

Abstract
In neurological disease and diabetes, the unfolded protein response (UPR) has been investigated for years, while its function in heart disease is less well understood. All three branches of the UPR are involved in ischaemia/reperfusion and can either protect or impair heart function. Recently, UPR has been found to play a role in arrhythmogenesis during human heart failure, and blocking UPR has an antiarrhythmic effect. This review will discuss the rationale for and challenges to targeting UPR in heart disease.
Keywords: activating transcription factor 6α, glucose-regulated protein-78, heart failure, inositol-requiring ER-to-nucleus signal kinase 1, ischaemia, protein kinase-like ER kinase
1. Introduction
The endoplasmic reticulum (ER) is the location for protein translation, folding and assembling before trafficking to the cytosol and plasma membrane. The ER lumen is an oxidative environment with the highest Ca2+ concentration within the cell. These two conditions are crucial for proper protein folding. The oxidative environment favours the formation of disulfide bonds to convey the tertiary and quaternary structure of proteins, and Ca2+ is essential for Ca2+-dependent molecular chaperones to interact with intermediate states of protein folding. When the ER homeostasis is disturbed, unfolded or misfolded proteins accumulate in the ER lumen, triggering the unfolded protein response (UPR).
The UPR is highly conserved in evolution and is initially an adaptive response. Nevertheless, when prolonged ER stress occurs, the UPR can lead to apoptosis. Unfolded protein accumulation is sensed by glucose-regulated protein-78 (Grp78), which will dissociate from three UPR sensors in response to unfolded protein. These sensors are the double-stranded RNA (dsRNA)-activated protein kinase-like ER kinase (PERK), inositol-requiring ER-to-nucleus signal kinase 1 (IRE1), and activating transcription factor 6α (ATF6α). Grp78 dissociation triggers oligomerization and auto-phosphorylation of UPR sensors and leads to their activation. The activated UPR sensors initiate signalling transduction for expression of genes that restore the protein folding capacity in the ER and for generation of chaperone proteins that participate in the ER-associated degradation (ERAD). The chaperones include Grp78, Grp94 and calreticulin, and so on. They play important roles in the ERAD process, by binding to the misfolded or unfolded protein, and by preventing the protein from further transit and secretion. Instead, the complex of chaperone–unfolded protein exits the ER and degrades in cytosol, alleviating the ER stress. The UPR also attenuates protein synthesis by preventing translation of mRNAs. PERK and IRE1 play major roles in this process.
The three UPR sensors have their own signalling pathways either to increase protein folding ability or to decrease protein synthesis and loading of the ER. The downstream effectors are phosphorylated translation initiation factor 2α (eIF2α) and ATF4 for PERK, spliced X-box binding protein 1 (sXBP1) for IRE1, and the cleaved N-terminus of ATF6α (ATF6N) for ATF6α, respectively (Figure 1). On the other hand, there is also crosstalk among the three branches. For example, the downstream effectors of PERK activation, eIF2α and ATF4, can regulate IRE1 and ATF6α. For example, phosphorylation of eIF2α leads to NF-κB activation, which induces downregulation of sXBP1 mRNA levels via microRNA-30c-2* in the IRE1 signalling pathway. ATF4 activates the ATF6α branch through enhancing ATF6α expression and translocation to the Golgi. This interplay may make the UPR more redundant and more complicated to target pharmacologically. Besides redundancy of the three UPR branches, each branch appears to have its special roles as well. For example, the IRE1 branch effector sXBP1 regulates lipid biosynthetic enzymes [1] and ERAD proteins, and the ATF6 branch plays major roles on promoting the UPR target genes expression.
Figure 1. A scheme of the UPR with its sensors, chaperones, effectors and targeting genes.
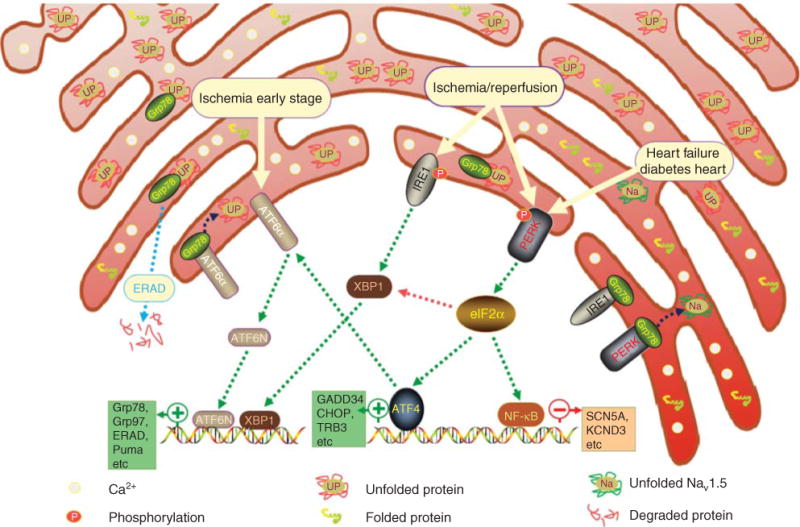
The ATF6α branch is activated in early stage of ischaemia. PERK and IRE1 are activated in ischaemia/reperfusion. PERK is also activated in HF and diabetic cardiomyopathy. Green dotted arrows indicate activation, red dotted arrows indicate inhibition and dark blue dotted arrows indicate dissociation from UPR sensors and binding to the unfolded protein.
ATF6α: Activating transcription factor 6α; HF: Heart failure; IRE1: inositol-requiring ER-to-nucleus signal kinase 1; PERK: Protein kinase-like ER kinase; UPR: Unfolded protein response.
2. Why target the UPR in heart disease?
In cardiomyocytes, the sarco-ER (SER) is pivotal not only for general cellular function but also for myocyte contractility. Heart disease has been associated with activation of the UPR (Figure 1). The SER stress that is common in heart disease can induce UPR activation, which can cause reduced expression of essential proteins that will affect cell function and even cell apoptosis when the UPR activation is prolonged. By targeting the UPR, we may be able to mitigate some of the effects of heart disease.
2.1 UPR in heart failure
Stimuli, such as oxidative stress, hypoxia and ischaemic insult that are common in heart failure (HF), induce the ER stress with accumulation of unfolded proteins and trigger the UPR. Activated PERK and IRE1 branches of the UPR are reported in human failing hearts as evidenced by increased mRNA levels of ATF4/CHOP and Grp78/sXBP1. In animal models of HF, Grp78 and CHOP are elevated significantly [2]. Recently, we have reported aberrant mRNA splicing of SCN5A that encodes cardiac Na+ channels (Nav1.5) in human HF. The truncated mRNA variants encode non-functional channel proteins that are trapped in the ER and activate PERK [3]. The lack of functional channels and PERK-dependent degradation of the full-length SCN5A mRNA contributes to the reduced membrane expression of Nav1.5 protein. The reduction of full-length functional Nav1.5 leads to a decreased INa and consequently decreased conduction velocity [3]. Once activated, the effect of UPR is not specific to Nav1.5. Similar effects of UPR are observed with the cardiac K+ channel Kv4.3, suggesting that UPR may play a crucial role in downregulation of multiple cardiac genes in HF.
The downregulation of these ion channel genes can contribute to arrhythmic risk. Cardiac Kv4.3 channel conducts Ito, which is the main contributor to the plateau Phase II of the cardiac action potential and responsible for early repolarisation. Reduced Ito reported in HF and diabetic heart disease can increase membrane resistance and cause shortening of the cardiac action potential duration, which is thought to contribute to diastolic dysfunction and lead to delayed after-depolarisations, an arrhythmic mechanism.
2.2 UPR in ischaemia
In ischaemia, all three UPR branches are activated, and they induce cell apoptosis that contributes to cardiomyocyte loss. Thus, these branches might be targets to prevent cell loss in ischaemia. When cardiomyocytes experience prolonged ischaemia, Grp78, XBP1, ATF4 and eIF2α, and CHOP are all elevated at the mRNA and protein levels, indicating activation of the IRE1 and PERK branches of the UPR. In hypoxic cardiomyocytes, Glembotski’s group demonstrated activation of the IRE1 branch with enhanced levels of Grp78 and XBP1 [4]. Later, the same group reported that the ATF6α branch of the UPR can be activated in ischaemia and inactivated upon reperfusion [5]. This study suggested that the ATF6α branch plays an inducible role during ischaemia that may affect preconditioning during reperfusion.
Expression of Tribbles 3 (TRB3, an intracellular pseudokinase), downstream of PERK and ATF4/CHOP, is significantly elevated in myocardial infarction [6]. Transgenic mice with heart tissue-specific overexpression of TRB3 show pathological cardiac remodelling after myocardial infarction [6]. An ER stress response gene and downstream of PERK, p53-upregulated modulator of apoptosis (PUMA) induces apoptosis when overexpressed in cardiomyocytes [7]. Knock out of PUMA shows protective effects on myocytes death from ischaemia/reperfusion in vivo [7]. These results indicate that PUMA plays a deleterious role in ischaemia/reperfusion, and inhibition of PUMA may be beneficial for myocardial infarction and HF.
3. Targeting the UPR in heart disease
3.1 Expected effects of targeting UPR
In heart disease, most of the protein alterations are downregulations. It is interesting to speculate about what percentage of the downregulation is the result of the UPR. If many are, then targeting the UPR may have multiple salutary effects. It is possible that by targeting the UPR, the expression of essential proteins will be elevated to maintain normal cell function. For example, downregulation of Nav1.5 and Kv4.3 in HF has shown to result from PERK activation. If a PERK inhibitor could be used to restore the channel protein levels, arrhythmic risk might be improved. Alternatively, targeting the UPR might decrease cell apoptosis and improve cardiomyopathy.
3.2 How do you target the UPR?
Possible strategies to affect the UPR in heart disease include decreasing unfolded proteins, preventing the UPR sensors from activation (i.e., preventing Grp78 dissociation from the UPR sensors) or inhibiting the activated UPR sensors and effectors. In the case of the cardiac Nav1.5 channel, decreasing unfolded protein could be undertaken by inhibiting abnormal mRNA splicing mediated by upregulation of RBM25 and LUC7L3 [8].
Grp78 overexpression may be used to prevent UPR sensor activation. Grp78 overexpression can elevate binding probability of Grp78 to the UPR sensors and, therefore, prevent the activation. Without UPR activation, nascent proteins could be translated for regular cell function and survival. Meanwhile, Grp78 overexpression can increase the binding of Grp78 to unfolded/misfolded proteins to accelerate ERAD. Therefore, therapies targeting Grp78 overexpression may be helpful for heart disease and other diseases with over-activated UPR. As an example, overexpression of Grp78 has been reported to attenuate hypoxia-mediated cardiomyocyte death [9]. In addition, overexpression of Grp94 protects cardiomyocytes against oxidative stress [10].
Inhibition of PERK, ATF6α, IRE1 or the downstream effectors (eIF2α/ATF4, ATF6N and XBP1) with specific inhibitors is also a strategy to prevent the deleterious effects of UPR. Although there is no specific inhibitor reported up to date, Minamino et al. discuss drugs targeting the UPR or the ER stress stimuli that may form the basis of compounds used in human heart disease [11]. Human anti-PERK short hairpin RNAmir has been used in vitro to block PERK activation in human-induced pluripotent stem cell-derived cardiomyocytes [3]. Recently, GSK2606414, a small-molecule oral agent, has been found to halt brain cell death by blocking PERK phosphorylation in vivo and has shown efficacy in prion-mediated disease associated with aggregation of misfolded protein in the ER [12]. This compound has been proposed as a new treatment for Alzheimer’s disease. Some limitations of GSK2606414 include weight loss and diabetes. Moreover, the drug is specific for PERK inhibition [12]. Therefore, it has no effects on diseases with activation of the other two UPR branches, ATF6α and IRE1. ROS overproduction can be up- or downstream of the UPR signalling. Antioxidants such as thiols and butylated hydroxyanisole have been reported to attenuate the UPR via the PERK branch.
3.3 Is it safe to target the UPR?
The UPR is an important process in normal cell function. Homozygous knockout mouse models of the UPR sensors and effectors have shown potentially damaging effects [13]. For instance, complete knockouts of XBP1 and IRE1α are embryonic lethal with incomplete development of the heart and blood vessels. PERK knockout leads to diabetes mellitus and growth retardation in mice. ATF6 knockout induces liver steatosis, hypoglycaemia and insulin resistance. Therefore, partial or temporary inhibition of the UPR sensors or organ specific gene therapy may be the safest alternative for targeting the UPR.
Timing and targeting of the UPR inhibition may be critical. The UPR has protective effects in certain circumstances. For example, activation of the ATF6α branch before ischaemia reduces myocardial tissue damage during ischaemia/reperfusion [14]. On the other hand, apelin-13 protects the heart from ischaemia-induced ER stress by inhibition of the PERK/CHOP branch [15]. In general, it may be that early ER stress is adaptive and protective, whereas sustained ER stress leads to apoptotic or necrotic cell death. One strategy may be to develop biomarkers to assess which UPR pathways are activated and their degree of activation. For example, circulating Nav1.5 splicing variants may be correlative with cardiac PERK activation.
4. Expert opinion
It seems clear that there may be times when modulating the UPR may be of clinical advantage. For example, in HF, UPR activation contributes to arrhythmic risk and inhibiting UPR may decrease arrhythmic risk. Nevertheless, the UPR can have salutary effects, and complete, whole body or indefinite inhibition may have untoward side effects. These limitations may be overcome with more knowledge of the system, more precisely targeting agents or time-limited treatment approaches. Overall, UPR inhibition seems like a potentially fruitful investigative line in novel therapeutics for cardiac disease.
Acknowledgments
The authors were supported by National Institutes of Health (NIH) Grants RO1 HL104025 (SCD), HL106592 (SCD), and a Veterans Affairs MERIT grant BX000859 (SCD). Dudley is an inventor on the following patent applications 1) PCT/US2012/020564. Scn5a splice variants for use in methods relating to sudden cardiac death and need for implanted cardiac defibrillations and 2) PCT/US2010/034271, drug targets for prevention of arrhythmia in heart disease.
Footnotes
Declaration of interest
The authors have no other relevant affiliations or financial involvement with any organisation or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
Bibliography
Papers of special note have been highlighted as either of interest (•) or of considerable interest (••) to readers.
- 1.Reimold A, Iwakoshi N, Manis J, et al. Plasma cell differentiation requires the transcription factor XBP-1. Nature. 2001;412:300–7. doi: 10.1038/35085509. [DOI] [PubMed] [Google Scholar]
- 2.Isodono K, Takahashi T, Imoto H, et al. PARM-1 is an endoplasmic reticulum molecule involved in endoplasmic reticulum stress-induced apoptosis in rat cardiac myocytes. PLoS One. 2010;5:e9746. doi: 10.1371/journal.pone.0009746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gao G, Xie A, Zhang J, et al. Unfolded protein response regulates cardiac sodium current in systolic human heart failure. Circ Arrhythm Electrophysiol. 2013;6:1018–24. doi: 10.1161/CIRCEP.113.000274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4•.Thuerauf DJ, Marcinko M, Gude N, et al. Activation of the unfolded protein response in infarcted mouse heart and hypoxic cultured cardiac myocytes. Circ Res. 2006;99:275–82. doi: 10.1161/01.RES.0000233317.70421.03. This study demonstrates a deleterious effect of IRE1 activation by ischaemia. [DOI] [PubMed] [Google Scholar]
- 5.Doroudgar S, Thuerauf DJ, Marcinko MC, et al. Ischemia activates the ATF6 branch of the endoplasmic reticulum stress response. J Biol Chem. 2009;284:29735–45. doi: 10.1074/jbc.M109.018036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Avery J, Etzion S, DeBosch BJ, et al. TRB3 function in cardiac endoplasmic reticulum stress. Circ Res. 2010;106:1516–23. doi: 10.1161/CIRCRESAHA.109.211920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nickson P, Toth A, Erhardt P. PUMA is critical for neonatal cardiomyocyte apoptosis induced by endoplasmic reticulum stress. Cardiovasc Res. 2007;73:48–56. doi: 10.1016/j.cardiores.2006.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gao G, Xie A, Huang SC, et al. Role of RBM25/LUC7L3 in abnormal cardiac sodium channel splicing regulation in human heart failure/clinical perspective. Circulation. 2011;124:1124–31. doi: 10.1161/CIRCULATIONAHA.111.044495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pan YX, Lin L, Ren AJ, et al. HSP70 and GRP78 Induced by endothelin-1 pretreatment enhance tolerance to hypoxia in cultured neonatal rat cardiomyocytes. J Cardiovasc Pharmacol. 2004;44(Suppl 1):S117–20. doi: 10.1097/01.fjc.0000166234.11336.a9. This study shows evidence that overexpression of Grp78 can play a protective role in hypoxic cardiomyocytes. [DOI] [PubMed] [Google Scholar]
- 10.Vitadello M, Penzo D, Petronilli V, et al. Overexpression of the stress-protein Grp94 reduces cardiomyocyte necrosis due to calcium overload and simulated ischemia. FASEB J. 2003;17(8):923–5. doi: 10.1096/fj.02-0644fje. [DOI] [PubMed] [Google Scholar]
- 11•.Minamino T, Komuro I, Kitakaze M. Endoplasmic reticulum stress as a therapeutic target in cardiovascular disease. Circ Res. 2010;107:1071–82. doi: 10.1161/CIRCRESAHA.110.227819. This study establishes efficacy of an oral agent, GSK2606414, to inhibit UPR in prion-mediated disease. [DOI] [PubMed] [Google Scholar]
- 12.Moreno JA, Halliday M, Molloy C, et al. Oral treatment targeting the unfolded protein response prevents neurodegeneration and clinical disease in prion-infected mice. Sci Transl Med. 2013;5:206ra138. doi: 10.1126/scitranslmed.3006767. [DOI] [PubMed] [Google Scholar]
- 13.Cornejo V, Pihán P, Vidal R, Hetz C. Role of the unfolded protein response in organ physiology: lessons from mouse models. IUBMB Life. 2013;65:962–75. doi: 10.1002/iub.1224. [DOI] [PubMed] [Google Scholar]
- 14•.Martindale JJ, Fernandez R, Thuerauf D, et al. Endoplasmic reticulum stress gene induction and protection from ischemia/reperfusion injury in the hearts of transgenic mice with a tamoxifen-regulated form of ATF6. Circ Res. 2006;98:1186–93. doi: 10.1161/01.RES.0000220643.65941.8d. This study demonstrates that activated ATF6 plays a protective role in ischaemia/reperfusion. [DOI] [PubMed] [Google Scholar]
- 15.Tao J, Zhu W, Li Y, et al. Apelin-13 protects the heart against ischemia-reperfusion injury through inhibition of ER-dependent apoptotic pathways in a time-dependent fashion. Am J Physiol Heart Circ Physiol. 2011;301:H1471–86. doi: 10.1152/ajpheart.00097.2011. [DOI] [PubMed] [Google Scholar]