

Abstract
Parkinson’s disease (PD) patients have excessive iron depositions in substantia nigra (SN). Neuroinflammation characterized by microglial activation is pivotal for dopaminergic neurodegeneration in PD. However, the role and mechanism of microglial activation in iron-induced dopaminergic neurodegeneration in SN remain unclear yet. This study aimed to investigate the role and mechanism of microglial β-nicotinamide adenine dinucleotide phosphate oxidase 2 (NOX2) activation in iron-induced selective and progressive dopaminergic neurodegeneration. Multiple primary midbrain cultures from rat, NOX2+/+ and NOX2−/− mice were used. Dopaminergic neurons, total neurons, and microglia were visualized by immunostainings. Cell viability was measured by MTT assay. Superoxide (O2·−) and intracellular reactive oxygen species (iROS) were determined by measuring SOD-inhibitable reduction of tetrazolium salt WST-1 and DCFH-DA assay. mRNA and protein were detected by real-time PCR and Western blot. Iron induces selective and progressive dopaminergic neurotoxicity in rat neuron–microglia–astroglia cultures and microglial activation potentiates the neurotoxicity. Activated microglia produce a magnitude of O2·− and iROS, and display morphological alteration. NOX2 inhibitor diphenylene iodonium protects against iron-elicited dopaminergic neurotoxicity through decreasing microglial O2·− generation, and NOX2−/− mice are resistant to the neurotoxicity by reducing microglial O2·− production, indicating that iron-elicited dopaminergic neurotoxicity is dependent of NOX2, a O2·−-generating enzyme. NOX2 activation is indicated by the increased mRNA and protein levels of subunits P47 and gp91. Molecules relevant to NOX2 activation include PKC-σ, P38, ERK1/2, JNK, and NF-ΚBP65 as their mRNA and protein levels are enhanced by NOX2 activation. Iron causes selective and progressive dopaminergic neurodegeneration, and microglial NOX2 activation potentiates the neurotoxicity. PKC-σ, P38, ERK1/2, JNK, and NF-ΚBP65 are the potential molecules relevant to microglial NOX2 activation.
Keywords: Dopaminergic neurodegeneration, Iron, Microglial activation, Neuroinflammation, β-nicotinamide adenine dinucleotide phosphate oxidase 2, Parkinson’s disease, Mechanism
Introduction
Parkinson’s disease (PD) is a common neurodegenerative disease, generally affecting population over 55 years old. Mechanisms for the accelerated loss of dopaminergic neurons in substantia nigra pars compacta (SNpc) remain enigmatic. Neuroinflammation featured by microglial activation is demonstrated by a wealth of evidence as an engine driving progressive dopaminergic neurodegeneration [1]. Microglia are the resident immune cells in the brain. Under physical condition, microglia play protective role on the brain through immunosurveillance and removal of cell debris. However, microglia become sensitive to disturbances in homeostasis of the brain and are readily activated during most neuropathological conditions [2]. In PD, microglia become deleterious and cause neuronal damage when they are activated by lots of exogenous factors, including 1-methyl- 4-phenyl-1, 2,3, 6-tetrahydropyridine [3], lipopolysaccharide [4], paraquat [5], and rotenone [6], and endogenous factors, including neuromelanin [7], aggregated [8] and mutant α-synuclein isoforms [9], 6-hydroxydopamine [10], matrix metalloproteinase-3 [11], μ-Calpain [12], and diesel exhaust particles [13]. Iron serves as an exogenous factor when it is uptaken excessively, and an endogenous factor when its metabolism is disturbed.
Iron is particularly pivotal in neurotransmission, myelination, and neuronal metabolism in the brain. In PD patients, an elevated iron level in substantia nigra (SN) has been documented ([14]). Total iron in SN is increased for 225 % in PD patients [15]. Iron level in SN may reflect a predilection to dopaminergic neurodegeneration and associated movement disorders. Furthermore, biochemical analysis indicates that iron deposition is mainly in SNpc of total SN [16], suggesting an important role of iron deposition in PD since SNpc bears the brunt of PD pathology. Iron deposition in SNpc may be resulted from the excessive uptake [17], abnormal metabolism [17], iron transportation and binding-related gene mutations [18], and compromised blood–brain barrier offering an entrance for iron from peripheral to central system [19].
In living organisms, iron is present as either reduced Fe2+ or oxidized Fe3+ state. In normal subjects, the ration of Fe3+:Fe2+ in SNpc is 2:1 [20]. However, the ratio is shifted to 1:2 in PD patients [20]. Iron accumulation in PD can be contributed by the changes of a number of iron-related proteins [21], among which, ferritin levels have been found to be decreased in postmortem PD brains [22, 23]. We also find that ferritin levels in the serum of PD patients are significantly declined comparing with control subjects (unpublished data). Ferritin, particularly, ferritin heavy chain performs the major function of converting higher toxic Fe2+ into lower toxic Fe3+ for the rapid absorption and utilization of iron. Accordingly, reduction of ferritin decreases its competence of converting Fe2+ into Fe3+, leading to the increased ration of Fe2+:Fe3+ in PD patients comparing with normal individuals. The general redox state of the cell may have an overall bearing on the ratios of Fe3+:Fe2+ in the cell. In our previous study, we found that aggregated α-synuclein produced a magnitude of O2·− [24], which can provoke Haber-Weiss reaction, causing excessive generation of Fe2+ [25].
Based on the findings that excessive Fe2+ deposits in SNpc and neuroinflammation featured by microglial activation servers as a driving force of dopaminergic neurodegeneration [1], we hypothesize that when dopaminergic neurons in SNpc are degenerated, the dead neurons may release Fe2+ into extracellular space and interact with surrounding cells, including dopaminergic neurons, microglia, and astroglia. Will Fe2+ damage the remaining dopaminergic neurons? If yes, will microglia, the major players of neuroinflammation, participate in Fe2-mediated dopaminergic neurotoxicity? What are the underlying mechanisms? In this study, we investigated the role and mechanism of microglial activation in Fe2+-mediated selective and progressive dopaminergic neurotoxicity using multiple well-established primary midbrain cultures [26], and particularly explored the key role and relevant molecules of β-nicotinamide adenine dinucleotide phosphate oxidase 2 (NOX2), a superoxide (O2·−)-generating enzyme, in microglial activation by using NOX2 knock out (NOX2−/−) mice and inhibitor.
Methods
Reagent
Materials relating cell cultures are from Invitrogen (USA). Polyclonal anti-tyrosine hydroxylase (TH), anti-complement receptors 3 (OX-42), anti-neuron-specific nuclear protein (Neu N), anti-P38, anti-extracellular signal-regulated kinase1/2 (ERK1/2), anti-c-Jun N-terminal kinase (JNK), anti-nuclear factor-ΚBP65 (NF-ΚB P65), and anti-protein kinase C-σ (PKC-σ) antibodies are from R&D Systems (USA). Vectastain ABC kit and biotinylated secondary antibodies are from Vector Laboratories (USA). FeCl2 is from Alfa Aesar (UK). Fluorescence probe 2′,7′-dichlorodi-hydrofluorescein (DCFH-DA), Leu-Leu methyl ester hydrobromedia (LME), superoxide dismutase (SOD), 2-(4 lodophenyl)-3-(4-nitrophenyl)-5-(2,4-disulfophenyl)-2H-tetrazo1ium (WST-1), diphenylene iodonium (DPI), and dimethyl sulfoxide (DMSO) are from Sigma-Aldrich (USA). 3-(4,5-dimethyl-thiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) is from Dojindo (Japan). SYBR green PCR master mix is from Applied Biosystems (UK).
Animals
SD rats are from Peking Vital River Laboratory and Centre of Experimental Animal of Peking University. C57 BL/6J (NOX2 wild type, NOX2+/+) and NOX2−/− mice are from Jackson Laboratories (Bar Harbor, Maine, USA). Breeding schedules were designed to achieve accurately timed pregnancy of 14±0.5 days for rats and 13±0.5 days for mice. Housing, breeding, and experimental use of the animals were performed in strict accordance with the National Institutes of Health guidelines.
Primary Midbrain Neuron–Microglia–Astroglia Cultures
Rat primary midbrain neuron–microglia–astroglia cultures were prepared from the brains of embryonic day 14±0.5 days of SD rats and 13±0.5 days of mice. Ventral midbrain tissues were removed and dissociated. Cells were seeded at culture plate precoated with poly-D-lysine and maintained at 37 °C incubator with humidified atmosphere of 5 % CO2 and 95 % air. Seven-day cultures were used for treatment. The composition of the cultures included 48 % astroglia, 11 % microglia, and 40 % neurons, in which 2–3 % was TH-ir dopaminergic neurons. Immunostainings were performed 7 days after treatment (see Supplemental Material, p. 1)
Primary Midbrain Neuron–Astroglia Cultures
Rat primary midbrain neuron–astroglia cultures were obtained by suppressing microglial proliferation with 1.5 mM LME 24 h after seeding neuron–microglia–astroglia cultures. Three days later, cultures were changed back to maintenance medium and used for treatment 7 days after seeding. The composition of the cultures included 54 % astroglia, 1 % microglia, and 45 % neurons. Immunostainings were performed 7 days after treatment [9].
Primary Microglial Cultures
Primary microglial cultures were prepared from the whole brains of pups from 1-day-old SD rats, NOX2+/+ and NOX2−/− mice. Briefly, brain tissues were triturated after removing meninges and blood vessels. Cells were seeded in a culture flask. After a confluent monolayer of glia were obtained, microglia were shaken off and seeded for measuring extracellular superoxide (O2·−), intracelluar reactive oxygen species (iROS), gene and protein expressions of NOX2 subunits P47 and gp91, PKC-σ, P38, ERK1/2, JNK, and NF-ΚBP65 (see Supplemental Material, p. 1).
Dopaminergic Cell Line
Dopaminergic cell line (MN9D) were seeded at 1×107 in a 25 cm2 culture flask with DMEM/F12 added with 5 mL fetus bovine serum. After a confluent monolayer of dopaminergic cell line had been obtained, 0.125 % pancreatin/0.01 % EDTA was used to detach the cell and then seeded in a 96-well plate at 3×104/well for 24 h. Cell viability was detected by using MTT when cells were treated with 5, 25, and 100 μM Fe2+ for 24 h.
Treatment
One millimolar Fe2+ was freshly prepared as a stock solution with PBS and diluted to the desired concentrations of 5, 25, and 100 μM in the treatment medium. Multiple cultures and dopaminergic cell lines were treated with vehicle and Fe2+ at 5, 25, and 100 μM in a final volume of 1 mL/well.
Immunostainings
Formaldehyde-fixed neuron–microglia–astroglia cultures were treated with 1 % hydrogen peroxide followed by sequential incubation with blocking solution, primary and biotinylated secondary antibodies and ABC reagents. Color was then developed and images were recorded. TH-ir dopaminergic neurons in a well, Neu N-ir total neurons and OX-42-ir microglia in nine representative areas per well were visualized under microscope at ×100 magnification (see Supplemental Material, p. 2).
Detection of Cell Viability
Dopaminergic cells was seeded followed by the treatment with 5, 25, and 100 μM Fe2+ and incubation for 24 h. MTT was then added and incubated for 6 h. Finally, DMSO was added to dissolve the formazan into a purple solution, which absorbance was quantified at wavelength of 570 nm. The formazan production was measured by the increased absorbance.
O2− Assay
Rat primary microglial cultures were seeded for 24 h, washed two times with Hank’s balanced salt solution (HBSS), received Fe2+ treatment followed by WST-1 in HBSS with or without SOD. The absorbance at 450 nm was read for a period of 40 min. O2·− production was measured by the increased absorbance in 30 min and expressed as the percentage of control group (see Supplemental Material, p. 2).
iROS Assay
Rat primary microglial cultures were seeded for 24 h, washed for two times with HBSS, received DCFH-DA, and were incubated for 2 h followed by Fe2+ treatment. Fluorescence intensity was measured at 485 nm for excitation and 530 nm for emission. iROS production was measured by the increased absorbance in 2 h (see Supplemental Material, p. 3).
RT and Real-Time PCR
Fe2+-induced gene expressions of microglial NOX2 subunits P47 and gp91, PKC-σ, P38, ERK1/2, JNK, and NF-ΚBP65 after Fe2+ treatment were detected by RT and real-time PCR. The sequences of the forward and reverse primers were as followed: β-actin: 5′-GAGACCTTCAACACCCCAGC-3′ (F), 5′-ATGTCACGCACGATTTCCC-3′ (R); P47: 5′-ACCT GAAGCTGCCCAATGAC-3′ (F), 5′-ATGGCCCGATAGGT CTGAAG-3′ (R); gp91: 5′-GACAGATTTGCTCTGCACAAGGT-3′ (F), 5′-AGGAGAGGTTGTGTGCACCATAG-3′ (R); PKC-σ: 5′-TCCCCGCCATCCGAGCAACA-3′ (F), 5′-TTGCGGCATCTCTCCGCCAG-3′ (R); P38 5′-TGGCTCGGCACACTGATGAC-3′ (F), 5′-CCGGTCAACAGCTCAGCCAT-3′ (R); ERK 1/2: 5′-GACCCTGAGCACGACCACAC-3′ (F), 5′-ATAGGCCGGTTGGAGAGCAT-3′ (R); JNK: 5′-CCCACCACCAAAGATCCCTG-3′ (F), 5′-TGACAGACGGCGAAGACGAC-3′ (R); NF-ΚBP65: 5′-GCCGGCCTCGGGACAAACAG-3′ (F), 5′-TCGCCAGAGGCGGAAATGCG-3′ (R). Total RNA was isolated with Trizol reagent, followed by purification, reversed transcription with MuLV reverse transcriptase and oligo-dT primers and real-time PCR analysis using SYBR green PCR master mix. The relative differences in expressions between groups were determined using cycle time (Ct) values as follows: the Ct values for the genes of interest were first normalized with β-actin of the same sample, then the relative differences between control and Fe2+-treated groups were calculated and expressed as relative increases, setting control as 100 %. Assuming that the Ct value was reflective of the initial starting copy and that there was 100 % efficiency, a difference of one cycle was equivalent to a twofold difference in starting copy. Standard curve analysis was performed and used for the calculation.
Western Blot
Primary microglial cultures treated with Fe2+ were detached by scraping in sampling buffer. NOX2 subunits P47 and gp91, PKC-σ, P38, ERK1/2, JNK, and NF-ΚBP65 were isolated via acid extraction adjusted to equal protein concentrations and separated by 4–12 % polyacrylamide–0.1 % SDS miningels, then transferred onto a PVDF membrane. Blots were incubated with primary antibody, then probed with a horseradish peroxidase-conjugated secondary antibody. Detection was performed by using ECL kit.
Statistical Analysis
Three wells were used in each treatment, which was considered as an independent experiment. Standard errors were calculated from three independent experiments. Results were expressed as the mean ± SE. Statistical significance was assessed with an analysis of one-way ANOVA, with the freedom representing the numbers of experiments minus one, followed by Bonferroni’s t test using SPSS20.0 software. A value of P <0.05 was considered statistically significant.
Results
Fe2+ Causes Progressive and Selective Dopaminergic Neurotoxicity
In rat primary midbrain neuron–microglia–astroglia cultures treated with Fe2+ at 5, 25, and 100 μM, the number of TH-ir dopaminergic neurons is significantly reduced 1, 4, and 7 day (s) after Fe2+ treatment, in a time-dependent manner (Fig. 1a). Morphologically, dopaminergic neurons show smaller cell bodies, reduced cytoplasmic TH staining, and fewer and shorter dendrites 7 days after Fe2+ treatment, in a dose-dependent manner comparing with control group (Fig. 1b).
Fig. 1.
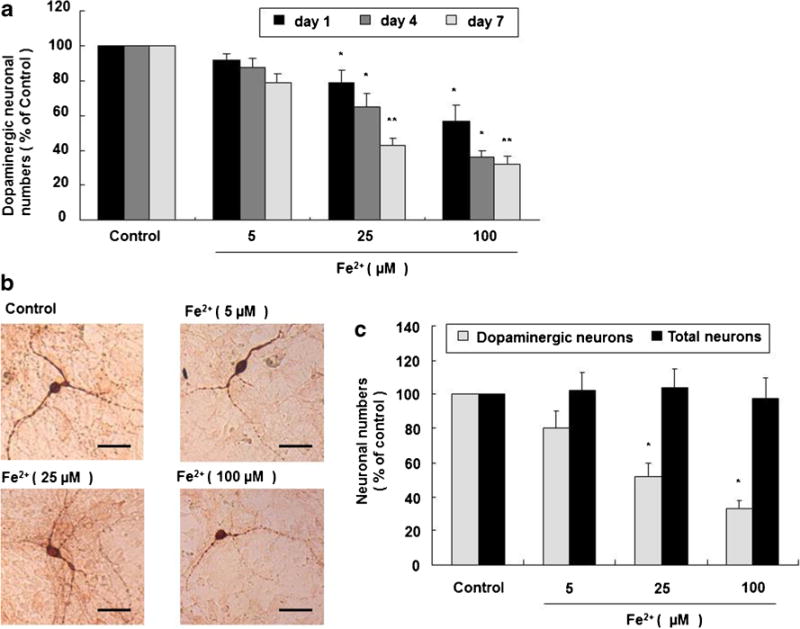
Fe2+ causes selective and progressive dopaminergic neurotoxicity. Rat primary midbrain neuron–microglia–astroglia cultures were seeded in a 24-well plate at 5×105/well and treated with 5, 25, and 100 μM Fe2+. One, 4, and 7 day (s) after treatment, the number of dopaminergic neurons was visualized after TH staining. Results are expressed as a percentage of the vehicle-treated control group and are the mean ± SE from three independent experiments in triplicate (a). *P <0.05 and **P <0.01 for Fe2+-treated groups vs. vehicle-treated control group. Representative microscopic images are shown for TH-ir dopaminergic neurons (TH staining, ×100 magnification). Scale bar = 100 μm (b). Rat primary midbrain neuron–microglia–astroglia cultures were seeded in a 24-well plate at 5×105/well and treated with 5, 25, and 100 μM Fe2+. Seven days later, the numbers of dopaminergic neurons and total neurons were visualized after TH and Neu N staining, respectively. Results are expressed as a percentage of the vehicle-treated control group and are the mean ± SE from three independent experiments in triplicate (c). *P <0.05 for TH-ir dopaminergic neurons vs. Neu N-ir total neurons
In rat primary midbrain neuron–microglia–astroglia cultures treated with Fe2+ at 5, 25, and 100 μM for 7 days, Neu N-ir total neurons are largely preserved comparing with TH-ir dopaminergic neurons (Fig. 1c).
Microglia Potentiates Fe2+-Induced Dopaminergic Neurotoxicity
Dopaminergic cell line was treated with 5, 25, and 100 μM Fe2+ for 24 h and cell viability was detected by MTT assay. Cell viabilities in 25 and 100 μM Fe2+-treated groups are strikingly decreased comparing with control group (Fig. 2a). Rat primary midbrain neuron–microglia–astroglia and neuron–astroglia cultures were simultaneously treated with Fe2+ at 5, 25, and 100 μM. Seven days later, the loss of TH-ir dopaminergic neurons in rat primary neuron–astroglia cultures is remarkably spared comparing with midbrain neuron–microglia–astroglia (Fig. 2b).
Fig. 2.
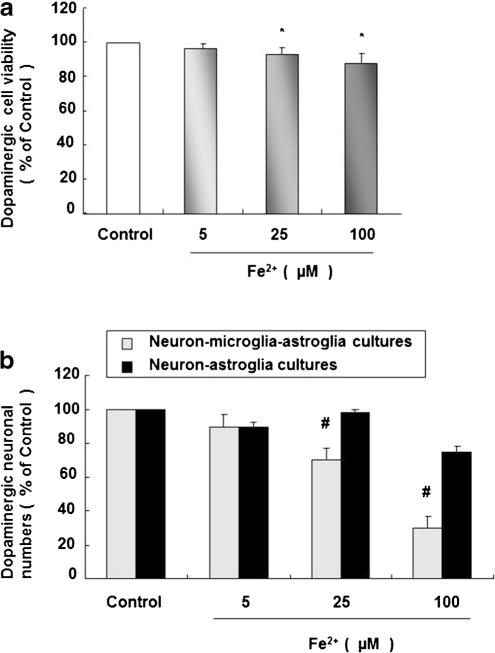
Microglia facilitates Fe2+-induced dopaminergic neurotoxicity. Dopaminergic cell line was seeded in a 96-well plate at 3×104/well for 24 h followed by the Fe2+ treatment with 5, 25, and 100 μM. Twenty-four hours later, cell viability was detected by MTT assay (a). Rat primary midbrain neuron–microglia–astroglia cultures and neuron–astroglia cultures were seeded in a 24-well plate at 5×105/well and treated with Fe2+ at 5, 25, and 100 μM. Seven days later, the number of dopaminergic neurons was visualized after TH staining (b). Results are expressed as a percentage of the vehicle-treated control group and are the mean ± SE from three independent experiments in triplicate. *P <0.05 for Fe2+-treated groups vs. vehicle-treated control group, #P <0.05 for neuron–microglia–astroglia cultures vs. neuron–astroglia cultures
Fe2+ Elicits Progressive Microglial Activation
In rat primary microglia cultures, 25 and 100 μM Fe2+ induces notable generation of extracellular O2·− 30 min later (Fig. 3a) and marked production of iROS 2 h later (Fig. 3b). In rat primary midbrain neuron–microglia–astroglia cultures, numbers of OX-42-ir microglia are prominently enhanced 1, 4, and 7 day (s) after Fe2+ treatment at 25 and 100 μM comparing with control group (Fig. 3c). Morphologically, activated microglia are indicated by the enlarged cell bodies, irregular shape, and intensified OX-42 staining (Fig. 3d).
Fig. 3.
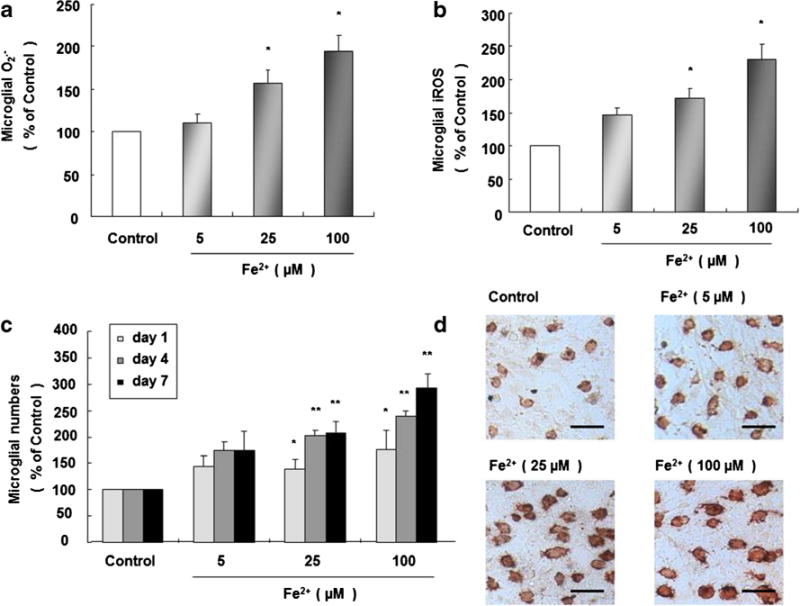
Fe2+ causes sustained microglial activation. Rat microglia cultures were seeded in a 96-well plate at 1×105/well for 24 h, and 5, 25, and 100 Fe2+-induced microglial productions of extracellular O2·− (a) and iROS (b) were determined 30 min and 2 h later. Rat primary midbrain neuron–microglia–astroglia cultures were seeded in a 24-well plate at 5×105/well, and treated with Fe2+ at 5, 25, and 100 μM. One, 4, and 7 day (s) after the treatment, the number of microglia was visualized and morphology was observed after OX-42 staining (c). *P <0.05 and **P <0.01 for Fe2+-treated groups vs. vehicle-treated control group. Representative microscopic images of microglia are shown (OX-42 staining, ×100 magnification) scale bar =100 μm (d)
NOX2−/− Mice Are More Resistant to Fe2+-Induced Dopaminergic Neurotoxicity Due to Less Microglial Activation
Primary microglia cultures derived from NOX2+/+ and NOX2−/− mice were treated with Fe2+ at 25 and 100 μM. Microglia derived from NOX2+/+ mice robustly elevate the levels of O2·− 30 min later (Fig. 4a) and iROS 2 h later (Fig. 4b), respectively, comparing with that derived from NOX2−/− mice. Primary midbrain neuron–microglia–astroglia cultures derived from NOX2+/+ and NOX2−/− mice were treated with Fe2+ at 25 and 100 μM. Seven days later, the number of activated microglia in NOX2+/+ mice is evidently increased comparing with that in NOX2−/− mice (Fig. 4c), and the number of dopaminergic neurons in NOX2+/+ mice is prominently decreased comparing with that in NOX2−/− mice (Fig. 4d). These results indicate that the number of microglia and microglial productions of O2·− and iROS in NOX2−/− mice were less than that in NOX2+/+ mice after Fe2+ treatment, accordingly, dopaminergic neurons derived from NOX2−/− mice are more resistant to Fe2+-induced neurotoxicity than that from NOX2+/+ mice.
Fig. 4.
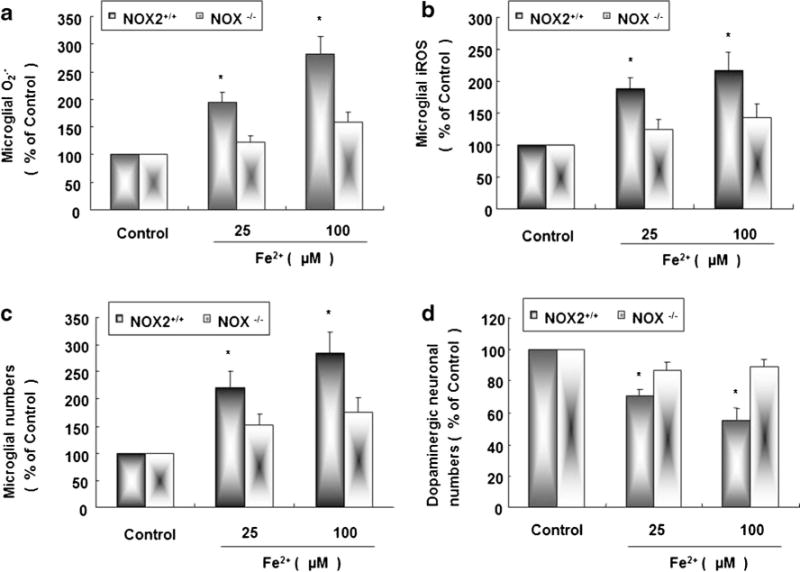
NOX2−/− mice are more resistant to Fe2+-induced dopaminergic neurotoxicity due to the less microglial activation. Microglial cultures derived from NOX2+/+ and NOX2−/− mice were seeded in a 96-well plate at 1×105/well for 24 h and 25 and 100 Fe2+-induced microglial production of extracellular O2·− (a) and iROS (b) were measured 30 min and 2 h after Fe2+ treatment, respectively. Primary midbrain neuron–microglia–astroglia cultures prepared from NOX2+/+ and NOX2−/− mice were seeded in a 24-well plate at 5×105/well and then treated with 25 and 100 μM Fe2+ for 7 days, and the numbers of microglia (c) and dopaminergic neurons (d) were visualized after OX-42 and TH staining, respectively. Results are expressed as a percentage of the vehicle-treated control group and are the mean ± SE from three independent experiments in triplicate. *P <0.05 for NOX2+/+ mice vs. NOX2−/− mice
Microglial NOX2 Inhibitor Inhibits Fe2+-Induced Microglial Activation and Protects Dopaminergic Neuronal Death
In rat primary microglia cultures, the levels of O2·− and iROS are remarkably enhanced 30 min and 2 h after 100 μM Fe2+ treatment, respectively. Pretreatment with DPI, a potential NOX2 inhibitor, at 0.01 and 0.1 μM 30 min before Fe2+ stimulation conspicuously decreases the levels of O2·− and iROS; 0.1 μM DPI alone shows no significant effect on the productions of both iROS and O2·− (Fig. 5a, b).
Fig. 5.
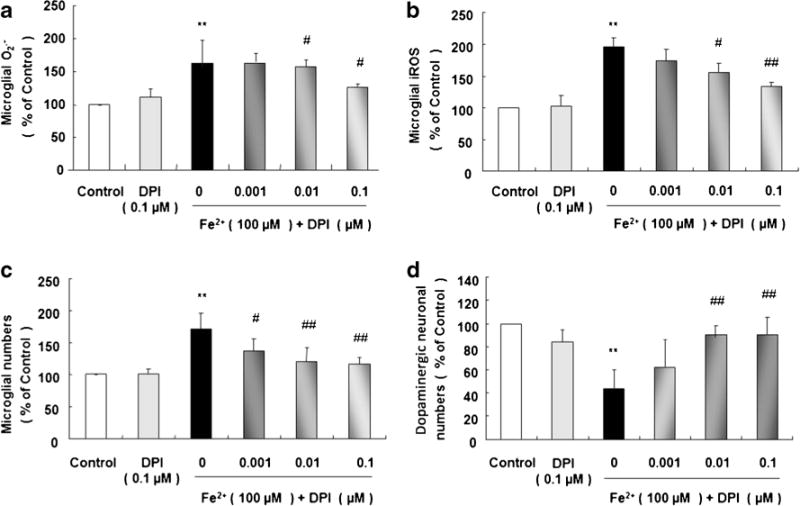
Microglial NOX2 inhibitor, DPI, decreases Fe2+-induced microglial activation and protects dopaminergic neurons. Rat microglia cultures were seeded in a 96-well plate at 1×105/well for 24 h. Effect of NOX2 inhibitor DPI (0.001, 0.01, and 0.1 μM, added 30 min before Fe2+ treatment) on 100 μM Fe2+-induced production of extracellular O2·− (a) and iROS (b) were measured 30 min and 2 h later, respectively. Rat primary midbrain neuron–microglia–astroglia cultures were seeded in a 24-well plate at 5×105/well and treated with Fe2+ at 5, 25, and 100 μM. Seven days later, the numbers of microglia (c) and dopaminergic neurons (d) were visualized after OX-42 and TH staining, respectively. Results are expressed as a percentage of the vehicle-treated control group from three independent experiments in triplicate. **P <0.01 for 100 μM Fe2+-treated group vs. vehicle-treated control group; #P <0.05 and ##P <0.01 for DPI pre-treatment groups vs. Fe2+-treated group
In rat primary midbrain neuron–microglia–astroglia cultures, the number of microglia is greatly enhanced and TH-ir dopaminergic neurons is highly reduced 7 days after 100 μM Fe2+ treatment. Pretreatment with DPI at 0.01 and 0.1 μM significantly decreases the number of activated microglia (Fig. 5c) and restores dopaminergic neuronal survival (Fig. 5d); 0.1 μM DPI alone presents no significant effect on the numbers of microglia and dopaminergic neurons (Fig. 5c, d).
Fe2+ Enhances Expressions of NOX2 Cytoplasm Subunit P47 and Cell Membrane Subunit gp91
In rat primary microglia cultures treated with Fe2+ at 25 and 100 μM, mRNA levels of P47 and gp91 are observably enhanced 15 min later (Fig. 6a) and protein levels of P47 and gp91 were markedly increased 20 min later comparing with control group (Fig. 6b and c).
Fig. 6.
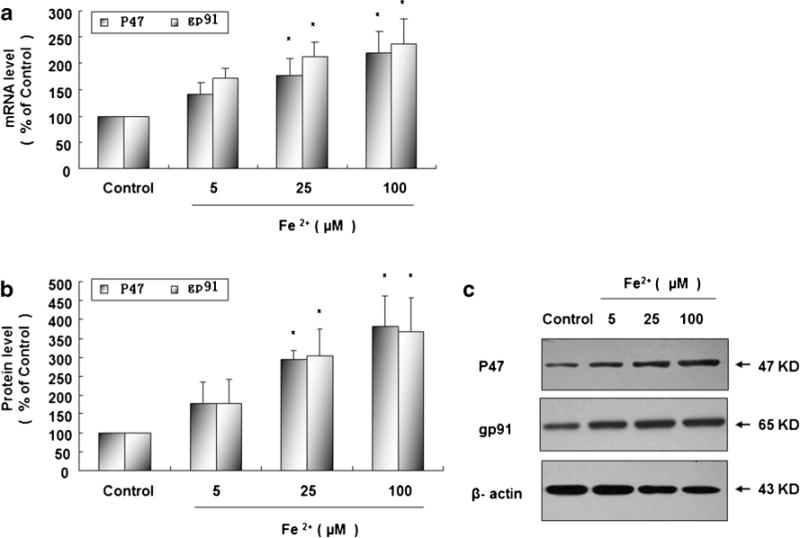
Fe2+ enhances mRNA and protein expressions of NOX2 cytoplasm subunit P47 and cell membrane subunit gp91. Rat microglia cultures were seeded in a 6-well plate at 2×106/well for 24 h and then treated with Fe2+ at 5, 25, and 100 μM. Fifteen minutes later, mRNA expressions of NOX2 cytoplasm subunit P47 and cell membrane subunit gp91 were measured by real-time PCR (a); 20 min later, protein expressions of P47 and gp91 were measured by Western blot (b). Results are expressed as a percentage of the vehicle-treated control group and are the mean ± SE from three independent experiments in triplicate. *P <0.05 for Fe2+-treated group vs. vehicle-treated control group. Representative autographs are shown (c)
Fe2+ Elevates Expressions of PKC-δ, P38, ERK1/2, JNK, and NF-κBP65
Rat primary microglia cultures were treated with Fe2+ at 5, 25, and 100 μM. Fe2+ causes significant expressions of mRNA and protein of following factors at different time points: PKC-δ at 5 min (Fig. 7a, f), mRNA of P38, ERK1/2, and JNK at 5 min (Fig. 7b–d), phosphorylation of P38, ERK1/2, and JNK at 10 min (Fig. 7b–d) and NF-κBP65 at 3 h (Fig. 7e, f). mRNA and protein levels of P38, ERK1/2, and JNK are significantly increased upon Fe2+ stimulation at 25 and 100 μM (Fig. 7b, c, d, and g). mRNA level of JNK is also dramatically enhanced after exposure to 5 μM Fe2+ (Fig. 7d).
Fig. 7.
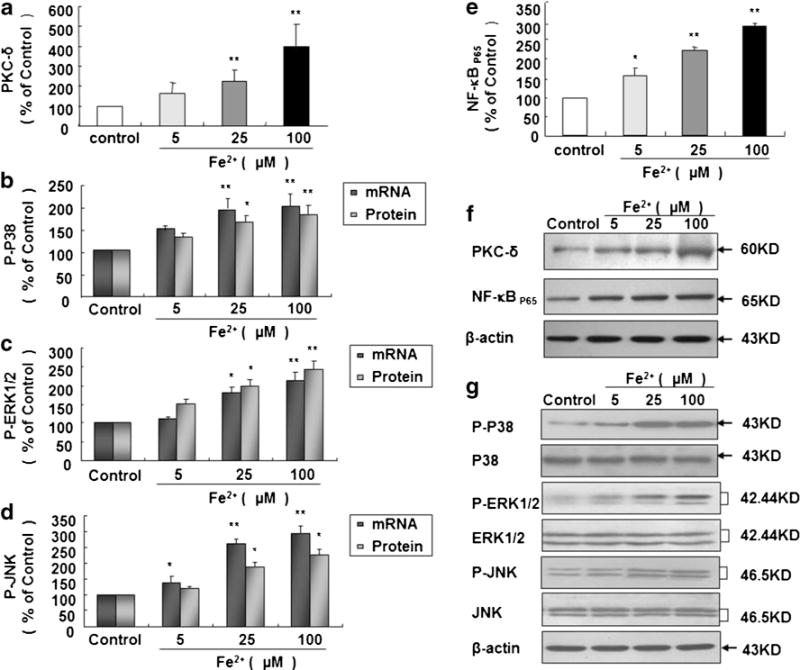
Fe2+ enhances expressions of PKC-δ, P38, ERK1/2, JNK, and NF-κB65. Rat microglia cultures were seeded in a 6-well plate at 2×106/well for 24 h and then treated with Fe2+ at 5, 25, and 100 μM. Five minutes later, PKC-δ level was detected by Western blot (a), and mRNA levels of P38 (b), ERK1/2 (c) and JNK (d) were detected by real-time PCR; 10 min later, phosphorylation levels of P38 (b), ERK1/2 (c), and JNK (d) were detected by Western blot; 3 h later, NF-κBP65 level was detected by Western blot (e). Results are expressed as a percentage of the vehicle-treated control group and are the mean ± SE from three independent experiments in triplicate. *P <0.05 and **P <0.01 for Fe2+-treated group vs. vehicle-treated control group. Representative autographs are shown (f, g)
Discussion
Iron transport and storage is a tightly regulated process, however, elevated exposure to extracellular iron in either normal condition or PD brain are reported. In normal condition, the major iron transporter protein in the body is transferrin, however, only about 30 % of all circulating transferrin units are occupied by iron [27]. Meanwhile, iron can bind to a series of small ligands with low molecular weight, such as citrate and ascorbate ions, released from astrocytes, which is called non-transferrin bound iron (NTBI). Investigator finds the presence of NTBI in the extracellular fluids [28] and the affinity of iron to small ligands is considerably smaller than that to transferrin [29], leading to a proportion of free iron in the extracellular space. Moreover, ferroportin, which mediates the export pathway and allows ferrous iron to be transported out of the cell [30], has been identified in both neurons [31] and astrocytes [32], contributing to ferrous iron in the extracellular space. Furthermore, neurons are thought to be devoid of ferritin in SNpc, in which more iron is exported instead of stored inside neurons [28]. In pathological condition of PD, it is characterized by the loss of dopaminergic neurons in SNpc with accompanying neuromelanin pigment in neuropil and pigment-laden activated macrophages [33], suggesting that neuromelanin is released from dead dopaminergic neurons. Additionally, several studies have demonstrated the existence of extracellular Lewy bodies, the pathological hallmark of PD, and aggregates immunoreactive to α-synuclein in SNpc [2, 34], indicating that aggregated α-synuclein is released from dead dopaminergic neurons. Interestingly, iron depositions have been found in Lewy bodies in PD brains [35], implying that iron may be released from dead dopaminergic neurons together with aggregated α-synuclein.
Iron has been shown to be toxic on dopaminergic neurons [36], however, most of studies mainly focus on iron-induced direct neurotoxicity [37], and less involved the role of glia, particularly microglia, the major player of neuroinflammation. Will iron in the extracellular space interact with surrounding microglia and propagate dopaminergic neurotoxicity? Here, we investigated the role and underlying mechanism of microglial activation in Fe2+-induced selective and progressive dopaminergic neurodegeneration.
Rat primary midbrain neuron–microglia–astroglia cultures with the composition similar to human is a well-established and useful tool for investigating the mechanism of dopaminergic neurodegeneration in PD. We firstly found that Fe2+ induced progressive dopaminergic neurotoxicity because the number of dopaminergic neurons was gradually abolished with time extending after Fe2+ treatment. Furthermore, Fe2+ caused damage to the morphology of dopaminergic neurons indicated by the shrinking cell bodies, reduced cytoplasmic TH staining and fewer and shorter dendrites. Thus, Fe2+ induces progressive dopaminergic neurotoxicity.
Neu N is a marker of all neurons. However, it is mainly used for evaluating non-dopaminergic neurons since dopaminergic neurons only account for 2–3 % of total neurons in SNpc. While Fe2+ elicited dramatic dopaminergic neurotoxicity in rat primary midbrain neuron–microglia–astroglia cultures, we failed to see the similar neurotoxicity in Neu N-ir total neurons. Hence, Fe2+-elicited neurotoxicity is selective to dopaminergic neurons, supporting a close correlation between iron deposition in SNpc and PD [38].
We then tested whether Fe2+-induced neurotoxicity was due to its direct effect on dopaminergic neurons or indirect effect via microglial activation. We observed that Fe2+ slightly decreased cell viability, implying that Fe2+ is mildly toxic to dopaminergic neurons and glia may potentiate the neurotoxicity. Which sort of glia, microglia or astroglia, is critical for Fe2+-potentiated neurotoxicity? Based on the finding that Fe2+ produces severer loss of dopaminergic neurons in rat neuron–microglia–astroglia cultures than that in neuron–astroglia cultures, we figure out a pivotal role of microglia in the enhanced neurotoxicity. Thus, microglia are promoters of Fe2+-induced dopaminergic neurotoxicity.
Strong evidences indicate that activated microglia secrete a myriad of neuroinflammatory factors, including ROS, nitrogen species, cytokines, prostaglandins, and chemokines, etc. [39, 40], which causes subsequent neurotoxicity. How do microglia potentiate dopaminergic neuronal damage after Fe2+ exposure? Here, we found that microglia produced a large amount of neurotoxic ROS, including extracellular O2·− and iROS, in response to Fe2+ exposure. Extracellular O2·− is the earliest factor produced after activation of NOX2, the key O2·−-producing enzyme in brain microglia [41]. One of the potential mechanisms mediating toxic effect of O2·− is through forming strong toxic intermediate like peroxinitrite by NO [42]. O2·− released from microglia displays autocrine effect to further enhance the expression of neuroinflammatory factors, including TNF-α [43] and PGE2 [8] through formation of hydrogen peroxide [44, 45]. Thus, early O2·− generation by activated NOX2 is critical in Fe2+-mediated dopaminergic neurotoxicity. iROS serves as the second messenger in neuroinflammatory event [26]. Among the multiple sources of iROS, NOX2-generating O2·− contributes more than 60 % of total iROS [46]. iROS triggers NF-ΚB expression and produces downstream neuroinflammatory factors. The production of each neuroinflammatory factor may not be sufficient, however, neuroinflammatory factors may work together and amplify the toxic effect of each other, potentiating dopaminergic neurotoxicity (data not shown). Thus, iROS greatly contributes to Fe2+-provoked dopaminergic neurotoxicity. Morphologically, Fe2+ causes continuous microglial activation demonstrated by the increased number of microglia with time extending, enlarged cell bodies, irregular cell shape, and intensified OX-42 staining. Accordingly, we conclude that microglial activation serves as an engine driving dopaminergic neurodegeneration and runs through the whole degenerative process.
NOX family is composed of NOX1–5 and dual oxidase 1–2 [41]. NOX2 is mainly distributed in SN, striatum, hippocampus, and cortex [47]. In these regions, scavenger receptor and macrophage antigen-1 receptor that can identify and remove external subjects are mainly expressed in microglia but not in astroglia and neurons. Upon varies of stimuli, O2·− generated by NOX2 in microglia are far more than that in astroglia and neurons, for example, in LPS in vitro model, it is NOX2 activation in microglia but not in astroglia and neurons, that elicits dopaminergic neurotoxicity; microglia derived from NOX2+/+ mice produce more O2·− and neuroinflammatory factors than that from NOX2−/− mice [46, 48]. Results from this investigation demonstrate that microglia are the major sources of NOX2-produced O2·− after stimulation and Fe2+-induced progressive and selective dopaminergic neurodegeneration is microglial NOX2-dependent. Additionally, a potential NOX2 inhibitor, DPI, is used for confirming the role of NOX2 in Fe2+-produced dopaminergic neurodegeneration. In addition to the finding similar to that reported by Casarejos showing that DPI decreases paraquat-elicited O2·− production ([49]), further decline of iROS, reduction of activated microglia, and subsequent restoration of dopaminergic neurons by DPI elucidate that Fe2+-potentiated dopaminergic neurotoxicity is in a microglial NOX2-dependent manner.
We then investigated the potential molecular mechanism of Fe2+-induced NOX2 activation. In all NOX2 subunits, cytoplasm subunit P47 and the membrane subunit gp91 are pivotal for NOX2 activation [50]. In resting state, they are located in cytoplasm and membrane separately. Upon stimulation, P47 translocates to membrane and combines with gp91, during which O2− was produced.
Involvement of subunits of P47 and gp91 in NOX2 activation after Fe2+ treatment was illustrated by the strikingly upregulated gene expressions and subsequent protein expressions of both P47 and gp91 subunits. These results reveal the participation of microglial NOX2 in Fe2+-elicited dopaminergic neurotoxicity.
Multiple signaling pathways, including PKC, MAPKs, and NF-ΚB are reported to be associated with neurodegeneration and neuroinflammation [51]. PKC family is greatly involved in PD, among which PKC-δ is pivotal in microlgial NOX2 activation [52]. PKC-δ increases P47 expression and causes translocation of P47 from cytoplasm to membrane [52]. Inhibition of 70 % PKC-δ expression decreases NOX2 activities for more than 90 % [53]. ROS is not only inhibited by DPI, but also by GF109203X, a PKC-δ inhibitor [54]. Hence, PKC-δ may be a potential molecule for microglial NOX2 activation. We found that PKC-δ level was significantly enhanced as early as 5 min after Fe2+ exposure, implying that PKC-δ is an earliest biomarker relevant to microglial NOX2 activation induced by Fe2+.
MAPKs, including P38, ERK1/2, and JNK, are activated by environmental factors and inflammagens [24, 55], and participate in the expressions of numerous proteins highly relating to neuroinflammation, and particularly, are closely associated with NOX2 activation [56]. We found that both mRNA and protein expressions of P38, ERK1/2, and JNK were significantly increased upon Fe2+ stimulation for 5 and 10 min respectively, which were prior to that of P47 and gp91, disclosing that MAPKs are the signaling molecules of Fe2+−induced microglial NOX2 activation.
NF-ΚB is a nuclear transcription factor mediating the production of neuroinflammatory factors [57]. NF-ΚB activation can be inhibited by DPI [57], implying a potential link between NF-ΚB and NOX2. Additionally, NF-κB has crosstalks with MAPKs and PKC [58]. We found that NF-ΚBp65 level was significantly elevated 3 h after Fe2+ treatment, thus speculate that NOX2-generated O2·− after Fe2+ treatment enters microglia and enhances iROS level, which triggers expression of NF-ΚBp65 and generations of neuroinflammatory factors, and propagates dopaminergic neurodegeneration.
Although mRNA and protein expressions of NOX2 subunits P47 and gp91, PKC-σ, P38, ERK1/2, JNK, and NF-kBp65 are found to be involved in NOX2 activation in this rodent in vitro model, genetics and gene expression of rodents may differ from humans. Thus, results from investigation may not be extrapolated from rodents to humans. Further study needs to be conducted regarding mRNA and protein expressions of these molecules or other relevant molecules in cerebrospinal fluid of PD patients with excessive iron depositions in SN by using magnetic resonance susceptibility weighted imaging.
In summary, we show for the first time that iron induces selective and progressive dopaminergic neurotoxicity, which is potentiated by microglia. Iron activates microglia by producing magnitude of O2·− and iROS and inducing morphological alteration. NOX2−/− mice are resistant to iron-induced neurotoxicity by reducing O2·− production, and potential NOX2 inhibitor DPI protects against iron-elicited dopaminergic neurotoxicity through decreasing O2·− generation, implying that iron-provoked dopaminergic neurotoxicity is microglial NOX2-dependent. NOX2 activation by iron is indicated by the increased mRNA and protein expressions of its subunits P47 and gp91. Molecules relevant to iron-induced NOX2 activation include PKC-σ, MAPKs (P38, ERK1/2, and JNK), and NF-ΚBP65 as their mRNA and protein expressions are all elevated. Thus, over exposure to iron may be relevant to PD. Inhibition of iron-induced neuroinflammation characterized by microglial NOX2 activation may be a novel target of drug development for PD.
Supplementary Material
Acknowledgments
This work is supported by the National Basic Research Program of China (2011CB504100); the National Natural Science Foundation of China (81071015, 30770745, and 81030062), the Natural Science Foundation of Beijing, China (7082032), High Level Technical Personnel Training Project of Beijing Health System, China (2009-3-26), Excellent Personnel Training Project of Beijing, China (20071D0300400076), Capital Clinical Characteristic Application Research (Z121107001012161), Important National Science and Technology Specific Projects (2011ZX09102-003-01), Key Project of Beijing Natural Science Foundation (kz200910025001), and Basic-Clinical Research Cooperation Funding of Capital Medical University (10JL49). We thank Dr. Yang Du for her efforts of editing the manuscript.
Footnotes
Electronic supplementary material
The online version of this article (doi:10.1007/s12035-013-8586-4) contains supplementary material, which is available to authorized users.
Contributor Information
Wei Zhang, Email: ttyyzw@163.com, Department of Neurology, Beijing Tiantan Hospital, Capital Medical University, Beijing 100050, China; Parkinson’s Disease Center of Beijing Institute of Brain Disorders, Beijing 100053, China; Beijing Key Laboratory on Parkinson’s Disease, Beijing 100053, China.
Zhao-fen Yan, Department of Neurology, Beijing Tiantan Hospital, Capital Medical University, Beijing 100050, China.
Jun-hua Gao, Department of Neurology, Beijing Tiantan Hospital, Capital Medical University, Beijing 100050, China.
Li Sun, Department of Neurology, Beijing Tiantan Hospital, Capital Medical University, Beijing 100050, China.
Xi-yan Huang, Department of Neurology, Beijing Tiantan Hospital, Capital Medical University, Beijing 100050, China.
Zhuo Liu, Department of Neurology, Beijing Tiantan Hospital, Capital Medical University, Beijing 100050, China.
Shu-yang Yu, Department of Neurology, Beijing Tiantan Hospital, Capital Medical University, Beijing 100050, China.
Chen-Jie Cao, Department of Neurology, Beijing Tiantan Hospital, Capital Medical University, Beijing 100050, China.
Li-jun Zuo, Department of Neurology, Beijing Tiantan Hospital, Capital Medical University, Beijing 100050, China.
Ze-Jie Chen, Department of Neurology, Beijing Tiantan Hospital, Capital Medical University, Beijing 100050, China.
Yang Hu, Department of Neurology, Beijing Tiantan Hospital, Capital Medical University, Beijing 100050, China.
Fang Wang, Department of Neurology, Beijing Tiantan Hospital, Capital Medical University, Beijing 100050, China.
Jau-shyong Hong, Neuropharmacology Section, Laboratory of Pharmacology and Chemistry, National Institute of Environmental Health Sciences/National Institutes of Health, Research Triangle Park, North Carolina 27709, USA.
Xiao-min Wang, Department of Physiology, Capital Medical University, Beijing 100069, China; Department of Neurobiology, Capital Medical University, Beijing 100069, China; Key Laboratory for Neurodegenerative Disorders of the Ministry of Education, Beijing 100069, China; Beijing Institute for Brain Disorders, Beijing 100069, China.
References
- 1.Block ML, Zecca L, Hong JS. Microglia-mediated neurotoxicity: uncovering the molecular mechanisms. Nat Rev Neurosci. 2007;8:57–69. doi: 10.1038/nrn2038. [DOI] [PubMed] [Google Scholar]
- 2.McGeer PL, Itagaki S, Boyes BE, McGeer EG. Reactive microglia are positive for HLA-DR in the substantia nigra of Parkinson’s and Alzheimer’s disease brains. Neurology. 1988;38:1285–1291. doi: 10.1212/wnl.38.8.1285. [DOI] [PubMed] [Google Scholar]
- 3.Zhang W, Wang T, Qin L, Gao HM, Wilson B, Ali SF, et al. Neuroprotective effect of dextromethorphan in the MPTP Parkinson’s disease model: role of NADPH oxidase. FASEB J. 2004;18:589–591. doi: 10.1096/fj.03-0983fje. [DOI] [PubMed] [Google Scholar]
- 4.Jung BD, Shin EJ, Nguyen XK, Jin CH, Bach JH, Park SJ, et al. Potentiation of methamphetamine neurotoxicity by intrastriatal lipopolysaccharide administration. Neurochem Int. 2010;56:229–244. doi: 10.1016/j.neuint.2009.10.005. [DOI] [PubMed] [Google Scholar]
- 5.Gao HM, Hong JS, Zhang W, Liu B. Synergistic dopaminergic neurotoxicity of the pesticide rotenone and inflammogen lipopolysaccharide: relevance to the etiology of Parkinson’s disease. J Neurosci. 2003;23:1228–1236. doi: 10.1523/JNEUROSCI.23-04-01228.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Miller RL, Sun GY, Sun AY. Cytotoxicity of paraquat in microglial cells: involvement of PKCdelta- and ERK1/2-dependent NADPH oxidase. Brain Res. 2007;1167:129–139. doi: 10.1016/j.brainres.2007.06.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zecca L, Zucca FA, Albertini A, Rizzio E, Fariello RG. A proposed dual role of neuromelanin in the pathogenesis of Parkinson’s disease. Neurology. 2006;67:S8–S11. doi: 10.1212/WNL.67.7_suppl_2.S8. [DOI] [PubMed] [Google Scholar]
- 8.Zhang W, Wang T, Pei Z, Miller DS, Wu X, Block ML, et al. Aggregated alpha-synuclein activates microglia: a process leading to disease progression in Parkinson’s disease. FASEB J. 2005;19:533–542. doi: 10.1096/fj.04-2751com. [DOI] [PubMed] [Google Scholar]
- 9.Zhang W, Dallas S, Zhang D, Guo JP, Pang H, Wilson B, et al. Microglial PHOX and Mac-1 are essential to the enhanced dopaminergic neurodegeneration elicited by A30P and A53T mutant alpha-synuclein. Glia. 2007;55:1178–1188. doi: 10.1002/glia.20532. [DOI] [PubMed] [Google Scholar]
- 10.Bernstein AI, Garrison SP, Zambetti GP, O’Malley KL. 6-OHDA generated ROS induces DNA damage and p53- and PUMA-dependent cell death. Mol Neurodegener. 2011;6:2. doi: 10.1186/1750-1326-6-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cho Y, Son HJ, Kim EM, Choi JH, Kim ST, Ji IJ, et al. Doxycycline is neuroprotective against nigral dopaminergic degeneration by a dual mechanism involving MMP-3. Neurotox Res. 2009;16:361–371. doi: 10.1007/s12640-009-9078-1. [DOI] [PubMed] [Google Scholar]
- 12.Levesque S, Wilson B, Gregoria V, Thorpe LB, Dallas S, Polikov VS, et al. Reactive microgliosis: extracellular micro-calpain and microglia-mediated dopaminergic neurotoxicity. Brain. 2010;133:808–821. doi: 10.1093/brain/awp333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Block ML, Wu X, Pei Z, Li G, Wang T, Qin L, et al. Nanometer size diesel exhaust particles are selectively toxic to dopaminergic neurons: the role of microglia, phagocytosis, and NADPH oxidase. FASEB J. 2004;18:1618–1620. doi: 10.1096/fj.04-1945fje. [DOI] [PubMed] [Google Scholar]
- 14.Arreguin S, Nelson P, Padway S, Shirazi M, Pierpont C. Dopamine complexes of iron in the etiology and pathogenesis of Parkinson’s disease. J Inorg Biochem. 2009;103:87–93. doi: 10.1016/j.jinorgbio.2008.09.007. [DOI] [PubMed] [Google Scholar]
- 15.Berg D, Hochstrasser H. Iron metabolism in Parkinsonian syndromes. Mov Disord. 2006;21:1299–1310. doi: 10.1002/mds.21020. [DOI] [PubMed] [Google Scholar]
- 16.Dexter DT, Carayon A, Javoy-Agid F, Agid Y, Wells FR, Daniel SE, et al. Alterations in the levels of iron, ferritin and other trace metals in Parkinson’s disease and other neurodegenerative diseases affecting the basal ganglia. Brain. 1991;114(Pt 4):1953–1975. doi: 10.1093/brain/114.4.1953. [DOI] [PubMed] [Google Scholar]
- 17.Powers KM, Smith-Weller T, Franklin GM, Longstreth WT, Jr, Swanson PD, Checkoway H. Dietary fats cholesterol and iron as risk factors for Parkinson’s disease. Parkinsonism Relat Disord. 2009;15:47–52. doi: 10.1016/j.parkreldis.2008.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Barbeito AG, Garringer HJ, Baraibar MA, Gao X, Arredondo M, Nunez MT, et al. Abnormal iron metabolism and oxidative stress in mice expressing a mutant form of the ferritin light polypeptide gene. J Neurochem. 2009;109:1078. doi: 10.1111/j.1471-4159.2009.06028.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kortekaas R, Leenders KL, van Oostrom JC, Vaalburg W, Bart J, Willemsen AT, et al. Blood–brain barrier dysfunction in parkinsonian midbrain in vivo. Ann Neurol. 2005;57:176–179. doi: 10.1002/ana.20369. [DOI] [PubMed] [Google Scholar]
- 20.Riederer P, Rausch WD, Schmidt B, Kruzik P, Konradi C, Sofic E, et al. Biochemical fundamentals of Parkinson’s disease. Mt Sinai J Med. 1988;55:21–28. [PubMed] [Google Scholar]
- 21.Hare D, Ayton S, Bush A, Lei P. A delicate balance: iron metabolism and diseases of the brain. Front Aging Neurosci. 2013;5:34. doi: 10.3389/fnagi.2013.00034.eCollection2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dexter DT, Carayon A, Vidailhet M, Ruberg M, Agid F, Agid Y, et al. Decreased ferritin levels in brain in Parkinson’s disease. J Neurochem. 1990;55:16–20. doi: 10.1111/j.1471-4159.1990.tb08814.x. [DOI] [PubMed] [Google Scholar]
- 23.Werner CJ, Heyny-Von Haussen R, Mall G, Wolf S. Proteome analysis of human substantia nigra in Parkinson’s disease. Proteome Sci. 2008;6:8. doi: 10.1186/1477-5956-6-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang X, Dong F, Ren J, Driscoll MJ, Culver B. High dietary fat induces NADPH oxidase-associated oxidative stress and inflammation in rat cerebral cortex. Exp Neurol. 2005;191:318–325. doi: 10.1016/j.expneurol.2004.10.011. [DOI] [PubMed] [Google Scholar]
- 25.Kelleher P, Pacheco K, Newman LS. Inorganic dust pneumonias: the metal-related parenchymal disorders. Environ Health Perspect. 2000;108(Suppl 4):685–696. doi: 10.1289/ehp.00108s4685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang W, Phillips K, Wielgus AR, Liu J, Albertini A, Zucca FA, et al. Neuromelanin activates microglia and induces degeneration of dopaminergic neurons: implications for progression of Parkinson’s disease. Neurotox Res. 2011;19:63–72. doi: 10.1007/s12640-009-9140-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Aisen P, Enns C, Wessling-Resnick M. Chemistry and biology of eukaryotic iron metabolism. Int J Biochem Cell Biol. 2001;33:940–959. doi: 10.1016/S1357-2725(01)00063-2. [DOI] [PubMed] [Google Scholar]
- 28.Moos T, Morgan EH. The metabolism of neuronal iron and its pathogenic role in neurological disease: review. Ann N Y Acad Sci. 2004;1012:14–26. doi: 10.1196/annals.1306.002. [DOI] [PubMed] [Google Scholar]
- 29.Leitner DF, Connor JR. Functional roles of transferrin in the brain. Biochim Biophys Acta. 2012;1820:393–402. doi: 10.1016/j.bbagen.2011.10.016. [DOI] [PubMed] [Google Scholar]
- 30.Donovan A, Lima CA, Pinkus JL, Pinkus GS, Zon LI, Robine S, et al. The iron exporter ferroportin/Slc40a1 is essential for iron homeostasis. Cell Metab. 2005;1:191–200. doi: 10.1016/j.cmet.2005.01.003. [DOI] [PubMed] [Google Scholar]
- 31.Abboud S, Haile DJ. A novel mammalian iron-regulated protein involved in intracellular iron metabolism. J Biol Chem. 2000;275:19906–19912. doi: 10.1074/jbc.M000713200. [DOI] [PubMed] [Google Scholar]
- 32.Dringen R, Bishop GM, Koeppe M, Dang TN, Robinson SR. The pivotal role of astrocytes in the metabolism of iron in the brain. Neurochem Res. 2007;32:1884–1890. doi: 10.1007/s11064-007-9375-0. [DOI] [PubMed] [Google Scholar]
- 33.Lowe JS, Leigh N. Disorders of movement and system degenerations. In: Graham DI, Lantos PL, editors. Greenfield’s Neuropathology. 7th. Vol. 2. London: Arnold; 2002. pp. 325–430. [Google Scholar]
- 34.Yamada T, McGeer PL, McGeer EG. Lewy bodies in Parkinson’s disease are recognized by antibodies to complement proteins. Acta Neuropathol. 1992;84:100–104. doi: 10.1007/BF00427222. [DOI] [PubMed] [Google Scholar]
- 35.Castellani RJ, Siedlak SL, Perry G, Smith MA. Sequestration of iron by Lewy bodies in Parkinson’s disease. Acta Neuropathol. 2000;100:111–114. doi: 10.1007/s004010050001. [DOI] [PubMed] [Google Scholar]
- 36.Lv Z, Jiang H, Xu H, Song N, Xie J. Increased iron levels correlate with the selective nigral dopaminergic neuron degeneration in Parkinson’s disease. J Neural Transm. 2011;118:361–369. doi: 10.1007/s00702-010-0434-3. [DOI] [PubMed] [Google Scholar]
- 37.Chew KC, Ang ET, Tai YK, Tsang F, Lo SQ, Ong E, et al. Enhanced autophagy from chronic toxicity of iron and mutant A53T alpha-synuclein: implications for neuronal cell death in Parkinson disease. J Biol Chem. 2011;286:33380–33389. doi: 10.1074/jbc.M111.268409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wypijewska A, Galazka-Friedman J, Bauminger ER, Wszolek ZK, Schweitzer KJ, Dickson DW, et al. Iron and reactive oxygen species activity in parkinsonian substantia nigra. Parkinsonism Relat Disord. 2010;16:329–333. doi: 10.1016/j.parkreldis.2010.02.007. [DOI] [PubMed] [Google Scholar]
- 39.Dutta G, Zhang P, Liu B. The lipopolysaccharide Parkinson’s disease animal model: mechanistic studies and drug discovery. Fundam Clin Pharmacol. 2008;22:453–464. doi: 10.1111/j.1472-8206.2008.00616.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tansey MG, Goldberg MS. Neuroinflammation in Parkinson’s disease: its role in neuronal death and implications for therapeutic intervention. Neurobiol Dis. 2010;37:510–518. doi: 10.1016/j.nbd.2009.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Infanger DW, Sharma RV, Davisson RL. NADPH oxidases of the brain: distribution, regulation, and function. Antioxid Redox Signal. 2006;8:1583–1596. doi: 10.1089/ars.2006.8.1583. [DOI] [PubMed] [Google Scholar]
- 42.Stewart VC, Heales SJ. Nitric oxide-induced mitochondrial dysfunction: implications for neurodegeneration. Free Radic Biol Med. 2003;34:287–303. doi: 10.1016/S0891-5849(02)01327-8. [DOI] [PubMed] [Google Scholar]
- 43.Qin L, Wu X, Block ML, Liu Y, Breese GR, Hong JS, et al. Systemic LPS causes chronic neuroinflammation and progressive neurodegeneration. Glia. 2007;55:453–462. doi: 10.1002/glia.20467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.McGeer PL, McGeer EG. Glial reactions in Parkinson’s disease. Mov Disord. 2008;23:474–483. doi: 10.1002/mds.21751. [DOI] [PubMed] [Google Scholar]
- 45.Ouchi Y, Yagi S, Yokokura M, Sakamoto M. Neuroinflammation in the living brain of Parkinson’s disease. Parkinsonism Relat Disord. 2009;15(Suppl 3):S200–S204. doi: 10.1016/S1353-8020(09)70814-4. [DOI] [PubMed] [Google Scholar]
- 46.Block ML. NADPH oxidase as a therapeutic target in Alzheimer’s disease. BMC Neurosci. 2008;9(Suppl 2):S8. doi: 10.1186/1471-2202-9-S2-S8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tammariello SP, Quinn MT, Estus S. NADPH oxidase contributes directly to oxidative stress and apoptosis in nerve growth factor-deprived sympathetic neurons. J Neurosci. 2000;20:RC53. doi: 10.1523/JNEUROSCI.20-01-j0006.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Qin L, Liu Y, Wang T, Wei SJ, Block ML, Wilson B, et al. NADPH oxidase mediates lipopolysaccharide-induced neurotoxicity and proinflammatory gene expression in activated microglia. J Biol Chem. 2004;279:1415–1421. doi: 10.1074/jbc.M307657200. [DOI] [PubMed] [Google Scholar]
- 49.Casarejos MJ, Menendez J, Solano RM, Rodriguez-Navarro JA, de Yebenes JG, Mena MA. Susceptibility to rotenone is increased in neurons from parkin null mice and is reduced by minocycline. J Neurochem. 2006;97:934–946. doi: 10.1111/j.1471-4159.2006.03777.x. [DOI] [PubMed] [Google Scholar]
- 50.Sorce S, Krause KH. NOX enzymes in the central nervous system: from signaling to disease. Antioxid Redox Signal. 2009;11:2481–2504. doi: 10.1089/ARS.2009.2578. [DOI] [PubMed] [Google Scholar]
- 51.Miller RL, James-Kracke M, Sun GY, Sun AY. Oxidative and inflammatory pathways in Parkinson’s disease. Neurochem Res. 2009;34:55–65. doi: 10.1007/s11064-008-9656-2. [DOI] [PubMed] [Google Scholar]
- 52.Zhao X, Xu B, Bhattacharjee A, Oldfield CM, Wientjes FB, Feldman GM, et al. Protein kinase Cdelta regulates p67phox phosphorylation in human monocytes. J Leukoc Biol. 2005;77:414–420. doi: 10.1189/jlb.0504284. [DOI] [PubMed] [Google Scholar]
- 53.Bey EA, Xu B, Bhattacharjee A, Oldfield CM, Zhao X, Li Q, et al. Protein kinase C delta is required for p47phox phosphorylation and translocation in activated human monocytes. J Immunol. 2004;173:5730–5738. doi: 10.4049/jimmunol.173.9.5730. [DOI] [PubMed] [Google Scholar]
- 54.Min KJ, Pyo HK, Yang MS, Ji KA, Jou I, Joe EH. Gangliosides activate microglia via protein kinase C and NADPH oxidase. Glia. 2004;48:197–206. doi: 10.1002/glia.20069. [DOI] [PubMed] [Google Scholar]
- 55.Murphy LO, Blenis J. MAPK signal specificity: the right place at the right time. Trends Biochem Sci. 2006;31:268–275. doi: 10.1016/j.tibs.2006.03.009. [DOI] [PubMed] [Google Scholar]
- 56.Bardwell L. Mechanisms of MAPK signalling specificity. Biochem Soc Trans. 2006;34:837–841. doi: 10.1042/BST0340837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Flohe L, Brigelius-Flohe R, Saliou C, Traber MG, Packer L. Redox regulation of NF-kappa B activation. Free Radic Biol Med. 1997;22:1115–1126. doi: 10.1016/S0891-5849(96)00501-1. [DOI] [PubMed] [Google Scholar]
- 58.Yang S, Yang J, Yang Z, Chen P, Fraser A, Zhang W, et al. Pituitary adenylate cyclase-activating polypeptide (PACAP) 38 and PACAP4-6 are neuroprotective through inhibition of NADPH oxidase: potent regulators of microglia-mediated oxidative stress. J Pharmacol Exp Ther. 2006;319:595–603. doi: 10.1124/jpet.106.102236. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.