

Abstract
The natural product paracaseolide A is a tetracyclic dilactone containing six adjacent stereocenters. It has an unprecedented skeleton and occupies unique structural space among the >200,000 characterized secondary metabolites. Six different research groups have reported a chemical synthesis of this compound, five of which used a thermal, net Diels–Alder [4+2] cycloaddition and dehydration at 110 °C to access the target by dimerization of a simple butenolide precursor. Here we report that this dimerization proceeds under much milder conditions and with a different stereochemical outcome than previously recognized. This can be rationalized by invoking a bis-pericyclic transition state. Furthermore, we find that spontaneous epimerization, necessary to correct the configuration at one key stereocenter, is viable and that natural paracaseolide A is racemic. Together these facts point to the absence of enzymatic catalysis (i.e., Diels–Alderase activity) in the cycloaddition and strongly suggest that a non-enzyme-mediated dimerization is the actual event by which paracaseolide A is produced in Nature.
Paracaseolide A (1a), a secondary metabolite first reported by Guo and coworkers in 2011,1 was isolated from the stem bark of Sonneratia paracaseolaris, a mangrove plant that was collected in Guangdong Province, China. It is reported to inhibit the action of the biologically relevant phosphatases CDC25B and PTP1B, kinases involved in cell cycle regulation (IC50 of 6.4 μM)1,2 and signal transduction in the insulin pathway (IC50 of 1.5 μM),3 respectively. Compound 1a has a fascinating, skeletally unique structure that quickly captured the attention of synthetic chemists. Our analysis led us to hypothesize that the carbon skeleton in 1a is assembled in Nature by a spontaneous, non-enzyme-mediated4 (here, Diels–Alderase-free) dimerization of the simple α-dodecenylbutenolide 3a as well as a facile epimerization event of the resulting Diels–Alder (DA) adduct. In fact, 3a was proposed as the possible biosynthetic precursor to 1a by Guo et al. The relevance of Diels–Alderase enzymatic action (or lack thereof) in biosynthesis5,6 remains a timely topic.7,8 Other biosynthetically relevant, spontaneous Diels–Alder dimerizations have been proposed and demonstrated; notable examples include those leading to carpanone9,10 and the endiandric acids.11,12
In the short period of time since the structure of paracaseolide A was revealed,1 researchers in five different laboratories have reported a total synthesis of 1a involving the same final key step.3,13,14,15,16 Namely, dimerization of the butenolide 3a (Fig. 1) accompanied by dehydration has been repeatedly used to produce 1a. In every instance 3a was heated to 110 °C to effect this overall transformation. In every instance the researchers initially rationalized the reaction to occur via an endo orientation of the two reacting monomers (see Fig. 1) and via the intermediacy of the endo Diels–Alder dimer 2. (In this manuscript, endo and exo refer to the relative orientation between the carbonyl group on the dienophilic component and the internal carbons of the 1,3-butadiene derivative in the cycloaddition transition state geometry and products derived therefrom.) This necessitated the supposition of a subsequent epimerization at C5 in the cyclohexenyl ring of 2 in order to account for the final formation of 1a. It is noteworthy that attempts to interconvert 5-epi-1a (not shown) with 1a (and vice-versa) by two groups of workers have been unsuccessful.14,15 These mechanistic and stereochemical dilemmas have recently been explored computationally by the research groups of Ganguly, Khan, and Mehta, leading those workers to conclude that a “stepwise mechanism [leading to 5-epi-2 (not shown)] is the likely pathway” and that “a concerted [4+2] cycloaddition and post-DA epimerization” is less likely.17
Figure 1. Structure and previous syntheses of paracaseolide A.
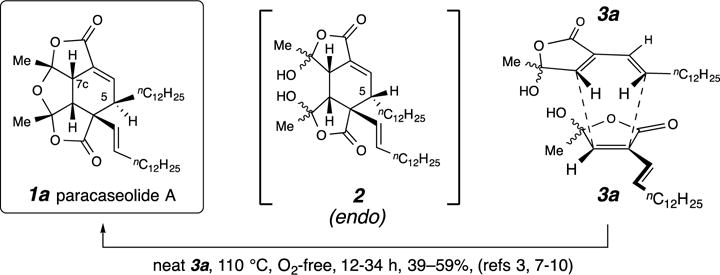
In several previous studies (refs 3, 7–10) concomitant thermal dimerization and dehydration of the monomeric alkenylbutenolide 3a was used as the key step. This cycloaddition has been presumed to proceed through the intermediacy of the endo-Diels–Alder adduct 2.
Results and Discussion
As reported here, our studies of this key Diels–Alder reaction reveal considerably different reaction energetics and stereochemical features to those reported by previous investigators. Namely, we show that dimerization of the alkenylbutenolide 3a [as well as its truncated analog 3b (Fig. 2)] occurs under much milder conditions than have been previously used. Moreover, the reaction proceeds, surprisingly, through an exo approach of the two reactants, as discussed in detail below. (Throughout this document compound structure numbers ending in “a” all contain n-dodecyl groups that correspond to the substituents present in the natural product; the structure numbers ending in “b” all contain a methyl in place of the n-dodecyl group and comprise a series of unnatural, truncated analogs.)
Figure 2. Strategy and methods for the synthesis of paracaseolide A and a truncated (methyl-containing) analogue.
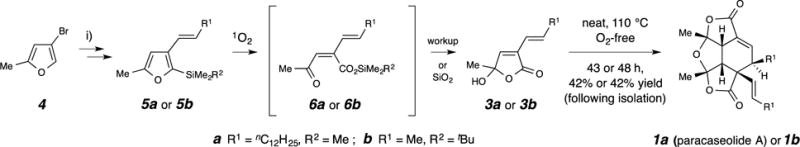
Paracaseolide A (1a) was prepared from 3a3,13,14,15,16 and the truncated analogue 1b from 3b by heating the neat compound in an oxygen-free-atmosphere. The monomers 3a and 3b were prepared by a two-step or four-step sequence, respectively, from 4-bromo-2-methylfuran.
Following similar strategies to those used by others,13,14,15 we synthesized the butenolide monomers 3a and 3b by the concise routes shown in Fig. 2, commencing with 4-bromo-2-methylfuran (4). The key step is the conversion of the silylated furan 5a/b to the racemic butenolide hemiacylal 3a/b with singlet oxygen, a reaction that proceeds by spontaneous cleavage of an initial endoperoxide to the intermediate O-silylated keto acid18 6a/b (observable by 1H NMR analysis). Desilylation provides 3a/b in which only the closed hemiacylal (or hydroxybutenolide) form was detected (>99%,1H NMR analysis in CD3OD). We too observed that heating 3a converted it to 1a, whose spectral data were in excellent accord with those reported.1,3,13,14,15,16,19
Insights gained using unnatural, truncated analogs (i.e., the “b” series of methyl-bearing compounds)
We used the simpler methyl-bearing analog 3b to gain useful insights that guided our later experiments with the n-dodecyl-containing 3a. We first verified that it behaved similarly to 3a when heated to 110 °C as a neat sample under nitrogen. That is, dimer 1b, a side-chain truncated analog of 1a, was formed as the dominant product (Fig. 2). More excitingly, we observed that when 3b, a liquid, was held as a neat sample, a new product appeared under much milder conditions (35 °C) than had been reported for 3a. The resulting dimer had not yet dehydrated. A crystalline sample of that new material was subjected to single crystal X-ray analysis, which revealed it to be the Diels–Alder adduct 7b (Fig. 3a). This dimerization occurs with a very high level of regio- and stereoselectivity. Most notably, the relative configuration among carbons 5/5a/7b/7c indicated that the [4+2] cycloaddition had proceeded by a pathway in which the diene had approached the dienophile with an exo orientation with respect to the carbonyl group on the dienophile (Fig. 3b).
Figure 3. The stereochemical outcome of spontaneous butenolide dimerization.
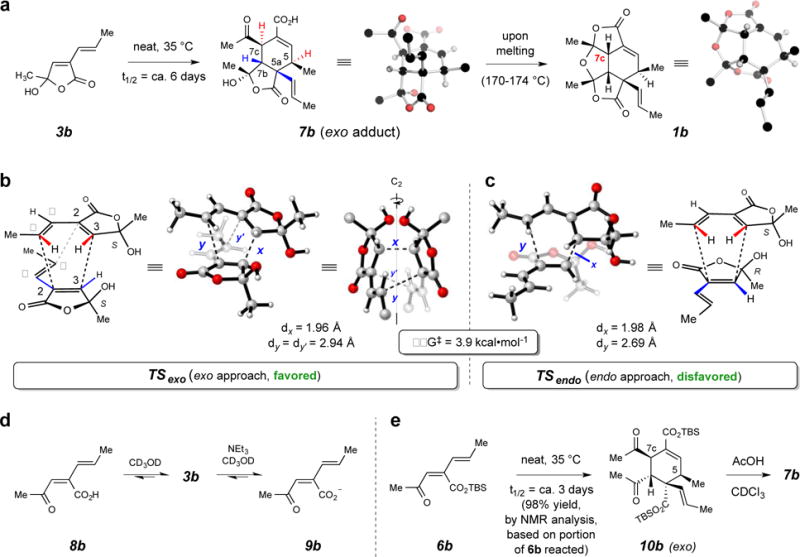
a, Facile dimerization of 3b cleanly produces the exo adduct 7b. b/c, Exo vs. endo modes of approach in the dimerization of alkenylbutenolide 3b; the computed TS structures [DFT, SMD/M06-2X/6-311+G(d,p) in 2-butanol] favor the C2-symmetric TSexo over TSendo. d, Neutral keto acid 8b prefers the closed hemiacylal 3b, but weak base promotes conversion of 3b to the ring-opened carboxylate 9b. e, tert-Butyldimethylsilyl (TBS) ester 6b readily dimerizes to 10b [97% yield, as judged by 1H NMR analysis based on the portion of remaining, unreacted 6b (ca. 10%); see pp S66–S69 Supplementary Information], the exo configuration of which was established by its conversion to the diacid 7b.
In an attempt to understand this stereochemical outcome, we performed density functional theory [DFT; SMD/M06-2X/6-311+G(d,p) in 2-butanol] calculations to evaluate the relative energies of transition state structures corresponding to the exo vs. the endo modes of dimerization of 3b (Fig. 3b vs. Fig. 3c). Consistent with the experimental observation, the lowest energy transition state (TS) structure for all possible (see Supplementary Information for details) exo modes of dimerization is favored by 3.9 kcal·mol−1 relative to the overall lowest energy for an endo approach of the two reactants. Moreover, the computed free energy of activation for the exo dimerization of 3b to 7b was 25.2 kcal·mol−1. That this value is in reasonably good agreement with the observed dimerization rate provides validity to the computational methodology. The overall reaction of two molecules of 3b to the initial bis-acylal-containing dimer, which subsequently ring-opens to the more stable keto acid 7b, is computed to be exergonic by 14.0 kcal•mol−1, suggesting that the dimerization should not be significantly reversible under the experimental conditions. We also admixed dimer 7b with the “real” dimeric diacid 7a (formed in analogous fashion, see Fig. 4a below) and incubated that neat sample at 35 °C for 7 days. Analysis by ESI-MS gave no evidence of any mixed dimer, which presumably would have formed had there been any appreciable reversibility back to 3b and 3a.
Figure 4. Conversion of the exo-Diels-Alder product to the natural product.
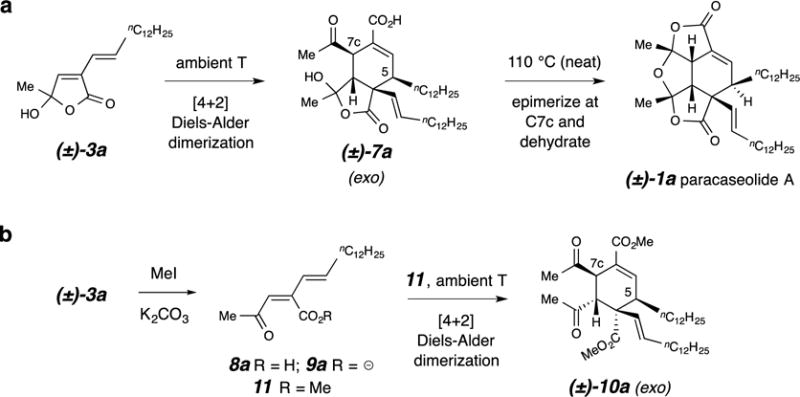
a, Ambient temperature dimerization of butenolide 3a produces a racemate of the exo adduct 7a; thermal dehydration requires additional heating. b, The acyclic methyl ester derivative of 3a, the ketoester 11, also dimerizes in a highly diastereoselective fashion at ambient temperature to produce the exo adduct 10a.
How can one rationalize this atypical preference for an exo over an endo approach for a Diels–Alder reaction (i.e., TSexo rather than TSendo)? It is enlightening to recognize that the geometry of the lowest energy TS (TSexo, Fig. 3b) is C2-symmetric, as reflected, for example, by the identical distance between carbons C2–Cβ in both pairs of reactants, (cf. y and y′ in Fig. 3b). This is an example of a bis-pericyclic process,20 passing through a TS in which the two possible modes of cycloaddition, [4+2] vs. [2+4], have fully merged. Subsequent (and degenerate) bifurcation,21 in which the partial bonding in TSexo between one or the other of the two C2–Cβ pairs is forfeited, forms the product 7b. This type of symmetrical TS structure was first invoked to account for Diels–Alder dimerizations of simple 4π-systems like acrolein22 and cyclopentadiene.20 More recently, a C2-symmetric, bis-pericyclic TS assembly has also been used to rationalize the stereochemical outcome of the self-dimerization of a complex orthoquinol in a synthesis of the secondary metabolite aquaticol.23 The extra stabilization that attends a bis-pericyclic TS has been nicely explained as its being “admirably suited to take advantage of the maximum accumulation of unsaturated centers”.20
We also established that the dimer 7b was a competent precursor to the paracaseolide A analog 1b. When a neat sample of crystalline 7b was melted (mp 170–174 °C, with sweating), the resulting cooled substance had cleanly been transformed to 1b. Moreover, holding crystalline 7b well below its melting point (110 °C) resulted in its virtually quantitative conversion to crystalline 1b. It should be noted that the stereochemical outcome of this transformation requires a change in the configuration at C7c, a point that is further discussed below.
The dimer 7b (Fig. 3a) contains a ring-opened keto acid in its “northern” region in both the solid state (X-ray) as well as in solution (1H NMR). This observation led us to wonder about the role of the analogous ring-opened “tautomer” of the precursor hemiacylal 3b as a participant in the Diels–Alder reaction itself. The 1H NMR spectrum of 3b in CD3OD showed no clear evidence for any of the isomeric keto acid 8b (Fig. 3d). However, when this solution was treated with slightly over one equivalent of the weak base Et3N, the NMR spectral data indicated that the Et3NH+ salt of the ring-opened carboxylate 9b was now the dominant species (Fig. 3d).24
These observations raised the question of the relevance of a ring-opened keto acid like 8b or carboxylate like 9b as a potential reactant in the dimerization involved in the biosynthesis of paracaseolide A (1a) itself. In particular, it might be expected that either of 8b or 9b, each having an alkene bearing two electron-withdrawing groups, would be a more reactive dienophile than the butenolide 3b. Previous workers have not considered the Diels–Alder reactivity of an open-chain tautomer like 8b or 9b. We used the TBS ester 6b, the direct product of singlet oxygen oxidation of furan 5b (Fig. 2), to probe this possibility. Interestingly, a neat sample of this acyclic analog of 3b dimerized to give, highly selectively, 10b with a rate about two times faster than that of 3b (Fig. 3e). The relative configuration of 10b was securely established by its conversion to 7b upon removal of both TBS esters. The structure of 10b indicated that the dimerization of 6b had also occurred through an approach involving an exo orientation of the two reacting molecules.
Extrapolation to the natural set of compounds (i.e., the “a” series)
We then examined the ambient temperature behavior of the ‘parent’ hydroxybutenolide 3a. By extrapolation, the fact that 3b dimerized exclusively to the exo adduct 7b implied that 7a should be formed in the dimerization of 3a that ultimately gives rise to (±)-paracaseolide A (1a). Recall that, in contrast, it is the endo adduct 2 (Fig. 1) that has been previously invoked by those who have delineated a structure for the initial Diels–Alder dimer.13,14,15 Indeed, when held as a highly concentrated sample at ambient temperature, 3a was observed to slowly but cleanly dimerize to the exo adduct 7a (Fig. 4a, below). This transformation is much faster (ca. 2% conversion per day at 21 °C) than what is implied by every previous experiment, all of which were carried out at 110 °C for 12–34 hours. Presumably these more forcing conditions were used because only the formation of the final dehydrated product 1a was being monitored. The relative configuration of 7a was confidently assigned in view of its many analogous 1H NMR spectroscopic features vis-à-vis those of its lower homolog 7b. In turn, this revised stereochemical outcome now requires adjustment of the configuration at C7c in 7a in order to ultimately arrive at the proper relative configuration present in 1a. This contrasts with previous presumptions that epimerization at C5 was necessary, a consequence of the incorrect assumption that the structure of the initial DA adduct was the endo intermediate 2.
We have also examined the potential of an open-chain analog of the hemiacylal 3a to undergo spontaneous [4+2] cycloaddition. A neat sample of the methyl ester 11 (obtained by treatment of 3a with MeI/K2CO3/acetone) also slowly dimerized at room temperature to give the exo adduct 10a in a highly diastereoselective process (≤3% of any other single compound was observed). The rate of this reaction was slightly faster than that of the dimerization of 3a itself. The structure of 10a was assigned on the basis of analogy, NOE (nuclear Overhauser enhancement) analysis, and 1H NMR spectroscopic comparisons with 10b. Because the methyl ester in 11 (as well as the TBS ester in the analogous open-chain analog 6b, Fig. 3e) represents a significant perturbation of the structure of the ring-open keto acid 8a (or its carboxylate 9a), we believe that drawing a definitive conclusion about the relative reactivities of the open vs. closed isomers of the acid 8a (or its carboxylate 9a, which might well be the species most relevant to the natural event enroute to 1a in the mangrove) vs. the hemiacylal 3a is unwarranted.
Facile exchange of the C7c-hydrogen atom—a path to spontaneous epimerization
But how might epimerization at C7c in 7a (or 7b) occur? Quite serendipitously, we made a remarkable observation, first using the truncated analog 7b–h. When this compound was held in methanol-d4 at room temperature, the C7c-proton spontaneously exchanged, quite cleanly, with a deuterium atom from the solvent to give 7b–d (t1/2 3.2 days at room temperature, Fig. 5a; also page S74 in Supplementary Information)! One rationale for this exchange is by way of intramolecular formation of the zwitterion 12 (Fig. 5b) followed by internal removal of the allylic C7c-proton by the proximal carboxylate to produce the enol 13. Intervention of a hydroxyl group, specifically the MeO–D bond here, as a proton shuttle to aid this process can also be envisioned. This interconversion of 7 with 13 constitutes an intramolecular, self-organocatalyzed, keto-enol tautomerization. Rapid solvent exchange of RCOOH to RCOOD in 13 followed by the microscopic reverse would return the observed mono-deuterated analog 7b–d. Importantly, this facile process provides access to an intermediate having a planar, sp2-hydribized C7c carbon atom, protonation of which from the β-face would effect the requisite C7c-epimerization leading to the relative configuration mandatory for final dehydration to 1.
Figure 5. Epimerization of the Diels-Alder adducts may occur spontaneously.
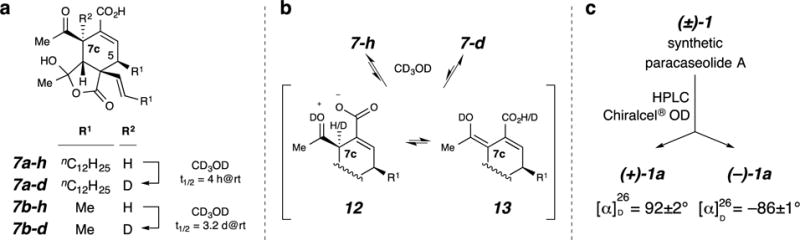
a, b, Mild, spontaneous H/D exchange at C7c in dimers 7a and 7b indicates that epimerization of the configuration of C7c is facile, perhaps via intermediates like 12 and 13. c, Each enantiomer of 1a has a specific optical rotation value that is much larger than that reported for the natural sample of paracaseolide A (1a), suggesting that the natural substance is racemic in nature.
The analogous adduct 7a–h (Fig. 4a) underwent even more facile H-D exchange at C7c to give 7a–d when dissolved in CD3OD (t1/2 ca. 4 h at room temperature). The same experiment using the dimethyl ester 10a (Fig. 4b) showed that this substrate had a much slower rate of deuteration at C7c (ca. 35% after 10 days), clearly suggesting a catalytic role for the carboxylic acid in 7a–h (cf. 12 and 13 in Fig. 5b). To judge if that process was unimolecular or bimolecular in nature, we prepared a CD3OD solution containing both 7a–h and 10a. After 12 hours, ESI-MS analysis indicated that the former had undergone exchange to the extent of ca. 75% whereas exchange in 10a was not detected. This argues against a bimolecular process in which a carboxylic acid in one molecule promotes the enolization and exchange of H7c alpha to the acetyl group in a second molecule of 7a–h or 10a.
Is paracaseolide A produced in the mangrove as a racemate?
Finally, we return to a key issue that is highly germane to the question of whether the spontaneous dimerization of diene 3a is the true biosynthetic event involved in the biosynthesis of paracaseolide A (1a). If this reaction is not catalyzed by an enzyme, one would expect the isolated natural sample of 1a to be racemic. The natural substance was reported to have a specific rotation of [α]24D = +0.9.1 In our view, particularly in light of minor resonances in the 1H NMR spectrum that indicate the presence of contaminants in that sample, the small value of this rotation is within experimental error of zero. To interrogate this point further, we resolved a sample of our synthetic (±)-1a on a Chiralcel® OD HPLC column (Fig. 5c). (Our attempts to obtain a small amount of the natural sample of paracaseolide A to analyze by this method were unsuccessful.) Essentially equal amounts (4.5 and 4.4 mg) of each enantiomer were recovered following semi-preparative-scale separation. The rotation values measured for the faster and the slower eluting enantiomers were +92° and −86°, respectively. This evidence strongly suggests that the natural material, like the synthetic, is racemic. This, in turn, is further support of our hypothesis that a Diels–Alderase is neither required nor responsible for the biosynthesis of paracaseolide A (1a).
Conclusion
Immediately upon contemplating the structure of paracaseolide A and in parallel with our interpretation of the fact that the natural material showed a near-zero value for its specific rotation,1 we hypothesized that its biosynthesis proceeded by spontaneous dimerization of the hydroxybutenolide 3a. To test this required that we examine the chemical reactivity of 3a to learn if it is sufficiently innately active to produce an adduct like 2 or 7a. Our conviction in that hypothesis led us to undertake the experiments reported here, through which we have established: (i) that the DA dimerization of 3a (or the isomeric ring-opened keto acid 8a) is sufficiently energetically accessible to be biosynthetically relevant, (ii) that this DA dimerization is exo selective, unlike what previous workers have presumed, (iii) using DFT computational studies, that this stereoselectivity can be explained by a bis-pericyclic pathway involving a C2-symmetric TS geometry, (iv) that facile H/D exchange of the C7c-hydrogen atom in 7a–h shows that the obligate planarization of C7c is a feasible process and enables a pathway for the required epimerization at C7c in 7a–h, and (v) that the natural sample of paracaseolide A is, in all likelihood, racemic—a fact incompatible with its formation under the action of a Diels–Alderase in Sonneratia paracaseolaris. Collectively, these observations lend substantial support to the idea that a spontaneous DA dimerization of the butenolide 3a is the key biosynthetic event leading to paracaseolide A—conceptualization that, until now, has been obscured by biased, but incorrect, assumptions that the dimerization required high temperature and occurred with either endo selectivity or by a stepwise mechanism.
Methods
HPLC separation of the antipodes of 1a and specific rotation measurements
A racemic (and synthetic) sample of paracaseolide A (1a, 14 mg) was dissolved in a mixture of CH2Cl2 (50 μL) and hexanes (100 μL). A Chiralcel® OD column (4.5 mm d × 250 mm l) was pre-equilibrated with 9:1 hexanes:2-propanol. The enantiomers of the racemic paracaseolide A were separated (4 injections) to give, in the order of elution, 4.5 mg of the (+)-enantiomer and 4.4 mg of the (−)-enantiomer. Each was then dissolved in HPLC grade CHCl3 (1.5 mL, ≥99.8%, amylene stabilized, no ethanol present) and transferred to a 1 mL polarimeter cell. For each enantiomer, 5 measurements (10 scans in each measurement) were made and averaged to give a specific rotation value of +92±2 for the first enantiomer and −86±1 for the second enantiomer. Measurements were made on a Rudolph Research Analytical (Autopol III) polarimeter.
All new synthetic compounds have been fully characterized; see Supplementary Information for preparation procedures, line listings of spectroscopic data, and copies of 1H and 13C NMR spectra.
Supplementary Material
Acknowledgments
T. W. acknowledges with appreciation the support of a Wayland E. Noland Fellowship and a University of Minnesota Graduate School Doctoral Dissertation Fellowship. The computational aspects of this work were performed with hardware and software resources available through the University of Minnesota Supercomputing Institute (MSI). Some graphical images were created using CYLview.25 We appreciate receiving guidance from a reviewer who encouraged us to explore in greater computational depth the Diels–Alder dimerization, which led to the identification of the fully symmetrical, bis-pericyclic transition state structure. Financial support for the research was provided by the National Cancer Institute of the National Institutes of Health (NIH) (CA76497). NMR spectra were recorded on an instrument purchased with support from the NIH Shared Instrumentation Grant program (S10OD011952).
Footnotes
Author Contributions
T. W. and T. R. H. conceived and designed the experiments, analyzed the data, and co-wrote the paper. T. W. performed the experiments.
Additional Information
Supplementary information is available in the online version of the paper.
References
- 1.Chen X-L, Liu H-L, Li J, Xin G-R, Guo Y-W. Paracaseolide A, first α-alkylbutenolide dimer with an unusual tetraquinane oxa-cage bislactone skeleton from Chinese mangrove Sonneratia paracaseolaris. Org Lett. 2011;13:5032–5035. doi: 10.1021/ol201809q. [DOI] [PubMed] [Google Scholar]
- 2.Lammer C, Wagerer S, Saffrich R, Mertens D, Ansorge W, Hoffmann I. The cdc25B phosphatase is essential for the G2/M phase transition in human cells. J Cell Sci. 1998;111:2445–2453. doi: 10.1242/jcs.111.16.2445. [DOI] [PubMed] [Google Scholar]
- 3.Yin J-P, Tang C-L, Gao L-X, Ma W-P, Li J-Y, Li Y, Li J, Nan F-J. Design and synthesis of paracaseolide A analogues as selective inhibitors of protein tyrosine phosphatase 1B inhibitors. Org Biomol Chem. 2014;12:3441–3445. doi: 10.1039/c4ob00214h. [DOI] [PubMed] [Google Scholar]
- 4.Gravel E, Poupon E. Biogenesis and biomimetic chemistry: Can complex natural products be assembled spontaneously? Nat Prod Rep. 2010;27:32–56. doi: 10.1039/b911866g. [DOI] [PubMed] [Google Scholar]
- 5.Stocking EM, Williams RM. Chemistry and biology of biosynthetic Diels–Alder reactions. Angew Chem Int Ed. 2003;42:3078–3115. doi: 10.1002/anie.200200534. [DOI] [PubMed] [Google Scholar]
- 6.Hideaki Oikawa H, Tokiwano T. Enzymatic catalysis of the Diels–Alder reaction in the biosynthesis of natural products. Nat Prod Rep. 2004;21:321–352. doi: 10.1039/b305068h. [DOI] [PubMed] [Google Scholar]
- 7.Kim AHJ, Ruszczycky MW, Choi S-H, Liu Y-N, Liu H-W. Enzyme-catalyzed [4+2] cycloaddition is a key step in the biosynthesis of spinosyn. Nature. 2011;473:109–112. doi: 10.1038/nature09981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hashimoto T, Hashimoto J, Teruya K, Hirano T, Shin-Ya K, Ikeda H, Liu H-w, Nishiyama M, Kuzuyama T. Biosynthesis of versipelostatin: Identification of an enzyme-catalyzed [4+2]-cycloaddition required for macrocyclization of spirotetronate-containing polyketides. J Am Chem Soc. 2015;137:572–575. doi: 10.1021/ja510711x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brophy GC, Mohandas J, Slaytor M, Sternhell S, Watson TR, Wilson LA. Novel lignans from a Cinnamomum sp. from Bougainville. Tetrahedron Lett. 1969;10:5159–5162. doi: 10.1016/s0040-4039(01)88911-5. [DOI] [PubMed] [Google Scholar]
- 10.Chapman OL, Engel MR, Springer JP, Clardy JC. The total synthesis of carpanone. J Am Chem Soc. 1971;93:6696–6698. [Google Scholar]
- 11.Bandaranayake WM, Banfield JE, Black D. St. C. Postulated electrocyclic reactions leading to endiandric acid and related natural products. J Chem Soc Chem Comm. 1980:902–903. [Google Scholar]
- 12.Nicolaou KC, Petasis NA, Zipkin RE, Uenishi J. The endiandric acid cascade. Electrocyclizations in organic synthesis 1 Stepwise, stereocontrolled total synthesis of endiandric acids A and B. J Am Chem Soc. 1982;104:5555–5557. [Google Scholar]
- 13.Noutsias D, Vassilikogiannakis G. First total synthesis of paracaseolide A. Org Lett. 2012;14:3565–3567. doi: 10.1021/ol301481t. [DOI] [PubMed] [Google Scholar]
- 14.Vasamsetty L, Khan FA, Mehta G. Total synthesis of a novel oxa-bowl natural product paracaseolide A via a ‘putative’ biomimetic pathway. Tetrahedron Lett. 2013;54:3522–3525. [Google Scholar]
- 15.Giera DS, Stark CBW. Total synthesis of (±)-paracaseolide A and initial attempts at a Lewis acid mediated dimerization of its putative biosynthetic precursor. RSC Adv. 2013;3:21280–21284. [Google Scholar]
- 16.Boukouvalas J, Jean M-A. Streamlined biomimetic synthesis of paracaseolide A via aerobic oxidation of a 2-silyloxyfuran. Tetrahedron Lett. 2014;55:4248–4250. [Google Scholar]
- 17.Vasamsetty L, Sahu D, Ganguly B, Khan FA, Mehta G. Total synthesis of novel bioactive natural product paracaseolide A and analogues: Computational evaluation of a ‘proposed’ biomimetic Diels–Alder reaction. Tetrahedron. 2014;70:8488–8497. [Google Scholar]
- 18.Adam W, Rodriguez A. Intramolecular silyl migration in the singlet oxygenation of 2-methyl-5-trimethylsilylfuran. Tetrahedron Lett. 1981;22:3505–3508. [Google Scholar]
- 19.Guney T, Kraus GA. Total synthesis of paracaseolide A. Org Lett. 2013;15:613–615. doi: 10.1021/ol303447r. [DOI] [PubMed] [Google Scholar]
- 20.Caramella P, Quadrelli P, Toma L. Unexpected bispericyclic transition structure leading to 4+2 and 2+4 cycloadducts in the endo dimerization of cyclopentadiene. J Am Chem Soc. 2002;124:1130–1131. doi: 10.1021/ja016622h. [DOI] [PubMed] [Google Scholar]
- 21.Ess DH, Wheeler SE, Iafe RG, Xu L, Çelebi-Ölçüm N, Houk KN. Bifurcations on potential energy surfaces of organic reactions. Angew Chem Int Ed. 2008;47:7592–7601. doi: 10.1002/anie.200800918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Toma L, Romano S, Quadrelli P, Caramella P. Merging of 4+2 and 2+4 cycloaddition paths in the regiospecific dimerization of methacrolein. A case of concerted crypto-diradical cycloaddition. Tetrahedron Lett. 2001;42:5077–5080. [Google Scholar]
- 23.Gagnepain J, Castet F, Quideau S. Total synthesis of (+)-aquaticol by biomimetic phenol dearomatization: Double diastereofacial differentiation in the Diels–Alder dimerization of orthoquinols with a C2-symmetric transition state. Angew Chem. 2007;119:1555–1557. doi: 10.1002/anie.200604610. [DOI] [PubMed] [Google Scholar]
- 24.Miles WH, Duca DG, Selfridge BR, Palha De Sousa CA, Hamman KB, Goodzeit EO, Freedman JT. Amine-catalyzed epimerization of γ-hydroxybutenolides. Tetrahedron Lett. 2007;48:7809–7812. [Google Scholar]
- 25.CYLview, 1.0b; Legault, C. Y., Université de Sherbrooke, 2009 (http://www.cylview.org).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.