

ABSTRACT
In 1994 in the Journal of Cell Science, Hennekes and Nigg reported that changing valine to arginine at the endoproteolytic cleavage site in chicken prelamin A abolishes its conversion to lamin A. The consequences of this mutation in an organism have remained unknown. We now report that the corresponding mutation in a human subject leads to accumulation of prelamin A and causes a progeroid disorder. Next generation sequencing of the subject and her parents' exomes identified a de novo mutation in the lamin A/C gene (LMNA) that resulted in a leucine to arginine amino acid substitution at residue 647 in prelamin A. The subject's fibroblasts accumulated prelamin A, a farnesylated protein, which led to an increased percentage of cultured cells with morphologically abnormal nuclei. Treatment with a protein farnesyltransferase inhibitor improved abnormal nuclear morphology. This case demonstrates that accumulation of prelamin A, independent of the loss of function of ZMPSTE24 metallopeptidase that catalyzes processing of prelamin A, can cause a progeroid disorder and that a cell biology assay could be used in precision medicine to identify a potential therapy.
KEY WORDS: Lamin, Nuclear envelope, Progeria, Mandibuloacral dysplasia, ZMPSTE24
Highlighted article: We describe a mutation in the human lamin A/C gene that destroys the ZMPSTE24 cleavage site and causes a progeroid disorder.
INTRODUCTION
Nuclear lamins are intermediate filament proteins that form the nuclear lamina on the inner aspect of the inner nuclear membrane (Gerace et al., 1978; Fisher et al., 1986; McKeon et al., 1986). The human lamin A/C gene (LMNA) encodes lamin A and lamin C (Lin and Worman, 1993). Lamin A is synthesized as a precursor, prelamin A, which undergoes a series of processing reactions to generate the mature protein (Sinensky et al., 1994; Young et al., 2005). First, protein farnesyltransferase catalyzes the addition of the farnesyl moiety to the cysteine residue of a C-terminal CAAX motif (CSIM in prelamin A). The terminal three amino acids are then cleaved, and isoprenylcysteine carboxyl methyltransferase catalyzes methylation of the farnesylcysteine. Zinc metallopeptidase ZMPSTE24 then recognizes the farnesylated protein and catalyzes its endoproteolytic cleavage, which removes the last 15 amino acids, including the farnesylcysteine α-methyl ester. Farnesylation is necessary for recognition by ZMPSTE24. Prelamin A is a transient intermediate and does not normally accumulate in cells.
Genetic mutations leading to defective processing of prelamin A have been shown to cause progeroid disorders. De novo dominant splice-donor site mutations in exon 11, generating a truncated prelamin A protein lacking the ZMPSTE24 cleavage site, cause Hutchinson–Gilford progeria syndrome (HGPS) (De Sandre-Giovannoli et al., 2003; Eriksson et al., 2003). Homozygous mutations leading to loss of function of ZMPSTE24 cause the neonatal lethal progeroid disorder restrictive dermopathy (Moulson et al., 2005; Navarro et al., 2005). Compound heterozygous mutations in ZMPSTE24 generating variants with reduced enzymatic activity also cause progeroid disorders, including mandibuloacral dysplasia with type B lipodystrophy (Agarwal et al., 2003; Shackleton et al., 2005; Barrowman et al., 2012b). In these conditions, a farnesylated prelamin A variant or unprocessed farnesylated prelamin A accumulates in cells. However, disease-causing alterations of ZMPSTE24 lead to loss of its other function in clearing the translocon (Ast et al., 2016). Hence, there is no human pathology reported so far in which ZMPSTE24-catalyzed cleavage of prelamin A is the only cellular defect.
In 1994, Hennekes and Nigg showed that changing a valine residue to an arginine at the endoproteolytic cleavage site in chicken prelamin A abolishes its conversion to lamin A (Hennekes and Nigg, 1994). Michaelis and colleagues subsequently demonstrated in cultured cells that had been transfected with prelamin A constructs that the ZMPSTE24-catalyzed cleavage of the corresponding human variant, in which a leucine is changed to arginine (L647R), is similarly blocked (Mallampalli et al., 2005; Barrowman et al., 2012a). Overexpression of this human prelamin A variant in transfected cells also induces abnormal nuclear morphology (Mallampalli et al., 2005). The pathophysiological significance of this variant, if any, has remained unknown. We now describe an individual with a progeroid disorder who is heterozygous for the LMNA L647R mutation. We further examine the consequence of this mutation in the individual's cells that express the variant protein at levels that induce pathology, and we demonstrate the use of a cell biology assay in precision medicine to identify a potential therapy.
RESULTS AND DISCUSSION
Progeroid disorder phenotype
The individual was a 17-year-old woman from the Dominican Republic referred for evaluation of short stature. Her height was 150 cm (<3rd centile), weight 36.7 kg (<3rd centile) and occipitofrontal circumference 51 cm (<3rd centile). She was the 2.7-kg product of a full-term uncomplicated pregnancy and delivered to a 23-year-old primigravida mother. Her postnatal growth was poor but cognitive development was normal. She reached menarche at 13 years and has since had regular menstrual cycles. Her parents and two brothers were of normal height and generally healthy.
On physical examination, the individual was small for her age and microcephalic. She had a slightly thinning anterior hairline, micrognathia and dental crowding. Skin examination revealed diffusely distributed hypopigmented patches, mild subcutaneous lipoatrophy, mottled pigmentation and prominent depigmented patches with perifollicular repigmentation on her elbows (Fig. 1A). Her skin was taut and dry, and there were sclerodermatous changes over the lower abdomen and proximal thighs. A shaved biopsy of the right posterior upper leg revealed thickened bundles of collagen aligned parallel to the skin surface and crowded in the reticular dermis, as well as loss of dendritic cells upon immunostaining for CD34, consistent with morphea (localized scleroderma or thickened skin). The individual's hands had mottled cutaneous pigmentation, nail dystrophy and several short fingers, including the index finger of the left hand (Fig. 1B). A radiograph of the left hand showed acro-osteolysis (resorption of the distal phalanx) of the second finger, tapering of the distal phalanx of the first digit, mild irregularity of the tufts of the third and fourth phalanges, and mild soft tissue swelling of the distal phalanges (Fig. 1C). There was also acro-osteolysis of the first, second, third and fifth distal phalanges of the right hand. Radiographs of the long bones and skull were normal, but the feet showed multiple well-defined lucencies, some with sclerotic margins, indicating abnormal bone resorption or deposition (Fig. 1D).
Fig. 1.
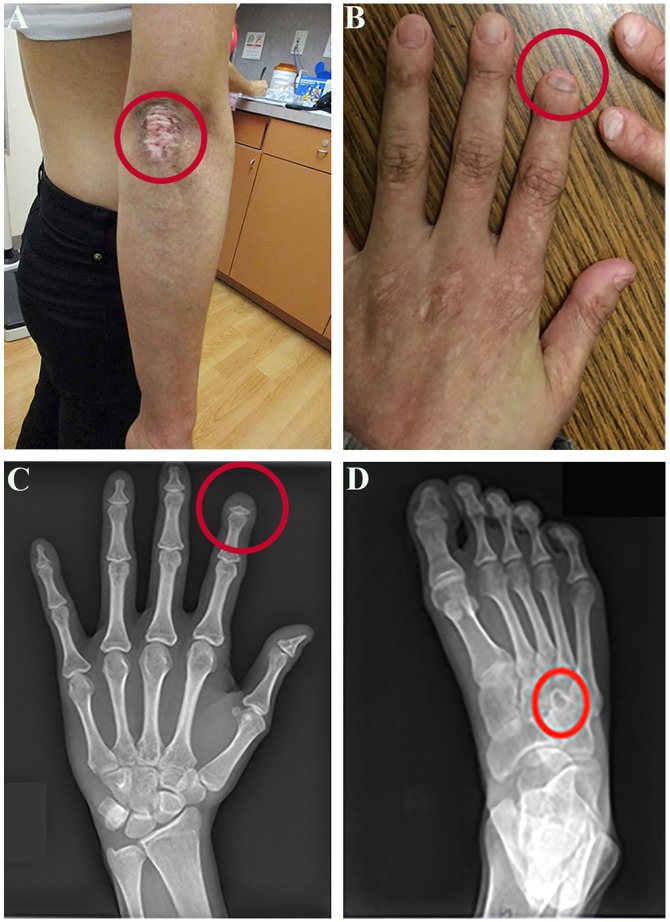
Phenotype of the affected individual. (A) Photograph of the individual's right arm showing a prominent depigmented patch. (B) Photograph of the individual's hands showing mottled cutaneous pigmentation, nail dystrophy and a short index finger on the left hand. (C) Radiograph of the individual's left hand showing acro-osteolysis of the distal second phalanx, tapering of the distal aspect of the distal phalanx of the first digit and mild irregularity of the tuft of the third and fourth phalanges. (D) Radiograph of right foot showing well-defined lucencies, some with sclerotic margins. A red circle surrounds the abnormality in each panel. The individual provided written consent to publish these photographs.
Complete blood cell count showed microcytic anemia. Serum antinuclear antibodies were present at a titer of 1:80 but anti-double-stranded-DNA, anti-SS-A/Ro, anti-SCL-70 and anti-centromere antibodies were not detected, essentially excluding systemic lupus erythematosus, systemic sclerosis and limited systemic sclerosis as diagnoses. Serum glucose, C-peptide, hemoglobin A1C, triglycerides and cholesterol levels were normal, indicating a lack of insulin resistance or disorders of lipid metabolism. Echocardiography showed a normal left ventricular ejection fraction of 55% to 60% and stress echocardiography showed no ischemic changes at 81% of the target heart rate, suggesting no coronary artery obstructive disease.
Based on the collective clinical examination, we conclude that the individual had skin and bone abnormalities typical of mandibuloacral dysplasia and a modest decrease in subcutaneous fat but did not have metabolic abnormalities that can occur with lipodystrophy, such as insulin resistance and hypertriglyceridemia. There is phenotypic overlap between mandibuloacral dysplasia and HGPS (Agarwal et al., 2003; Shackleton et al., 2005; Barrowman et al., 2012b). Similar skin, bone and fat abnormalities can occur in HGPS. However, the individual we describe was affected to a much lesser extent than children with HGPS and showed no apparent coronary artery disease at 18 years of age. Most children with HGPS die from cardiovascular disease in their second decade (Merideth et al., 2008).
Pathologic LMNA mutation that abolishes prelamin A processing
Whole exome sequencing revealed a heterozygous T>G transversion at nucleotide 1940 of LMNA (Chr1:156,108,520; GRCh37/hg19; Transcript: LMNA-001 ENST00000368300) that resulted in a L647R (Protein: ENSP00000357283) amino acid substitution (Fig. 2A) [based on Ensembl Release 83 annotation (http://useast.ensembl.org/index.html)]. The mutation was de novo as it was not present in the mother or father, who each shared approximately 50% of their rare (<1% population frequency) polymorphisms with the individual. Residue 647 is at the endoproteolytic site where ZMPSTE24-catalyzed cleavage of farnesylated prelamin A occurs. A change from a hydrophobic to a basic amino acid at this residue is predicted to reduce the ability of ZMPSTE24 to catalyze the cleavage of prelamin A (Fig. 2B). Immunoblotting of protein extracts from the individual's fibroblasts using antibodies against lamin A and lamin C demonstrated abnormal accumulation of a more slowly migrating protein that is not usually present in normal cells but that is detectable in normal cells if prelamin A processing is blocked by treatment with a protein farnesyltransferase inhibitor (FTI), consistent with it being prelamin A (Fig. 2C, left panel). This more slowly migrating protein was confirmed to be prelamin A by immunoblotting with a specific antibody against prelamin A (Fig. 2C, right panel). The individual's fibroblasts, but not cells from a control individual, were further shown to accumulate prelamin A by performing immunofluorescence microscopy using a specific antibody against prelamin A, whereas an antibody against lamin A/C labeled both normal fibroblasts and those from the individual (Fig. 2D).
Fig. 2.

Identification of the LMNA L647R mutation predicted to destroy the ZMPSTE24 cleavage site in prelamin A and accumulation of prelamin A in the individual's fibroblasts. (A) Readout of next-generation sequencing and the presence of a heterozygous T>G transversion that results in a L647R amino acid substitution in prelamin A. (B) The L647R amino acid substitution is predicted to abolish the endoproteolytic cleavage of farnesylated-carboxymethylated (represented in green) prelamin A that is catalyzed by ZMPSTE24 (represented by scissors). (C) Immunoblots of protein extracts from fibroblasts of the individual (patient) and fibroblasts from a normal individual (normal) with or without treatment with an FTI (FTI-277). Blots were probed with an anti-lamin A/C antibody that recognized prelamin A, lamin A and lamin C (left) or an antibody that specifically recognized prelamin A (right). Labeling with an antibody against GAPDH is shown as a loading control. Migrations of molecular mass standards are indicated at the left of each blot. (D) Immunofluorescence micrographs of fibroblasts from a normal subject (top panels) and the individual (bottom panels) labeled with antibodies that specifically recognized prelamin A (green), lamin A/C (red) and the overlay of these signals (yellow).
This LMNA L647R mutation corresponds to the first amino acid substitution experimentally introduced into chicken prelamin A to demonstrate that this residue is essential for prelamin A processing (Hennekes and Nigg, 1994). The affected individual's cells accumulated unprocessed prelamin A (with a single amino acid change), analogous to what occurs in subjects with mandibuloacral dysplasia or atypical progeroid disorders that are caused by compound heterozygous mutations in ZMPSTE24 and lead to partial loss of the enzyme activity. ZMPSTE24 also functions in clearing the translocon, and disease-associated mutations lead to loss of this function (Ast et al., 2016). The current case demonstrates that the selective loss of ZMPSTE24-catalyzed cleavage of prelamin A, distinct from any defect in translocon unclogging, can cause a progeroid disorder.
Prelamin A has also been reported to accumulate in cells of subjects with autosomal recessive mandibuloacral dysplasia with type A lipodystrophy caused by the LMNA R527H mutation (Novelli et al., 2002; Capanni et al., 2005). The mechanism responsible for prelamin A accumulation in this disorder is unknown. However, fibroblasts from a subject with a progeroid disorder and a cysteine residue substituted for arginine at prelamin A residue 644, which biochemical assays indicate is crucial for efficient ZMPSTE24-catalyzed cleavage, do not accumulate prelamin A (Toth et al., 2005). In HGPS, a truncated prelamin A variant accumulates (Eriksson et al., 2003; Goldman et al., 2004). In both the affected individual discussed here and in HGPS individuals, one LMNA allele is mutated and the other is normal. One hypothesis regarding the more severe phenotype in HGPS is that the truncated prelamin A variant in that disease lacks 50 amino acids, whereas in the individual we describe, there is only a single amino acid substitution. Hence, loss of those additional amino acids could lead to more profound defects. We caution however that we are comparing only one individual to the published literature on HGPS.
Abnormal nuclear morphology and reversal with an FTI
Abnormal nuclear morphology is a characteristic of fibroblasts from individuals with alterations in lamin A and lamin C. We therefore carefully examined the affected individual's fibroblasts, which express prelamin A at a level that causes pathology, by performing immunofluorescence microscopy using antibodies against lamin A/C. There was a significant decrease in the percentage of cells with normal nuclear morphology compared to control cells, and the percentage of cells with normal nuclear morphology further decreased with passage number in culture (Fig. 3A,B). This also occurs with fibroblasts from subjects with HGPS (Goldman et al., 2004). The individual we describe also had fewer fibroblasts with morphologically abnormal nuclei than subjects with HGPS, in which the percentage of fibroblasts with abnormal nuclei can reach 50% by passage 13 in culture (Goldman et al., 2004). Various nuclear shape abnormalities were observed in the individual's fibroblasts (Fig. S1). Treatment of cultures with an FTI that blocks protein farnesylation partially reverses the abnormal nuclear morphology in fibroblasts from human subjects with HGPS or ZMPSTE24 loss-of-function mutations (Capell et al., 2005; Toth et al., 2005). Similarly, treatment with an FTI reverses abnormal nuclear morphology in transiently transfected HeLa cells overexpressing a green fluorescent protein fusion of prelamin A with the L647R amino acid substitution (Mallampalli et al., 2005). We therefore treated cultured fibroblasts from the individual with an FTI. When the individual's fibroblasts were treated with an FTI, there were significantly more cells with normal nuclear morphology compared to those treated with placebo (Fig. 3C,D). FTI treatment also generated some abnormal ‘donut’-shaped nuclei, as previously described (Verstraeten et al., 2011).
Fig. 3.

Abnormal nuclear morphology in the individual's fibroblasts and effects of FTI treatment. (A) Immunofluorescence micrographs of the individual's cultured fibroblasts labeled with anti-lamin-A/C antibodies after passage 4 (P4), 8 (P8), 13 (P13) and 16 (P16); control fibroblasts are also shown at passage 16 [Ctr (P16)]. Arrows indicate representative morphologically abnormal nuclei. (B) Percentages of normal-shaped nuclei in cultures from individual fibroblasts after each indicated passage and for control cells at passage 16. (C) Immunofluorescence micrographs of the individual's cultured fibroblasts labeled with an anti-lamin-A/C antibody after passage 16 (P16) and treated with either the placebo DMSO or FTI for 72 hours. Arrows indicate representative morphologically abnormal nuclei. (D) Percentages of normal-shaped nuclei in cultures of the affected individual's fibroblasts at passage 16 after treatment with placebo (DMSO) or FTI. For quantification, nuclei in 200–800 cells of 5–10 microscopic fields per sample were scored as having normal or abnormal morphology by an independent observer with experience in cell biology that was blind to the experiment. This person was unaware of cell passage number and drug treatment. Information about features counted as abnormal, including representative images, is provided in Fig. S1. Values in B,D are means±s.e.m.; *P<0.05, **P<0.01 (two-tailed Student's t-test with unequal variance). The bar at the top of panel B indicates the differences between the individual's cells at P4, P8, P13 and P16 compared to control.
Farnesylated prelamin A is undetectable in normal cells but accumulates if ZMPSTE24 activity is reduced. In HGPS, the prelamin A variant lacking the ZMPSTE24 cleavage site also remains farnesylated. Evidence that the farnesyl modification of these proteins contributes to pathogenesis comes from studies in model mice that lack ZMPSTE24 or that express the HGPS lamin A variant, where treatment with an FTI improves their progeroid phenotypes (Fong et al., 2006; Yang et al., 2006; Capell et al., 2008). A clinical trial of an FTI has even been carried out in children with HGPS (Gordon et al., 2012). Treatment with statins and aminobisphosphonates has been shown to have similar beneficial effects in mice that lack ZMPSTE24 (Varela et al., 2008). FTI treatment improved the abnormal nuclear morphology in fibroblasts from the individual described in this study. This raises the possibility of treating the individual with drugs that block protein farnesylation and demonstrates the use of a cell biology assay in precision medicine.
MATERIALS AND METHODS
Exome sequencing
Genomic DNA was isolated from whole blood. Agilent Sureselect (V.5 +UTR) capture and 100 base pair paired-end high-throughput sequencing (Illumina HiSeq2500) were used for next-generation whole exome sequencing. Alignment and mutation calling were performed using the NextGENe software (version 2.3.4) (Softgenetics). Mutation filtering and annotation were performed by the New-York-State-approved in-house-developed pipeline and reviewed as part of the clinical workflow for constitutional clinical exome sequencing in the laboratory of Personalized Genomic Medicine at Columbia University Medical Center.
Cells and cell culture
The Columbia University Medical Center Institutional Review Board approved the protocol to obtain dermal fibroblasts and informed consent was obtained for skin biopsy. Control primary fibroblasts were from a healthy 45-year-old woman (obtained from the Progeria Research Foundation) and used at the same passage number as the affected individual's fibroblasts. Cells were cultured in Chang medium D (Irvine Scientific) and showed no evidence of contamination. In some experiments, cells were treated with 2.5 µM FTI-277 (Sigma) dissolved in dimethyl sulfoxide (DMSO) or DMSO alone, as described previously (Wang et al., 2012).
Immunoblotting and immunofluorescence microscopy
Materials and methods for protein extraction, electrophoresis, immunoblotting, immunofluorescence microscopy and FTI treatment of cells were as those described previously (Wang et al., 2012). Primary antibodies were rabbit anti-lamin A/C (H110, sc-20681, Santa Cruz) used at a dilution of 1:2000 for immunofluorescence microscopy or 1:6000 for immunoblotting, mouse anti-GAPDH (6C5, AM4300, Ambion) used at a dilution of 1:3000 and rat anti-prelamin-A [Lee et al., 2010; generously provided by Drs Loren Fong and Stephen G. Young, University of California, Los Angeles, CA] used at a dilution of 1:3000. Data were analyzed using Microsoft Excel; P-values were calculated using two-tailed Student t-test with unequal variance.
Acknowledgements
We thank the individual in this study for consenting to participate.
Footnotes
Competing interests
Y.W., U.L.-K., K.A.-Y., J.E.S., C.Ö., J.-Y.S., L.N.C. and G.G.G. have no competing interests to declare. J.T.L. is currently an employee of AstraZeneca and anticipates owning equity in the company. P.L.N. is currently an employee of MNG Laboratories and owns equity in the company. H.J.W. is a member of the scientific advisory board of AlloMek Therapeutics, which is developing a preclinical drug to treat individuals with disease caused by LMNA mutations, and owns equity in the company.
Author contributions
Y.W., U.L.-K. and H.J.W. designed the research; Y.W., C.Ö. and J.-Y.S. performed experiments; Y.W., C.Ö., J.-Y.S., L.N.C., G.G.G., P.L.N. and H.J.W. analyzed data; U.L.-K. identified the study individual, U.L.-K., K.A.-Y., J.E.S. and J.T.L. cared for and phenotyped the study individual; J.T.L. obtained fibroblasts from the study individual, P.L.N. established the platform for exome sequencing and analysis; H.J.W. wrote the manuscript and all the other authors contributed to parts of it.
Funding
Supported by the Eunice Kennedy Shriver National Institute of Child Health and Human Development as part of the National Institutes of Health [grant number R01HD070713] to G.G.G. and H.J.W. Deposited in PMC for release after 12 months.
Supplementary information
Supplementary information available online at http://jcs.biologists.org/lookup/suppl/doi:10.1242/jcs.187302/-/DC1
References
- Agarwal A. K., Fryns J.-P., Auchus R. J. and Garg A. (2003). Zinc metalloproteinase, ZMPSTE24, is mutated in mandibuloacral dysplasia. Hum. Mol. Genet. 12, 1995-2001. 10.1093/hmg/ddg213 [DOI] [PubMed] [Google Scholar]
- Ast T., Michaelis S. and Schuldiner M. (2016). The protease Ste24 clears clogged translocons. Cell 164, 103-114. 10.1016/j.cell.2015.11.053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrowman J., Hamblet C., Kane M. S. and Michaelis S. (2012a). Requirements for efficient proteolytic cleavage of prelamin A by ZMPSTE24. PLoS ONE 7, e32120 10.1371/journal.pone.0032120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrowman J., Wiley P. A., Hudon-Miller S. E., Hrycyna C. A. and Michaelis S. (2012b). Human ZMPSTE24 disease mutations: residual proteolytic activity correlates with disease severity. Hum. Mol. Genet. 21, 4084-4093. 10.1093/hmg/dds233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capanni C., Mattioli E., Columbaro M., Lucarelli E., Parnaik V. K., Novelli G., Wehnert M., Cenni V., Maraldi N. M., Squarzoni S. et al. (2005). Altered pre-lamin A processing is a common mechanism leading to lipodystrophy. Hum. Mol. Genet. 14, 1489-1502. 10.1093/hmg/ddi158 [DOI] [PubMed] [Google Scholar]
- Capell B. C., Erdos M. R., Madigan J. P., Fiordalisi J. J., Varga R., Conneely K. N., Gordon L. B., Der C. J., Cox A. D. and Collins F. S. (2005). Inhibiting farnesylation of progerin prevents the characteristic nuclear blebbing of Hutchinson-Gilford progeria syndrome. Proc. Natl. Acad. Sci. USA 102, 12879-12884. 10.1073/pnas.0506001102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capell B. C., Olive M., Erdos M. R., Cao K., Faddah D. A., Tavarez U. L., Conneely K. N., Qu X., San H., Ganesh S. K. et al. (2008). A farnesyltransferase inhibitor prevents both the onset and late progression of cardiovascular disease in a progeria mouse model. Proc. Natl. Acad. Sci. USA 105, 15902-15907. 10.1073/pnas.0807840105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Sandre-Giovannoli A., Bernard R., Cau P., Navarro C., Amiel J., Boccaccio I., Lyonnet S., Stewart C. L., Munnich A., Le Merrer M. et al. (2003). Lamin A truncation in Hutchinson-Gilford progeria. Science 300, 2055 10.1126/science.1084125 [DOI] [PubMed] [Google Scholar]
- Eriksson M., Brown W. T., Gordon L. B., Glynn M. W., Singer J., Scott L., Erdos M. R. Robbins C. M., Moses T. Y., Berglund P. et al. (2003). Recurrent de novo point mutations in lamin A cause Hutchinson-Gilford progeria syndrome. Nature 423, 293-298. 10.1038/nature01629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher D. Z., Chaudhary N. and Blobel G. (1986). cDNA sequencing of nuclear lamins A and C reveals primary and secondary structural homology to intermediate filament proteins. Proc. Natl. Acad. Sci. USA 83, 6450-6454. 10.1073/pnas.83.17.6450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fong L. G., Frost D., Meta M., Qiao X., Yang S. H., Coffinier C. and Young S. G. (2006). A protein farnesyltransferase inhibitor ameliorates disease in a mouse model of progeria. Science 311, 1621-1623. 10.1126/science.1124875 [DOI] [PubMed] [Google Scholar]
- Gerace L., Blum A. and Blobel G. (1978). Immunocytochemical localization of the major polypeptides of the nuclear pore complex-lamina fraction. Interphase and mitotic distribution. J. Cell Biol. 79, 546-566. 10.1083/jcb.79.2.546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldman R. D., Shumaker D. K., Erdos M. R., Eriksson M., Goldman A. E., Gordon L. B., Gruenbaum Y., Khuon S., Mendez M., Varga R. et al. (2004). Accumulation of mutant lamin A causes progressive changes in nuclear architecture in Hutchinson-Gilford progeria syndrome. Proc. Natl. Acad. Sci. USA 101, 8963-8968. 10.1073/pnas.0402943101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon L. B., Kleinman M. E., Miller D. T., Neuberg D. S., Giobbie-Hurder A., Gerhard-Herman M., Smoot L. B., Gordon C. M., Cleveland R., Snyder B. D. et al. (2012). Clinical trial of a farnesyltransferase inhibitor in children with Hutchinson-Gilford progeria syndrome. Proc. Natl. Acad. Sci. USA 109, 16666-16671. 10.1073/pnas.1202529109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hennekes H. and Nigg E. A. (1994). The role of isoprenylation in membrane attachment of nuclear lamins. A single point mutation prevents proteolytic cleavage of the lamin A precursor and confers membrane binding properties. J. Cell Sci. 107, 1019-1029. [DOI] [PubMed] [Google Scholar]
- Lee R., Chang S. Y., Trinh H., Tu Y., White A. C., Davies B. S. J., Bergo M. O., Fong L. G., Lowry W. E. and Young S. G. (2010). Genetic studies on the functional relevance of the protein prenyltransferases in skin keratinocytes. Hum. Mol. Genet. 19, 1603-1617. 10.1093/hmg/ddq036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin F. and Worman H. J. (1993). Structural organization of the human gene encoding nuclear lamin A and nuclear lamin C. J. Biol. Chem. 268, 16321-16326. [PubMed] [Google Scholar]
- Mallampalli M. P., Huyer G., Bendale P., Gelb M. H. and Michaelis S. (2005). Inhibiting farnesylation reverses the nuclear morphology defect in a HeLa cell model for Hutchinson-Gilford progeria syndrome. Proc. Natl. Acad. Sci. USA 102, 14416-14421. 10.1073/pnas.0503712102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKeon F. D., Kirschner M. W. and Caput D. (1986). Homologies in both primary and secondary structure between nuclear envelope and intermediate filament proteins. Nature 319, 463-468. 10.1038/319463a0 [DOI] [PubMed] [Google Scholar]
- Merideth M. A., Gordon L. B., Clauss S., Sachdev V., Smith A. C. M., Perry M. B., Brewer C. C., Zalewski C., Kim H. J., Solomon B. et al. (2008). Phenotype and course of Hutchinson-Gilford progeria syndrome. N. Engl. J. Med. 358, 592-604. 10.1056/NEJMoa0706898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moulson C. L., Go G., Gardner J. M., van der Wal A. C., Smitt J. H. S., van Hagen J. M. and Miner J. H. (2005). Homozygous and compound heterozygous mutations in ZMPSTE24 cause the laminopathy restrictive dermopathy. J. Invest. Dermatol. 125, 913-919. 10.1111/j.0022-202X.2005.23846.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navarro C. L., Cadiñanos J., De Sandre-Giovannoli A., Bernard R., Courrier S., Boccaccio I., Boyer A., Kleijer W. J., Wagner A., Giuliano F. et al. (2005). Loss of ZMPSTE24 (FACE-1) causes autosomal recessive restrictive dermopathy and accumulation of lamin A precursors. Hum. Mol. Genet. 14, 1503-1513. 10.1093/hmg/ddi159 [DOI] [PubMed] [Google Scholar]
- Novelli G., Muchir A., Sangiuolo F., Helbling-Leclerc A., D'Apice M. R., Massart C., Capon F., Sbraccia P., Federici M., Lauro R. et al. (2002). Mandibuloacral dysplasia is caused by a mutation in LMNA-encoding lamin A/C. Am. J. Hum. Genet. 71, 426-431. 10.1086/341908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shackleton S., Smallwood D. T., Clayton P., Wilson L. C., Agarwal A. K., Garg A. and Trembath R. C. (2005). Compound heterozygous ZMPSTE24 mutations reduce prelamin A processing and result in a severe progeroid phenotype. J. Med. Genet. 42, e36 10.1136/jmg.2004.029751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinensky M., Fantle K., Trujillo M., McLain T., Kupfer A. and Dalton M. (1994). The processing pathway of prelamin A. J. Cell Sci. 107, 61-67. [DOI] [PubMed] [Google Scholar]
- Toth J. I., Yang S. H., Qiao X., Beigneux A. P., Gelb M. H., Moulson C. L., Miner J. H., Young S. G. and Fong L. G. (2005). Blocking protein farnesyltransferase improves nuclear shape in fibroblasts from humans with progeroid syndromes. Proc. Natl. Acad. Sci. USA 102, 12873-12878. 10.1073/pnas.0505767102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varela I., Pereira S., Ugalde A. P., Navarro C. L., Suárez M. F., Cau P., Cadiñanos J., Osorio F. G., Foray N., Cobo J. et al. (2008). Combined treatment with statins and aminobisphosphonates extends longevity in a mouse model of human premature aging. Nat. Med. 14, 767-772. 10.1038/nm1786 [DOI] [PubMed] [Google Scholar]
- Verstraeten V. L. R. M., Peckham L. A., Olive M., Capell B. C., Collins F. S., Nabel E. G., Young S. G., Fong L. G. and Lammerding J. (2011). Protein farnesylation inhibitors cause donut-shaped cell nuclei attributable to a centrosome separation defect. Proc. Natl. Acad. Sci. USA 108, 4997-5002. 10.1073/pnas.1019532108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y., Östlund C., Choi J. C., Swayne T. C., Gundersen G. G. and Worman H. J. (2012). Blocking farnesylation of the prelamin A variant in Hutchinson-Gilford progeria syndrome alters the distribution of A-type lamins. Nucleus 3, 452-462. 10.4161/nucl.21675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang S. H., Meta M., Qiao X., Frost D., Bauch J., Coffinier C., Majumdar S., Bergo M. O., Young S. G. and Fong L. G. (2006). A farnesyltransferase inhibitor improves disease phenotypes in mice with a Hutchinson-Gilford progeria syndrome mutation. J. Clin. Invest. 116, 2115-2121. 10.1172/JCI28968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young S. G., Fong L. G. and Michaelis S. (2005). Prelamin A, Zmpste24, misshapen cell nuclei, and progeria--new evidence suggesting that protein farnesylation could be important for disease pathogenesis. J. Lipid Res. 46, 2531-2558. 10.1194/jlr.R500011-JLR200 [DOI] [PubMed] [Google Scholar]