

Abstract
The Blue Obelisk Movement (http://www.blueobelisk.org/) is the name used by a diverse Internet group promoting reusable chemistry via open source software development, consistent and complimentary chemoinformatics research, open data, and open standards. We outline recent examples of cooperation in the Blue Obelisk group: a shared dictionary of algorithms and implementations in chemoinformatics algorithms drawing from our various software projects; a shared repository of chemoinformatics data including elemental properties, atomic radii, isotopes, atom typing rules, and so forth; and Web services for the platform-independent use of chemoinformatics programs.
1. Introduction
While the past 20 or 30 years of development in chemoinformatics has created a plethora of published software systems and algorithms for solving chemical problems, little effort has been spent in providing the community with open components and data, to be reused and improved by communal efforts. Bioinformatics, with its much younger history, adopted the principles taught by success stories of the open source movement in general, and Linux in particular, from the very beginning. Recent years, however, have seen the emergence of open tools and databases also in chemical informatics.50400bb00001−50400bb00004 These draw on the existing ideas of independent peer review and scientific collaboration, mixed with “open source” software development paradigms. Community involvement, including assessments, suggestions, critiques, and rapid evolution, is a core component of these efforts. The benefits of open source software have been discussed in great detail by Eric Raymond in his seminal work The Cathedral and the Bazaar and following works.50400bb00005 The Open Source Initiative (OSI) summarizes: “Open source promotes software reliability and quality by supporting independent peer review and rapid evolution of source code. To be OSI certified, the software must be distributed under a license that guarantees the right to read, redistribute, modify, and use the software freely.”50400bb00006
In the beginning, most scientific software was free. It was so difficult to port that scientists did not bother about licenses—one was delighted if someone else could get it working on another machine. But the 1980s saw the value of chemical informatics and the need to “productize” it. Much of this was meritorious, as it brought informatics into the classroom and the research lab and helped pay for some chemistry research, but it also had hidden costs, which we are now facing today. In particular, costs include non-interoperability and centralized control of informatics.
Now, several open chemistry and chemoinformatics projects (Table 1) have pooled forces to enhance interoperability between these tools in a movement we call “The Blue Obelisk” (BO). The name originates from an informal meeting place in San Diego, California, during the American Chemical Society 2005 Spring National Meeting (see Figure 1) and was coined by one of the authors. Because contributors to the component projects live around the world, few had met in person—instead collaborating and meeting via the Internet.
Table 1.
Current Blue Obelisk Projects
| project | URL | principal authors |
| CML, JUMBO12 | http://cml.sf.net/ | P.M.-R., H.R. |
| JChemPaint13 | http://jchempaint.sf.net/ | C.S., E.L.W. |
| Jmol | http://jmol.sf.net/ | M.T.H., E.L.W. |
| NMRShiftDB3 | http://www.nmrshiftdb.org/ | C.S. |
| JOElib | http://joelib.sf.net/ | J.W. |
| Kalzium | http://edu.kde.org/kalzium/ | Carsten Niehaus |
| Octet | http://octet.sf.net/ | Rich Apodaca |
| Open Babel | http://openbabel.sf.net/ | G.R.H. |
| QSAR | http://qsar.sf.net/ | E.L.W., R.G., C.S., J.W. |
| The Chemistry Development Kit1 | http://cdk.sf.net/ | E.L.W., C.S. |
| WWMM | http://wwmm.sf.net/ | P.M.-R. |
Figure 1.

Where it all began. The Blue Obelisk in San Diego, California, at the 2005 American Chemical Society meeting.
We identify three core areas for the Blue Obelisk Movement:
• Open Source. One can use other people's code without further permission, including changing it for one's own use and distributing it again.
• Open Standards. One can find visible community mechanisms for protocols and communicating information. The mechanisms for creating and maintaining these standards cover a wide spectrum of human organizations, including various degrees of consent. We have been heavily influenced by the mantra of the Internet Engineering Task Force: “rough consensus and running code”.
• Open Data. One can obtain all data in the public domain when wanted and reuse it for whatever purpose. This is an underused term, which we are resurrecting. It is independent of “open access” and has relevance to “closed access” as well.
As outlined above, these areas are independent of the concept of “open access” to read publications freely. Instead, the three points focus on access to the scientific data, algorithms, and implementations themselves, rather than the formatted manuscript. In particular, we believe that these concepts strongly continue the spirit of communal peer review and reproducibility at the heart of modern scientific research.
It is well-known in software development that 80% of the costs are caused by maintaining software and not by the initial implementation.50400bb00007 This holds both for the in-house development in pharmaceutical companies and the development for commercial chemoinformatics suppliers. Besides judging software by its standardized functional quality, it can also be compared on the basis of its long-term stability and interoperability. Openly standardized algorithms and chemical information can help to reduce the maintenance costs, because developers can reuse available modules or test their tools against open source software and open data. This reduces the risk for both the “buy” and “build” strategies for software implementation. We agree with De Lano50400bb00008 that the try-before-buy paradigm for open source software does not necessarily require open standards. Open specifications for standard algorithms such as kekulization,50400bb00009 chirality coding,50400bb00010 and atom typing,50400bb00011 however, are indispensable in academic chemoinformatics research to build better, more stable, and more reproducible chemical information systems.
In this contribution, we outline several examples for how the Blue Obelisk projects address this need: a shared dictionary of algorithms and implementations in chemoinformatics algorithms drawing from our various software projects and a shared repository of chemoinformatics data including elemental properties, atomic radii, isotopes, atom typing rules, a set of Web-based chemoinformatics services, and the process of providing open algorithms and data. All of these projects were developed with continual community involvement, an open standardization process, and provide open data to key chemoinformatics processes. Anyone can take part; we welcome those in commercial organizations, academia, government, and so forth, and contributions come as code, compilations of data and molecules, testing, and more.
2. The Importance of Open Specifications for Algorithms and Data
The World Wide Web as it is used today is a collection of linked HTML pages and other data formats. Whenever there is chemical or other scientific knowledge or data published via this mechanism, it is often difficult or impossible to discover, because it lacks the semantics that would help machines—the only practical way to harvest information “from the Internet”—to identify and classify it. Recognizing this lack, Tim Berners-Lee introduced the concept he termed the “Semantic Web”. The Semantic Web is a mesh of information linked up in such a way as to be easily processable by machines, on a global scale. One can think of it as being an efficient way of representing data on the World Wide Web, or as a globally linked database. An analogy of the Semantic Web, projected onto the currently heavily researched idea of creating global networks of computational resources, so-called Grids, are the Semantic Grids. A Semantic Web, and even more a Semantic Grid, is predicated on the supply of information and services without requiring the user to know the details of how the resource was obtained. The “users”, who may be humans or robots, request precise services but should be unconcerned exactly how or where they originate. For example, the calculation of a molecular property might depend on a precise method but should not, in principle, depend on the actual program used, its version, the operating system, and the machine involved.
We note that many chemical calculations are described in an imprecise manner. For example, “molecular weight” is an imprecise term, and the result of an algorithm returning this cannot be regarded as precise. The IUPAC Gold Book14 describes
Relative molecular mass, Mr: ratio of the mass of a molecule to the unified atomic mass unit. This is sometimes called the molecular weight or relative molar mass.
Relative molar mass: molar mass divided by 1 g mol-1 (the latter is sometimes called the standard molar mass).
Unified atomic mass unit: non-SI unit of mass (equal to the atomic mass constant), defined as 1/12 of the mass of a carbon-12 atom in its ground state and used to express masses of atomic particles, u≈ 1.660 540 2(10) × 10-27 kg.
However, “molar mass” does not occur as a term. These appear to refer to the mass of a single molecule, not to the properties of a bulk sample. However, atomic masses include the concept of “average” as in
Relative atomic mass (atomic weight), Ar: the ratio of the average mass of the atom to the unified atomic mass unit. See also standard atomic weight.
There are at least two algorithms that could be used to obtain the “molecular mass”:
• sum the average masses of all the atoms in the molecule (the normal “molecular weight”)
• sum the precise masses of the most frequent isotopes in the molecule (giving the “high-resolution molecular mass”). Even this latter method is imprecise because, in mass spectroscopy, it relates to ions, and presumably, the mass of the ionizing electron(s) should be accounted for.
Moreover, the actual values of atomic weights vary between program systems. We have frequently observed variations in molecular weights between different authorities—often at the second decimal place.
Current practice does not constrain any of this. Many chemoinformatics and computational chemistry papers use data resources which are not available to reviewers and readers and algorithms which are not portable or distributed. It is a matter of trust rather than verification whether such work is accepted by the community. We believe it is essential that computational chemistry is able to provide the basic scientific tenet of reproducibility—if a scientist repeats the work in an article they should be able to duplicate the result. This is simple, in principle: computers should run reliably, and if the same data are given to the same algorithm, identical results should be obtained. However, it is surprisingly difficult to assert that the “same” method is being used. Wirth15 observed that “Data Structures + Algorithms = Programs”. We can amend this to “known validated data resources + known validated algorithms = validated Web resources.”
There is relatively little practice of public validation of data resources and certification of algorithms in the field of chemistry, but without this, a global chemical semantic web is difficult to implement. This article explores the basis for such interoperability and outlines a working proof of concept. We hope that, in the long-term, appropriate bodies such as IUPAC and other learned societies might come to oversee this practice; until then, the Blue Obelisk can be seen as an informal, neutral mechanism to which those interested in open semantics can contribute.
An interoperable chemical approach requires at least the following communally agreed upon components in its architecture (in no particular order): terminology, data typing, extensible data structures, conformance specification and tools, links and references, namespaces, and metadata for provenance and discoverability.
Syntactic support for all of these is provided by Chemical Markup Language (CML)16 and other XML namespaces (XHTML, MathML, etc.). This article is largely concerned with how the semantic containers for terminology, data, and algorithms are populated. There is also an important need for machine-enforceable behavior, which may also benefit from inheritance mechanisms but is not discussed in this work.
Our design and practice is heavily influenced by the practice and specifications from the International Union of Crystallography (IUCr). For the past three decades, the IUCr, through its Data Commission and other bodies, has actively developed communal practice for the interchange of data. One of us (P.M.-R.) has been associated with the Committee for the Maintenance of the CIF Standard (COMCIFS) project for a decade. The crystallographic information file (CIF) is the latest design of the IUCr's semantically rich data structures and is fully described in this journal and the recently published Volume G of the Int. Tab. The primary approach is through dictionaries, each of which can describe a subdomain (e.g., core, macromolecules, powder diffraction, publications, etc.). Any valid crystallographic data must conform to one or more dictionaries. The dictionaries are similarly constrained by a dictionary definition language (DDL) which is also recursively conformant.
The groundbreaking DDL and CIF specifications are the major vehicle for publications of crystallographic information, both textual and numeric. The community has developed software for validation and processing; though, the full power of the DDL is only recently becoming realized. DDL and CIF predated XML by a decade and are almost isomorphic to XML Schema (XSD) and XML in their architecture. CIF dictionaries traditionally describe the human-readable meaning of a term, together with its structure and constraints (cardinality, lexical form, numeric range, enumerations, etc.).
This architecture can reasonably be considered an ontology for the hard sciences. Because the semantics of crystallography have been well-understood for many decades, much of the ontology, including the algorithms, can be “hard-coded.”
More recently, through the dREL specification, the IUCr has started to add machine-enforceable semantics into their dictionaries.
Chart 1 shows a typical CIF dictionary entry using the starDDL approach (courtesy of Prof. S. R. Hall and Dr. N. Spadaccini). This specification is being actively considered by the IUCr's COMCIFS committee.
Chart 1.

Example of a CIF Dictionary Entry
Much of this example is self-explanatory. description.text (within ; ... ;) is the human-readable meaning, where there are references to other dictionary items. _type.container, _type.value, and _units.code correspond to 〈scalardataType=“float” units=“daltons”〉 in CML. The enumeration.range term describes a non-negative integer (e.g., xsd:nonNegativeInteger in XML Schema). The main enhancement is the machine-readable semantics in the method.* loop_. In this loop, a piece of code, based on Python and extended in the dRel language, describes the precise algorithm for the evaluation of the atomic mass of the cell. It defines a mass, initially zero, and a list of atom_types in the data object (the CIF). The atom_types have subfields number_in_cell (provided by the author) and atomic_mass (from a lookup table provided by IUCr). The sum of the atomic masses of all of the atoms is returned as _cell.atomic_mass, the identification of the dictionary entry.
These dictionaries are now compilable and executable in a proof-of-concept system.50400bb00017 They are powerful enough to allow the complete calculation of many crystallographic quantities (e.g., structure factors from atomic sites and form factors). The code can be run directly as Python, in Java through Jython, and compiled into other languages through the JJTree compiler.
This type of approach has great benefits for chemistry. Many of the BO algorithms (e.g., hundreds of JUMBO18 methods) are sufficiently simple to be documented as machine-enforceable semantics. The dictionary approach enforces communal semantics for objects (e.g., through Octet); for example, a molecule contains atoms and bonds which can provide dRel-like iterators.
There may be concerns about using a procedural language rather than a functional one (e.g., Scheme or LISP). We believe that the approach above is easily implemented and can run in a wide range of environments. It has the benefit of synergy with the code and systems developed in crystallography.
Note that the approach also contains a precise identification of, and therefore retrieval of, algorithms. Thus, _cell.atomic_mass.EVALUATION is a precise pointer to a defined algorithm. The BO approach is informed by this architecture; though, the precise syntax and semantics use XML-based approaches rather than CIF.
3. The Blue Obelisk Dictionary
The Blue Obelisk Chemoinformatics Dictionary is our effort of defining a standard set of chemoinformatics algorithms.50400bb00019 If a software project implements one of these algorithms, they can refer to this dictionary. By using unique identifiers, the dictionary allows using Web search engines, like Google.com, to find implementations for an algorithm in the dictionary. A similar dictionary has been developed for QSAR descriptors previously.50400bb00020
3.1. The Dictionary. The dictionary uses the following technologies: Scientific, Technical, and Medical Markup Language (STMML; http://www.xml-cml.org/stmml/) is used as a general container, and Mathematical Markup Language (MathML) is used to contain mathematical formulas. Likewise, scalable vector graphics (SVG) could be used to add graphics to the dictionary; though, this is currently not used. References are contained in BibTeXML, an extended markup language for managing bibliographies. The full source of the latest XML source for the dictionary can be retrieved from ref (50400bb00021).
The XML document is accompanied by an XML Schema document that encompasses the used XML languages. This allows XML-aware editors to syntactically validate the document and filter out syntax errors in either of the three XML languages.
Each entry in the dictionary has an associated identifier (id), which is unique throughout the XML document. When XML namespace technologies are used, a worldwide unique identifier can be composed that uniquely points to the entry in the dictionary. For example, by defining a namespace http://qsar.sourceforge.net/dicts/blue-obelisk with a related prefix blue-obelisk, one can uniquely point to an entry describing a Kabsch algorithm to align two molecules (id=alignmentKabsch)22 within this namespace by referring to blue-obelisk:alignmentKabsch.
Chart 2 is an example of an entry currently used in the Blue Obelisk dictionaries. In this example, an entry is defined for an algorithm that finds the smallest set of smallest rings, given a molecular graph. BibTeXML is used using the bibtex namespace prefix to cite the article in which the algorithm was described. The entry has a bit of meta content using the Dublin Core standard, for which the namespace uses the prefix dc. Additionally, a classification is made (into the area of graph theory), and a related entry is mentioned.
Chart 2.

Example of an XML Dictionary Entry
Extensible stylesheet language transformation (XSLT) is used to transform the XML source code into an XHTML document which can be displayed by a MathML-aware Web browser, like Mozilla Firefox.
3.2. Finding Implementations. The Blue Obelisk Movement agreed on using the same namespace prefix, that is, blue-obelisk, allowing Web pages for specific software projects to cite entries in the dictionary. Links from those pages currently must be made explicitly, but having the citations on those pages allows a Web search engine to easily find software projects that implement a specific algorithm. The XHTML Web page generated from the XML source of the dictionary contains, for each entry, a link to Google.com that shows available implementations of that algorithm (see Figure 2). This setup provides a powerful tool to find software that implements published algorithms.
Figure 2.
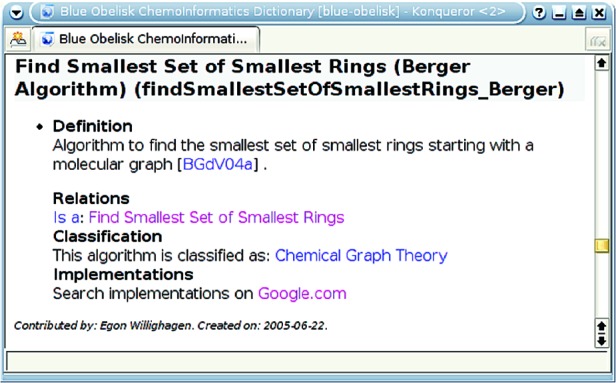
Screen shot of the XHTML output of the Blue Obelisk Chemoinformatics Dictionary showing the “Search implementations on Google.com” feature.
At the time of writing, CDK and Jmol each provide a Web page that cites and links to individual Blue Obelisk Chemoinformatics Dictionary entries.50400bb00023,50400bb00024 The Open Babel project has also included links to the dictionary in its developer documentation and is in the process of producing a complete index of entries as a separate Web page. All of the projects are continuing to add entries to the dictionary for common algorithms.
4. The Blue Obelisk Repository
Because many chemoinformatics projects rely on accurate atomic and molecular data such as atomic masses, isotopes, electronegativities, van der Waals radii, covalent radii, and so on, we have initiated a repository of a standard set of chemoinformatics data, building on the processes involved in the dictionary mentioned above.
Conventional standards bodies, such as IUPAC, have established a variety of published data, particularly on isotopes, atomic masses, elemental abundances, element symbols and names, and so on. Many chemoinformatics algorithms, however, rely on other data which may not have a clear-cut definition. For example, there is no obvious way to specify a van der Waals radius—not all elements are perfectly spherical, and multiple definitions exist including those taken from crystal structures, gas-phase measurements, and molecular mechanics force fields.50400bb00025−50400bb00029
To address these issues, the Blue Obelisk Movement has established the Blue Obelisk Data Repository.50400bb00030 Software can use and refer to this repository when it needs standardized data for a wide range of chemical properties and other facts, of which an overview is given in Table 2. It is anticipated that, over the next year, the repository will considerably increase in the amount of available data.
Table 2.
Current Content of the Data Repository, with a Few of the Used Sources.
| property type | property | sources |
| physical properties | isotope abundances | |
| isotope masses | 31 | |
| atomic masses | 32 | |
| ionization energies | ||
| chemical properties | affinities radii | 33 |
| electronegativities | ||
| element densities | ||
| discovery | year of discovery | |
| name and etymology | ||
| other | atom type definitions | |
| 2D and 3D coloring schemes |
The repository uses CML and dictionaries to allow the explicit markup of data types, units, and the experimental errors, as well as metadata like bibliographic sources, creation dates, and indications of authority. An example entry in the Blue Obelisk Data Repository is presented in Chart 3 and lists properties for hydrogen. For example, it states that the ionization energy is 13.5984 eV and that the mass is 1.007 94 amu. It does not explicitly state which mass is meant but refers, for the definition, to the Blue Obelisk Dictionary (see Section 3).
Chart 3.

Example of a Blue Obelisk Data Repository Entry
5. Web Services
The preceding material has described how chemoinformatics data can be managed and accessed in a collaborative manner. Another aspect of collaboration is the use of distributed functionality, that is, the use of function implementations that are not necessarily on the local machine. An example of this type of approach is the use of Web services. Though Web-based applications are ubiquitous, they are generally full-fledged applications that are monolithic in nature. The term Web services refers to functionality that can be accessed over the Internet in a programmatic manner. In the context of chemoinformatics, this means that a programmer can access functions, which, for example, calculate binary fingerprints, over the Internet without having to understand what language the underlying function is written in or whether the function is up-to-date. Of course, this implies that the calling mechanism for the given function is well-defined and that the maintainer has kept it up-to-date. This approach is useful on a smaller scale, say, at the organizational level. The advantage of having Web-based services implies that updates and modifications can be made on a single server, rather than requiring updates on individual machines.
We have used the CDK to provide Web services for molecular similarity and descriptor calculations, available at http://blue.chem.psu.edu/rajarshi/code/java/cdkws.html. Access to these services can be programmatic (using the SOAP34 protocol) or by a Web-based interface which simply calls the service and presents the results. Since the algorithms are well-documented and the calling mechanism is well-defined, the service provides a relatively transparent method to obtain chemoinformatics functionality in a distributed manner.
The downside of Web service functionality is that the user does not have control. This can be a problem if the service is not documented, but at the same time, it can be an advantage in that it relieves the user of the maintenance of yet another library. Furthermore, with the advent of open source and open data, a user is free to investigate the inner workings of a Web service if he or she so wishes. This would allow the user to ensure that the Web service does indeed do what it advertises. Once again, this depends on the fact that the maintainer of the Web service actually assigns an open license to the Web service (in terms of access as well as code). Clearly, increased usage of Web services is dependent on the transparency of the service. That is, a user must be able to ensure that a Web service does indeed do what it says and should be able to rely on the provider of the service. We believe that the open principles underlying the Blue Obelisk Movement are conducive to the development of transparent Web services which provide easy access to a variety of functionalities in a distributed manner.
6. Social Aspects
It has been mentioned previously that the Blue Obelisk Movement is a communal effort. Given the three goals of the movement, it is obvious why such an endeavor must be a community effort rather than that of an individual. In this sense, the Blue Obelisk Movement characterizes the nature of open source development in general and serves as an example of how this mode of development can be applied to problems in the field of chemical algorithms, standards, and data. A striking feature of the Blue Obelisk Movement is the wide variety of contributors to the individual projects that make up the movement. Contributors range from full professors to graduate students to commercial employees. The contributions themselves range from things as large as entire programs or frameworks to things as small as small amounts of data (e.g., to the data repository) or bug reports. However, it should be understood that, though a bug report may appear to be a minor contribution compared to a whole framework, each contribution plays a vital role in the communal development and peer review of these projects.
At the same time, it is important to realize that open source efforts represented by the Blue Obelisk movement do not always involve renumeration. Thus, in many cases, the contributors work on the respective projects in their spare time. This leads to the situation where some areas in a project do not get as much attention as others, simply because it has not caught the attention of a contributor or because of a lack of expertise among the contributors. In many cases, contributions to these projects are the result of a developer having “an itch” that needed to be “scratched.” Thus, compared to commercial projects, it may appear that the projects represented by the Blue Obelisk movement lack in certain areas. Given the open nature of these projects, it is a simple matter for anybody with the interest and expertise to contribute to such an area, thus filling the gap.
The above discussion paints a picture of many people contributing whatever they feel like. Naturally, this would lead one to think of a chaotic development process. How is all this managed? This is an important question because the contributors to the Blue Obelisk projects are located all over the world. Furthermore, most projects are large enough that a single person cannot always manage the contributions from a large user community.
The fundamental mechanism for distributed communal development is mailing lists, that is, via e-mail. Mailing lists are the mode by which the majority of decisions are made by the community for a given project, both in terms of use and development. Decisions are made by consensus; although, sometimes, the “benevolent dictator” model of development is followed. Mailing lists also serve as archives of discussion, in addition to the use of traditional Web pages and collaborative Web pages (Wiki) for the development of documentation. A more real-time mode of communication is the use of Internet relay chat, which allows multiple people to “convene” in a virtual room and communicate in real time. In general, this is restricted to text, but current instant messaging services allow for the use of both audio- and video-based communication. This type of interaction is very fruitful, because contributors can discuss current problems and decisions in real time as they are working on the projects themselves.
These methods represent approaches to communication between the contributors. But how are the contributions (such as code or documents) themselves managed? Once again, this is a very important question because multiple people will be working on a program or document, and manually managing individual contributions does not scale for projects of even moderate size. The workhorses for managing actual contributions are version control systems such as CVS or Subversion. These allow multiple contributors to submit changes to a program file or a document to a centrally located repository. If multiple contributors make changes to the same document, the system allows them to intelligently merge the resultant conflicts. These systems also allow developers to track changes and essentially view the “history” of a project. Workflows and Web services can also be used in the development process, and the utility of such types of applications has been mentioned previously.
Many of the Blue Obelisk projects make use of services provided by Sourceforge.net, which is a community effort to provide open source projects with a set of tools and functionality for efficient code maintenance and communication. The site supports a number of features such as CVS, mailing lists, bug trackers, and so on, all of which are freely available to open source projects.
Clearly, current Internet-based technology allows for easy and efficient management of contributions to the various Blue Obelisk projects from contributors located all over the world. In a sentence, the Blue Obelisk Movement is an example of the use of open source technology and methods to customize tools and social practices for the development of chemical information services.
7. Conclusion
We have described a communal effort to realize interoperability in chemical informatics, which we call the Blue Obelisk Movement, named after the first meeting place of our community. The BO Movement currently consists of more than 10 open source and open data projects all related to chemoinformatics. We identify concepts and algorithms, codify them in a collaborative dictionary, and link them to concrete implementations in Blue Obelisk projects and beyond to make them machine-searchable. We have started a public repository of chemical data of general interest, including data for chemical elements and isotopes (boiling points, colors, electron affinities, masses, covalent radii, etc.), definitions of atom types, and more. All of the data is augmented with documentation, citations of origin, and a bibliography. We are working on a system of Web services to provide access to chemoinformatics functionality without the knowledge of the details of the individual implementation and without the need to master the installation and programming interface of yet another chemoinformatics library. We emphasize that this work in progress, because of its emphasis on interoperability, has a value beyond that of open source and open data efforts. While standardization efforts in chemistry have a long history, modern computing and data processing, the Internet, and the World Wide Web have, for the first time, created the possibility of effortlessly searchable and reusable data and computer programs. Thus, this article addresses the “old guard” of developers and asks them to contribute their wisdom and their work. The result can be the survival of a work of a lifetime which otherwise might not survive the emeritation or the next sale of the company. This article is also addressed to newcomers asking them to adopt the ideas of open data and software from the very beginning. We welcome those in commercial organizations. What is prized is contributions that help support the communal vision (e.g., Raymond35). Our approach is not incompatible with commercial systems; though, the preservation of authorship moral rights is taken very seriously.
Acknowledgments
The authors thank all of the members of the Blue Obelisk subprojects for their continuing support. Rich Apodaca's work on creating a standardized chemoinformatics API are especially acknowledged. Each is a result of open community involvement. P.M.-R. and C.S. are grateful for financial support by the German Academic Exchange Service (DAAD) and the British council. We express our thanks to contributions to the Blue Obelisk data repository by the Carsten Niehaus and the Kalzium teams, which were started after the submission of this article and influenced revisions to the manuscript.
References
- Steinbeck C.; Han Y. Q.; Kuhn S.; Horlacher O.; Luttmann E.; Willighagen E. The Chemistry Development Kit (CDK): An open-source Java library for chemo- and bioinformatics. J. Chem. Inf. Comput. Sci. 2003, 43, 493–500. 10.1021/ci025584y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Open Babel Chemical File Format Conversion Package. http://openbabel.sourceforge.net/ (accessed Feb 2005).
- Steinbeck C.; Kuhn S.; Krause S. J. Chem. Inf. Comput. Sci. 2003, 43, 1733–1739. 10.1021/ci0341363. [DOI] [PubMed] [Google Scholar]
- JOELib − A Java Based Computational Chemistry Package. http://joelib.sourceforge.net/ (accessed Feb 2005).
- Raymond E. S.The Cathedral and the Bazaar, 1st ed.; O'Reilly and Associates, Inc.: Sebastopol, CA 95472, 1999.
- The Open Source Initiative (OSI). http://www.opensource.org/ (accessed Sep 2005).
- Weaver D. C. Build vs. Buy vs. Both. Pharm. Discovery 2005, 42–43. [Google Scholar]
- DeLano W. L. The case for open-source software in drug discovery. Drug Discovery Today 2005, 10, 213–217. 10.1016/S1359-6446(04)03363-X. [DOI] [PubMed] [Google Scholar]
- Miličević A.; Nikolić S.; Trinajstić N. Coding and Ordering Kekulé Structures. J. Chem. Inf. Comput. Sci. 2004, 44, 415–421. [DOI] [PubMed] [Google Scholar]
- Aires-de Sousa J.; Gasteiger J.; Gutman I.; Vidović D. Chirality Codes and Molecular Structure. J. Chem. Inf. Comput. Sci. 2004, 44, 831−836. 10.1021/ci030410h [DOI] [PubMed] [Google Scholar]
- Labute P. On the Perception of Molecules from 3D Atomic Coordinates. J. Chem. Inf. Model. 2005, 45, 215–221. 10.1021/ci049915d. [DOI] [PubMed] [Google Scholar]
- Murray-Rust P.; Rzepa H Chemical Markup, XML, and the World-Wide Web. 2. Information Objects and the CMLDOM. J. Chem. Inf. Comput. Sci. 2001, 41, 1113–1123. 10.1021/ci000404a. [DOI] [PubMed] [Google Scholar]
- Steinbeck C.; Krause S.; Willighagen E. JChemPaint - Using the Collaborative Forces of the Internet to Develop a Free Editor for 2D Chemical Structures. Molecules 2000, 5, 93–98. 10.3390/50100093. [DOI] [Google Scholar]
- McNaught A. D.; Wilkinson A.. Compendium of Chemical Terminology, 2nd ed.; Blackwell Science, Inc.: Malden, MA, 1997.
- Wirth N.Algorithms + Data Structures = Programs; Prentice Hall: Upper Saddle River, NJ, 1976.
- Murray-Rust P.; Rzepa H Chemical Markup, XML, and the World-Wide Web. 1. Basic Principles. J. Chem. Inf. Comput. Sci. 1999, 39, 928–942. 10.1021/ci990052b. [DOI] [Google Scholar]
- Hall S.; Spadaccini N.; du Boulay D.; Castleden I. Semantics for Scientific Data: Smart Dictionaries as Ontologies. http://www.biomedchem.uwa.edu.au/our_people/homepages/hall/swwspaper (accessed Sep 2005).
- JUMBO, A Full Kit for Building CML Schemas. http://wwmm.ch.cam.ac.uk/moin/Jumbo4_2e6 (accessed Sept 2005).
- Hoppe C.; Murray-Rust P.; Steinbeck C.; Willighagen E. Blue Obelisk ChemoInformatics Dictionary. http://qsar.sourceforge.net/dicts/blue-obelisk/index.xhtml. (accessed Dec 2005).
- Steinbeck C.; Hoppe C.; Kuhn S.; Floris M.; Guha R.; Willighagen E. Recent Developments of the Chemistry Development Kit (CDK) − An Open-Source Java Library for Chemo- and Bioinformatics. Curr. Pharm.Des. 2005, in press. [DOI] [PubMed]
- The Blue Obelisk Dictionary of Algorithms. http://cvs.sourceforge.net/viewcvs.py/qsar/bo-dicts/blue-obelisk.xml?view=markup (accessed Dec 2005).
- Kabsch W. A Solution for the Best Rotation to Relate Two Sets of Vectors. Acta Crystallogr., Sect. A 1976, 32, 922–923. 10.1107/S0567739476001873. [DOI] [Google Scholar]
- CDK Reference to the The Blue Obelisk Dictionary of Algorithms. http://wiki.jmol.org/JmolBlueObelisk (accessed Dec 2005).
- Jmol Reference to the The Blue Obelisk Dictionary of Algorithms. http://almost.cubic.uni-koeln.de/cdk/cdk_top/docu/dictrefindex/ (accessed Dec 2005).
- Zefirov Y. V. Van der Waals radii and current problems of their application. Russ. J. Inorg. Chem. 2001, 46, 568–572. [Google Scholar]
- Batsanov S. S. Van der Waals radii of elements. Inorg. Mater. 2001, 37, 871–885. 10.1023/A:1011625728803. [DOI] [Google Scholar]
- Allinger N. L.; Zhou X. F.; Bergsma J. Molecular Mechanics Parameters. THEOCHEM 1994, 118, 69–83. 10.1016/S0166-1280(09)80008-0. [DOI] [Google Scholar]
- Bondi A. van der Waals Volumes and Radii. J. Phys. Chem. 1964, 68, 441–451. 10.1021/j100785a001. [DOI] [Google Scholar]
- Pauling L.The Nature of the Chemical Bond, 3rd ed.; Cornell University: Ithaca, NY, 1960.
- Hutchison G. R.; Murray-Rust P.; Steinbeck C.; Willighagen E. Blue Obelisk ChemoInformatics Data Repository. http://www.blueobelisk.org/repos/blueobelisk/ (accessed Sep 2005).
- Wapstra A.; Audi G.; Thibault C. The AME2003 atomic mass evaluation (I). Evaluation of input data, adjustment procedures. Nucl. Phys. A 2003, 729, 129. 10.1016/j.nuclphysa.2003.11.002. [DOI] [Google Scholar]
- Loss R. Atomic weights of the elements 2001 (IUPAC Technical Report). Pure Appl. Chem. 2003, 75, 1107–1122. 10.1351/pac200375081107. [DOI] [Google Scholar]
- Andersen T.; Haugen H.; Hotop H. Atomic weights of the elements 2001 (IUPAC Technical Report). J. Phys. Chem. Ref. Data 1999, 28, 1511–1533. 10.1063/1.556047. [DOI] [Google Scholar]
- Simple Object Access Protocol. http://www.w3.org/TR/soap/ (accessed on Aug 2005).
- Homesteading the Noosphere. http://en.wikipedia.org/wiki/Homesteading_the_Noosphere (accessed Sep 2005).