

ABSTRACT
The Rb tumor suppressor is conserved in Drosophila, and its inactivation can lead to cell proliferation or death depending on the specific cellular context. Therefore, identifying genes that affect the survival of Rb-mutant cells can potentially identify novel targets for therapeutic intervention in cancer. From a genetic screen in Drosophila, we identified synthetic lethal interactions between mutations of fly Rb (rbf) and the ESCRT-0 components stam and hrs. We show that inactivation of ESCRT-0 sensitizes rbf-mutant cells to undergo apoptosis through inhibition of EGFR signaling and accumulation of Hid protein. Mutation of stam inhibits EGFR signaling upstream of secreted Spi and downstream of Rhomboid expression, and causes Rhomboid protein to accumulate in the abnormal endosomes labeled with both the early and late endosomal markers Rab5 and Rab7. These results reveal that ESCRT-0 mutants inhibit EGFR signaling by disrupting Rhomboid endosomal trafficking in the ligand-producing cells. Because ESCRT-0 also plays crucial roles in EGFR downregulation after ligand binding, this study provides new insights into how loss of ESCRT-0 function can either increase or decrease EGFR signaling.
KEY WORDS: Rhomboid, Stam, Hrs, EGFR signaling, Rbf, Apoptosis
Summary: Inactivation of ESCRT-0 complex promotes Rb-mutant cell death by disrupting Rhomboid endosomal trafficking and inhibiting EGFR signaling.
INTRODUCTION
The retinoblastoma tumor suppressor (Rb) is a member of the Rb family and is often inactivated in cancers. This family of proteins functions by binding to a large number of cellular proteins, particularly the E2F family of transcription factors (Morris and Dyson, 2001). There are three subgroups of E2F proteins in mammalian cells: the activating E2F1-3, the repressive E2F4-5 and Rb-independent E2F6-8 (Attwooll et al., 2004; Trimarchi and Lees, 2002). The Rb and E2F proteins are highly conserved but much simpler in Drosophila melanogaster (Du and Pogoriler, 2006; Gordon and Du, 2011a; van den Heuvel and Dyson, 2008). There is only one activating E2F protein (dE2F1), one repressive E2F protein (dE2F2) and two Rb-family proteins (RBF and RBF2) in flies. RBF, which interacts with both dE2F1 and dE2F2, functions like the mammalian Rb protein and regulates cell proliferation, differentiation and apoptosis in Drosophila (Du, 2000; Moon et al., 2005; Sukhanova et al., 2011; Tanaka-Matakatsu et al., 2009). By contrast, RBF2, which interacts with dE2F2 but not dE2F1, does not cause obvious defects in cell proliferation, apoptosis or differentiation (Stevaux et al., 2005). The much simpler and yet highly conserved Rb and E2F pathway between Drosophila and mammalian systems prompted us to take advantage of the fly system to study this pathway.
The Drosophila developing eye provides a model system to identify genes that modulate the proliferation, differentiation or apoptosis of rbf-inactivated cells (Gordon et al., 2013; Li et al., 2010; Steele et al., 2009; Tanaka-Matakatsu et al., 2009; Zhang et al., 2014). Photoreceptor differentiation in the developing eye initiates in the morphogenetic furrow (Treisman, 2013). Anterior to the morphogenetic furrow, cells are undifferentiated and asynchronously proliferating. Expression of Ato, which is induced by retinal determination factors and bHLH protein Daughterless just anterior to the morphogenetic furrow (Tanaka-Matakatsu and Du, 2008; Tanaka-Matakatsu et al., 2014, 2015; Zhang et al., 2006), is progressively restricted to small clusters and eventually to the individual R8 precursors within the morphogenetic furrow (Sun et al., 1998). The R8 precursors express the membrane protease Rhomboid (Rho) and release EGFR ligands to activate EGFR signaling, which regulates stepwise retinal differentiation and cell proliferation, and promotes cell survival in posterior eye discs (Baker and Yu, 2001; Dominguez et al., 1998). There are four EGF ligands, representing two different classes – the TGF-α ligands Spitz (Spi), Gurken (Grk) and Keren (Krn), and the neuregulin-like ligand Vein (Vn) (Neuman-Silberberg and Schupbach, 1993; Reich and Shilo, 2002; Rutledge et al., 1992; Schnepp et al., 1996). The main ligand of EGFR in the Drosophila eye disc is Spi (Freeman, 1994), which is synthesized as a transmembrane pro-protein (mSpi) (Schweitzer et al., 1995). The post-transcriptional processing of Spi involves the transport of mSpi out of the endoplasmic reticulum (ER) through the chaperone Star (Lee et al., 2001; Tsruya et al., 2002), the palmitoylation of Spi at its N-terminal cysteine residue by the membrane bound O-acyltransferase Rasp (Miura et al., 2006) and the cleavage of Spi by the membrane protease Rhomboid (Urban et al., 2001). In addition to its effect on Spi, Rhomboid can also cleave Star and regulate the level of Spi secretion (Tsruya et al., 2007). In Drosophila, Rhomboid expression is dynamically regulated, whereas other components of EGFR signaling are ubiquitously expressed. Therefore, the expression pattern of Rhomboid determines the location of the active EGFR ligand release and EGFR signaling activation. Termination of EGFR signaling is regulated at multiple levels, which includes the induction of negative-feedback regulators such as Argos (Aos) and the induction of receptor downregulation involving the Endosomal Sorting Complex Required for Transport (ESCRT) machinery (ESCRT-0 to ESCRT-III) (Katzmann et al., 2002; Williams and Urbe, 2007).
Because the consequences of Rb inactivation, including cell proliferation or cell death, are influenced by additional cell intrinsic factors and extrinsic survival signaling, identification of genes that modulate the proliferation or apoptosis of Rb-inactivated cells in vivo will provide new insights into the regulatory mechanisms and potentially identify novel targets for cancer intervention (Gordon and Du, 2011b). Interestingly, inactivation of RBF in the Drosophila developing eye causes increased apoptosis mostly in the morphogenetic furrow area (Du, 2000), suggesting the presence of regulatory pathways that affect cell death or survival induced by Rb inactivation. In this manuscript, we characterize several mutants that inactivate ESCRT-0 and that induce cell death in synergy with Rb inactivation.
RESULTS
Mutations of ESCRT-0 components stam and hrs promote apoptosis in rbf-mutant clones
From genetic screening on chromosome 2L to identify genes that are important for the survival of rbf-null cells, we isolated three mutations 19, 4-19-3 and 5-14-3. Although significant amounts of tissue were observed in rbf, 19, 4-19-3 or 5-14-3 single-mutant clones in adult fly eyes (Fig. 1A–C,E; Fig. S1E, white patches), combining rbf mutation with any of these novel alleles showed little double-mutant tissue (Fig. 1D,F; Fig. S1F, white patches). These observations suggest that these mutations promote the elimination of rbf-mutant cells during eye development.
Fig. 1.

Mutation of stam or hrs induces cell death in synergy with rbf mutation and promotes the elimination of double-mutant clones in adult eyes. (A–F) Representative pictures of adult eyes with clones of wild-type control (A). (B–F) rbf, stam and hrs single- or double-mutant clones are shown. Mutant clones are marked by lack of red pigment. (G–R) Levels of apoptosis in 3rd instar eye discs (G–K′) or wing discs (L–P′) with rbf, stam or hrs single- or double-mutant clones are shown. Mutant clones are marked by lack of GFP, and an antibody to detect cleaved Caspase-3 (C3) was used to detect apoptosis. Yellow arrows point to mutant clones. The level of apoptosis in mutant clones located in the posterior of eye discs and wing discs was quantified, shown in Q and R respectively. Data are mean±s.d. The number of discs quantified for each genotype was: rbf, n=7; stam19, n=6; rbf stam19, n=8; hrs4-19-3, n=7; rbf hrs4-19-3, n=6. Similar results were observed in three independent experiments. Asterisks indicate a statistically significant difference (P<0.0001, Student's t-test) between double- and single-mutant clones, and the white scale bars indicate 100 µm. In this and all the subsequent figures, eye discs are orientated dorsal up and posterior to the right; different genotype mutant clones are indicated. The complete genotypes of the flies analyzed are detailed in Table S1.
To directly test if the observed loss of double-mutant clones in adult eyes correlates with increased apoptosis in developing eye discs, 3rd-instar eye discs from single- or double-mutant clones were stained with an antibody against activated Caspase-3. As shown previously, rbf-mutant cells (GFP-negative clones) exhibited increased apoptosis near the morphogenetic furrow with little apoptosis detected in rbf clones posterior to the morphogenetic furrow (Fig. 1G, yellow arrow). In addition, single-mutant clones of 19 (Fig. 1H,H′), 4-19-3 (Fig. 1J,J′) or 5-14-3 (Fig. S2D,D′) showed very low levels of Caspase-3 staining. However, significantly increased Caspase-3 staining in posterior eye discs was observed in rbf 19 (Fig. 1I,I′), rbf 4-19-3 (Fig. 1K,K′) and rbf 5-14-3 (Fig. S2E,E′) double-mutant clones (Fig. 1Q; Fig. S2F; P<0.0005 between the double-mutant clones and each of the corresponding single-mutant clones, Student's t-test). These results suggest that the 19, 4-19-3 and 5-14-3 mutations induce greater levels of cell death in synergy with rbf mutation (hereafter referred to as synergistic cell death) in posterior eye discs, which is correlated with loss of the double-mutant tissue in adult eyes. To further determine whether the synergistic apoptosis observed is limited to the developing eye discs, we further characterized the apoptosis of the single- and double-mutant clones in developing wing discs. Synergistic cell death was also observed in the rbf 19, rbf 5-14-3 and rbf 4-19-3 double-mutant clones (Fig. 1L–R; Fig. S2J–L; P<0.0005 between the double-mutant clones, and each of the corresponding single-mutant clones, Student's t-test). Therefore, these mutations promote the apoptosis of rbf-mutant cells in multiple tissues.
Mutations 4-19-3 and 5-14-3 were found to be in the same complementation group, whereas mutation 19 was distinct. Recombination and deficiency mapping showed that the mutations responsible for the phenotypes described above in 4-19-3 and 5-14-3 mutants, and those in the 19 mutant were mapped to genomic regions 23A and 32B1–32B4, respectively. Whole-genome sequencing of the 4-19-3 and 19 mutants, and of the FRT40 control, was performed to identify the specific gene mutations that caused the observed phenotypes. A single G to A mutation in hepatocyte growth factor-regulated tyrosine kinase substrate (Hrs) was found in 4-19-3, which caused a Glu264 to Lys substitution in the ubiquitin-binding domain, which is highly conserved and important to mediate the interaction between Hrs and ubiquitylated cargo (Lloyd et al., 2002). A single T to A mutation in Signal Transducing Adaptor Molecule (Stam) was found in 19, which caused an amino acid change from Trp269 to Arg in the conserved Src3 homology (SH3) domain of the Stam protein.
Hrs and Stam interact with each other to form the ESCRT-0 complex, which plays important roles in sorting ubiquitylated cargo in endosomes (Williams and Urbe, 2007). To further demonstrate that mutations 19 and 4-19-3 are alleles of stam and hrs, respectively, we obtained previously generated stam and hrs alleles. Both stam3297 and stam2896 (Chanut-Delalande et al., 2010) failed to complement the 19 mutation. In addition, stam3297 mutation also induced synergistic cell death with rbf mutation (Fig. S2A–C,F–I,L) and caused the loss of double-mutant tissue in adult eyes (Fig. S1A–D). Similarly, both hrs4-19-3 and hrs5-14-3 failed to complement the hrsD28 null allele (Lloyd et al., 2002). In addition, hrsD28 mutation also induced synergistic cell death with rbf mutation (Fig. S2L–O). Furthermore, eye-specific knockdown of Rbf and Stam, or Rbf and Hrs using RNA interference (RNAi) constructs dramatically reduced the size of adult eyes, whereas RNAi against each of the single genes only exhibited moderate effects (Fig. S1G–L). Taken together, these results demonstrate that inactivation of Hrs and Stam promotes the apoptosis of rbf-mutant cells and causes the loss of double-mutant cells in adult tissues.
dE2F1 activity and increased Hid levels contribute to synergetic cell death in rbf and stam double-mutant tissues
In Drosophila eye discs, cell death in rbf-mutant tissue is observed near the morphogenetic furrow, which requires dE2F1 activity and is mediated by increased Hid expression (Du, 2000; Li et al., 2010; Moon et al., 2005; Tanaka-Matakatsu et al., 2009; Zhang et al., 2014). We examined the effects of rbf and stam on the levels of Hid protein. Increased levels of Hid protein were detected in rbf-mutant clones near the morphogenetic furrow but not in the posterior (Fig. 2A,A′). Although mutation of stam alone did not substantially affect Hid protein levels in eye discs (Fig. 2B,B′), substantially increased Hid protein levels were observed in rbf stam19 double-mutant clones, both near to the morphogenetic furrow and in the posterior eye disc (Fig. 2C,C′). The increased Hid protein in rbf stam double-mutant clones in the posterior eye discs correlated with the observed synergistic cell death there (Fig. 1G–I), suggesting that the increase of Hid protein contributes to the cell death. To directly test this possibility, we generated the rbf stam double-mutant clones in the hid138 mutant background (Tanaka-Matakatsu et al., 2009). hid138 mutation completely blocked cell death in rbf stam double-mutant clones (Fig. 2D–E′,G; P<0.0001, Student's t-test).
Fig. 2.

Synergistic cell death of rbf stam double-mutant clones depends on Hid and dE2F1 activity. (A–C′) Hid protein levels in rbf and stam single- and double-mutant clones were determined by staining with an antibody against Hid. Similar results were observed in different eye discs for each genotype. Mutant clones are marked by lack of GFP; yellow arrows point to mutant clones in the posterior of eye discs. (D–F′) Mutation of hid or de2f1 blocks synergistic apoptosis in rbf stam double-mutant clones. Blue and yellow arrows point to mutant clones in the morphogenetic furrow and posterior of eye discs, respectively. The levels of apoptosis in mutant clones located in the posterior of eye discs were quantified, and the means±s.d. are shown in G. The number of discs quantified for each genotype was: rbf stam19, n=6; rbf stam19 hid138, n=7; rbf stam19 de2f1, n=6. Results were repeated in three independent experiments. Asterisks indicates a statistically significant difference (P<0.0001, Student's t-test) between triple- and double-mutant clones. The complete genotypes of the flies analyzed are detailed in Table S1. Scale bars: 50 µm.
We further determined whether the observed synergistic cell death in rbf stam double-mutant clones requires deregulated dE2F1 activity. We examined the level of cell death in the de2f1i2/de2f1729 background. de2f1i2 encodes a dE2F1 protein with a deletion of the transactivation domain and de2f1729 is a P-element insertion mutant that behaves in a manner similar to that of the null (Duronio et al., 1995; Royzman et al., 1997). de2f1i2/de2f1729 mutants are viable and can rescue the lethality of rbf-null mutants (Du, 2000). Significantly reduced cell death was observed in rbf stam double-mutant clones in the de2f1i2/de2f1729 background (Fig. 2D–G, P<0.0001, Student's t-test). Therefore, deregulated dE2F1 transcriptional activity contributes to the synergistic cell death of rbf stam double-mutant cells. Taken together, these results indicate that dE2F1 activity and increased Hid protein level in rbf stam double-mutant clones both contribute to the observed synergistic cell death.
stam and hrs mutations impair EGFR signaling
EGFR signaling, which is active in the posterior eye disc, plays important roles in inhibiting apoptosis, at least in part, through MAPK-mediated phosphorylation and degradation of Hid protein (Baker and Yu, 2001; Bergmann et al., 1998). Mutations of ESCRT-0 components have been shown to either increase or decrease EGFR signaling in different settings (Chanut-Delalande et al., 2010; Lloyd et al., 2002; Miura et al., 2008). To determine whether mutation of stam or hrs affects EGFR signaling in our system, we used aos–lacZ reporter to determine the transcription of argos, a negative regulator as well as target of EGFR signaling in Drosophila. Although expression of the aos–lacZ reporter was not substantially affected in rbf single-mutant clones (Fig. 3A,A′), substantially reduced aos–lacZ reporter expression was observed in stam or hrs single-mutant clones, as well as in rbf stam or rbf hrs double-mutant clones (Fig. 3B–E′; Fig. S3A–D). To further confirm that ESCRT-0 loss reduces EGFR signaling, we examined the effect of stam mutations on the level of phosphorylated ERK (dpERK; also known as Rolled) (Gabay et al., 1997). Significantly decreased dpERK staining in the posterior of eye discs was observed in the stam19 as well as stam3297 single-mutant clones (Fig. 3G,G′; Fig. S3E, yellow arrows). The rbf stam double-mutant clones showed a similar reduction in dpERK staining, whereas mutation of rbf did not significantly affect dpERK staining in posterior eye discs (Fig. 3F–H; Fig. S3F, yellow arrows). Furthermore, the previously characterized hrsD28 null mutant, which exhibited increased EGFR signaling in the embryos, also showed reduced dpERK levels in posterior eye discs (Fig. S3G). Therefore, mutation of ESCRT-0 complex components reduced both the EGFR-signaling-mediated MAPK activation and its transcription output in eye discs. It should be pointed out that some mutant cells at the clone border exhibited high dpERK levels (Fig. 3G–H′; Fig. S3E,G), suggesting that the inhibition of EGFR signaling through mutations of stam or hrs might not be completely cell autonomous.
Fig. 3.

stam and hrs mutations decrease activation of EGFR signaling in the posterior of eye discs and wing discs. (A–E′) The effects of rbf, stam and hrs single or double mutants on the levels of the EGFR-signaling target aos–lacZ in the posterior of eye discs are shown. (F–H′) The effects of rbf and stam single- or double-mutant clones on dpERK levels in posterior eye discs are shown. Mutant clones are marked by the lack of GFP, and yellow arrows point to eye disc mutant clones in the posterior. White arrows in G–H′ point to some mutant cells adjacent to wild-type tissue that has high dpERK levels. Similar results were observed for each genotype. (I–K) stam mutation significantly decreases EGFR signaling in L3 primordia and near the wing margin area, but only weakly affects EGFR signaling in the L4 primordia. (I) Wild-type pattern of EGFR signaling in late 3rd instar wing discs is shown by the aos–lacZ reporter. aos-lacZ expression was observed in the wing margin area and in the L3, L4 and L5 primordia. (J,J′) stam-mutant clones are marked by the lack of GFP. (K) stam MARCM clones are marked by GFP expression. Mutation of stam significantly decreases aos–lacZ levels in the L3 primordia (yellow arrows in J–K, the strong aos–lacZ signals in J′ are in GFP-positive wild-type cells) and in the wing margin area (blue arrows in J). By contrast, substantial levels of the aos–lacZ reporter in L4 primordia were still expressed in stam-mutant clones (J,J′, white arrow). The complete genotypes of the flies analyzed are detailed in Table S1. Scale bars: 50 µm.
Because the R8 photoreceptor, the first photoreceptor neuron identified, is the main source of EGFR ligand for the recruitment of additional photoreceptors in developing eye discs, we further characterized the effect of stam or hrs mutations on photoreceptor differentiation. We found that flies possessing stam19, the hrs-null mutation hrsD28 or the rbf hrsD28 double mutation did not exhibit changes in R8 differentiation, as shown by the normal onset of the R8 marker Senseless (Sens) (Fig. S4A,C,E). These results are consistent with the previous report that hrs mutation does not affect R8 differentiation (Miura et al., 2008). In contrast, although stam19 only slightly delayed additional photoreceptor differentiation, hrsD28 single and rbf hrsD28 double mutants exhibited a more substantial delay in photoreceptor differentiation (Fig. S4B,D,F). Because EGFR signaling is required for the differentiation of all photoreceptor cells except R8, these observations suggest that the hrs and stam mutations decrease, but do not completely block, EGFR signaling and that the reduced EGFR signaling leads to delayed recruitment of additional photoreceptors after R8 differentiation.
We further characterized the effect of stam mutation on EGFR signaling activity in wing discs. In wild-type late 3rd instar wing discs, aos–lacZ expression was observed in cells adjacent to the wing margin and in the L3, L4 and L5 primordia (Fig. 3I). Mutation of stam substantially inhibited aos–lacZ levels in cells adjacent to the wing margin area (Fig. 3J,J′, blue arrows) and in L3 primordia (Fig. 3J–K, yellow arrows). By contrast, substantial levels of aos–lacZ in L4 primordia were still expressed in stam-mutant clones (Fig. 3J,J′, white arrows). The neuregulin-like EGFR ligand Vein plays important roles during wing development (Schnepp et al., 1996). Because Argos expression in the L4 primordia (but not in other regions) of 3rd instar wing discs is significantly decreased in vn1 mutants (Wessells et al., 1999), Vein is likely to play an important role in the activation of EGFR signaling in L4 primordia, whereas Spi is likely to play a major role in EGFR signaling activation in most other regions. Therefore, these results support the possibility that stam mutation preferentially reduces Spi- but not Vein-induced EGFR signaling activation. Because MAPK activity plays a crucial role in regulating Hid-mediated cell death (Bergmann et al., 1998), these results suggest that decreased EGFR–MAPK signaling in mutant clones of ESCRT-0 components contributes to the synergistic cell death observed with rbf mutation.
Mutation of stam blocks EGFR signaling upstream of EGFR activation and downstream of Rhomboid expression
In developing eye discs, EGFR signaling activation is mediated by the membrane-tethered ligands (such as Spi) that require processing by the membrane protease Rhomboid, which is expressed in the developing photoreceptors. To determine whether decreased EGFR–MAPK signaling mediates synergistic cell death of rbf stam double mutants, we used the Mosaic Analysis with a Repressible Cell Marker (MARCM) system to express Rhomboid, secreted Spi, activated EGFR or activated Ras (RasV12) in double-mutant clones. rbf stam double-mutant MARCM clones were positively labeled with GFP, and they showed considerable levels of Caspase-3 staining (Fig. 4A,A′). Expression of RasV12 significantly decreased the Caspase-3 staining in rbf stam double-mutant clones (Fig. 4B,B′,H; P<0.0001, Student's t-test). Similarly, expression of a constitutively activated form of EGFR (EGFRCA) or secreted Spi in the double-mutant clones of rbf stam19 also significantly decreased Caspase-3 staining (Fig. 4C–D′,H, P<0.0001, Student's t-test). In contrast, Rhomboid expression failed to decrease Caspase-3 staining in rbf stam19 double-mutant clones (Fig. 4G–H, P=0.9) even though Rhomboid expression blocked cell death in rbf single-mutant clones near the morphogenetic furrow (Fig. 4E–F′). These results suggest that stam mutation blocks EGFR signaling at a step that is upstream of secreted Spi and activated EGFR receptor but downstream of Rhomboid expression.
Fig. 4.
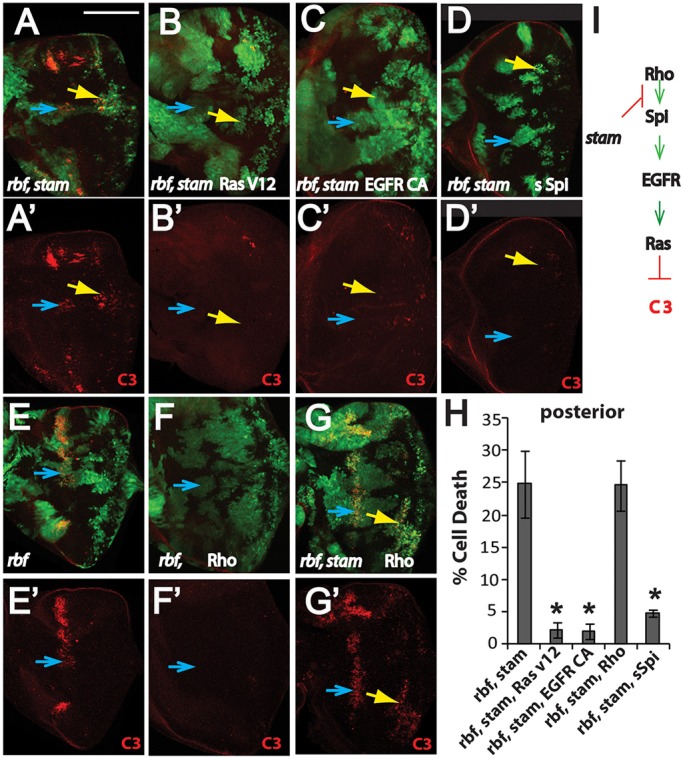
Synergistic cell death of rbf stam double mutants is suppressed by expression of activated Ras, activated EGFR and secreted Spi but not by Rhomboid expression. (A–Gʹ) rbf stam (A–Dʹ,G,Gʹ) and rbf (E–Fʹ) MARCM clones are marked by GFP expression, and apoptotic cells are identified by Caspase-3 (C3) staining shown in red. Apoptosis in the morphogenetic furrow and posterior are indicated by blue and yellow arrows, respectively. (H) The levels of apoptosis in the indicated genotypes of rbf stam double-mutant MARCM clones located in the posterior of eye discs were quantified. The mean±s.d. are shown. The number of discs quantified for each genotype was: rbf stam19, n=8; rbf stam19 rasv12, n=6; rbf stam19 EGFRCA, n=6; rbf stam1 sSpi, n=9; rbf stam19 rho, n=7. Asterisks indicate statistically significant difference (P<0.0001, Student's t-test) between triple- and double-mutant clones. Similar results were observed in three independent experiments. (I) A diagram that summarizes where stam mutation blocks EGFR signaling activation. The complete genotypes of the flies analyzed are detailed in Table S1. Scale bar: 100 µm.
We further characterized the effect of stam mutation on Rhomboid- or EGFRCA-induced aos–lacZ and MAPK activation. Rhomboid expression in wild-type eye discs induced precocious aos–lacZ expression and dpERK staining anterior to the morphogenetic furrow (Fig. 5A–A″,C–C″). In contrast, Rhomboid expression in stam-mutant clones failed to induce precocious aos–lacZ or dpERK staining anterior to the morphogenetic furrow (Fig. 5B–B″,D–D″). In addition, the aos–lacZ and dpERK levels in posterior eye discs were still reduced in stam-mutant clones even with Rhomboid expression (Fig. 5B″,D″), which is similar to that of the stam-mutant clones (Fig. 3B,G). Therefore stam mutation blocks Rhomboid-induced EGFR signaling activation. In contrast, stam mutation did not affect EGFRCA-induced aos–lacZ or dpERK levels in either the anterior or posterior of eye discs (Fig. 5E–H″). Furthermore, null alleles of the ESCRT-0 mutants stam2896 and hrsD28 did not affect EGFRCA-induced aos–lacZ levels (Fig. S3H–J). These results show that stam mutation inhibits EGFR signaling upstream of EGFR activation and downstream of Rhomboid expression.
Fig. 5.

Effect of stam mutation on EGFR signaling induced by Rhomboid, secreted Spi or activated EGFR. Rhomboid (Rho) expression in wild-type discs (A–A″ and C–C″) induces precocious aos–lacZ expression (A–A″) and dpERK (C–C″) anterior to the morphogenetic furrow. In contrast, Rhomboid expression in stam MARCM clones (marked by GFP) failed to induce precocious aos–lacZ (B–B″) or dpERK levels (D–D″). By contrast, expression of activated EGFR induces precocious aos–lacZ (E–F″) and dpERK levels (G–H″) in both the wild-type background (E–E″,G–G″) and the stam MARCM clones (F–F″,H–H″). (I–J″) Expression of secreted Spi in the wild-type background (I–I″) and in stam MARCM clones (J–J″). sSpi expression induces substantial precocious aos–lacZ expression both within the sSpi-expressing cells (marked by GFP) and in cells adjacent to the sSpi-expressing cells in either the wild-type background (I–I″) or in stam MARCM clones (J–J″). White arrows in I′–I″ and J′–J″ point to background aos–lacZ levels in anterior eye discs. Multiple discs were examined and representative results for each genotype are shown. The complete genotypes of the flies analyzed are detailed in Table S1. Yellow arrows point to Rho, sSpi or EGFRCA MARCM clones. Panels labeled ′ and ″ correspond to the zoomed in image of the boxed areas. The white dashed lines outline the MARCM clones. Scale bars: 50 µm.
Because Rhomboid is required for Spi processing, we tested the effect of stam mutation on secreted Spi (sSpi) (Schweitzer et al., 1995) using the MARCM approach. Expression of sSpi strongly increased aos–lacZ levels both in sSpi-expressing clones as well as in cells surrounding the clone, probably owing to the diffusion of the sSpi ligand (Fig. 5I–I″, compare with the background levels pointed by white arrows). It is interesting to note that sSpi induced stronger aos–lacZ levels in cells surrounding the clones, probably owing to the induction of receptor downregulation and negative-feedback regulation in the presence of excess levels of sSpi. Importantly, sSpi expression still induced ectopic aos–lacZ expression in stam clones (Fig. 5J–J″, yellow arrows, compare with the background aos–lacZ levels pointed by white arrows), and mutation of stam did not significantly affect sSpi-induced aos–lacZ levels either within the clone or in cells surrounding the clones (Fig. 5I–J″). These observations indicate that stam mutation does not block the release of sSpi or its activation of EGFR signaling. Taken together, our results show that stam mutation blocks EGFR signaling downstream of Rhomboid expression and upstream of secreted Spi.
Mutation of stam causes accumulation of Rhomboid in abnormal endosomal compartments
Rhomboid has been shown to localize to the endosomes (Tsruya et al., 2007). In wild-type eye discs, Rhomboid is expressed in the posterior and can be detected by an antibody against Rhomboid as endosome-localized small spots (Sturtevant et al., 1996; Sukhanova et al., 2011) (see also Fig. 6F,G). Interestingly, larger and brighter Rhomboid spots were observed in stam- or hrs-mutant clones (Fig. 6B,C). The effect of stam or hrs mutation on Rhomboid is not due to alteration of expression because levels of the enhancer trap Rhomboid–lacZ were unaffected (Fig. 6D,E). Because ESCRT-0 affects endosomal trafficking, these observations raised the possibility that hrs and stam mutation interferes with normal Rhomboid endosomal transport. Consistent with this possibility, the larger and brighter Rhomboid spots were also observed in clones of ESCRT-I, ESCRT-II and ESCRT-III mutants (Fig. S4G–I′). In addition, the ESCRT-I–ESCRT-III mutants also exhibited substantially decreased EGFR signaling, as shown by reduced aos–lacZ levels (Fig. S4J–L′). These observations suggest that inactivation of any of the ESCRT complexes blocks normal Rhomboid endosomal trafficking and inhibits EGFR signaling. Consistent with this, RNAi of ESCRT-I components also decreases dpERK levels, although ESCRT-I inactivation inhibits dpERK levels downstream of EGFR activation (Miura et al., 2008). It is possible that although all the ESCRT complexes can block Rhomboid trafficking and inhibit EGFR signaling, ESCRT-I and ESCRT-II can also block EGFR signaling activation downstream of receptor activation.
Fig. 6.

stam and hrs mutations cause abnormal localization of Rhomboid in endosomes. (A–E) Effects of stam and hrs mutations on Rhomboid (Rho) protein levels (A–Cʹ) and on Rhomboid–lacZ enhancer trap expression (D–E) are shown. Yellow arrows point to mutant clones, which are marked by the lack of GFP. (F–I″) Colocalization study of Rhomboid with YFP-tagged Rab5 (F–F″ and H–H″) or Rab7 (G–G″ and I–I″) in wild-type (WT) eye discs (F–G″) or eye discs with stam-mutant clones (H–I″). White arrowheads point to Rhomboid that did not show colocalization, and yellow arrowheads point to Rhomboid that showed colocalization with Rab5 or Rab7. (J–J‴) Colocalization study of Rhomboid and membrane Spi (mSpi) expressed in R8 cells (using the Sca–Gal4 driver; Sca-mSpi) in a stam-mutant clone (indicated by yellow arrowheads) and in adjacent wild-type tissues (indicated by white arrowheads). Multiple discs were examined, and representative results for each genotype are shown. Mutant clones in H–J‴ are marked by the lack of β-galactosidase staining (shown in blue). The complete genotypes of the flies analyzed are detailed in Table S1. Scale bars: 50 µm (A); 10 µm (F). The white dashed lines outline the stam-mutant clones.
We used the early-endosome marker Rab5 and the late-endosome marker Rab7 to further characterize the effect of stam mutation on Rhomboid localization. Previous reports have shown that overexpressed Rhomboid is localized in the Rab4, Rab14 and Rab7 endosomes but rarely in the Rab5 endosomes (Tsruya et al., 2007; Yogev et al., 2010). Consistent with this, we found that endogenous Rhomboid protein rarely colocalized with Rab5 but partially colocalized with Rab7 (Fig. 6F–G″). hrs mutation has been shown to cause enlarged endosomes and block multivesicular body (MVB) formation (Lloyd et al., 2002). Interestingly, in stam-mutant clones, Rhomboid accumulated in enlarged endosomes that often had the early-endosome marker Rab5 and late-endosome marker Rab7 (Fig. 6H–I″). Therefore, mutation of ESCRT-0 perturbs the endosome compartment and traps Rhomboid in the abnormal endosomes.
The localization of Rhomboid to the abnormal endosomes in stam-mutant clones can potentially block EGFR signaling by preventing Rhomboid and Spi from localizing to the same compartment. To test this possibility, we used the Sca–Gal4 driver to express membrane Spi (mSpi) in the developing R8 cells in order to determine colocalization between mSpi and endogenous Rho. Colocalization of mSpi and Rhomboid could be detected both in wild-type cells as well as in stam-mutant clones (Fig. 6J, white and yellow arrows). Therefore, stam mutation does not prevent Spi and Rhomboid from colocalizing to the same endosomal compartment. It is possible that the abnormal endosomes prevent the colocalized Spi from being trafficked to the location at which it is required to activate EGFR signaling.
DISCUSSION
In this report, we show that inactivation of the ESCRT-0 components stam or hrs induced synergistic cell death with rbf-inactivation by inhibiting EGFR signaling. In addition, stam and hrs mutants delay photoreceptor differentiation after R8 determination. Because EGFR signaling is required for the differentiation of all the photoreceptors except R8 (Dominguez et al., 1998), it is likely that the reduced EGFR signaling in stam and hrs mutants also contributes to the delayed photoreceptor differentiation phenotypes. It is worth pointing out that the photoreceptor differentiation delay phenotype of hrs or stam is reminiscent of that of rno, which also inhibits EGFR signaling (Sukhanova et al., 2011). However, because mutation of rno blocks EGFR signaling in the nucleus downstream of MAPK activation, rno mutation does not inhibit MAPK activity, and rbf rno mutation does not result in synergistic cell death (Steele et al., 2009; Sukhanova et al., 2011). The lack of synergistic cell death despite substantial differentiation delay in rbf rno double mutants suggests that the synergistic cell death of rbf stam cells is likely to be due to reduced MAPK activity in the posterior eye disc rather than to the indirect effect of delayed photoreceptor differentiation. However, because stam and hrs mutants have been shown to affect the localization of a large number of receptors in different signaling pathways, the possibility that other pathways also contribute to the synergistic cell death of rbf stam double mutants cannot be excluded.
Previously, mutations of axin, gig and Drosophila Tsc1 (dTsc1) have also been reported to induce synergistic cell death with rbf (Gordon et al., 2013; Hsieh et al., 2010; Li et al., 2010; Zhang et al., 2014). Interestingly, although synergistic cell death of rbf stam double-mutant cells is restricted in posterior eye discs, synergistic cell death in rbf axin, rbf gig or rbf dTsc1 double-mutant clones is more prominent in the anterior proliferating region of the eye disc. The distinct patterns of synergistic cell death observed in different double-mutant clones are mediated by different mechanisms. The synergistic cell death in rbf axin, rbf gig and rbf dTsc1 double-mutant cells is mediated by excessive cellular stress and increased dE2F1 levels (Gordon et al., 2013; Hsieh et al., 2010; Li et al., 2010; Zhang et al., 2014), whereas synergistic cell death of rbf stam or rbf hrs mutants in posterior eye discs is mediated by reduced EGFR signaling. In addition, mutation of groucho, which causes de-repression of Rhomboid expression and precocious EGFR signaling activation in eye discs (Zhang and Du, 2015), can suppress cell death in rbf mutants near the morphogenetic furrow (our unpublished results). The dependency of rbf-mutant cell apoptosis in the morphogenetic furrow and posterior eye disc on EGFR signaling suggests that this system can potentially be used to identify additional modulators of EGFR signaling.
Previous reports have shown that mutation of hrs enhances EGFR signaling owing to a block in endosomal trafficking that results in EGFR degradation in the developing embryos (Lloyd et al., 2002). By contrast, stam and hrs mutations have also been shown to inhibit EGFR signaling in eye and wing discs (Chanut-Delalande et al., 2010; Miura et al., 2008), but the mechanism is unknown. Here, we show that stam and hrs mutations inhibit EGFR signaling by blocking the normal endosomal trafficking of Rhomboid. Furthermore, we show that stam mutation leads to the accumulation of Rhomboid in enlarged endosomes that were positive for the early-endosome marker Rab5 and the late-endosome marker Rab7.
A recent report has shown that unprocessed Spi can be observed at the apical surface of the polarized epithelia in the presence of Star, but such Spi is inactive owing to the localization of the receptor in the basal lateral region (Steinhauer et al., 2013). In that report, it is suggested that Spi is endocytosed, cleaved by Rhomboid in endosomes and released to the basal lateral region to activate EGFR signaling (Steinhauer et al., 2013). Alternatively, Spi can potentially enter the Rhomboid-containing endosomes from Golgi and be released to the appropriate location after processing by Rhomboid (Yogev et al., 2010). Because Rhomboid localizes both in the Rab4-type fast recycling endosomes and in the Rab14-positive endosomes that mediate trafficking between Golgi and endosomes (Yogev et al., 2010), it is possible that both mechanisms contribute to the pool of active Spi ligand in vivo. This would be consistent with the observation that inactivation of either Rab4 or Rab14 does not result in any discernible phenotype, but inactivation of Rab11 inhibits EGFR signaling in developing eye discs (Yogev et al., 2010). Interestingly, expression of membrane Spi in R8 cells revealed that stam mutation does not block colocalization of membrane Spi and Rhomboid (Fig. 6J). It is possible that Spi that is localized in the abnormal endosomes cannot be properly exported in order to activate EGFR signaling, even if it is cleaved by Rhomboid in the same compartment.
In addition to regulating Rhomboid trafficking and active ligand release, which positively regulates EGFR signaling, ESCRT proteins can also negatively regulate EGFR signaling through receptor downregulation after ligand binding (Fischer et al., 2006; Dobrowolski et al., 2012). Indeed, hrs mutants have been shown to exhibit decreased receptor downregulation and increased EGFR signaling in embryos (Lloyd et al., 2002). In contrast, our results show that mutation of stam or hrs decreases EGFR signaling in imaginal discs. Therefore, stam and hrs mutations can potentially either decrease or increase EGFR signaling, depending on specific tissues or developmental stages. Although the exact mechanism of the differential effects of stam and hrs mutants on EGFR signaling has not been established, there are a number of possibilities. One is that activation of EGFR signaling by different ligands can potentially contribute to the differential effects of hrs mutation on EGFR signaling. There are four different EGFR ligands in flies: Spi, Grk, Vein and Keren. Vein is unique in that it is a secreted soluble EGFR ligand, whereas the others are membrane tethered. Therefore, EGFR activation by Vein is likely to be independent of Rhomboid and Star, and localized expression of Vein contributes to the localized activation of EGFR signaling (Schnepp et al., 1996). Interestingly, although Vein has been shown to play important roles in the induction of EGFR signaling activation in L4 primordia, but not in other regions of 3rd instar wing disc (Wessells et al., 1999), we found that stam mutation only weakly decreased aos–lacZ upregulation in the L4 primordia but substantially inhibited aos–lacZ in other wing disc regions (Fig. 3J). These results are consistent with the possibility that stam mutation preferentially reduces Spi- but not Vein-induced EGFR signaling activation. In addition, the membrane tethered EGFR ligands can differ in their dependence on processing by Rhomboid and Star proteins. For example, although Spi is strictly dependent on Rhomboid and/or Star to activate EGFR, Krn can undergo low-level cleavage and activate EGFR independent of Rhomboid and/or Star (Reich and Shilo, 2002). Another possibility is that in some tissues that are not polarized, EGFR signaling can potentially be activated by the unprocessed ligands exported by Star (Steinhauer et al., 2013). In this case, Stam and Hrs mainly regulate EGFR downregulation after ligand binding. Finally, the level of activation of EGFR signaling is likely to be influenced by the specific cellular background, feedback regulation, ligand levels, assay time, etc. For example, it is somewhat unexpected that higher EGFR signaling levels are induced in cells surrounding the sSpi-expressing clones than those in the sSpi-expressing cells (Fig. 5I). Because sSpi can still be palmitoylated, which enhances membrane association and restricts diffusion, sSpi-expressing cells are expected to be exposed to higher concentrations of sSpi ligands than the surrounding cells. It is possible that at the very high concentrations of sSpi, EGFR signaling is no longer limited by the level of sSpi ligand but is limited by the availability of EGFR, which is potentially depleted by the excess sSpi ligand in the overexpressing clones. Therefore, under this specific circumstance, higher levels of sSpi induce lower levels of EGFR signaling activation when assayed several days after sSpi clone induction.
MATERIALS AND METHODS
Drosophila stocks and genetics
Fly stocks used in this study were: rbf15aΔ (Zhang et al., 2014), hid138 (Tanaka-Matakatsu et al., 2009), de2f1i2 (Royzman et al., 1997), de2f1rm729 (Duronio et al., 1995), stam3297, stam2896 (Chanut-Delalande et al., 2010), hrsD28 (BL54574), aos–lacZ (BL2513), rho–lacZ (Freeman et al., 1992), UAS–Rho (Zhang and Du, 2015), UAS–sSpi (Miura et al., 2006), UAS–mSpi–GFP (Yogev et al., 2010), UAS–stam RNAi (BL35016), UAS–hrs RNAi (BL28026), UAS–Rbf RNAi (BL36744), UAS–YFP.Rab5 (BL24616), UAS–YFP.Rab7 (BL23270), UAS–EGFRCA (BL9533), UAS–rasv12 (BL4847), Vps28D2 (BL39624), Vps22 (BL39631), Vps32 (BL39623), Sca–gal4(BL6479), Long GMR–Gal4 (BL8121).
Flies were cultured at 25°C on standard cornmeal-yeast-agar medium. Double-mutant clones of rbf and stam (or hrs) were generated in a manner similar to that described previously (Tanaka-Matakatsu et al., 2009; Zhang et al., 2014) except that an RBF genomic rescue construct RBF-G3 inserted on chromosome 2L was used. This RBF rescue construct completely rescues the viability and fertility of rbf-null mutants. The exact genotypes used in this study are detailed in Table S1.
Genetic screen for mutations that modulate the phenotypes of rbf mutants
To identify mutations on chromosome 2L that can modulate the phenotypes of rbf, ethyl methanesulfonate (EMS) was used to generate mutants, which were then screened, in a manner similar to that described previously (Tanaka-Matakatsu et al., 2009). Isogenized w; p{ry+, neoFRT40A} males were used for mutagenesis, rbf15aΔ,w, eyFLP; p{ry+, neoFRT40A} p{w+, Ubi-GFP} p{w+, rbf-G3} and w, eyFLP; p{ry+,neoFRT40A} p{w+, Ubi-GFP} stocks were used for screening and an rbf dependence test. Mutations that gave rbf-dependent phenotypes were further mapped with a chromosome 2L deficiency kit from Bloomington, and the precise mutations were identified by whole-genome sequencing.
Whole-genome sequencing and alignment of sequencing reads
Genomic DNA from homozygous mutants of 19, 4-19-3 and isogenized FRT40A controls were prepared and sequenced. The raw reads were filtered with fastx-tools (http://hannonlab.cshl.edu/fastx_toolkit/commandline.html) to remove the low quality reads which 50% bases under the quality score 20. The reads were further trimmed into 80 bp length with quality scores larger than 20. The filtered reads were mapped to FlyBase r5.33 using BWA (version 0.6.1) with no more than three mismatches. The reads that showed unique differences were further analyzed in order to identify mutations.
Mutation detection
The consensus sequences were generated by SAMtools (version 0.1.18) with a quality score of 22 based on the published report (Blumenstiel et al., 2009). All repeat regions were masked by RepeatMasker. SAMtools and BCFtools were used to call single nucleotide polymorphisms (SNPs), and the maximum read depth for each mutation site was less than 100 (SNPs with super high coverage might be caused by copy number variation). For each pairing of wild-type sample and mutant sample, the mutation site was called twice. First, wild-type was used as reference to identify the mutation sites in the mutant sample. Second, the mutant sample was used as a reference to identify the ‘mutation sites’ in the wild-type sample. A candidate mutation was retained if it could be found using the above two steps. All mutations were annotated with SnpEff, which reports the non-synonymous mutation sites. Combining the mapping data with these analyses allowed the identification of phenotype-causing mutations in 19 and 4-19-3.
Immunostaining
Immunostaining was performed at room temperature. Larval imaginal discs were dissected in 1× PBS, fixed with 4% formaldehyde in PBS for 30 min, washed twice with 1× PBS with 0.3% Triton-X100 (PBST), blocked in blocking solution (PBST plus 10% normal goat serum) for 1 h and incubated with primary antibody in blocking solution overnight at 4°C. Primary antibodies were: rabbit anti-activated-Caspase-3 (C3, 1:300 from Cell Signaling, catalog #9661S), rabbit anti-Rhomboid (Rho, 1:500, gift from Dr. Ethan Bier; Sturtevant et al., 1996), rabbit anti-dpErk (1:400, Cell Signaling, catalog #4370S), mouse anti-β-galactosidase (1:100, Developmental Studies Hybridoma Bank, catalog #40-1a) and anti-Hid (1:500, gift from Dr. Don Ryoo; Ryoo et al., 2004). Following incubation with primary antibody, samples were washed three times (10 min each) in PBST, incubated with secondary antibodies from Jackson ImmunoResearch (1:400) for 1 h and washed three times with PBST. Samples were mounted in 70% glycerol with 1,4-diazabicyclo[2.2.2]octane (DABCO) at 12.5 mg/ml. Imaging was performed with the Zeiss Axio observer microscope with ApoTome using the AxioCam CCD camera controlled by Zeiss Axiovision software.
Quantification of cell death levels in developing imaginal discs
Cell death (%) was determined as described previously (Li et al., 2010) by calculating the percentage of the clone area (pixels) that exhibited above background levels of Caspase-3 signal by using the histogram function in Photoshop. The background level of Caspase-3 signal was determined as the level that was equal to or below 99% of the Caspase-3 signal in the wild-type tissues that exhibited no apoptosis. The average and standard deviation of the percentage cell death for a disc of each genotype was then determined and compared for at least six imaginal discs. Two-way Student's t-test was used to determine the significance of statistical differences between different genotypes.
Acknowledgements
We would like to thank Drs Jessica Treisman, Ben-Zion Shilo, Matthew Freeman, Marcus Affolter, Ethan Bier and Don Ryoo for providing fly stocks and reagents. We thank the Bloomington Stock Center (National Institutes of Health funded P40OD018537) for providing fly stocks and the Developmental Studies Hybridoma Bank (DSHB; created by the National Institute of Child Health and Human Development of the National Institutes of Health and maintained at The University of Iowa) for providing antibodies. We would also like to thank Dr Gabe Gordon for reading the manuscript and members of the Du lab for many discussions.
Footnotes
Competing interests
The authors declare no competing or financial interests.
Author contributions
Z.S. and W.D. designed the experiments and wrote the manuscript. Z.S. collected data presented in this manuscript. T.Z., Z.S., X.P. and X.L. performed the genetic screen and the genetic mapping studies. L.Y. and Z.Z. performed whole-genome sequencing and mutation identification.
Funding
This work was supported by grants from National Institutes of Health [grant numbers CA149275 and GM074197] to W.D.; and from the National Natural Science Foundation of China [grant number 31271398, 91131012] to Z.Z. Deposited in PMC for release after 12 months.
Data availability
Whole-genome sequencing data for the indicated mutants have been deposited at http://gsa.big.ac.cn with the following accession numbers: FRT40A control, CRR006280; stam mutation (19), CRR006281; Hrs mutation (4-19-3), CRR006282.
Supplementary information
Supplementary information available online at http://jcs.biologists.org/lookup/suppl/doi:10.1242/jcs.182261/-/DC1
References
- Attwooll C., Lazzerini Denchi E. and Helin K. (2004). The E2F family: specific functions and overlapping interests. EMBO J. 23, 4709-4716. 10.1038/sj.emboj.7600481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker N. E. and Yu S.-Y. (2001). The EGF receptor defines domains of cell cycle progression and survival to regulate cell number in the developing Drosophila eye. Cell 104, 699-708. 10.1016/S0092-8674(01)00266-5 [DOI] [PubMed] [Google Scholar]
- Bergmann A., Agapite J., McCall K. and Steller H. (1998). The Drosophila gene hid is a direct molecular target of Ras-dependent survival signaling. Cell 95, 331-341. 10.1016/S0092-8674(00)81765-1 [DOI] [PubMed] [Google Scholar]
- Blumenstiel J. P., Noll A. C., Griffiths J. A., Perera A. G., Walton K. N., Gilliland W. D., Hawley R. S. and Staehling-Hampton K. (2009). Identification of EMS-induced mutations in Drosophila melanogaster by whole-genome sequencing. Genetics 182, 25-32. 10.1534/genetics.109.101998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chanut-Delalande H., Jung A. C., Baer M. M., Lin L., Payre F. and Affolter M. (2010). The Hrs/Stam complex acts as a positive and negative regulator of RTK signaling during Drosophila development. PLoS ONE 5, e10245 10.1371/journal.pone.0010245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobrowolski R. and De Robertis E. M. (2012). Endocytic control of growth factor signalling: multivesicular bodies as signalling organelles. Nat. Rev. Mol. Cell Biol. 13, 53-60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Domínguez M., Wasserman J. D. and Freeman M. (1998). Multiple functions of the EGF receptor in Drosophila eye development. Curr. Biol. 8, 1039-1048. 10.1016/S0960-9822(98)70441-5 [DOI] [PubMed] [Google Scholar]
- Du W. (2000). Suppression of the rbf null mutants by a de2f1 allele that lacks transactivation domain. Development 127, 367-379. [DOI] [PubMed] [Google Scholar]
- Du W. and Pogoriler J. (2006). Retinoblastoma family genes. Oncogene 25, 5190-5200. 10.1038/sj.onc.1209651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duronio R. J., O'Farrell P. H., Xie J. E., Brook A. and Dyson N. (1995). The transcription factor E2F is required for S phase during Drosophila embryogenesis. Genes Dev. 9, 1445-1455. 10.1101/gad.9.12.1445 [DOI] [PubMed] [Google Scholar]
- Fischer J. A., Eun S. H. and Doolan B. T. (2006). Endocytosis, endosome trafficking, and the regulation of Drosophila development. Annu. Rev. Cell Dev. Biol. 22, 181-206. 10.1146/annurev.cellbio.22.010605.093205 [DOI] [PubMed] [Google Scholar]
- Freeman M. (1994). The spitz gene is required for photoreceptor determination in the Drosophila eye where it interacts with the EGF receptor. Mech. Dev. 48, 25-33. 10.1016/0925-4773(94)90003-5 [DOI] [PubMed] [Google Scholar]
- Freeman M., Kimmel B. E. and Rubin G. M. (1992). Identifying targets of the rough homeobox gene of Drosophila: evidence that rhomboid functions in eye development. Development 116, 335-346. [DOI] [PubMed] [Google Scholar]
- Gabay L., Seger R. and Shilo B.-Z. (1997). In situ activation pattern of Drosophila EGF receptor pathway during development. Science 277, 1103-1106. 10.1126/science.277.5329.1103 [DOI] [PubMed] [Google Scholar]
- Gordon G. M. and Du W. (2011a). Conserved RB functions in development and tumor suppression. Protein Cell 2, 864-878. 10.1007/s13238-011-1117-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon G. M. and Du W. (2011b). Targeting Rb inactivation in cancers by synthetic lethality. Am. J. Cancer Res. 1, 773-786. [PMC free article] [PubMed] [Google Scholar]
- Gordon G. M., Zhang T., Zhao J. and Du W. (2013). Deregulated G1-S control and energy stress contribute to the synthetic-lethal interactions between inactivation of RB and TSC1 or TSC2. J. Cell Sci. 126, 2004-2013. 10.1242/jcs.121301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsieh T.-C., Nicolay B. N., Frolov M. V. and Moon N.-S. (2010). Tuberous sclerosis complex 1 regulates dE2F1 expression during development and cooperates with RBF1 to control proliferation and survival. PLoS Genet. 6, e1001071 10.1371/journal.pgen.1001071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katzmann D. J., Odorizzi G. and Emr S. D. (2002). Receptor downregulation and multivesicular-body sorting. Nat. Rev. Mol. Cell Biol. 3, 893-905. 10.1038/nrm973 [DOI] [PubMed] [Google Scholar]
- Lee J. R., Urban S., Garvey C. F. and Freeman M. (2001). Regulated intracellular ligand transport and proteolysis control EGF signal activation in Drosophila. Cell 107, 161-171. 10.1016/S0092-8674(01)00526-8 [DOI] [PubMed] [Google Scholar]
- Li B., Gordon G. M., Du C. H., Xu J. and Du W. (2010). Specific killing of Rb mutant cancer cells by inactivating TSC2. Cancer Cell 17, 469-480. 10.1016/j.ccr.2010.03.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lloyd T. E., Atkinson R., Wu M. N., Zhou Y., Pennetta G. and Bellen H. J. (2002). Hrs regulates endosome membrane invagination and tyrosine kinase receptor signaling in Drosophila. Cell 108, 261-269. 10.1016/S0092-8674(02)00611-6 [DOI] [PubMed] [Google Scholar]
- Miura G. I., Buglino J., Alvarado D., Lemmon M. A., Resh M. D. and Treisman J. E. (2006). Palmitoylation of the EGFR ligand Spitz by Rasp increases Spitz activity by restricting its diffusion. Dev. Cell 10, 167-176. 10.1016/j.devcel.2005.11.017 [DOI] [PubMed] [Google Scholar]
- Miura G. I., Roignant J.-Y., Wassef M. and Treisman J. E. (2008). Myopic acts in the endocytic pathway to enhance signaling by the Drosophila EGF receptor. Development 135, 1913-1922. 10.1242/dev.017202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moon N.-S., Frolov M. V., Kwon E.-J., Di Stefano L., Dimova D. K., Morris E. J., Taylor-Harding B., White K. and Dyson N. J. (2005). Drosophila E2F1 has context-specific pro- and antiapoptotic properties during development. Dev. Cell 9, 463-475. 10.1016/j.devcel.2005.08.015 [DOI] [PubMed] [Google Scholar]
- Morris E. J. and Dyson N. J. (2001). Retinoblastoma protein partners. Adv. Cancer Res. 82, 1-54. 10.1016/S0065-230X(01)82001-7 [DOI] [PubMed] [Google Scholar]
- Neuman-Silberberg F. S. and Schüpbach T. (1993). The Drosophila dorsoventral patterning gene gurken produces a dorsally localized RNA and encodes a TGF alpha-like protein. Cell 75, 165-174. 10.1016/S0092-8674(05)80093-5 [DOI] [PubMed] [Google Scholar]
- Reich A. and Shilo B.-Z. (2002). Keren, a new ligand of the Drosophila epidermal growth factor receptor, undergoes two modes of cleavage. EMBO J. 21, 4287-4296. 10.1093/emboj/cdf439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Royzman I., Whittaker A. J. and Orr-Weaver T. L. (1997). Mutations in Drosophila DP and E2F distinguish G1-S progression from an associated transcriptional program. Genes Dev. 11, 1999-2011. 10.1101/gad.11.15.1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutledge B. J., Zhang K., Bier E., Jan Y. N. and Perrimon N. (1992). The Drosophila spitz gene encodes a putative EGF-like growth factor involved in dorsal-ventral axis formation and neurogenesis. Genes Dev. 6, 1503-1517. 10.1101/gad.6.8.1503 [DOI] [PubMed] [Google Scholar]
- Ryoo H. D., Gorenc T. and Steller H. (2004). Apoptotic cells can induce compensatory cell proliferation through the JNK and the Wingless signaling pathways. Dev. Cell 7, 491-501. 10.1016/j.devcel.2004.08.019 [DOI] [PubMed] [Google Scholar]
- Schnepp B., Grumbling G., Donaldson T. and Simcox A. (1996). Vein is a novel component in the Drosophila epidermal growth factor receptor pathway with similarity to the neuregulins. Genes Dev. 10, 2302-2313. 10.1101/gad.10.18.2302 [DOI] [PubMed] [Google Scholar]
- Schweitzer R., Shaharabany M., Seger R. and Shilo B. Z. (1995). Secreted Spitz triggers the DER signaling pathway and is a limiting component in embryonic ventral ectoderm determination. Genes Dev. 9, 1518-1529. 10.1101/gad.9.12.1518 [DOI] [PubMed] [Google Scholar]
- Steele L., Sukhanova M. J., Xu J., Gordon G. M., Huang Y., Yu L. and Du W. (2009). Retinoblastoma family protein promotes normal R8-photoreceptor differentiation in the absence of rhinoceros by inhibiting dE2F1 activity. Dev. Biol. 335, 228-236. 10.1016/j.ydbio.2009.09.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinhauer J., Liu H. H., Miller E. and Treisman J. E. (2013). Trafficking of the EGFR ligand Spitz regulates its signaling activity in polarized tissues. J. Cell Sci. 126, 4469-4478. 10.1242/jcs.131169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevaux O., Dimova D. K., Ji J.-Y., Moon N. S., Frolov M. V. and Dyson N. J. (2005). Retinoblastoma family 2 is required in vivo for the tissue-specific repression of dE2F2 target genes. Cell Cycle 4, 1272-1280. 10.4161/cc.4.9.1982 [DOI] [PubMed] [Google Scholar]
- Sturtevant M. A., Roark M., O'Neill J. W., Biehs B., Colley N. and Bier E. (1996). The Drosophila rhomboid protein is concentrated in patches at the apical cell surface. Dev. Biol. 174, 298-309. 10.1006/dbio.1996.0075 [DOI] [PubMed] [Google Scholar]
- Sukhanova M. J., Steele L. J., Zhang T., Gordon G. M. and Du W. (2011). RBF and Rno promote photoreceptor differentiation onset through modulating EGFR signaling in the Drosophila developing eye. Dev. Biol. 359, 190-198. 10.1016/j.ydbio.2011.08.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y., Jan L. Y. and Jan Y. N. (1998). Transcriptional regulation of atonal during development of the Drosophila peripheral nervous system. Development 125, 3731-3740. [DOI] [PubMed] [Google Scholar]
- Tanaka-Matakatsu M. and Du W. (2008). Direct control of the proneural gene atonal by retinal determination factors during Drosophila eye development. Dev. Biol. 313, 787-801. 10.1016/j.ydbio.2007.11.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka-Matakatsu M., Xu J., Cheng L. and Du W. (2009). Regulation of apoptosis of rbf mutant cells during Drosophila development. Dev. Biol. 326, 347-356. 10.1016/j.ydbio.2008.11.035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka-Matakatsu M., Miller J., Borger D., Tang W.-J. and Du W. (2014). Daughterless homodimer synergizes with Eyeless to induce Atonal expression and retinal neuron differentiation. Dev. Biol. 392, 256-265. 10.1016/j.ydbio.2014.05.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka-Matakatsu M., Miller J. and Du W. (2015). The homeodomain of Eyeless regulates cell growth and antagonizes the paired domain-dependent retinal differentiation function. Protein Cell 6, 68-78. 10.1007/s13238-014-0101-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Treisman J. E. (2013). Retinal differentiation in Drosophila. Wiley Interdiscip. Rev. Dev. Biol. 2, 545-557. 10.1002/wdev.100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trimarchi J. M. and Lees J. A. (2002). Sibling rivalry in the E2F family. Nat. Rev. Mol. Cell Biol. 3, 11-20. 10.1038/nrm714 [DOI] [PubMed] [Google Scholar]
- Tsruya R., Schlesinger A., Reich A., Gabay L., Sapir A. and Shilo B.-Z. (2002). Intracellular trafficking by Star regulates cleavage of the Drosophila EGF receptor ligand Spitz. Genes Dev. 16, 222-234. 10.1101/gad.214202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsruya R., Wojtalla A., Carmon S., Yogev S., Reich A., Bibi E., Merdes G., Schejter E. and Shilo B.-Z. (2007). Rhomboid cleaves Star to regulate the levels of secreted Spitz. EMBO J. 26, 1211-1220. 10.1038/sj.emboj.7601581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urban S., Lee J. R. and Freeman M. (2001). Drosophila rhomboid-1 defines a family of putative intramembrane serine proteases. Cell 107, 173-182. 10.1016/S0092-8674(01)00525-6 [DOI] [PubMed] [Google Scholar]
- van den Heuvel S. and Dyson N. J. (2008). Conserved functions of the pRB and E2F families. Nat. Rev. Mol. Cell Biol. 9, 713-724. 10.1038/nrm2469 [DOI] [PubMed] [Google Scholar]
- Wessells R. J., Grumbling G., Donaldson T., Wang S.-H. and Simcox A. (1999). Tissue-specific regulation of vein/EGF receptor signaling in Drosophila. Dev. Biol. 216, 243-259. 10.1006/dbio.1999.9459 [DOI] [PubMed] [Google Scholar]
- Williams R. L. and Urbé S. (2007). The emerging shape of the ESCRT machinery. Nat. Rev. Mol. Cell Biol. 8, 355-368. 10.1038/nrm2162 [DOI] [PubMed] [Google Scholar]
- Yogev S., Schejter E. D. and Shilo B.-Z. (2010). Polarized secretion of Drosophila EGFR ligand from photoreceptor neurons is controlled by ER localization of the ligand-processing machinery. PLoS Biol. 8, e1000505 10.1371/journal.pbio.1000505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang T. and Du W. (2015). Groucho restricts rhomboid expression and couples EGFR activation with R8 selection during Drosophila photoreceptor differentiation. Dev. Biol. 407, 246-255. 10.1016/j.ydbio.2015.09.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang T., Ranade S., Cai C. Q., Clouser C. and Pignoni F. (2006). Direct control of neurogenesis by selector factors in the fly eye: regulation of atonal by Ey and So. Development 133, 4881-4889. 10.1242/dev.02669 [DOI] [PubMed] [Google Scholar]
- Zhang T., Liao Y., Hsu F.-N., Zhang R., Searle J. S., Pei X., Li X., Ryoo H. D., Ji J.-Y. and Du W. (2014). Hyperactivated Wnt signaling induces synthetic lethal interaction with Rb inactivation by elevating TORC1 activities. PLoS Genet. 10, e1004357 10.1371/journal.pgen.1004357 [DOI] [PMC free article] [PubMed] [Google Scholar]