

Abstract
Phormidolide (1) is a polyketide bearing a 16-membered macrolactone produced by a cultured filamentous marine cyanobacterium. Its complex structure is recognizably derived from a polyketide synthase pathway, but possesses unique and intriguing structural features that prompted interest to investigate its biosynthetic origin. Stable isotope incorporation experiments confirmed the polyketide nature of this compound. We further characterized the phormidolide gene cluster (phm) through genome sequencing followed by bioinformatic analysis. Two discrete trans-type acyltransferase (trans-ATs) ORFs along with KS-AT adaptor regions (ATd) within the PKS megasynthases, suggests that the phormidolide gene cluster is a trans-AT PKS. Insights gained from analysis of the mode of acetate incorporation and ensuing ketoreduction prompted our reevaluation of the stereochemistry of phormidolide hydroxyl groups located along the linear polyketide chain.
Keywords: phormidolide, polyketide synthase, trans-AT, biosynthesis, macrolactone
Graphical Abstract
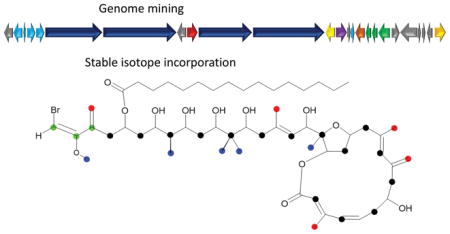
Integrating information gained from stable isotope incorporation studies with bioinformatic analysis of genome data, the biosynthetic pathway of phormidolide, a toxic cyanobacterial polyketide was identified and characterized. The biosynthesis occurs via a trans-AT system and features several interesting deviations from typical modular PKS pathways.
Introduction
Continuing advances in sequencing technology have led to a rapid increase in the number of sequenced cyanobacterial genomes, which in turn has led to an expansion in the tentative identification of the modular gene clusters responsible for the assembly and tailoring of numerous secondary metabolites.[1] One such secondary metabolite, phormidolide (1), is a brominated macrocylcic polyketide we isolated and characterized from a cyanobacterium collected from Sulawesi, Indonesia in 1995 and have sustained in laboratory culture.[2] Whereas the original work assigned the taxonomy of the producing organism to a Phormidium sp. based on filament morphology, subsequent 16S rRNA analysis led to its reassignment as a Leptolyngbya sp.[3] This compound showed strong toxicity to brine shrimp (1.5 μM) and possesses several unique structural features that involve a high level of modification of the carbon skeleton with hydroxy and pendant methyl groups, several points of unsaturation and a vinyl bromide functionality at one terminus of the molecule.
Preliminary inspection of the structure strongly suggested that 1 was a polyketide synthase-derived natural product. Polyketide synthases (PKSs) are large multifunctional enzyme systems that catalyze the formation of metabolites with carbon chains formed by ketide extensions of a “starter unit”, often acetyl CoA, propionyl CoA or more complex acyl CoAs. The extender units are most often malonyl CoA or methylmalonyl CoA derived from acetate and propionate, respectively. The main chain-building step is a decarboxylative condensation analogous to the chain elongation step of fatty acid biosynthesis. In PKSs the reactions of ketoreduction, dehydration and enoyl reduction that follow the condensation step are not obligate, such that a remarkable structural diversity ensues in the products.[4, 5]
Type I PKS enzymes are organized into modules wherein each module is used singly and catalyzes the addition and further modification of one extender unit to the growing polyketide chain. Thus, processing of the developing polyketide chain is done sequentially, similar to the operation of an assembly line, and the completed product is released and sometimes cyclized by a thioesterase (TE) at the pathway terminus. Predictions and inferences can be made through examination of gene sequences as they directly specify the structure of the product through the presence and functioning of ketoreductase (KR), dehydratase (DH) and enoyl reductase (ER) domains.[6–8] During the biosynthetic assembly process or following release, tailoring enzymes introduce modifications such as methylations, oxidations and halogenations to create a final structure. Ketoreductases generally control the configuration of the β-hydroxy group in a polyketide,[9] generating either an L- or D-configured alcohol.[10] Ketoreductases have also been shown to control the configuration of secondary methyls through epimerase activity and epimer-dependent reductase activity.[11]
Trans-AT PKS biosynthetic pathways represent an emerging class in the microbial world. In these systems, a separately encoded, discrete AT loads substrates onto the modular ACPs. Trans-AT systems are often observed to possess several deviations from cis-AT modular PKS systems including the splitting of modules between PKS open reading frames[12, 13], the presence of putatively inactive KS domains in the pathway[12–14] and β-branched methylation performed by a dedicated HMG-CoA synthase (HCS) cassette. [15–17] This cassette introduces a diversity of functionality, including methyl groups into growing polyketide chains at the unreduced C-3 position of the extended polyketide chain. The pendant methyl groups come from C-2 of an acetate unit in a Claisen-type condensation reaction; the branching acetate unit is subsequently decarboxylated and dehydrated to form a double bond, and can then be isomerized or reduced.[18] In trans-AT systems, duplicated or tandem ACP domains are often observed at β-branch points within modules of the PKS megasynthase architecture.[13, 17, 19]
In the present work, we characterized the phormidolide gene cluster as a trans-AT biosynthetic pathway using genome mining and bioinformatic analysis in addition to stable isotope feeding experiments. This allowed us to propose a model for phormidolide biosynthesis that is initiated by a three-carbon starter unit derived from primary metabolism and extended by 15 acetyl CoA extensions that are carried out by four PKS megasynthases. During construction of the carbon backbone, tailoring enzymes such as O- and C- SAM-methyl transferases, an iteratively engaged HCS cassette, and a halogenase complete the core framework. Appendage of the fatty acyl group is believed to occur post assembly by a fatty acyl CoA ligase. Additionally, a bioinformatic analysis suggested that a three-carbon starter unit, such as 1,3-bisphosphoglycerate, initiates the pathway. As a result of bioinformatic analysis of the KR domains within the pathway, we were motivated to re-examine the originally-deduced absolute configuration of 1. This involved formation of a tri-acetonide methyl ester derivative on which a Mosher’s ester analysis could be unambiguously performed.
Results and Discussion
[13C]-Acetate feeding experiments
Initial isotope feeding experiments utilized [1-13C] acetate, [2-13C] acetate and [1, 2-13C2] acetate. By 13C NMR, phormidolide isolated following [1-13C] acetate feeding showed 80–320% enhancements in carbons 1, 3, 5, 7, 9, 11, 13, 15, 17, 19, 21, 23, 25, 27 and 29 (cf. Table 1, Figure 1, and Figure S1 B) compared to naturally abundant phormidolide (Figure S1 A), indicating they all derive from C-1 of acetate. However, no enrichment was observed at C-31 of phormidolide, and revealed that the starter unit was not derived from a carbon-extended acetate unit, but rather, most likely arises from a three-carbon unit originating in primary metabolism. Similarly, 13C NMR analysis of phormidolide isolated from the [2-13C] acetate feeding studies showed that the backbone carbons 2, 4, 6, 8, 10, 12, 14, 16, 18, 20, 22, 24, 26, 28 and 30 are all derived from C2 of acetate (cf. Table 1, Figure 1 and Figure S1 C). This latter feeding experiment led to enrichment in the pendant methyl groups 34, 35, 36, 38 and 42, indicating that they are all derived from C2 of acetate through a presumed HMG-CoA synthase-like mechanism (cf. Table 1 Figure 1 and Figure S1 C). Next, feeding of Leptolyngbya sp. cultures with [1, 2-13C2] acetate confirmed the incorporation of intact acetate units in the polyketide backbone (Figure 1). Degeneracy of 13C NMR signals in phormidolide, along with many coupling constants being the same or nearly the same as a result of repetitive structural features, precluded analysis of the entire coupling pattern from the 1 D 13C NMR spectrum alone. However, a new ACCORD-ADEQUATE pulse sequence was devised which enabled inverse detection of the coupled 13C-13C pairs with enhanced sensitivity (Figure S2).[20] None of these acetate feeding experiments showed 13C incorporation at positions 31, 32 and 33 of phormidolide, and thus confirmed that the starter unit was not acetate-derived. This result excluded any Krebs cycle intermediate from being involved in the starter unit as these would have been labeled by [13C] acetate.
Table 1.
Relative enhancements of carbon resonances from phormidolide (1) biosynthetic feeding experiments.
| Position | Chemical Shift (ppm)a | [1-13C] acetate | [2-13C] acetate | 1Jcc values from[1, 213C2]acetate (Hz) | [CH3-13C] methionine |
|---|---|---|---|---|---|
| 1 | 167.5 | 20.6 | 0.6 | 77.4 | 0.0 |
| 2 | 118.3 | 0.2 | 9 | 77.4 | −0.1 |
| 3 | 152.1 | 32.8 | 1.3 | 52.1 | 0.5 |
| 4 | 134.1 | 1.0 | 9.2 | 52.1 | −0.2 |
| 5 | 132.6 | 12.8 | 0.4 | 41.2 | −0.3 |
| 6 | 44.1 | 0.6 | 11.8 | 41.4 | 0.1 |
| 7 | 73.1 | 16.9 | 0.6 | 38.3 | −0.2 |
| 8 | 43.8 | 0.3 | 10.0 | 38.3 | −0.1 |
| 9 | 141.5 | 15.4 | 0.2 | 54.4 | −0.2 |
| 10 | 132.4 | 1.2 | 5.5 | 54.4 | −0.4 |
| 11 | 133.4 | 17.5 | 0.7 | 41.4 | −0.1 |
| 12 | 48.3 | 0.2 | 10.2 | 41.4 | 0.0 |
| 13 | 76.7 | ||||
| 14 | 34.8 | 0.1 | 8.6 | 31.4 | −0.2 |
| 15 | 79.6 | 19.7 | 0.6 | 39.1 | −0.4 |
| 16 | 86.9 | 0.4 | 10.3 | 39.1 | −0.1 |
| 17 | 69.7 | 15.7 | 0.4 | 49.8 | −0.3 |
| 18 | 127 | −0.3 | 8.6 | 49.8 | −0.1 |
| 19 | 137.4 | 11.6 | 0.3 | 41.4 | −0.4 |
| 20 | 42.3 | 0.2 | 11 | 41.4 | 0.0 |
| 21 | 77.5 | 9.1 | 0.4 | 37.4 | −0.4 |
| 22 | 40.4 | −0.4 | 5.2 | 38.3 | −0.6 |
| 23 | 81.6 | 19.1 | 1.4 | 39.1 | 0.2 |
| 24 | 35.1 | −0.3 | 7.4 | 39.4 | −0.2 |
| 25 | 77.8 | 12.2 | 0.3 | 38.3 | −0.4 |
| 26 | 41.5 | 0.0 | 9.7 | 38.3 | −0.2 |
| 27 | 73.8 | 11.1 | 0.4 | 39.1 | −0.2 |
| 28 | 39.2 | −0.3 | 6.3 | 39.1 | −0.3 |
| 29 | 70.5 | 8.6 | 0.0 | 39.1 | −0.5 |
| 30 | 39.3 | −0.4 | 4.3 | 38.3 | −0.5 |
| 31 | 138.3 | 0.4 | 0.0 | −0.5 | |
| 32 | 158.4 | −0.2 | −0.1 | −0.7 | |
| 33 | 78.8 | 0.2 | 0.1 | −0.5 | |
| 34 | 13.9 | 0.5 | 9.0 | 39 | −0.3 |
| 35 | 113.8 | 1.2 | 16.7 | 71.3 | 0.6 |
| 36 | 16.8 | 0.0 | 5.8 | 42.2 | −0.4 |
| 37 | 21 | 1.7 | 0.8 | 10.9 | |
| 38 | 17.3 | 0.2 | 9.9 | 43.8 | 0.1 |
| 39 | 13.7 | 0.8 | 0.8 | 8.8 | |
| 40 | 21.6 | 0.6 | 0.2 | 6.5 | |
| 41 | 5 | 0.2 | 0.0 | 5.3 | |
| 42 | 122.1 | −0.4 | 3.5 | −0.5 | |
| 43 | 55.6 | 0.0 | 0.0 | 4.7 | |
| 44 | 173.7 | 8.6 | 0.2 | 57.5 | −0.5 |
Referenced to δCDCl3 at δ77.0. Bolded numbers indicate positions of 13C enrichment in phormidolide.
Figure 1.

Biosynthetic subunits of phormidolide (1). Results of feeding experiments using various stable isotope labeled substrates which demonstrated the origin of carbon atoms in phormidolide.
[1-13C, 18O2]-Acetate feeding
An additional feeding experiment of sodium [1-13C, 18O2] acetate was also performed, and the analysis of the 13C NMR spectrum revealed the expected upfield isotopic shifts of ca. 0.3 ppm[21] due to the presence of 18O incorporation at carbons C-1, C-15, C-29 and C-44 in phormidolide (1) (Figure S3). Smaller isotopic shifts (0.01–0.02 ppm) were present at carbons C-7, C-17, C-21, C-23, C-25 and C-27, which were initially difficult to observe. The furan oxygen bridging C-13 and C-16 did not show evidence for 18O incorporation, demonstrating that this oxygen atom is not acetate derived.
S-[Methyl-13C] methionine feeding experiment
In order to establish the origin of the four pendant methyl groups not labeled by [2-13C] acetate as well as the methoxy group, an S-[methyl-13C] methionine feeding experiment was performed. The NMR spectrum of phormidolide isolated from this treatment showed intense enrichment of carbon positions 37, 39, 40, 41 and 43, revealing that they are each derived from the S-methyl group of SAM (cf. Table 1, Figure 1 and Figure S4).
Except for the three-carbon starter unit, the origin of the entire backbone of phormidolide and all pendant methyl groups were thus established. A number of potential metabolic sources for the starter unit were fed, including [1-13C] pyruvate, [3-13C] alanine, [1-13C] propionate, [1-13C] glycine, [U-13C] glucose, and [1-13C] glyceric acid; (see Supporting Information). Only the [U-13C] glucose feeding experiment showed incorporation. However, the coupling pattern was consistent with a symmetric doubly labeled intermediate (see below) and not with intact incorporation of the three-carbon unit.
Identification and bioinformatic characterization of the phormidolide gene cluster
The Leptolyngbya sp. genome was sequenced using Illumina HISeq with DNA isolated from unicyanobacterial cultures. Assembly following binning of cyanobacterial sequences versus those derived from associated heterotrophic bacteria was accomplished with SPADes 3.0. This resulted in a draft genome of 8.5 MB with 47.39% GC content composed of 160 contigs mapping to 141 scaffolds, and an overall N50 of 301850. The program antiSMASH annotated 16 secondary metabolite gene clusters in the assembled cyanobacterial genome that were predicted to encode for: terpenes (2), bacteriocins (3), bacteriocin-NRPS (1), NRPS (1), NRPS-PKS (4), PKS (3) and others (2). As phormidolide is produced via a PKS pathway, there were three potential candidate gene clusters. One candidate cluster was 98.3 kb in length and had ORFs predicted to encode a HMG-CoA synthase, two enoyl-CoA hydratases, a fatty acid CoA ligase and a halogenase along with four large PKS ORFs and several carrier and transfer domains (Figure 2). These putative enzymes were consistent with those predicted to be involved in phormidolide biosynthesis. The individual PKS domains and remaining ORFs were thus annotated using Domain Enhanced Lookup Time Accelerated BLAST (DELTA-BLAST), which is useful in detecting protein homologs through annotating specific domains,[22] and aligned and compared to proteins with known functions (Table S1).
Figure 2.

Orientation of the phormidolide (1, phm) gene cluster. For further explanation of ORFs see Table S2.
The phormidolide PKS ORFs (phmE, F, H, and I) are predicted to encode megasynthases containing 17 ketosynthases (KS), 16 KS-AT adaptor regions (ATd), 10 ketoreductases (KR), three dehydratases (DH), four methyltransferases (MT), one of which is an O-methyltransferase (O-MT), twenty-one acyl carrier proteins (ACP), four enoyl-CoA hydratases (ECH), one FkbH-like domain (FkbH), one pyran synthase, and one NRPS-like condensation domain (C). Inserted after phmE and phmF there is an ORF encoding for a flavin-dependent monooxygenase (Figure 2). At the 5′ end of the cluster four ORFs (phmA-D) exist that are predicted to code for a discrete KS (PhmA), a discrete ACP (PhmB) and two discrete AT proteins (PhmC and PhmD). Following the PKS megasynthases are eight ORFs encoding tailoring and transfer proteins. The first (phmJ) encodes a halogenase with 62% amino acid identity and 77% similarity to JamD in the jamaicamide biosynthetic cluster. [18] In jamaicamide, this halogenase is predicted to form an alkynl bromide[18] whereas in phormidolide PhmJ is predicted to form a vinyl bromide moiety. The phmK gene is predicted to encode a hydrolase while phmL and phmM encode a phosphopantetheinyl transferase and fatty acid CoA-ligase, respectively. The next three ORFs (phmN, O, P) are predicted to code for proteins associated with an HCS cassette; and is comprised of two enoyl-CoA hydratases and an HMG-CoA synthase, respectively. The phmQ gene is predicted to code for a phosphoenolpyruvate synthase.
DELTA-BLAST searching revealed that the predicted phormidolide loading module contains an FkbH domain. FkbH-like enzymes are members of the HAD phosphatase superfamily that have been experimentally shown to shuttle glycolytic intermediates, specifically 1,3-bisphosphoglycerate, into PKS pathways.[23, 24] FkbH-like enzymes have two C-terminal domains necessary for starter unit processing and tethering of the glyceryl-S-ACP: a phosphatase domain and acyltransferase domain. The FkbH-like domain in the phormidolide pathway possesses both of these domains and shares the consensus sequence DXDX(T/V) with other FkbH-like enzymes as well as the active site D and C proposed to be involved in attachment and dephosphorylation of the phosphoglyceryl moiety (Figure 3A).[24] While the isotope-labeled feeding studies described above, which included [1-13C] glycerate, were not able to specifically identify this precursor, [U-13C] glucose was incorporated (Figures S5). However, the coupling pattern was consistent with a symmetric doubly labeled intermediate being incorporated in both orientations, due to couplings observed between C-31 and C-32 (63.6 Hz) and separately between C-32 and C-33 (93.5 Hz). Thus, intact incorporation of a three carbon fragment deriving from [U-13C] glucose was not observed. Intact incorporation of a three-carbon unit is expected to give rise to a complex coupling pattern at C-32 instead of two separate and easily identifiable 13C-13C couplings. One possible explanation for this incorporation pattern is provided by the high activity reported for enzymes in the Calvin cycle under lighted growth conditions and results in CO2 fixation from air.[25, 26] In this process, an unlabeled carbon atom ultimately deriving from atmospheric CO2 would be introduced next to two labeled 13C atoms coming from [U-13C] glucose or [U-13C] glycerol. This three carbon unit, in which two atoms are 13C-labeled and one is not, is predicted to proceed through a symmetrical intermediate, such that scrambling of the intact 13C incorporation occurs between C-31/C-32 and C-32/C-33. Thus, it is conceivable that the starter unit for phormidolide is a three-carbon unit derived from glucose such as phosphoenolpyruvate, or 1,3-bisphosphoglycerate, but intact incorporation from labeled precursors cannot be observed due to intrinsic characteristics of cyanobacterial primary metabolism. This could be further explored through expression of the phormidolide loading module and assessment of its substrate specificity via biochemical assays.
Figure 3.

Sequence alignments of conserved domains of the deduced amino acid sequence of genes in the phormidolide gene cluster. A) Domain alignment of FkbH-like phosphatases. PhmE FkbH domain contains the DXDX(T/V) consensus sequence * and catalytic aspartate (black triangle) and cysteine (black arrow) proposed to dephosphorylate and attach the phosphoglyceryl starter unit. Numbering based on the Tmn16 (AB193609) sequence. OzmB, ABA39082; TtmE, DQ088168; FkbH, AAF86387. B) Domain alignment of 17 KS domains from Phm pathway. The conserved sequence with catalytic His HGTGT is denoted by *, which is absent from predicted non-elongating KS0 (KS-9, KS-17). C) AT domain alignment of trans-ATs from the pederin (PedC, AAS47559; PedD, AAS47563) and bryostatin (BryP, ABM63531) pathways, a cis-AT from the jamaicamide A pathway (JamE, AAS98777) and a predicted AT-docking domain from the leinamycin (Lnm, AAN85523) pathway. Predicted trans-ATs PhmC and PhmD align well with trans- and cis-ATs, while the predicted AT-docking domain (PhmE-AT1) is truncated. The malonate-specific motif GHS[LVIFAM]G is denoted by *. D) ACP domain alignment. The active site sequence GXDS is denoted by * and the conserved tryptophan in ACPs associated with β-branching is identified with a black arrow. A “B” denotes ACPs predicted to be involved in β-branched methylation. E) KR domain alignment, including the conserved D1758 in B-type KRs. The conserved NADPH binding motif CGGXGXXG is denoted by *. The conserved motif HXAXXXXD is denoted by * and a black arrow indicates an amino acid motif of B-type KRs that generates the D-3-hydroxyacyl moiety. F) Phm DH and PS domains aligned to DH and PS domains from sorangicin (SorE, ADN68480; SorG, ADN68482). The conserved DH active-site motif HXXXGXXXXP is denoted by *.
While 15 KS domains are predicted for the complete biosynthesis of phormidolide, the PKS megasynthase ORFs include seventeen of these domains. However, two are predicted to be non-elongating KS domains (KS0). These latter domains are putatively non-functional and have been observed in many polyketide natural products including oocydin[12] and bacillaene.[14] The predicted KS0 domains in the phormidolide pathway are the most divergent in their percent identity compared to the 15 predicted elongating KSs, and further, they lack the catalytic His residue in the conserved motif HGTGT;[13] this residue is present in all 15 of the putatively elongating KS domains in the pathway (Figure 3B).
The 16 KS-AT adaptor (ATd) regions within the PKS megasynthases lack the conserved malonate-specific motif GHS[LVIFAM]G[27] that is present in PhmC and PhmD (Figure 3C). Moreover, PhmC and PhmD show 34.7% and 52.1% identity to the discrete trans-ATs PedC and PedD in the pederin biosynthetic gene cluster[15] as well as 33.5% and 47.5% identity to the biochemically characterized discrete trans-ATs BryP2 and BryP1 in the bryostatin gene cluster, respectively.[28] By contrast, the ATd regions show only 6–8% amino acid identity to full length cis-type AT and trans-AT domains but show 27% sequence identity to the previously described ATd regions from the leinamycin biosynthetic pathway.[29] The discrete trans-ATs (PhmC and PhmD) are predicted to possess the required acyltransferase activity for phormidolide biosynthesis. The methyltransferases encoded by phmF show the conserved motif for SAM-binding GXGXG,[30] while BLAST searching identified the putative O-methyltransferase in PhmE as a member of the Fkb family.[31] All ACP domains show the active site sequence GXDS[32] except for the stand-alone ACP encoded by phmB (GXNS). The ACPs associated with β-branching show a conserved tryptophan residue which identifies these ACPs for participation with HCS cassette enzymes (Figure 3D).[33] DELTA-BLAST searches classified each of the 10 KR domains as belonging to subgroup 2 with the characteristic Rossmann fold.[34] The NADPH binding motif GGXGXXG was present in all KRs except for two that showed GGXGXXA and GAXGXXG sequences, respectively (Figure 3E). The DH domains encoded by phmH and phmI show the conserved active-site motif HXXXGXXXXP,[35] while the DH in phmE shows HXXXTXXXXP (Figure 3F). Initially, DELTA-BLAST analysis annotated two repeating DH domains in the second elongation module in phmH. However, further analysis of this second DH domain showed low sequence identity to the other DHs in the pathway (~19%) and an absence of the active-site glycine (Figure 3F). This second DH-like domain showed greater sequence identity and shared conserved regions with previously described pyran synthase (PS) domains (Figure 3F). Pyran synthases (PS) are DH-like domains that have been shown to assist in ring closure during polyketide biosynthesis[36] and have been annotated in the gene clusters of sorangicin,[13] pederin,[15] and bryostatin.[16] The PS domain is positioned in a modular fashion within phmH to facilitate furan ring formation during the biosynthetic process.
Enoyl-CoA hydratase pairs (ECH1 and ECH2) have been shown to catalyze the successive dehydration and decarboxylation of an HMG-type intermediate resulting in β-branch type methylation, such as in the biosynthesis of curacin A and jamaicamide A.[35, 37] In addition to the ECH1 (PhmN) and ECH2 (PhmO) proteins of the HCS cassette in the phm gene cluster, there are also two sets of modular ECH domains within the megasynthase genes, phmE and phmI. This organization is unusual, although not unprecedented, and has been observed in the gene clusters that produce oocydin,[12] pederin,[15] onnamide,[38] and psymberin.[39] PhmN shows 44.3% sequence identity to the ECH1 protein CurE in the curacin gene cluster,[35] while the modular ECH1 domains in phmE and phmI show 13.3% and 16.7% sequence identity to CurE, respectively.[35] PhmO shows 43.4% sequence identity to the ECH2 domain CurF,[35] while the modular ECH2 domains in phmE and phmI show 38% sequence identity to CurF. All three possess residues that are predicted to form an oxyanion hole which will accommodate the intermediate enolate anion formed during the β-branching process.[40] The biochemical role of the modular ECH domains versus that of the ECHs in the HCS cassette remains to be determined experimentally.
Proposed biosynthesis of phormidolide
The proposed biosynthesis of phormidolide generally follows an “assembly line” construction of the macrolactone carbon skeleton that is then modified by tailoring enzymes located at the 3′ end of the gene cluster (Figure 4). A 1,3-bisphosphoglycerate starter unit is predicted to be loaded onto the FkbH domain of PhmE where it is modified via dephosphorylation and transferred to the ACP domain as glyceryl-S-ACP. Upon covalent tethering, dehydration and O-methylation are predicted to occur. The next PKS module is presumed to extend the molecule by two carbon atoms, and the newly formed β-ketone functionality is then transformed by the HCS cassette to a branching vinyl group. Interestingly, this module contains two modular ECH domains, which align with dehydrating ECHs and decarboxylating ECHs, respectively.41 This module also has tandem ACPs, a feature which is commonly observed at sites where β-branching methylation occurs via the HCS cassette.[13, 42] The final PKS module in PhmE extends the molecule by another acetate residue and the ketone is reduced by a KR. The enzymes encoded by phmF add five additional acetate units (C-15 to C-24) that are each modified by KR domains and methylated at C-2 positions via SAM methylation (C-37, 39, 40 and 41), including a geminal dimethylation at C-22. The HCS cassette modifies the ketone in the penultimate module of PhmF; this module does not have modular ECH domains but does possess tandem ACP domains. PhmH begins with a KS0 domain and three elongation modules and adds C-9 to C-14. We predict that PhmG, a flavin-dependent monooxygenase, adds a hydroxy group to C-16 which is then predicted to add via a PS-domain assisted Michael addition to the C-12,13 double bond. The olefin is created by a dehydratase in the second elongating module in PhmH. This prediction is similar to the putative creation of the furan ring present in the haterumalide oocydin.[12] β-branched methylation putatively occurs via enzymes encoded by the HCS cassette in the final module of the PhmH. A second ACP is present, which has been observed in domains targeted by the HCS cassette. However, this second ACP is present on the ensuing megasynthase, phmI, which encodes four PKS modules that are predicted to contribute carbon atoms C-1 to C-8 in phormidolide. The first module is modified by the HCS cassette enzymes and contains two modular ECH domains; this contrasts with the final elongating module in PhmI which is also modified by the HCS cassette but does not contain any ECH domains. The next module in PhmI is predicted to contain a second non-elongating KS domain. A final domain in PhmI was annotated by DELTA-BLAST as an NRPS-like condensation domain, and we predict that this domain positions the hydroxy group at C-15 to attack the C-1 carbonyl to cyclize and release the molecule, in a similar fashion to the mechanism of cyclization and release proposed in the cyanobacterial lipopeptide apratoxin.[43]
Figure 4.

Model of phormidolide (1) biosynthesis with 1,3-bisphosphoglycerate as the starter unit. The proposed mechanism of the β-branched methylation by the HCS cassette is detailed only for the first branching event. Subsequent β-branching events are indicated by the presence of HCS cassette enzymes.
Following release of the initially formed carbon backbone of phormidolide, the molecule is putatively halogenated by PhmJ. BLAST analysis indicates that PhmJ is an FAD-binding halogenase which is predicted to oxidize Br− to Br+ which undergoes electrophilic addition to the terminal olefinic bond of the starter unit. Additionally, palmitic acid is predicted to be transferred to the hydroxyl group at C-29 by the products of phmL and phmM, genes that encode an Sfp-type 4′-phosphopantetheinyl transferase and fatty acid-CoA ligase, respectively. As discussed above, phmN, phmO, and phmP code for two ECHs and an HMG-CoA synthase that comprise the HCS cassette and add the β-branched methyl groups or their equivalents. Typically five proteins constitute the full HCS cassette. In addition to an HMG-CoA synthase and two enoyl-CoA hydratase domains, a discrete ketosynthase and discrete ACP are needed. Amino acid sequence analysis of PhmA showed that it lacked the catalytic cysteine necessary for Claisen condensation in elongating KSs. However, the catalytic histidine necessary for decarboxylation was present. PhmA showed 35.8% sequence identity to TaK in the myxovirescin pathway.[44] TaK lacks the catalytic cysteine for condensation, but was experimentally shown to possess decarboxylase activity.[45] Therefore, we predict that PhmA is a decaroboxylating KS (KSs). PhmB, a putative acyl carrier protein, showed 37.7% amino acid sequence identity to TaB in the myxovirescin pathway, which was shown experimentally to serve as the free thiolation domain for β-branch formation in myxovirescin.[45] Thus, we predict that the Mal-S-PhmB substrate is decarboxylated by PhmA to generate the nucleophilic Ac-S-PhmB units for β-branching in the phormidolide pathway and the full complement of proteins in the HCS cassette is present. Finally, PhmQ is a predicted phosphoenolpyruvate synthase and may be involved in forming the starter unit or a precursor.
Biosynthetic control of configuration
Ketoreductases are usually responsible for producing the configuration of the hydroxy groups and alkyl subunits in polyketides.[9, 10, 11] Sequence examination[46] and domain swapping studies of KR-types in the erythromycin biosynthetic pathway have defined KR domains that catalyze reductions resulting in hydroxy groups having either R or S stereochemistry.[47, 48] An LDD motif has been identified in B-type KRs that generate a D-3-hydroxyacyl moiety.[10, 46] It is the second aspartate in this motif (D1758) which is most predicative of B-type KRs.[10, 46] A-type KRs possess a conserved W motif and generate an L-3-hydroxyacyl moiety.[10] Sequence alignments of the KR domains in the phormidolide pathway identified that 9 of the 10 KRs contained the conserved D1758 residue present in B-type KRs, but none contained the LDD motif (Figure 3E). The KR performing the reduction at C-15 (KR-7) is the one KR that does not possess the D1758 residue of B-type KRs nor does it contain the W motif of A-type KRs. Thus, because the KRs in the phm pathway possessed features that were similar to both A- and B-type KRs from bacterial PKS pathways, the KR type could not be distinguished based on this metric. Phylogenetic analysis of the KRs in the phm pathway did not show a clear divergence between KR subtypes when compared to KRs from other microorganisms (data not shown). In the original report on the structural characterization of phormidolide, the absolute stereochemistry of C-7 was determined using a variable temperature NMR experiment of a methoxyphenyl acetic acid derivative.[2] This stereochemical information was relayed to the remainder of the structure using ROESY, NOE and J-based configuration analysis.[2] However, with these ambiguities in the types of KRs present in the phormidolide pathway, reassessment of the absolute configuration phormidolide was warranted. A triacetonide methyl ester derivative of phormidolide was generated which contained a single free secondary alcohol. This derivative was analyzed using a modified Mosher’s ester protocol.[49] Two equal portions of the tri-protected phormidolide derivative were acylated with R-(−)- and S-(+)-α-methoxy-α-(trifluoromethyl)phenylacetyl chloride (MTPA-Cl). This yielded the C-7 R-ester from (S)-MTPA-Cl and the C-7 S-ester from (R)-MTPA-Cl. The 1H NMR and TOCSY spectra of the diastereomeric MTPA esters were examined and 1H resonances were assigned for each ester in order to calculate Δ(δS-δR) values. The results of this analysis showed that protons H-35, H-4, H-5 and H-6 had negative Δ(δS-δR) values, while the protons H-7, H-8, H-36, and H-37 had positive values for a 7 R configuration, which supports the original assignment of Williamson and coworkers. Analysis of the KR domains for the various modules incorporating acetate into phormidolide showed all but one (KR-7, C-15 hydroxyl) to be of the B-type on the basis of possession of an aspartic acid residue at position 1758. This would predict the generation of D-hydroxy groups at positions 7, 17, 21, 23, 25, 27 and 29. However, from the previous analysis of Williamson et al, and the stereoanalysis performed in this current work, the C-7 alcohol (introduced by KR-9) has been shown to be of L-configuration, and hence, indicates that the Asp at 1758 is not definitive for forming D-alcohols. Additionally, experimentally validated A type KRs, PlmKR1 and AmpKR2 possess the D1758 residue but generate L-alcohols.[10] On this basis, the original stereoanalysis by Williamson et al. appears correctly formulated (Figure S17).
Conclusions
Utilizing stable isotope incorporation studies and bioinformatic analysis, we have characterized the phormidolide (1) biosynthetic pathway as a variant of the trans-AT PKS type. Trans-ATs have rarely been reported in cyanobacteria, with the first being recently discovered from a lichen-associated Nostoc sp. and coding for the biosynthesis of the polyketide ‘nosperin’.[50] The identification of a trans-AT system in a filamentous marine cyanobacterium raises interesting questions about the distribution of these pathways, especially since this Leptolyngbya sp. is free-living and not known to be involved in symbiotic associations. Moreover, this characterization of the phormidolide PKS pathway as belonging to the trans-AT PKSs suggests that this type of pathway may be responsible for the formation of other cyanobacterial polyketides, such as oscillariolide[51] the caylobolides[52, 53] and bastimolide.[54]
Experimental Section
General Procedures: All 13C NMR spectra were recorded in CDCl3 on a Bruker AM 400 NMR spectrometer operating at a 13C resonance frequency of 100.61 MHz and processed with 1.0 Hz line broadening (zgig Bruker pulse program). 1H NMR spectrum of phormidolide was recorded with residual CHCl3 as the internal standard (δC 77.0, δH 7.26) on a Varian Unity 500 MHz spectrometer. 1H NMR spectrum and 2D NMR spectra of phormidolide acetonide derivatives were obtained on a Bruker 600 MHz spectrometer equipped with a 1.7 mm MicroCryoProbe. Semi-preparative HPLC was carried out using a Waters 515 pump system equipped with a Waters 996 PDA. High resolution electrospray ionization mass spectra (HRESIMS) were obtained using an Agilent 1290 Infinity system with an Agilent 6530 Accurate Mass Q-TOF LC/MS.
Culture Conditions
The phormidolide-producing cyanobacterium was grown in flasks containing SWBG11 medium, at 28°C with a 16 hr light/8 hr dark regime (5.4 to 7.0 μEinsteins). For feeding experiments either one or two Fembach flasks with 1.5 L of media were used. After 64 or 70 days, cells were harvested by filtration and the biomass was frozen at −80 °C until extraction.
Extraction protocol
The biomass was ground with mortar and pestle in 2:1 CH2Cl2/MeOH, infused at room temperature for 40 min in the same solvent, then boiled 2 × 15 min in fresh solvent. The resulting extracts were combined and evaporated in vacuo yielding an organic extract.
Isolation of phormidolide
In a typical experiment the crude extract was then subjected to Vacuum Liquid Chromatography (VLC, using TLC grade silica gel) with a range of solvents (0 to 100% EtOAc/hexanes followed by MeOH wash). The fraction eluting with 100% EtOAc was further fractionated by NP-HPLC (dual Phenomenex Luna 10 μm silica, 2 × 250 mm × 4.6 mm) with a 60% hexanes/35% EtOAc/5% IPA solvent system, 4.5 mL/min flow rate to yield pure phormidolide (1).
[1-13C] Acetic acid feeding
Sodium [1-13C] acetate (45 mg total/flask) diluted 1:1 with unlabeled NaOAc was introduced in 2 culture flasks containing 1.5 L SWBG-11 on days 49, 51, 53 and the biomass was harvested on day 56 (8.07 g wet weight, 1.57 g dry weight). A total of 19 mgs of phormidolide (1) were isolated after extraction, VLC and HPLC from 103 mg of organic extract using the process described above.
[2-13C] Acetic acid feeding
Sodium [2-13C] acetate (45 mg total/flask) diluted 1:1 with unlabeled NaOAc was introduced into 2 culture flasks with 1.5 L SWBG-11 on days 49, 51, 53 and the biomass was harvested on day 56 (10.25 g wet weight, 1.69 g dry weight). A total of 22 mg of phormidolide (1) were isolated after extraction, VLC and HPLC from 120 mg of organic extract using the protocol described above.
Sodium [1, 2-13C2] acetate feeding
Sodium [1, 2-13C2] acetate (30 mg total/flask) diluted 1:2 with unlabeled NaOAc was introduced into 2 culture flasks containing 1.5 L SWBG-11 on days 54, 56, 58 and the biomass was harvested on day 61 (7.04 g wet weight, 1.47 g dry weight). A total of 17 mgs of phormidolide (1) were isolated after extraction, VLC and HPLC from 163 mg of organic extract. Two other label incubation periods, 16 days and 24 days, were also performed in addition to this 7 day experiment, with analogous results.
Sodium [1-13C, 18O2] acetate feeding
Sodium [1-13C, 18O2] acetate (50 mg total) diluted 1:1 with unlabeled sodium acetate was provided to one culture in 1.5 L SWBG-11 on days 70, 72, 75 and the biomass was harvested on day 79 (7.3 g wet weight, 1.2 g dry weight). Thirty milligrams of phormidolide (1) were isolated after extraction, VLC and HPLC from 144 mg organic extract.
S-[Methyl-13C] methionine feeding
S-[Methyl-13C] methionine (70 mg total/flask) was fed to 2 culture flasks containing 1.5 L SWBG-11 on days 49, 51, 53 and the biomass was harvested on day 56 (14.8 g wet weight, 1.67 g dry weight). A total of 28 mgs of phormidolide (1) were isolated after extraction, VLC and HPLC from 158 mg of organic extract.
Calculation of 13C enrichments from precursor feeding experiments
The relative enrichments from exogenously supplied isotopically labeled precursors were calculated as follows. For each experiment, carbon resonance intensities for the natural abundance and enriched sample were tabulated and the enriched sample was normalized to natural abundance C-43 for the labeled acetate feeding experiments and to C-1 for the S-[methyl-13C] methionine experiment. Enrichment was calculated as the normalized integral of the enriched sample minus the integral of the natural abundance sample divided by the integral of the natural abundance for each carbon resonance (enriched minus natural/natural).
Genome sequencing and bioinformatics
Genomic DNA was isolated from live cultures of the phormidolide-producer isolated from Indonesia (collection code: ISBN3-Nov-94-8) using a standard phenol: chloroform: isoamyl alcohol (PCI) extraction protocol. In brief, two grams (wet weight) of cultured biomass were rinsed with fresh SW BG-11 media and flash frozen in liquid nitrogen and ground to a fine powder using a pre-chilled mortar and pestle. The powder was resuspended in 10 volumes of lysis buffer (10 mM Tris pH 8, 0.1 M EDTA pH 8, 0.5% w/v SDS and 20 μg/mL RNase) and incubated at 37 °C for 30 min. Proteinase K was added (100 μg/mL final concentration) and samples were incubated at 50 °C for one hour. After cooling the samples to room temperature, one volume of equilibrated phenol (Life Technologies) was added and samples were mixed for 10 min. Phases were separated by centrifugation at 3300 rcf for 10 min, and the aqueous phase was removed to a new tube. The phenol extraction was repeated twice followed by a chloroform: isoamyl (24:1) (Life Technologies) extraction. DNA was precipitated and purified by ethanol precipitation. Genomic DNA was quantified using a NanoDrop (Thermo Scientific), qualified by gel electrophoresis and sequenced using Illumina HISeq. Reads were then assembled using SPAdes[55, 56] genome assembler with error corrections using BayesHammer[57] followed by scaffold assembly with Opera and binning by coverage and GC content. Sequencing resulted in a genome of 8,574,384 bp with 160 contigs of which 71 were greater than 1000 bp. The largest contig was 717,955 bp. Secondary metabolite biosynthetic gene clusters were annotated using antiSMASH.[58] Predicted functions and protein domain organization of the putative phormidolide cluster (located on a single contig of 99059 bp) were made using Geneious[59] and NCBI DELTA-BLAST. Multiple sequence alignments of pathway proteins and domains were constructed using ClustalW with MEGA v. 5.2.2.[60]
Preparation of phormidolide acetonide derivatives
A total of 19.4 mg of pure phormidolide (1, Figure S6) was reacted with 11.4 mg of p-toluenesulfonic acid (pTsOH) in 2.71 mL of 2, 2-dimethoxypropane for 24 h. The reaction was quenched with 5% NaHCO3 and the mixture was partitioned between CH2Cl2 and H2O. The organic layer was dried under N2 and the residue (16 mg) was purified using reversed-phase HPLC (Synergi Hydro-RP, 4 μ, 250 × 10 mm) eluting with 100% MeOH at a flow rate of 4 mL/min. The major compound was isolated and characterized as phormidolide diacetonide methyl ester (2) (3.5 mg, 18% yield): 1H NMR: see Figure S7–S9 and Table S2; HRESIMS m/z [M + Na]+ 1211.6945 (calcd. for C66H109BrO13, 12111.6944). The 3.5 mg of phormidolide diacetonide methyl ester (2) was reacted with 2 mg of pTsOH in 1 mL of acetone for 24 h. The reaction was quenched with 5% NaHCO3 and the mixture partitioned between CH2Cl2 and H2O. The organic layer was dried under N2 and the residue was purified using reversed-phase HPLC (Kinetex C18, 5 μ, 150 × 10 mm) eluting with 100% MeOH, 3 mL/min. The major compound was isolated and characterized as phormidolide triacetonide methyl ester (3) (0.8 mg): 1H NMR: See Figure S10–S12 and Table S3; HRESIMS m/z [M + Na]+ 1251.7237 (calcd. for C69H113BrO13, 1251.7257).
Preparation of phormidolide triacetonide methyl ester MPTA esters
Phormidolide triacetonide methyl ester (0.4 mg) was reconstituted in 1 mL CHCl3 in a 2 mL vial to which 3.1 equivalents of dry pyridine and 4 equivalents of of (S)-(−)-α-methoxy-α-(trifluoromethyl)phenylacetyl chloride were added. The vial was capped and the reaction was stirred for 24 h. The identical procedure was repeated with an equal amount of the phormidolide derivative and (R)-(+)-α-methoxy-α-(trifluoromethyl)phenylacetyl chloride. The reaction mixtures were quenched with H2O and extracted with CH2Cl2. The preparation was passed through a 50 mg silica SPE column eluting with EtOAc-hexanes (1:1). R ester (S13 and S14): partial 1H NMR (500 MHz, CDCl3) δ 5.72 (1H, m, H-2), 6.19 (1H, m, H-4), 6.18 (1H, m, H-5), 2.43 (1H, m, H-6a), 2.36 (1H, m, H6b), 5.31 (1H, m, H-7), 2.24 (1H, m, H-8a), 2.21 (1H, m, H-8b), 5.71 (1H, s, H-10), 2.42 (1H, m, H-12a); 2.40 (1H, m, H-12b), 3.96 (1H, m, H-13), 2.36 (1H, m, H-14a), 1.73 (1H, m, H-14b), 4.16 (1H, m, H-15) 3.70 (3H, s, H-34), 2.28 (3H, s, H35), 5.08 (1H, s, H-36a), 4.92 (1H, s, H-36b), 1.75 (3H, s, H-37); S-ester (S15 and S16): partial 1H NMR (500 MHz, CDCl3) δ 5.71 (1H, m, H-2), 6.17 (1H, ovlp, H-4), 6.17 (1H, ovlp, H-5), 2.38 (1H, m, H-6a) 2.34 (1H, m, H-6b); 5.64 (1H, m, H-7), 2.28 (1H, m, H-8a) 2.25 (1H, m, H-8b), 5.73 (1H, s, H-10), 2.55 (1H, m, H-12a), 2.43 (1H, m, H-12b), 3.97 (1H, m, H-13), 2.41 (1H, m, H-14a), 1.80 (1H, m, H-14b), 4.16 (1H, m, H-15), 3.70 (3H, s, H-34), 2.28 (3H, s, H-35), 5.08 (1H, s, H-36a), 4.99 (1H, s, H-36b), 1.80 (3H, s, H-37).
Supplementary Material
Scheme 1.
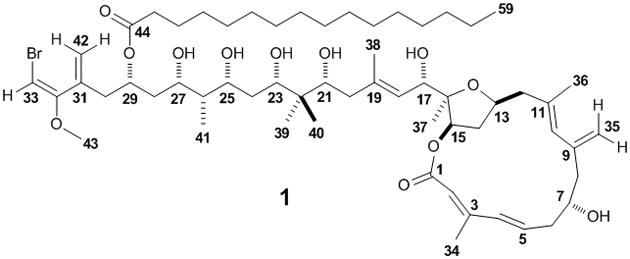
Structure of phormidolide (1).
Acknowledgments
This work was supported by NIH GM107550-03 and NIH CA108874. M.B. was supported by NRSA 5 T32 DA 7315-11. A.K. was supported by the Russian Science Foundation (grant 14-50-00069).
Footnotes
Accession number: The phormidolide (1) gene cluster has been deposited in GenBank with the accession number KT727016.
Supporting information for this article is available on the WWW under http://www.chembiochem.org or from the author.
References
- 1.Jones AC, Monroe EA, Eisman EB, Gerwick L, Sherman DH, Gerwick WH. Nat Prod Rep. 2010;27:1048–1065. doi: 10.1039/c000535e. [DOI] [PubMed] [Google Scholar]
- 2.Williamson RT, Boulanger A, Vulpanovici A, Roberts MA, Gerwick WH. J Org Chem. 2002;67:7927–7936. doi: 10.1021/jo020240s. [DOI] [PubMed] [Google Scholar]
- 3.Thornburg CC. PhD thesis. Oregon State University; USA: 2013. [Google Scholar]
- 4.Carreras CW, Pieper R, Khosla C. Topics Curr Chem. 1997;188:85–126. [Google Scholar]
- 5.Bentley R, Bennett JW. Annu Rev Microbiol. 1999;53:411–446. doi: 10.1146/annurev.micro.53.1.411. [DOI] [PubMed] [Google Scholar]
- 6.Hopwood DA. Chem Rev. 1997;97:2465–2498. doi: 10.1021/cr960034i. [DOI] [PubMed] [Google Scholar]
- 7.Katz L. Chem Rev. 1997;97:2557–2575. doi: 10.1021/cr960025+. [DOI] [PubMed] [Google Scholar]
- 8.Staunton J, Weissman KJ. Nat Prod Rep. 2001;18:380–416. doi: 10.1039/a909079g. [DOI] [PubMed] [Google Scholar]
- 9.Keatinge-Clay AT, Stroud RM. Structure. 2006;14:737–748. doi: 10.1016/j.str.2006.01.009. [DOI] [PubMed] [Google Scholar]
- 10.Bonnett SA, Whicher JR, Papireddy K, Florova G, Smith JL, Reynolds KA. Chem Biol. 2013;20:772–783. doi: 10.1016/j.chembiol.2013.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zheng J, Keatinge-Clay AT. Med Chem Commun. 2013;4:34–40. [Google Scholar]
- 12.Matilla MA, Stöckmann H, Leeper FJ, Salmond GP. J Biol Chem. 2012;287:39125–39138. doi: 10.1074/jbc.M112.401026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Irschik H, Kopp M, Weissman KJ, Buntin K, Piel J, Müller R. ChemBioChem. 2010;11:1840–1849. doi: 10.1002/cbic.201000313. [DOI] [PubMed] [Google Scholar]
- 14.Moldenhauer J, Chen XH, Borriss R, Piel J. Angew Chem Int Ed. 2007;46:8195–8197. doi: 10.1002/anie.200703386. [DOI] [PubMed] [Google Scholar]
- 15.Piel J. Proc Natl Acad Sci USA. 2002;99:14002–14007. doi: 10.1073/pnas.222481399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sudek S, Lopanik NB, Waggoner LE, Hildebrand M, Anderson C, Liu H, Patel A, Sherman DH, Haygood MG. J Nat Prod. 2007;70:67–74. doi: 10.1021/np060361d. [DOI] [PubMed] [Google Scholar]
- 17.El-Sayed AK, Hothersall J, Cooper SM, Stephens E, Simpson TJ, Thomas CM. Chem Biol. 2003;10:419–430. doi: 10.1016/s1074-5521(03)00091-7. [DOI] [PubMed] [Google Scholar]
- 18.Edwards DJ, Marquez BL, Nogle LM, McPhail K, Goeger DE, Roberts MA, Gerwick WH. Chem Biol. 2004;11:817–833. doi: 10.1016/j.chembiol.2004.03.030. [DOI] [PubMed] [Google Scholar]
- 19.Mattheus W, Gao LJ, Herdewijn P, Landuyt B, Verhaegen J, Masschelein J, Volckaert G, Lavigne R. Chem Biol. 2010;17:149–159. doi: 10.1016/j.chembiol.2010.01.014. [DOI] [PubMed] [Google Scholar]
- 20.Williamson RT, Marquez BL, Gerwick WH, Koehn FE. Magn Reson Chem. 2001;39:544–548. [Google Scholar]
- 21.Vederas JC. Nat Prod Rep. 1987;4:277–337. doi: 10.1039/np9870400277. [DOI] [PubMed] [Google Scholar]
- 22.Boratyn GM, Schäfer AA, Agarwala R, Altschul SF, Lipman DJ, Madden TL. Biol Direct. 2012;7:12. doi: 10.1186/1745-6150-7-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dorrestein PC, Van Lanen SG, Li W, Zhao C, Deng Z, Shen B, Kelleher BS. J Am Chem Soc. 2006;128:10386–10387. doi: 10.1021/ja0639362. [DOI] [PubMed] [Google Scholar]
- 24.Sun Y, Hong H, Gillies F, Spencer JB, Leadley PF. ChemBioChem. 2008;9:150–156. doi: 10.1002/cbic.200700492. [DOI] [PubMed] [Google Scholar]
- 25.Yang C, Hua Q, Shimizu K. Metab Eng. 2002;4:202–216. doi: 10.1006/mben.2002.0226. [DOI] [PubMed] [Google Scholar]
- 26.Tamoi M, Miyazaki T, Fukamizo T, Shigeoka S. Plant J. 2005;42:504–513. doi: 10.1111/j.1365-313X.2005.02391.x. [DOI] [PubMed] [Google Scholar]
- 27.Yadav G, Gokhale RS, Mohanty D. J Mol Biol. 2003;328:335–363. doi: 10.1016/s0022-2836(03)00232-8. [DOI] [PubMed] [Google Scholar]
- 28.Lopanik NB, Shields JA, Buchholz TJ, Rath CM, Hothersall J, Haygood MG, Håkansson K, Thomas CM, Sherman DH. Chem Biol. 2008;15:1175–1186. doi: 10.1016/j.chembiol.2008.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tang GL, Cheng YQ, Shen B. Chem Biol. 2004;11:33–45. doi: 10.1016/j.chembiol.2003.12.014. [DOI] [PubMed] [Google Scholar]
- 30.Wilkinson CJ, Frost EJ, Staunton J, Headlay PF. Chem Biol. 2001;8:1197–1208. doi: 10.1016/s1074-5521(01)00087-4. [DOI] [PubMed] [Google Scholar]
- 31.Singh S, McCoy JG, Zhang C, Bingman CA, Phillips GN, Thorson JS. J Biol Chem. 2008;283:22628–22636. doi: 10.1074/jbc.M800503200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.He J, Hertweck C. Chem Biol. 2003;10:1225–1232. doi: 10.1016/j.chembiol.2003.11.009. [DOI] [PubMed] [Google Scholar]
- 33.Buchholz TJ, Rath CM, Lopanik NB, Gardner NP, Håkansson K, Sherman DH. Chem Biol. 2010;17:1092–1100. doi: 10.1016/j.chembiol.2010.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Marchler-Bauer A, Derbyshire MK, Gonzales NR, Lu S, Chitsaz F, Geer LY, Geer RC, He J, Gwadz M, Hurwitz DI, Lanczycki CJ, Lu F, Marchler GH, Song JS, Thanki N, Wang Z, Yamashita RA, Zhang D, Zheng C, Bryant SH. Nucleic Acids Res. 2015;43:D222–D226. doi: 10.1093/nar/gku1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chang Z, Sitachitta N, Rossi JV, Roberts MA, Flatt PM, Jia J, Sherman DH, Gerwick WH. J Nat Prod. 2004;67:1356–1367. doi: 10.1021/np0499261. [DOI] [PubMed] [Google Scholar]
- 36.Pöplau P, Frank S, Morinaka BI, Piel J. Angew Chem Int Ed. 2013;52:13215–13218. doi: 10.1002/anie.201307406. [DOI] [PubMed] [Google Scholar]
- 37.Gu L, Wang B, Kulkarni A, Geders TW, Grindberg RV, Gerwick L, Håkansson K, Wipf P, Smith JL, Gerwick WH, Sherman DH. Nature. 2009;459:731–735. doi: 10.1038/nature07870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Piel J, Hui D, Wen G, Butzke D, Platzer M, Fusetani N, Matsunaga S. Proc Natl Acad Sci USA. 2004;101:16222–16227. doi: 10.1073/pnas.0405976101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fisch KM, Gurgui C, Heycke N, van der Sar SA, Anderson SA, Webb VL, Taudien S, Platzer M, Rubio BK, Robinson SJ, Crews P, Piel J. Nat Chem Biol. 2009;5:494–501. doi: 10.1038/nchembio.176. [DOI] [PubMed] [Google Scholar]
- 40.Geders TW, Gu L, Mowers JC, Liu H, Gerwick WH, Håkansson K, Sherman DH, Smith JL. J Biol Chem. 2007;282:35954–35963. doi: 10.1074/jbc.M703921200. [DOI] [PubMed] [Google Scholar]
- 41.Hamed RB, Batchelar ET, Clifton IJ, Schofield CJ. Cell Mol Life Sci. 2008;65:2507–2527. doi: 10.1007/s00018-008-8082-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.El-Sayed AK, Hothersall J, Cooper SM, Stephens E, Simpson TJ, Thomas CM. Chem Biol. 2003;10:419–430. doi: 10.1016/s1074-5521(03)00091-7. [DOI] [PubMed] [Google Scholar]
- 43.Grindberg RV, Ishoey T, Brinza D, Esquenazi E, Coates RC, Liu W, Gerwick L, Dorrestein PC, Pevzner P, Lasken R, Gerwick WH. PLOS One. 2011;6:e18565. doi: 10.1371/journal.pone.0018565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Simunovic V, Zapp J, Rachid S, Krug D, Meiser P, Müller R. ChemBioChem. 2006;7:1206–1220. doi: 10.1002/cbic.200600075. [DOI] [PubMed] [Google Scholar]
- 45.Calderone CT, Iwig DF, Dorrestein PC, Kelleher NL, Walsh CT. Chem Biol. 2007;14:835–846. doi: 10.1016/j.chembiol.2007.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Reid R, Piagentini M, Rodriguez E, Ashley G, Viswanathan N, Carney J, Santi DV, Hutchinson CR, McDaniel R. Biochemistry. 2003;42:72–79. doi: 10.1021/bi0268706. [DOI] [PubMed] [Google Scholar]
- 47.Cortes J, Wiesmann KE, Roberts GA, Brown MJ, Staunton J, Leadlay PF. Science. 1995;268:1487–1489. doi: 10.1126/science.7770773. [DOI] [PubMed] [Google Scholar]
- 48.Kao CM, McPherson M, McDaniel RN, Fu H, Cane DE, Khosla C. J Am Chem Soc. 1998;120:2478–2479. [Google Scholar]
- 49.Hoye T, Jeffrey CS, Shao F. Nat Protoc. 2007;2:2451–2458. doi: 10.1038/nprot.2007.354. [DOI] [PubMed] [Google Scholar]
- 50.Kampa A, Gagunashvili AN, Gulder TAM, Morinaka BI, Daolio MG, Miao VPW, Piel J, Andrésson OS. Proc Natl Acad Sci USA. 2013;110:E3129–E3137. doi: 10.1073/pnas.1305867110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Murakami M, Matsuda H, Makabe K, Yamaguchi K. Tetrahedron Lett. 1991;32:2391–2394. [Google Scholar]
- 52.MacMillan JB, Molinski TF. Organic Lett. 2002;4:1535–1538. doi: 10.1021/ol025759p. [DOI] [PubMed] [Google Scholar]
- 53.Salvador LA, Paul VJ, Luesch H. J Nat Prod. 2010;73:1606–1609. doi: 10.1021/np100467d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Shao C, Linington RG, Balunas MJ, Centeno A, Boudreau P, Zheng C, Engene N, Spadafora C, Mutka TS, Kyle DE, Gerwick L, Wang C, Gerwick WH. J Org Chem. 2015 doi: 10.1021/acs.joc.5b01264. [DOI] [PubMed] [Google Scholar]
- 55.Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, Lesin VM, Nikolenko SI, Pham S, Prjibelski AD, Pyshkin A, Sirotkin AV, Vyahhi N, Tesler G, Alekseyev MA, Pevner PA. J Comp Biol. 2012;19:455–477. doi: 10.1089/cmb.2012.0021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Nurk S, Bankevich A, Antipov D, Gurevich AA, Korobeynikov A, Lapidus A, Prjibelski A, Pyshkin A, Sirotkin A, Sirotkin Y, Stepanauskas R, Clingenpeel S, Woyke T, McLean J, Lasken R, Tesler G, Alekseyev MA, Pevzner PA. J Comp Biol. 2013;20:714–737. doi: 10.1089/cmb.2013.0084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Nikolenko S, Korobeynikov A, Alekseyev MA. BMC Genomics. 2013;14:S7. doi: 10.1186/1471-2164-14-S1-S7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Medema MH, Blin K, Cimermancic P, de Jager V, Zakrzewski P, Fischbach MA, Weber T, Takano E, Breitling R. Nucleic Acids Res. 2011;39:1–8. doi: 10.1093/nar/gkr466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kearse M, Moir R, Wilson A, Stones-Havas S, Cheung M, Sturrock S, Buxton S, Cooper A, Markowitz S, Duran C, Thierer T, Ashton B, Meintjes P, Drummond A. Bioinformatics. 2012;28:1647–1649. doi: 10.1093/bioinformatics/bts199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. Mol Biol Evol. 2011;28:2731–2739. doi: 10.1093/molbev/msr121. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.