both control siRNA and CFIm25 siRNA-treated cells was isolated and subjected to mRNA-seq analysis using Illumina High-Seq sequencing and analyzed using our recently described DaPars algorithm (Xia et al. 2014). In HeLa cells, we found no expression from the GLS2 gene (data not shown), whereas expression was observed for the GLS gene. In control siRNA-treated cells, the level of GAC isoform transcript is far more abundant than that of KGA isoform (Fig. 1B). After knockdown of CFIm25 we observed two significant changes. First, we found reduced splicing from exon 14 to the GAC-specific exon 15 (which is the terminal exon of the GAC isoform) and, concurrent with this change there is an increase in the splicing of exon 14 to the KGA-specific exon 15, leading to biogenesis of KGA isoform. The second distinct observation was a dramatic switch to the usage of a proximal poly(A) site within the KGA 3′UTR encoded by the KGA-specific terminal exon (exon 18), which resulted in a near total loss of read density throughout this region (Fig. 1B, bottom; close-up panels). The combination of these two changes led to an approximately sixfold increase in the mRNA abundance of the 3′UTR-shortened KGA isoform (compare 165 reads for Con. versus 928 for CFIm25 KD) and a significant reduction in the levels of the GAC isoform. The high degree of 3′UTR shortening was specific to the KGA 3′UTR as no detectable change in the relative read distribution throughout the GAC 3′UTR was found after CFIm25 knockdown. In support of the hypothesis that the alterations in GLS isoform expression were directly due to CFIm25 reduction, we took advantage of a deposited CFIm25 CLIP-seq data set published by Martin et al. (2012). We observed considerable density of CFIm25 iCLIP tags within both the GAC and KGA 3′UTRs (Fig. 1B). Importantly, we found a more striking enrichment of tags at the pPAS and the more distal poly(A) signal (dPAS) of the KGA 3′UTR while such significant enrichment was not as prominent for the GAC 3′UTR. Taken together, these results indicate that the KGA 3′UTR is subject to CFIm25-regulated APA while the GAC 3′UTR is not, and that CFIm25 levels impact the AS of the two GLS isoforms.
To further confirm the results observed with RNA-seq data, we designed a series of amplicons throughout the 3′ regions of GLS isoforms and measured the abundance of specific GLS mRNA isoforms before and after CFIm25 knockdown using quantitative RT-PCR (qRT-PCR). In agreement with the RNA-seq results, qRT-PCR analysis of GLS isoforms demonstrated that in control siRNA-treated cells there is a preference for the GAC over the KGA isoform, which is reversed after CFIm25 knockdown (Fig. 1C). Furthermore, we found no significant changes in the overall level of GLS transcripts as assessed using an amplicon targeting an upstream common sequence (exons 3–5), indicating that overall GLS transcription is unchanged upon CFIm25 depletion.
These results revealed two forms of CFIm25-related regulation of the GLS pre-mRNA: activation of GAC terminal exon inclusion and repression of the KGA pPAS usage. One possible model explaining CFIm25 function is that it governs PAS choice within the KGA 3′UTR and is also directly involved in activating GAC terminal exon definition. A second possibility is that CFIm25 plays no role in regulating inclusion of the GAC terminal exon. Rather, alternative splicing to the KGA exon 15 is preferred in HeLa cells, but in the presence of CFIm25, the distal KGA PAS is utilized, thus generating a longer 3′UTR that presumably contains destabilizing miRNA binding sites and/or other potential negative RNA elements, e.g., the ARE, to keep the corresponding long transcript unstable. As a result, the steady-state level of GAC transcript is more abundant than that of KGA transcript with a longer 3′UTR. This is consistent with the low levels of KGA transcript with a longer 3′UTR in control knockdown cells. After CFIm25 depletion, the switch from the KGA dPAS to its pPAS results in significant stabilization of the 3′UTR shortened transcript, thereby revealing the existing preferred splicing of exon 14 to KGA exon 15. While the mechanism of CFIm25 APA regulation is currently unknown, it is noteworthy that CFIm25 iCLIP tags are highly enriched at both proximal and distal PASs within the KGA 3′UTR (Fig. 1B). This bimodal distribution of CFIm25 iCLIP tags within the 3′UTR is recently found typical of CFIm25 regulated transcripts (Masamha et al. 2014). It is possible that CFIm25 may enforce PAS choice through the simultaneous activation of the distal site and the repression of the proximal site.
CFIm25 regulates GLS protein isoform expression and represses proximal poly(A) site usage of the KGA 3′UTR
To determine the impact of CFIm25 depletion on overall glutaminase protein expression and that of GLS two isoforms, we probed cell lysates from RNAi-treated cells with an antibody that recognizes both isoforms of GLS, or one that is GAC- or a KGA-specific. Using an antibody that recognizes both isoforms, we observed switching in the GLS isoforms after CFIm25 knockdown in HeLa cells that were treated with either the same siRNA used for RNA-seq or a second distinct siRNA. In agreement with both the qRT-PCR and the RNA-seq data, we observed a large increase in the levels of KGA protein and a decrease in the levels of GAC (Fig. 2A). To determine whether these effects are cell-type specific, we conducted the same knockdowns in the human embryonic kidney cell line 293T. The Western blot results (Fig. 2B) showed that in 293T cells, CFIm25 depletion also results in a similar dramatic switch in GLS isoform expression.
FIGURE 2.

Comparison of GLS isoform expression as it relates to CFIm25 expression. (A) Changes in the protein levels of total GLS and both the GAC and KGA isoforms were determined by Western blotting using isoform specific antibodies in HeLa cells after knockdown of CFIm25 by two different siRNAs (#1 and #2) and compared to control (Con.). (*) Represents a cross-reacting band of unknown identity. (B) Levels of GLS isoform expression in 293T cells were determined by Western blotting after knocking down CFIm25 using two different siRNAs. (C) Lysates were prepared from HeLa cells after cotransfecting RNAi resistant pcDNA6 Flag-CFIm25 (25*) or pcDNA6 Flag-GFP (Vec.) with either the control siRNA (Con.) or CFIm25 si#2 for 48 h, and the levels of CFIm25 and total GLS were determined by Western blotting. Tubulin was used as a loading control. (D) Following CFIm25 depletion by siRNA, qRT-PCR was performed to measure levels of miR23 and miR100, and the results were normalized to SNORD48. Data represented the average ± SD (n = 3) and t-tests were done relative to the control (Con.) samples. (E) Schematic of dual luciferase reporters where different 3′UTRs were cloned downstream from Renilla luciferase. Lengths of inserted 3′UTRs are shown in base pairs. (F) Results of luciferase assays that were normalized to Firefly luciferase expression in lysates from HeLa cells transfected with reporters shown in schematic. All t-tests were done relative to the control plasmid. (G) Results of luciferase assays of lysates transfected with reporters and either control siRNA or CFIm25 siRNA. In each case, individual reporter expression was normalized to control siRNA transfection. The t-test was done for each plasmid between the control and CFIm25 knockdown. The values used were P < 0.01 (**) and P < 0.001 (***).
To further support that the changes observed in GLS levels are a result of CFIm25 knockdown, we developed a Flag-tagged CFIm25 cDNA with conservative mutations in the cDNA so that the resulting CFIm25 transcript is resistant to a CFIm25-specific siRNA. Cotransfection of CFIm25 siRNA with the RNAi resistant CFIm25 cDNA plasmid resulted in depletion of endogenous CFIm25 and not the ectopically expressed Flag-CFIm25. The total KGA protein in Flag-CFIm25 rescued cells was at a very low level similar to that of the control (Fig. 2C), ruling out that our results are due to off-target siRNA effects. Finally, oncogenic levels of c-Myc are known to inhibit miR-23 transcription (Gao et al. 2009), which presumably allow KGA mRNA isoforms with a longer 3′UTR to evade gene silencing by miR-23. To ensure that CFIm25 knockdown in HeLa cells did not fortuitously reduce the levels of miR-23, we also measured the levels of this miRNA (with miR-100 levels as a control) by qRT-PCR after CFIm25 knockdown (Fig. 2D). There was no reduction in the levels of either miRNA after CFIm25 depletion, thus ruling out the possibility that decrease in miR-23 levels might be a contributing factor to the observed, highly up-regulated expression of KGA isoform.
Based upon the RNA-seq results before and after knock down of CFIm25, we found that there are three possible 3′UTRs for GLS mRNA transcripts: GAC, KGA that is full-length/long (KGA-L), and KGA that is truncated/short (KGA-S). It is noteworthy that the KGA-L contains both a pPAS and dPAS while the KGA-S only contains the pPAS. To determine the effects of each 3′UTR on protein expression, we utilized a dual luciferase reporter system with the individual 3′UTRs cloned downstream from Renilla luciferase. As a control, we also used a vector-derived 3′UTR containing the SV40 PAS (Fig. 2E). In each case, we included an additional ∼100 nucleotides (nt) downstream from the annotated PAS to ensure inclusion of a DSE (down-stream element), which is known to be required for efficient cleavage and polyadenylation. Strikingly, we observed that the GAC 3′UTR is sixfold more efficient to support expression of the heterologous luciferase reporter than the KGA-L 3′UTR (Fig. 2F). This is consistent with our recent report that the GAC 3′UTR is devoid of any predicted microRNA targeting sites (Xia et al. 2014). Moreover, we surprisingly found that luciferase reporters containing either the KGA-L or KGA-S 3′UTR are equally poor in supporting expression of the luciferase reporter. We expected that the KGA-L reporter would be expressed low because CFIm25-mediated repression of its pPAS ensures that any transcript synthesized will utilize the dPAS. Therefore, as is the case for the endogenous GLS gene, any KGA-L reporter mRNA synthesized will possess a long 3′UTR containing negative elements to keep the corresponding transcript unstable. However, we did not expect to observe low expression of the KGA-S reporter because these negative elements were removed. One possible explanation is that CFIm25 is still capable of repressing cleavage and polyadenylation of the KGA-S reporter even though a dPAS is not present. To test this idea, we repeated the same experiment both with and without depletion of CFIm25 by RNAi (Fig. 2G). In each case, luciferase levels of the CFIm25 knockdown were normalized to control knockdown. While only modest effect on luciferase expression was observed for control or GAC, both the KGA-L and KGA-S were markedly increased in response to CFIm25 knockdown. These results lend support to a model that when CFIm25 levels are reduced, the polyadenylation signal present in KGA-S is no longer repressed allowing for productive mRNA 3′ end processing.
CFIm25 and miR-23 repression of the KGA 3′UTR is independent and additive
It was previously shown that overexpression of c-Myc can cause transcriptional repression of miR-23, thereby increasing KGA isoform expression. To explore the relationship between miR-23 targeting of the KGA isoform and the 3′UTR shortening elicited by CFIm25 knockdown, we determined the exact position of proximal polyadenylation in CFIm25-depleted HeLa cells. The available, truncated KGA transcripts present within the Ensembl database do not contain the site of proximal polyadenylation necessitating its empirical identification. We performed 3′RACE with primers positioned within the KGA terminal exon to specifically amplify the KGA pPAS. Consistent with the RNA-seq analysis, only when we knocked down CFIm25 did we observe a PCR product of ∼500 nt (Fig. 3A, left panel). Sequencing of several individual PCR clones definitively mapped the KGA pPAS sequence to an AAUAAA located 178 nt downstream from the KGA stop codon while the site of cleavage and polyadenylation is located between positions +207 and +212 (Fig. 3A, right panel). This demonstrates that after CFIm25 knockdown, the KGA 3′UTR changes from its initial ∼2.6 kb length (long 3′UTR) to ∼210 nt (short 3′UTR). However, unexpectedly, the miR-23 targeting site is still present within the KGA short transcript downstream from the PAS and upstream of the poly(A) tail. To determine which of the two KGA 3′UTRs (L versus S) is still sensitive to miR-23 repression, we cotransfected dual luciferase reporters with either miR-23 mimics or antagomirs. Intriguingly, we observed that miR-23 mimics repress the KGA-S significantly but KGA-L only modestly (Fig. 3B). Similarly, miR-23 antagomirs derepress KGA-S effectively but KGA-L marginally (Fig. 3C). These results suggest that the ARE present in the KGA-L 3′UTR is dominant to repress KGA expression and that its removal through APA renders the KGA-S exquisitely sensitive to miR-23-dependent repression. Our results also suggest that the level of endogenous miR-23 in HeLa cells appears insufficient to elicit any profound silencing effect through KGA-L and -S 3′UTRs.
FIGURE 3.

Depletion of CFIm25 from 293T cells results in sensitization to glutamine withdrawal and slows cell growth. (A) Results of 3′RACE of RNA isolated from HeLa cells treated with either control siRNA or siRNA targeting CFIm25. Schematic depicting the position of the KGA proximal poly(A) site (AAUAAA), the miR23 target region (underlines), and the identified positions of pre-mRNA cleavage (arrows) through sequencing of multiple clones from the 3′RACE. (B) Luciferase assay results in cell lysates isolated from cells transfected with either KGA-L or KGA-S as well as control RNA mimic or miR-23 mimic. Results represent the average of three independent experiments with t-tests done for each plasmid in the presence of the control or miR23 mimic. (C) Same as in panel B with the exception that either control antagomir or miR-23 antagomirs were cotransfected with reporters. (D,E) Luciferase assay results as conducted in panels B,C with the exception that CFIm25 was also knocked down using siRNA. In both panels, data are normalized to the first condition and expressed as fold relative to that condition, and t-test comparisons are all relative to that first condition. The values used were P < 0.05 (*), P < 0.01 (**), and P < 0.001 (***).
To determine how CFIm25 levels and miR-23 impact KGA-S expression, we repeated these experiments both in the presence and absence of CFIm25 siRNA. We found that depletion of CFIm25 can potently de-repress KGA-S, likely through relieving the repression of pPAS usage and thus enhancing its 3′ end processing, but this 3′UTR is still capable of being repressed by miR-23 mimics (Fig. 3D). In agreement with these results, we found that miR-23 antagomirs were capable of de-repressing KGA-S even further after CFIm25 knockdown. Overall, these data demonstrate that the KGA 3′UTR is subject to CFIm25-mediated repression [likely through repression of the proximal poly(A) site usage] and additionally, any KGA-S transcript that is made is also under negative regulation by miR-23.
CFIm25 expression levels can modulate cellular sensitivity to glutaminase levels
Mounting evidence suggests that the GAC isoform of GLS (more so than the KGA isoform) may be a driver in glutamine-dependent tumor growth, making it an attractive chemotherapeutic target (Wang et al. 2010; Cassago et al. 2012). Our results thus far further support this model as the KGA 3′UTR is subject to negative regulation by CFIm25, miR-23, and a potent ARE. To explore this idea in the context of CFIm25 regulation, we developed siRNAs targeting unique regions of either the GAC or the KGA isoform and monitored their ability to elicit apoptosis. Consistent with the RNA-seq results, transfection of HeLa cells with GAC-specific siRNA caused significant reduction in GLS expression, whereas no effect was seen using KGA-specific siRNA (Fig. 4A). This is because HeLa cells normally express mainly GAC isoform with little expression of KGA isoform. Moreover, only the GAC-specific siRNA was able to elicit apoptosis, while the KGA-specific siRNA had no effect (Fig. 4B). Importantly, the apoptosis observed upon depletion of the GAC isoform by RNAi could be partially alleviated if CFIm25 expression was also reduced by RNAi, which in turn induced the appearance of KGA isoform expression (Fig. 4B). The lack of full rescue could be attributed to the slightly lower catalytic activity of the KGA isoform even though its expression is comparable to the levels of GAC in unperturbed HeLa cells. Consistently, apoptosis could be induced in HeLa cells after transfection with KGA-specific siRNA when CFIm25 was also knocked down, which was due to the induced GLS isoform switching (Fig. 4A,B). These results demonstrate that cellular sensitivity to GAC isoform depletion/inhibition can be circumvented at least in part through altered expression of GLS isoforms by modulating CFIm25 level. Moreover, they uncover possible distinct mechanisms that cancer cells have evolved in order to maintain GLS at certain cellular levels through isoform switching for survival.
FIGURE 4.

CFIm25 can modulate cellular sensitivity to GLS isoform-specific siRNA. (A) Western blot analysis of HeLa cell lysates from cells transfected with siRNA. In all cases, the total level of transfected siRNA was kept constant using control siRNA. (B) Results of Caspase assays of cell lysates isolated from cells transfected with siRNA as in panel A. The results are the average of three independent experiments. (C) Model of GLS post-transcriptional regulation. CFIm25 functions positively to promote GAC terminal exon definition but negatively to repress KGA proximal PAS recognition. The KGA 3′UTR is under repression by miR-23, which is itself under transcriptional repression by c-Myc. Finally, the KGA 3′UTR is under additional repression by at least an AU-rich element (ARE). The values used were P < 0.01 (**) and P < 0.001 (***).
Model of GLS isoform expression
The recent focus on cancer metabolism has shed new light on why altered expression of basic metabolic enzymes occurs in tumors (for review, see Vander Heiden et al. 2009). Based upon the results presented here and those of other investigators, we can envisage a multilevel model of GLS post-transcriptional regulation (Fig. 4C). The first component of this model was initially described by Curthoys and colleagues (Laterza et al. 1997; Laterza and Curthoys 2000b) where a potent ARE is present within the rat KGA 3′UTR that causes its destabilization. The second component of this model was determined by Gao et al. (2009) and is dependent on c-Myc overexpression to transcriptionally repress miR-23, thereby relieving gene silencing specifically of the KGA 3′UTR. As we have shown here, the KGA pPAS is also under repression of CFIm25, which is consistent with its function at other genes subject to APA (Masamha et al. 2014). Finally, we also show that CFIm25 is required to maintain inclusion of the GAC 3′UTR, suggesting that it may play a positive role in terminal exon definition. During the review of this manuscript, a study by Redis et al. (2016) was published showing that the noncoding RNA CCAT2 is capable of modulating CFIm25's function at the GAC exon. They show that CCAT2 possibly acts, in an allele-specific manner, as a scaffold to recruit CFIm25 to the GAC terminal exon to stimulate its usage. Those results provide a potential mechanism of how CFIm25 is functioning to activate GAC terminal exon inclusion and are consistent with what we show here. A function for the CFIm complex in AS has been suggested (Proudfoot 2011) but GLS represents the first case where within a single gene, an RNA processing factor functions positively at one terminal exon and negatively at another. Modulating the expression of the CCAT2 or tissue specific alteration of CFIm25 levels could mediate GLS isoform switching, as could posttranslational modifications within CFIm25. Indeed, CFIm25 has been found to be subject to several different modifications including tyrosine phosphorylation and lysine acetylation (Rush et al. 2005; Shimazu et al. 2007).
The redundant pathways, which lead to enhanced GLS expression regardless of the isoform, suggest that post-transcriptional regulation is a key component in its normal gene regulation and that mammalian cells have evolved intricate ways to modulate levels of GLS expression under varying physiological conditions. Indeed, it was previously shown that in the kidney, the presence of an ARE within the KGA 3′UTR helps recruitment of an ARE-binding protein, AUF1, resulting in the repression of GLS expression (Schroeder et al. 2006). Interestingly, under acidic conditions, AUF1 is displaced, which is followed by a significant increase in GLS protein expression (specifically the KGA isoform) allowing the kidney to produce the basic ions that buffer the blood (Laterza et al. 1997; Laterza and Curthoys 2000a,b).
In summary, our findings reveal a novel way of achieving glutamine sensitivity through CFIm25-mediated APA and AS, which is independent of c-Myc/miR-23 mediated regulation. Thus, they provide critical new insight into the molecular mechanism underlying post-transcriptional regulation of glutaminase. These results also illustrate the complex interplay of alternative splicing, APA, and mRNA decay within a single gene unit and exemplify the layered regulation that metabolic enzymes evolved to meet the changing cellular milieu.
MATERIALS AND METHODS
Tissue culture and caspase assays
Both cell lines (HeLa and 293T) were cultured in DMEM containing 10% FBS with penicillin and streptomycin at concentrations of 100 I.U/mL and 100 µg/mL, respectively. The apoptosis assays were carried out using the caspase-Glo 3/7-assay kit from Promega according to the manufacturer's protocol.
siRNA transfection and Western blot analysis
Lipofectamine 2000 (Invitrogen) was used for all transfections as previously described (Wagner and Garcia-Blanco 2002). The Control (Con.) siRNA sequence is available (Wagner and Garcia-Blanco 2002) and the following siRNAs targeting CFIm25 (SASI_Hs01_00146875 [si#1] and SASI_Hs01_00146877 [si#2]) were purchased from Sigma-Aldrich. To knockout GLS, we developed siRNAs specific for either the KGA or GAC 3′UTR. Their sequences were as follows; KGA 5′-GACAUGGAACAGCGGGACU and GAC 5′-CUAUGAAAGUCUCCAACAA. Western blot analysis was conducted as previously described (Albrecht and Wagner 2012) and the following antibodies from Proteintech labs were used in this study: anti-KGA (1:1000 dilution), anti-GAC (1:1000 dilution), and anti-CFIm25 (1:1000 dilution). The GLS antibody (1:2500) and anti-tubulin or anti-GAPDH that was used as the loading control were from Abcam and Ambion, respectively.
RNA isolation and RNA-seq analysis
Total RNA was isolated using TRIzol reagent (Life Sciences) and was submitted to LC Sciences for mRNA sequencing using the Illumina High-Seq 2000, which generated >3e8 mapped reads per sample.
qRT-PCR and miRNA qRT-PCR
For quantitative real time-PCR (qRT-PCR) to detect GLS, the mRNA sample was reverse transcribed using MMLV-RT (Invitrogen) according to the manufacturer's protocol to generate cDNA. qRT-PCR reactions were performed using Stratagene MxPro3000P (Agilent Technologies) and SYBRGREEN (Fermentas). The following amplicons were used to detect total GLS levels: Forward CAACATCAGATGGTGTCATGCTAGAC and Reverse AACAGACACACCCCACAAATCGGGACTG. Specific primers targeting specific regions present only in KGA or only in GAC isoform were also designed and used as follows: KGA specific Forward ACTGGAGATGTGTCTGCACTTCGAAG and Reverse CCAAAGTGCAGTGCTTCATCCATGGGAGTG and GAC specific Forward TTGGACTATGAAAGTCTCCAACAAGAAC and Reverse CCATTCTATATACTACAGTTGTAGAGATGTCCTC. All the reagents used for the miRNA PCR were from Exiqon. The miRCURY LNAUniversal RT microRNA cDNA synthesis kit was used to synthesize cDNA, and ExiLENT SYBR Green Master Mix was used for miRNA qRT-PCR according to the manufacturer's protocol. The primers for the following small RNAs were used: hsa-miR-23a-3p and hsa-miR-100-5p with SNORD48 (hsa) as a control.
3′RACE
Total RNA was isolated with TRIzol (Life Technologies) and was treated with DNase (Promega) according to the manufacturer's protocol. An oligo-(dT)-T7 primer (GGCCGGTAATACGACTCACTATAGT25) was used for cDNA synthesis with MMLV-RT. For the first PCR (20 cycles), the primers used were oligo dT-T7 and GLS-F1 (ACTGGAGATGTGTCTGCACTTCGAAG). Then, 2 µL of the first PCR product was used for the nested PCR (20 cycles) reaction, and the primers used were GLS-F2 (AGATTTGCTTTGTCAGCTATGGACATGGA) and T7 (GGCCTAATACGACTCACTATAG). PCR products were directly sequenced multiple times and also cloned and sequenced using Zero Blunt TOPO (Invitrogen).
Plasmids and RNAi rescue experiment
To generate the RNAi resistant 3× Flag-tagged CFIm25 plasmid, the cDNA containing the CFIm25 ORF wild-type sequence was cloned into pcDNA6 (Thermo Fisher Scientific). This wild-type plasmid was mutagenized by introducing silent mutations in the CFIm25-siRNA target sequence using the QuikChange Site-Directed Mutagenesis Kit (Stratagene) according to the manufacturer's instructions. The GFP ORF was cloned into the same plasmid to serve as a vector control. To perform the RNAi rescue experiment, 1 µg of plasmid was cotransfected with the siRNA for 48 h. For the luciferase assays, each 3′UTR was cloned downstream from the Renilla luciferase gene in the psicheck 2 (Promega) dual luciferase plasmid. HeLa cells were transfected with 100 ng of plasmid and luciferase assays were done using the Dual-Glo Luciferase Assay System (Promega) according to the manufacturer's protocol.
Antagomir and miR-23a mimic experiments
For the mi-R23a experiments, all the controls, mimics, and antagomirs were purchased from Sigma, i.e., hsa-miR-23a HM10406 and hsa miR-23a-3a HSTUD0406.
Statistics
Data were analyzed throughout this manuscript using the Student's independent t-test. The values used were P < 0.05 (*), P < 0.01 (**), and P < 0.001 (***).
DATA DEPOSITION
Raw sequence data have been deposited in the NCBI Gene Expression Omnibus under accession number GSE42420.
ACKNOWLEDGMENTS
We thank members of the Wagner and Shyu laboratories for helpful discussions and Dr. Phillip Carpenter for reviewing the manuscript. This work was supported by a Cancer Prevention and Research Institute of Texas grant to E.J.W. and A.-B.S. (RP100107) and E.J.W. (RP140800); in part by a Department of Defense grant to E.J.W. (W81XWH-11-1-0304); National Institutes of Health grants to A.-B.S. (GM046454), E.J.W. (CA167752 and CA166274); and an endowment from the Houston Endowment, Inc. (A.-B.S.). Work in the Li laboratory is funded by grants from the US Department of Defense (W81XWH-10-1-0501), CPRIT (RP150292), and National Institutes of Health (R01HG007538 and R01CA193466). C.P.M. acknowledges a US Department of Defense Visionary Postdoctoral Fellowship (W81XWH-12-1-0218).
Footnotes
Article published online ahead of print. Article and publication date are at http://www.rnajournal.org/cgi/doi/10.1261/rna.055939.116.
REFERENCES
- Albrecht TR, Wagner EJ. 2012. snRNA 3′ end formation requires heterodimeric association of integrator subunits. Mol Cell Biol 32: 1112–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aledo J, Gomez-Fabre P, Olalla L, Marquez J. 2000. Identification of two human glutaminase loci and tissue-specific expression of the two related genes. Mamm Genome 11: 1107–1110. [DOI] [PubMed] [Google Scholar]
- Beaudoing E, Gautheret D. 2001. Identification of alternate polyadenylation sites and analysis of their tissue distribution using EST data. Genome Res 11: 1520–1526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beaudoing E, Freier S, Wyatt JR, Claverie JM, Gautheret D. 2000. Patterns of variant polyadenylation signal usage in human genes. Genome Res 10: 1001–1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cassago A, Ferreira APS, Ferreira IM, Fornezari C, Gomes ERM, Greene KS, Pereira HM, Garratt RC, Dias SMG, Ambrosio ALB. 2012. Mitochondrial localization and structure-based phosphate activation mechanism of Glutaminase C with implications for cancer metabolism. Proc Natl Acad Sci 109: 1092–1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang TC, Yu D, Lee Y-S, Wentzel EA, Arking DE, West KM, Dang CV, Thomas-Tikhonenko A, Mendell JT. 2008. Widespread microRNA repression by Myc contributes to tumorigenesis. Nat Genet 40: 43–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CYA, Shyu AB. 1995. AU-rich elements: characterization and importance in mRNA degradation. Trends Biochem Sci 20: 465–470. [DOI] [PubMed] [Google Scholar]
- DeBerardinis RJ, Cheng T. 2010. Q's next: the diverse functions of glutamine in metabolism, cell biology and cancer. Oncogene 29: 313–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de la Rosa V, Campos-Sandoval JA, Martín-Rufián M, Cardona C, Matés JM, Segura JA, Alonso FJ, Márquez J. 2009. A novel glutaminase isoform in mammalian tissues. Neurochem Int 55: 76–84. [DOI] [PubMed] [Google Scholar]
- Elgadi KM, Meguid RA, Qian M, Souba WW, Abcouwer SF. 1999. Cloning and analysis of unique human glutaminase isoforms generated by tissue-specific alternative splicing. Physiol Genomics 1: 51–62. [DOI] [PubMed] [Google Scholar]
- Elkon R, Drost J, van Haaften G, Jenal M, Schrier M, Vrielink JA, Agami R. 2012. E2F mediates enhanced alternative polyadenylation in proliferation. Genome Biol 13: R59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman RC, Farh KKH, Burge CB, Bartel DP. 2009. Most mammalian mRNAs are conserved targets of microRNAs. Genome Res 19: 92–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao P, Tchernyshyov I, Chang TC, Lee YS, Kita K, Ochi T, Zeller KI, De Marzo AM, Van Eyk JE, Mendell JT, et al. 2009. c-Myc suppression of miR-23a/b enhances mitochondrial glutaminase expression and glutamine metabolism. Nature 458: 762–765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gruber AR, Martin G, Keller W, Zavolan M. 2012. Cleavage factor Im is a key regulator of 3′ UTR length. RNA Biol 9: 1405–1412. [DOI] [PubMed] [Google Scholar]
- Laterza OF, Curthoys NP. 2000a. Effect of acidosis on the properties of the glutaminase mRNA pH-response element binding protein. J Am Soc Nephrol 11: 1583–1588. [DOI] [PubMed] [Google Scholar]
- Laterza OF, Curthoys NP. 2000b. Specificity and functional analysis of the pH-responsive element within renal glutaminase mRNA. Am J Physiol Renal Physiol 278: F970–F977. [DOI] [PubMed] [Google Scholar]
- Laterza OF, Hansen WR, Taylor L, Curthoys NP. 1997. Identification of an mRNA-binding protein and the specific elements that may mediate the pH-responsive induction of renal glutaminase mRNA. J Biol Chem 272: 22481–22488. [DOI] [PubMed] [Google Scholar]
- Martin G, Gruber AR, Keller W, Zavolan M. 2012. Genome-wide analysis of pre-mRNA 3′ end processing reveals a decisive role of human cleavage factor I in the regulation of 3′ UTR length. Cell Rep 1: 753–763. [DOI] [PubMed] [Google Scholar]
- Masamha CP, Xia Z, Yang J, Albrecht TR, Li M, Shyu A-B, Li W, Wagner EJ. 2014. CFIm25 links alternative polyadenylation to glioblastoma tumour suppression. Nature 510: 412–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayr C, Bartel DP. 2009. Widespread shortening of 3′UTRs by alternative cleavage and polyadenylation activates oncogenes in cancer cells. Cell 138: 673–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oermann EK, Wu J, Guan KL, Xiong Y. 2012. Alterations of metabolic genes and metabolites in cancer. Semin Cell Dev Biol 23: 370–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porter LD, Ibrahim H, Taylor L, Curthoys NP. 2002. Complexity and species variation of the kidney-type glutaminase gene. Physiol Genomics 9: 157–166. [DOI] [PubMed] [Google Scholar]
- Proudfoot NJ. 2011. Ending the message: poly(A) signals then and now. Genes Dev 25: 1770–1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Redis Roxana S, Vela Luz E, Lu W, Ferreira de Oliveira J, Ivan C, Rodriguez-Aguayo C, Adamoski D, Pasculli B, Taguchi A, Chen Y, et al. 2016. Allele-specific reprogramming of cancer metabolism by the long non-coding RNA CCAT2. Mol Cell 61: 520–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rush J, Moritz A, Lee KA, Guo A, Goss VL, Spek EJ, Zhang H, Zha XM, Polakiewicz RD, Comb MJ. 2005. Immunoaffinity profiling of tyrosine phosphorylation in cancer cells. Nat Biotechnol 23: 94–101. [DOI] [PubMed] [Google Scholar]
- Sandberg R, Neilson JR, Sarma A, Sharp PA, Burge CB. 2008. Proliferating cells express mRNAs with shortened 3′ untranslated regions and fewer microRNA target sites. Science 320: 1643–1647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroeder JM, Ibrahim H, Taylor L, Curthoys NP. 2006. Role of deadenylation and AUF1 binding in the pH-responsive stabilization of glutaminase mRNA. Am J Physiol Renal Physiol 290: F733–F740. [DOI] [PubMed] [Google Scholar]
- Shimazu T, Horinouchi S, Yoshida M. 2007. Multiple histone deacetylases and the CREB-binding protein regulate pre-mRNA 3′-end processing. J Biol Chem 282: 4470–4478. [DOI] [PubMed] [Google Scholar]
- Takagaki Y, Seipelt RL, Peterson ML, Manley JL. 1996. The polyadenylation factor CstF-64 regulates alternative processing of IgM heavy chain pre-mRNA during B cell differentiation. Cell 87: 941–952. [DOI] [PubMed] [Google Scholar]
- Tian B, Hu J, Zhang H, Lutz CS. 2005. A large-scale analysis of mRNA polyadenylation of human and mouse genes. Nucleic Acids Res 33: 201–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vander Heiden MG, Cantley LC, Thompson CB. 2009. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science 324: 1029–1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner EJ, Garcia-Blanco MA. 2002. RNAi-mediated PTB depletion leads to enhanced exon definition. Mol Cell 10: 943–949. [DOI] [PubMed] [Google Scholar]
- Wang JB, Erickson JW, Fuji R, Ramachandran S, Gao P, Dinavahi R, Wilson KF, Ambrosio AL, Dias SM, Dang CV, et al. 2010. Targeting mitochondrial glutaminase activity inhibits oncogenic transformation. Cancer Cell 18: 207–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward Patrick S, Thompson Craig B. 2012. Metabolic reprogramming: a cancer hallmark even Warburg did not anticipate. Cancer Cell 21: 297–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wise DR, Thompson CB. 2010. Glutamine addiction: a new therapeutic target in cancer. Trends Biochem Sci 35: 427–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wise DR, DeBerardinis RJ, Mancuso A, Sayed N, Zhang XY, Pfeiffer HK, Nissim I, Daikhin E, Yudkoff M, McMahon SB, et al. 2008. Myc regulates a transcriptional program that stimulates mitochondrial glutaminolysis and leads to glutamine addiction. Proc Natl Acad Sci 105: 18782–18787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia Z, Donehower LA, Cooper TA, Neilson JR, Wheeler DA, Wagner EJ, Li W. 2014. Dynamic analyses of alternative polyadenylation from RNA-seq reveal a 3′-UTR landscape across seven tumour types. Nat Commun 5: 5274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan J, Marr TG. 2005. Computational analysis of 3′-ends of ESTs shows four classes of alternative polyadenylation in human, mouse, and rat. Genome Res 15: 369–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuneva M, Zamboni N, Oefner P, Sachidanandam R, Lazebnik Y. 2007. Deficiency in glutamine but not glucose induces MYC-dependent apoptosis in human cells. J Cell Biol 178: 93–105. [DOI] [PMC free article] [PubMed] [Google Scholar]